Immune Checkpoints as a Target for Colorectal Cancer Treatment




Abstract
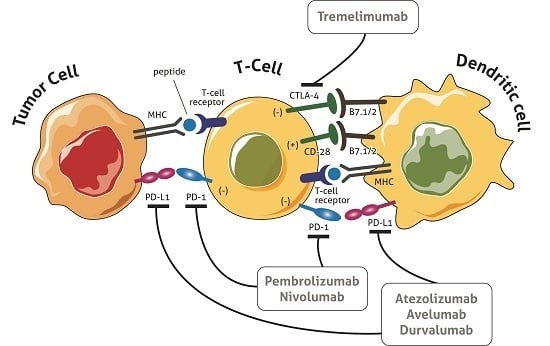
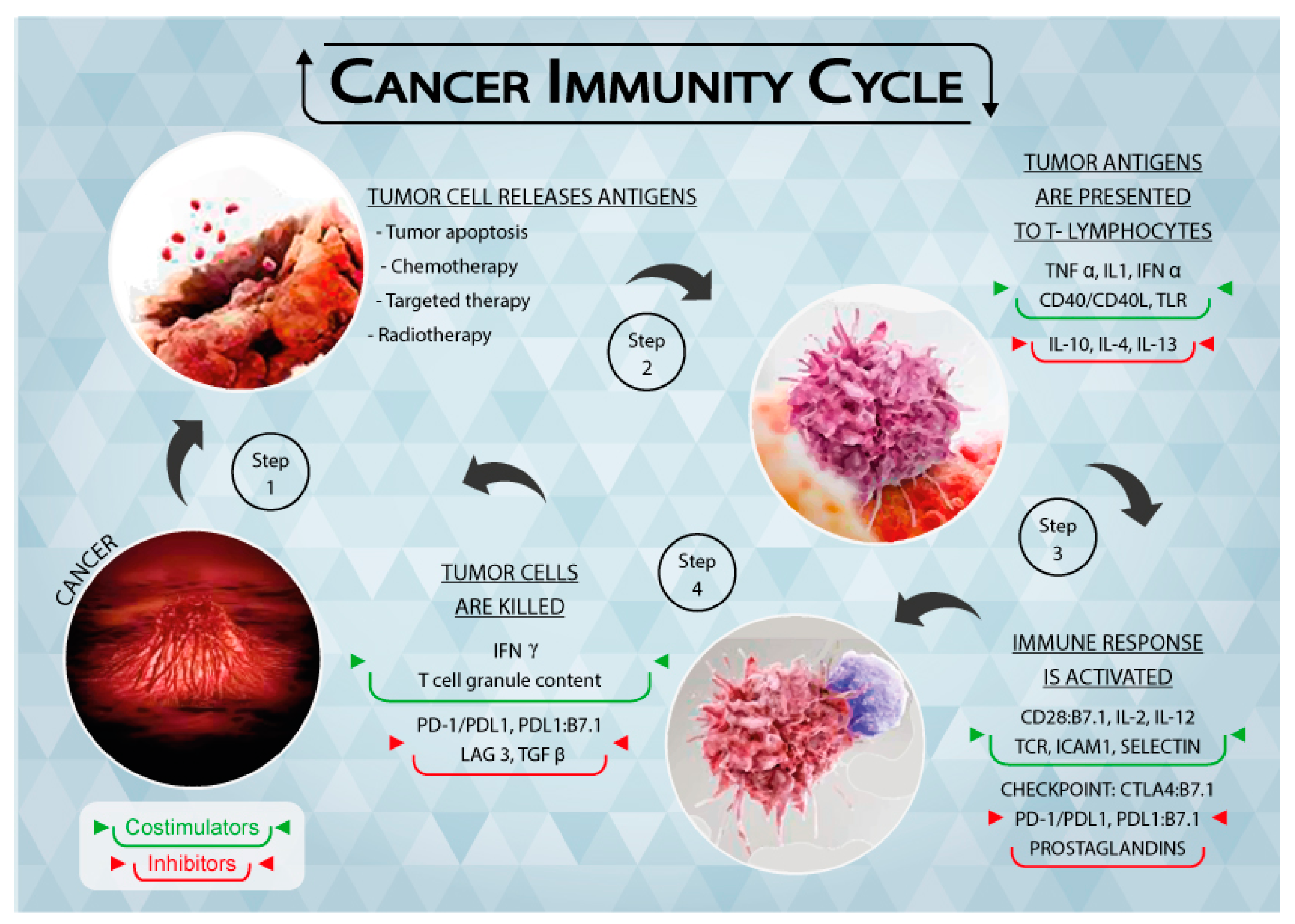
1. Introduction
2. The Immune System and the Tumor
3. Immune Checkpoints
4. Immunological Features of CRC
5. Clinical Results of Immune Checkpoint Inhibitors in mCRC
5.1. Cytotoxic T-Lymphocyte-Associated Antigen (CTLA)-4 Blockade
5.2. Programmed Death (PD1) Blockade
5.3. Programmed Death-Ligand 1 (PD-L1) Blockade
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CTLA-4 | Cytotoxic T-lymphocyte antigen 4 |
| PD1 | Programmed death-1 receptor |
| PD-L1 | Programmed death-1 receptor ligand |
| MMR | Mismatch repair |
| CRC | Colorectal cancer |
References
- Bilgin, B.; Sendur, M.A.; Bulent Akinci, M.; Sener Dede, D.; Yalcin, B. Targeting the PD-1 pathway: A new hope for gastrointestinal cancers. Curr. Med. Res. Opin. 2017, 33, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Pandya, P.H.; Murray, M.E.; Pollok, K.E.; Renbarger, J.L. The immune system in cancer pathogenesis: Potential therapeutic approaches. J. Immunol. Res. 2016, 2016, 4273943. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases—Elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Kyi, C.; Postow, M.A. Checkpoint blocking antibodies in cancer immunotherapy. FEBS Lett. 2014, 588, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Zhu, C.; Kuchroo, V.K. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 2017, 276, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune checkpoint blockade in cancer therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Shih, K.; Arkenau, H.T.; Infante, J.R. Clinical impact of checkpoint inhibitors as novel cancer therapies. Drugs 2014, 74, 1993–2013. [Google Scholar] [CrossRef] [PubMed]
- Naboush, A.; Roman, C.A.; Shapira, I. Immune checkpoint inhibitors in malignancies with mismatch repair deficiency: A review of the state of the current knowledge. J. Investig. Med. 2017, 65, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Krummel, M.F.; Allison, J.P. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J. Exp. Med. 1996, 183, 2533–2540. [Google Scholar] [CrossRef] [PubMed]
- Funt, S.A.; Page, D.B.; Wolchok, J.D.; Postow, M.A. CTLA-4 antibodies: New directions, new combinations. Oncology 2014, 28 (Suppl. S3), 6–14. [Google Scholar] [PubMed]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Chen, L. Inhibitory B7-family molecules in the tumour microenvironment. Nat. Rev. Immunol. 2008, 8, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Reiss, K.A.; Forde, P.M.; Brahmer, J.R. Harnessing the power of the immune system via blockade of PD-1 and PD-L1: A promising new anticancer strategy. Immunotherapy 2014, 6, 459–475. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Eder, J.P. Prospects for targeting PD-1 and PD-L1 in various tumor types. Oncology 2014, 28 (Suppl. 3), 15–28. [Google Scholar] [PubMed]
- Taube, J.M.; Anders, R.A.; Young, G.D.; Xu, H.; Sharma, R.; McMiller, T.L.; Chen, S.; Klein, A.P.; Pardoll, D.M.; Topalian, S.L.; et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci. Transl. Med. 2012, 4, 127ra37. [Google Scholar] [CrossRef] [PubMed]
- Haggar, F.A.; Boushey, R.P. Colorectal cancer epidemiology: Incidence, mortality, survival, and risk factors. Clin. Colon Rectal Surg. 2009, 22, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Jasperson, K.W.; Tuohy, T.M.; Neklason, D.W.; Burt, R.W. Hereditary and familial colon cancer. Gastroenterology 2010, 138, 2044–2058. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; de la Chapelle, A. Hereditary colorectal cancer. N. Engl. J. Med. 2003, 348, 919–932. [Google Scholar] [PubMed]
- Kastrinos, F.; Ojha, R.P.; Leenen, C.; Alvero, C.; Mercado, R.C.; Balmana, J.; Valenzuela, I.; Balaguer, F.; Green, R.; Lindor, N.M.; et al. Comparison of prediction models for Lynch Syndrome among individuals with colorectal cancer. J. Natl. Cancer Inst. 2015, 108, djv308. [Google Scholar] [CrossRef] [PubMed]
- Abdulovic, A.L.; Hile, S.E.; Kunkel, T.A.; Eckert, K.A. The in vitro fidelity of yeast DNA polymerase delta and polymerase epsilon holoenzymes during dinucleotide microsatellite DNA synthesis. DNA Repair 2011, 10, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, A.; Peltomaki, P.; Mecklin, J.P.; Jarvinen, H.; Salovaara, R.; Nystrom-Lahti, M.; de la Chapelle, A.; Aaltonen, L.A. Loss of the wild type MLH1 gene is a feature of hereditary nonpolyposis colorectal cancer. Nat. Genet. 1994, 8, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Popat, S.; Hubner, R.; Houlston, R.S. Systematic review of microsatellite instability and colorectal cancer prognosis. J. Clin. Oncol. 2005, 23, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Samowitz, W.S.; Curtin, K.; Ma, K.N.; Schaffer, D.; Coleman, L.W.; Leppert, M.; Slattery, M.L. Microsatellite instability in sporadic colon cancer is associated with an improved prognosis at the population level. Cancer Epidemiol. Biomark. Prev. 2001, 10, 917–923. [Google Scholar]
- Veigl, M.L.; Kasturi, L.; Olechnowicz, J.; Ma, A.H.; Lutterbaugh, J.D.; Periyasamy, S.; Li, G.M.; Drummond, J.; Modrich, P.L.; Sedwick, W.D.; et al. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc. Natl. Acad. Sci. USA 1998, 95, 8698–8702. [Google Scholar] [CrossRef] [PubMed]
- Timmermann, B.; Kerick, M.; Roehr, C.; Fischer, A.; Isau, M.; Boerno, S.T.; Wunderlich, A.; Barmeyer, C.; Seemann, P.; Koenig, J.; et al. Somatic mutation profiles of MSI and MSS colorectal cancer identified by whole exome next generation sequencing and bioinformatics analysis. PLoS ONE 2010, 5, e15661. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Quinn, E.; Hawkins, N.; Yip, Y.L.; Suter, C.; Ward, R. CD103+ intraepithelial lymphocytes—A unique population in microsatellite unstable sporadic colorectal cancer. Eur. J. Cancer 2003, 39, 469–475. [Google Scholar] [CrossRef]
- Banerjea, A.; Ahmed, S.; Hands, R.E.; Huang, F.; Han, X.; Shaw, P.M.; Feakins, R.; Bustin, S.A.; Dorudi, S. Colorectal cancers with microsatellite instability display mRNA expression signatures characteristic of increased immunogenicity. Mol. Cancer 2004, 3, 21. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Phillips, S.M.; Banerjea, A.; Feakins, R.; Li, S.R.; Bustin, S.A.; Dorudi, S. Tumour-infiltrating lymphocytes in colorectal cancer with microsatellite instability are activated and cytotoxic. Br. J. Surg. 2004, 91, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Saeterdal, I.; Bjorheim, J.; Lislerud, K.; Gjertsen, M.K.; Bukholm, I.K.; Olsen, O.C.; Nesland, J.M.; Eriksen, J.A.; Moller, M.; Lindblom, A.; et al. Frameshift-mutation-derived peptides as tumor-specific antigens in inherited and spontaneous colorectal cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 13255–13260. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Freeman-Mills, L.; Rayner, E.; Glaire, M.; Briggs, S.; Vermeulen, L.; Fessler, E.; Medema, J.P.; Boot, A.; Morreau, H.; et al. Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: A retrospective, pooled biomarker study. Lancet Gastroenterol. Hepatol. 2016, 1, 207–216. [Google Scholar] [CrossRef]
- Gatalica, Z.; Snyder, C.; Maney, T.; Ghazalpour, A.; Holterman, D.A.; Xiao, N.; Overberg, P.; Rose, I.; Basu, G.D.; Vranic, S.; et al. Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2965–2970. [Google Scholar] [CrossRef] [PubMed]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Inaguma, S.; Lasota, J.; Wang, Z.; Felisiak-Golabek, A.; Ikeda, H.; Miettinen, M. Clinicopathologic profile, immunophenotype, and genotype of CD274 (PD-L1)-positive colorectal carcinomas. Mod. Pathol. 2017, 30, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Droeser, R.A.; Hirt, C.; Viehl, C.T.; Frey, D.M.; Nebiker, C.; Huber, X.; Zlobec, I.; Eppenberger-Castori, S.; Tzankov, A.; Rosso, R.; et al. Clinical impact of programmed cell death ligand 1 expression in colorectal cancer. Eur. J. Cancer 2013, 49, 2233–2242. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liang, L.; Dai, W.; Cai, G.; Xu, Y.; Li, X.; Li, Q.; Cai, S. Prognostic impact of programed cell death-1 (PD-1) and PD-ligand 1 (PD-L1) expression in cancer cells and tumor infiltrating lymphocytes in colorectal cancer. Mol. Cancer 2016, 15, 55. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.Y.; Gore, I.; Fong, L.; Venook, A.; Beck, S.B.; Dorazio, P.; Criscitiello, P.J.; Healey, D.I.; Huang, B.; Gomez-Navarro, J.; et al. Phase II study of the anti-cytotoxic T-lymphocyte-associated antigen 4 monoclonal antibody, tremelimumab, in patients with refractory metastatic colorectal cancer. J. Clin. Oncol. 2010, 28, 3485–3490. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Drake, C.G.; Wollner, I.; Powderly, J.D.; Picus, J.; Sharfman, W.H.; Stankevich, E.; Pons, A.; Salay, T.M.; McMiller, T.L.; et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 2010, 28, 3167–3175. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.; Lonardi, S.; Leone, F.; McDermott, R.; Morse, M.; Wong, K.; Neyns, B.; Leach, J.; Alfonso, P.; Lee, J.; et al. Nivolumab in patients with DNA mismatch repair deficient/microsatellite instability high metastatic colorectal cancer: Update from CheckMate 142. J. Clin. Oncol. 2017, 35, 519. [Google Scholar] [CrossRef]
- Patnaik, A.; Kang, S.P.; Rasco, D.; Papadopoulos, K.P.; Elassaiss-Schaap, J.; Beeram, M.; Drengler, R.; Chen, C.; Smith, L.; Espino, G.; et al. Phase I study of pembrolizumab (MK-3475; Anti-PD-1 monoclonal antibody) in patients with advanced solid tumors. Clin. Cancer Res. 2015, 21, 4286–4293. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Muro, K.; Chung, H.C.; Shankaran, V.; Geva, R.; Catenacci, D.; Gupta, S.; Eder, J.P.; Golan, T.; Le, D.T.; Burtness, B.; et al. Pembrolizumab for patients with PD-L1-positive advanced gastric cancer (KEYNOTE-012): A multicentre, open-label, phase 1b trial. Lancet Oncol. 2016, 17, 717–726. [Google Scholar] [CrossRef]
- Nanda, R.; Chow, L.Q.; Dees, E.C.; Berger, R.; Gupta, S.; Geva, R.; Pusztai, L.; Pathiraja, K.; Aktan, G.; Cheng, J.D.; et al. Pembrolizumab in patients with advanced triple-negative breast cancer: Phase Ib KEYNOTE-012 study. J. Clin. Oncol. 2016, 34, 2460–2467. [Google Scholar] [CrossRef] [PubMed]
- Plimack, E.R.; Bellmunt, J.; Gupta, S.; Berger, R.; Chow, L.Q.; Juco, J.; Lunceford, J.; Saraf, S.; Perini, R.F.; O’Donnell, P.H. Safety and activity of pembrolizumab in patients with locally advanced or metastatic urothelial cancer (KEYNOTE-012): A non-randomised, open-label, phase 1b study. Lancet Oncol. 2017, 18, 212–220. [Google Scholar] [CrossRef]
- Diaz, L.; Marabelle, A.; Delord, J.; Shapira-Frommer, R.; Geva, R.; Peled, N.; Kim, T.; Andre, T.; van Cutsem, E.; Guimbaud, R.; et al. Pembrolizumab therapy for microsatellite instability high (MSI-H) colorectal cancer (CRC) and non-CRC. J. Clin. Oncol. 2017, 35, 3071. [Google Scholar]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Akbari, O.; Stock, P.; Singh, A.K.; Lombardi, V.; Lee, W.L.; Freeman, G.J.; Sharpe, A.H.; Umetsu, D.T.; Dekruyff, R.H. PD-L1 and PD-L2 modulate airway inflammation and iNKT-cell-dependent airway hyperreactivity in opposing directions. Mucosal Immunol. 2010, 3, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Fukuyama, S.; Eguchi-Tsuda, M.; Nakano, T.; Matsumoto, T.; Matsumura, M.; Moriwaki, A.; Kan-o, K.; Wada, Y.; Yagita, H.; et al. B7-DC induced by IL-13 works as a feedback regulator in the effector phase of allergic asthma. Biochem. Biophys. Res. Commun. 2008, 365, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.; Powderly, J.; Lieu, C.; Eckhardt, S.; Hurwitz, H.; Hochster, H.; Murphy, J.; Funke, R.; Rossi, C.; Wallin, J.; et al. Safety and efficacy of MPDL3280A (anti-PDL1) in combination with bevacizumab (bev) and/or FOLFOX in patients (pts) with metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2015, 33, 704. [Google Scholar] [CrossRef]
- Bendell, J.; Kim, T.; Goh, B.; Wallin, J.; Oh, D.; Han, S.; Lee, C.; Hellmann, M.; Desai, J.; Lewin, J.; et al. Clinical activity and safety of cobimetinib (Cobi) and atezolizumab in colorectal cancer (CRC). J. Clin. Oncol. 2016, 34, 3502. [Google Scholar]


{kind=link}
{kind=link}
| ClinicalTrials.gov Identifier | Agent | Trial | Patient Population | Phase | Primary Endpoint |
|---|---|---|---|---|---|
| NCT02860546 | Nivolumab | A study evaluating TAS-102 plus nivolumab in patients with MSS CRC | mCRC | 2 | irORR |
| NCT02060188 | Nivolumab | An investigational immunotherapy study of nivolumab and nivolumab in combination with other anti-cancer drugs in colon cancer that returned or spread (CheckMate142) | MSI/MSS mCRC | 2 | ORR |
| NCT02981524 | Pembrolizumab | Phase 2 Study of GVAX (With CY) and pembrolizumab in pMMR advanced colorectal cancer | MMR-p mCRC | 2 | ORR |
| NCT02437071 | Pembrolizumab | Assessment of the efficacy of pembrolizumab plus radiotherapy or ablation in metastatic colorectal cancer patients | mCRC | 2 | ORR |
| NCT02563002 | Pembrolizumab | Study of pembrolizumab (MK-3475) vs. standard therapy in patients with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) stage IV colorectal carcinoma (MK-3475-177/KEYNOTE-177) | mCRC | 3 | PFS |
| NCT01876511 | Pembrolizumab | Phase 2 Study of MK-3475 in patients with microsatellite instability (MSI) tumors | MSI/MSS mCRC | 2 | irORR/irPFS |
| NCT02788279 | Atezolizumab | A study to investigate efficacy and safety of cobimetinib plus atezolizumab and atezolizumab monotherapy vs. regorafenib in patients with metastatic colorectal adenocarcinoma | mCRC | 3 | OS |
| NCT02291289 | Atezolizumab | A multi-center study of biomarker-driven therapy in metastatic colorectal cancer | mCRC | 2 | PFS |
| NCT02992912 | Atezolizumab | Atezolizumab with stereotactic ablative radiotherapy in patients with metastatic tumors (SABR-PD-L1) | Metastatic tumors | 2 | PFS |
| NCT03050814 | Avelumab | Standard of care alone or in combination with Ad-CEA vaccine and avelumab in patients with previously untreated metastatic colorectal cancer (QUILT-2.004) | mCRC | 2 | 18mPD |
| NCT02870920 | Tremelimumab | Durvalumab and tremelimumab and best supportive care vs. best supportive care alone in patients with advanced colorectal adenocarcinoma refractory to standard therapies | mCRC | 2 | OS |
| NCT02227667 | MEDI4736 | Evaluation of the efficacy of MEDI4736 in immunological subsets of advanced colorectal cancer | mCRC | 2 | BRR |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Passardi, A.; Canale, M.; Valgiusti, M.; Ulivi, P. Immune Checkpoints as a Target for Colorectal Cancer Treatment. Int. J. Mol. Sci. 2017, 18, 1324. https://doi.org/10.3390/ijms18061324
Passardi A, Canale M, Valgiusti M, Ulivi P. Immune Checkpoints as a Target for Colorectal Cancer Treatment. International Journal of Molecular Sciences. 2017; 18(6):1324. https://doi.org/10.3390/ijms18061324
Chicago/Turabian StylePassardi, Alessandro, Matteo Canale, Martina Valgiusti, and Paola Ulivi. 2017. "Immune Checkpoints as a Target for Colorectal Cancer Treatment" International Journal of Molecular Sciences 18, no. 6: 1324. https://doi.org/10.3390/ijms18061324
APA StylePassardi, A., Canale, M., Valgiusti, M., & Ulivi, P. (2017). Immune Checkpoints as a Target for Colorectal Cancer Treatment. International Journal of Molecular Sciences, 18(6), 1324. https://doi.org/10.3390/ijms18061324