
Cutaneous Manifestations of Human and Murine Leishmaniasis

Abstract
:1. Introduction
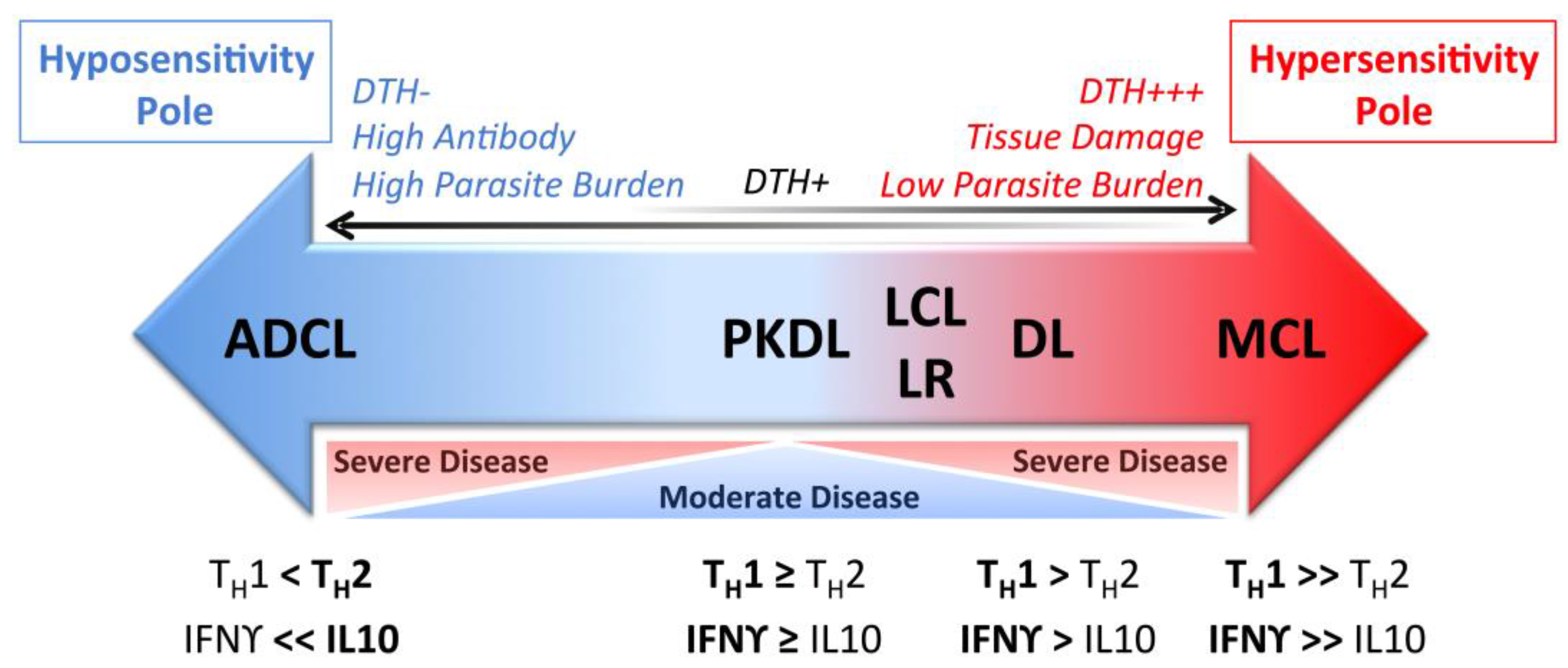
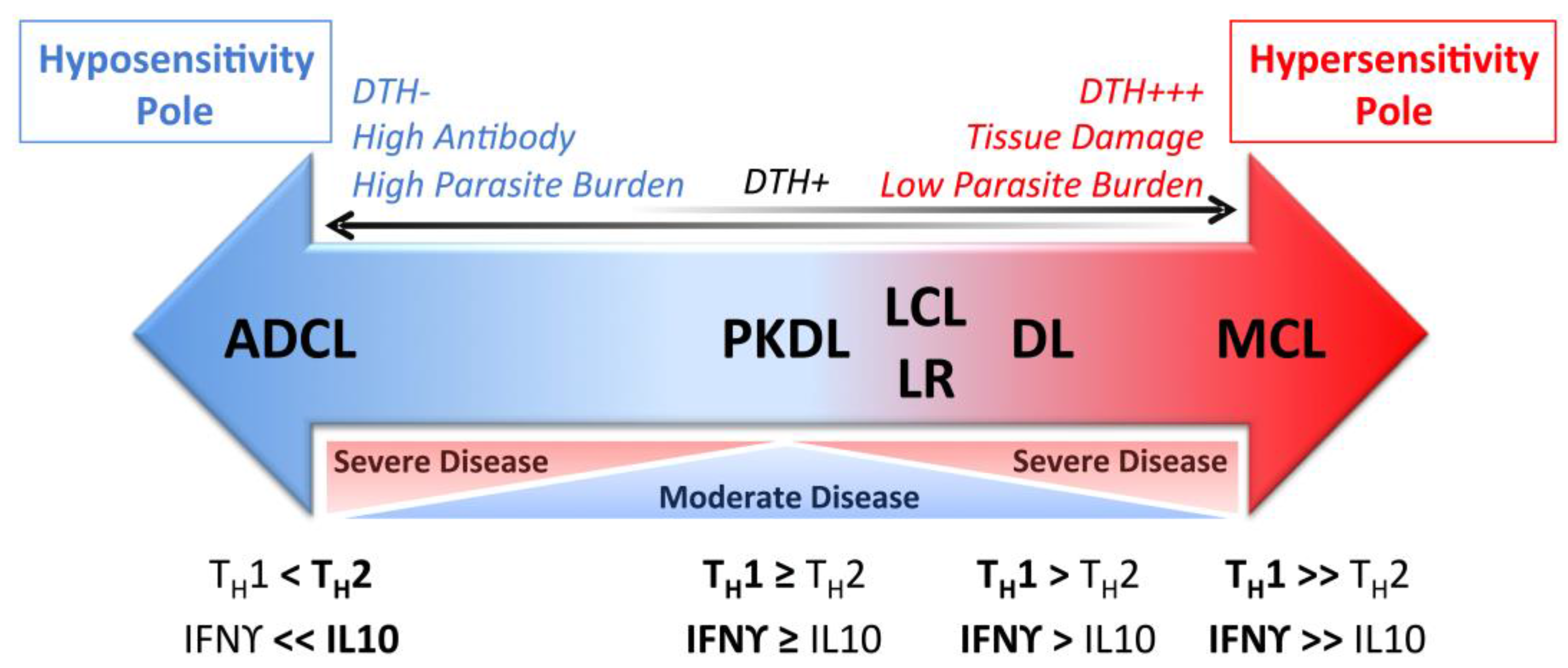
2. Nomenclature
3. Localized Cutaneous Leishmaniasis
4. Mucosal Leishmaniasis
5. Disseminated Leishmaniasis
6. Anergic Diffuse Cutaneous Leishmaniasis
7. Leishmaniasis Recidivans
8. Post Kala-Azar Dermal Leishmaniasis
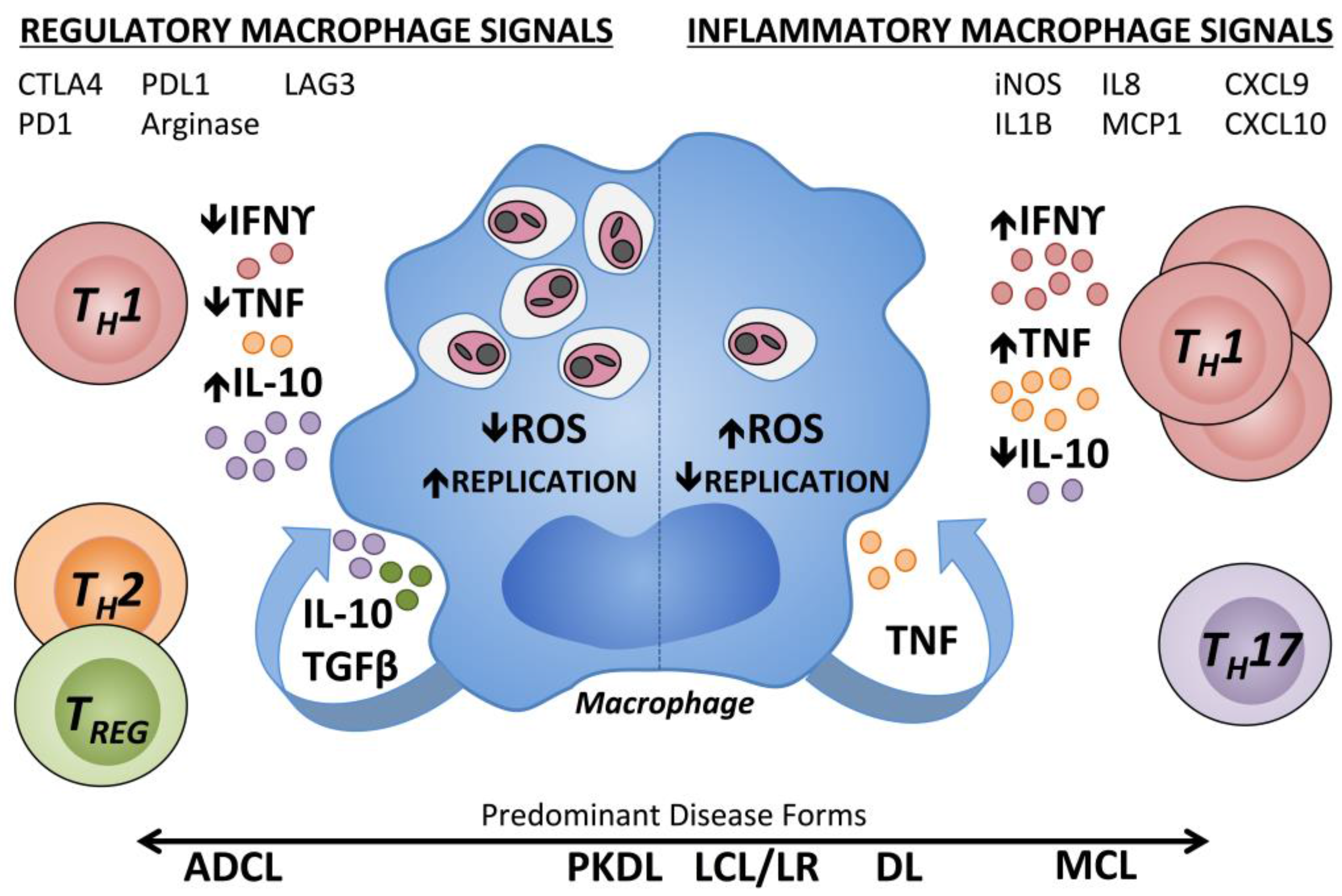
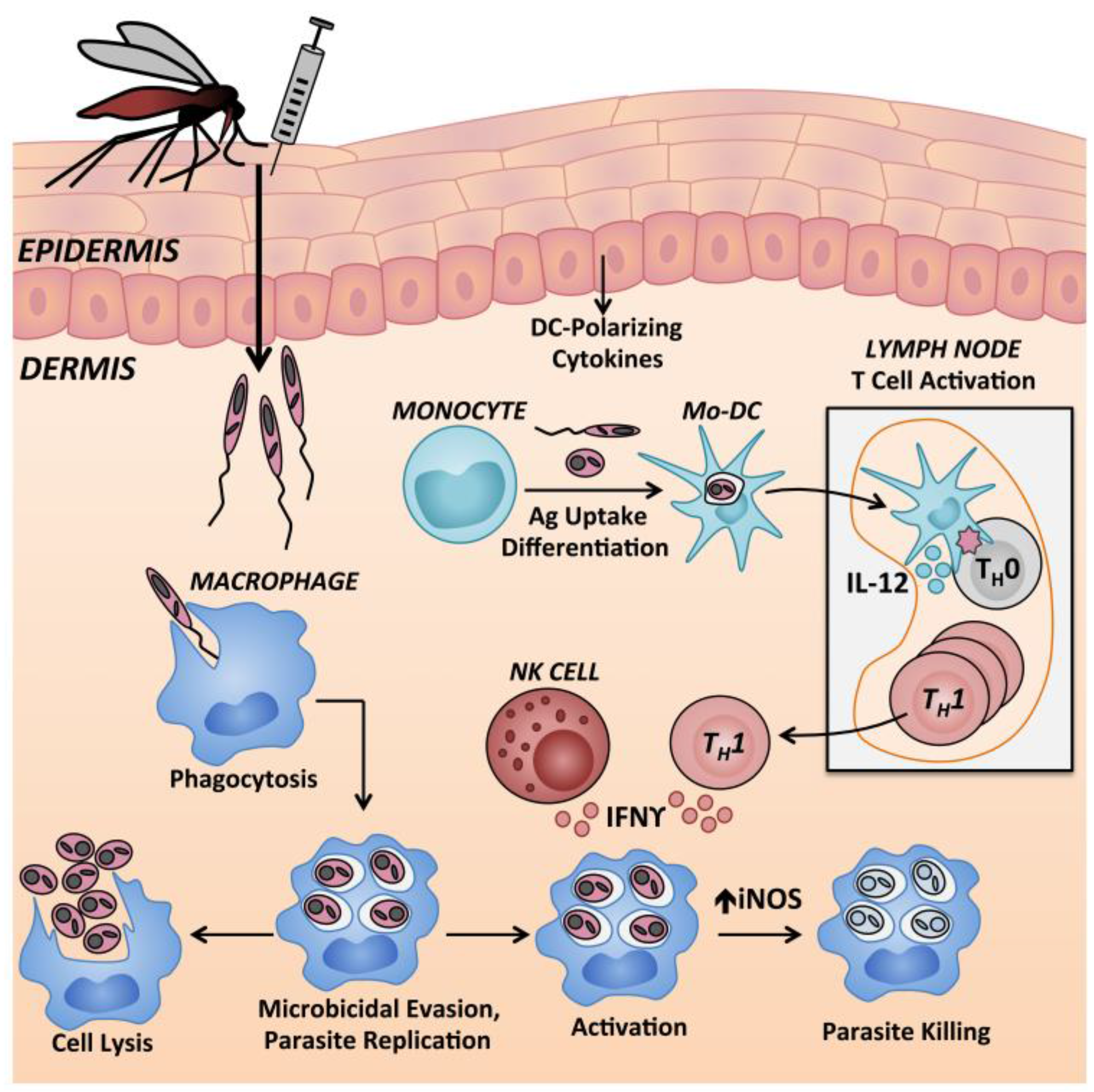
9. Cellular Determinants of the Immune Response in Murine Cutaneous Leishmaniasis
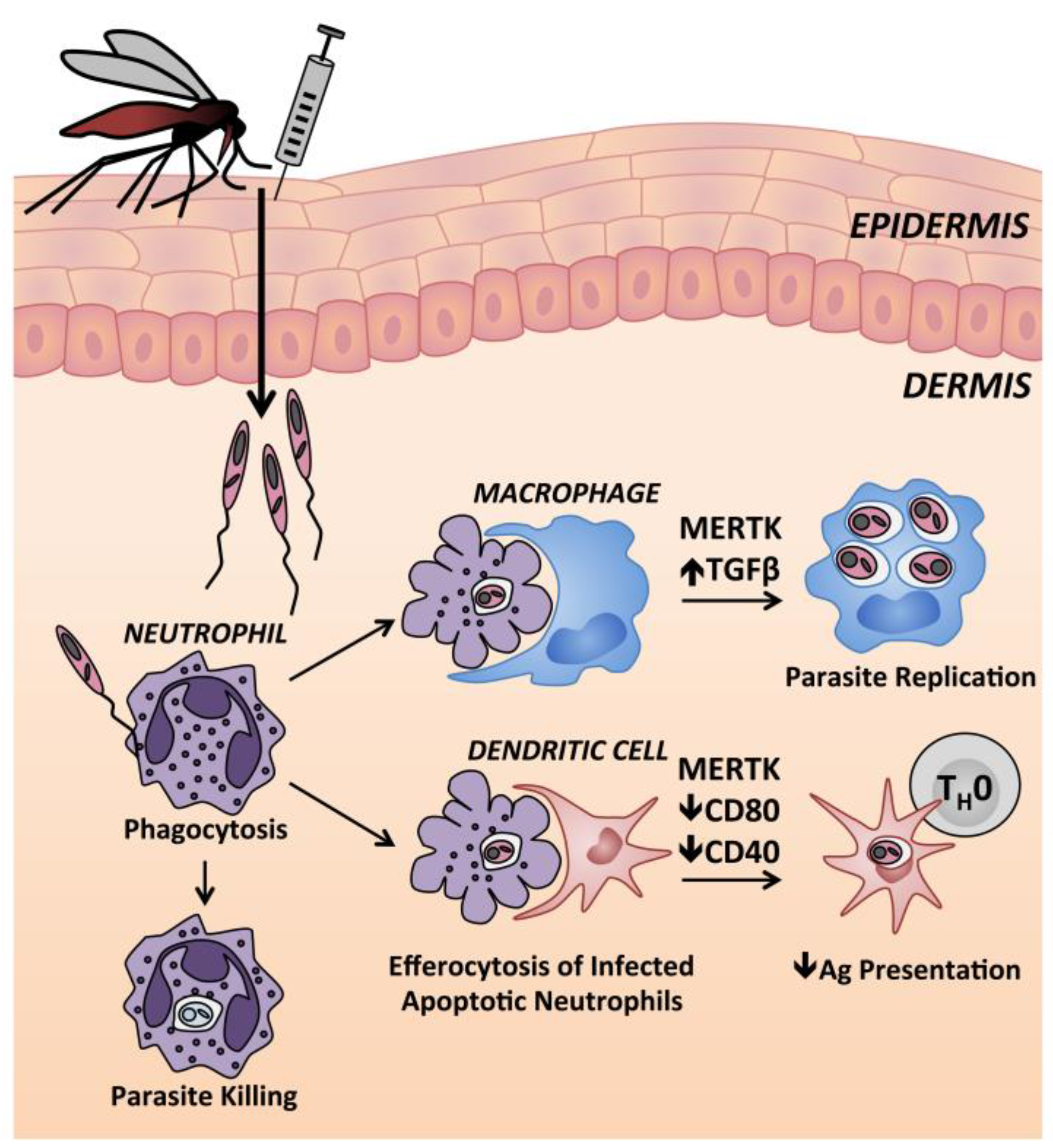
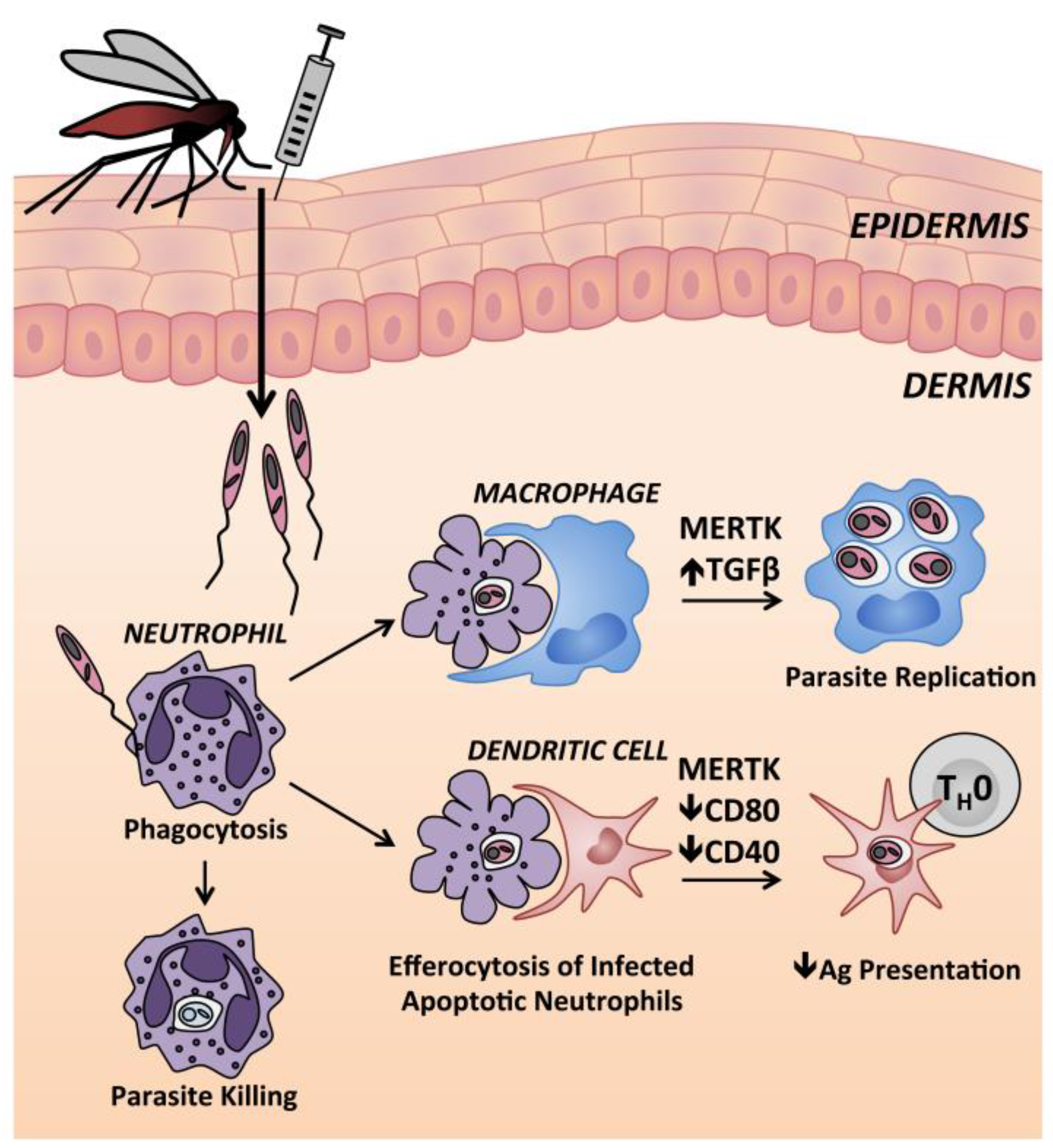
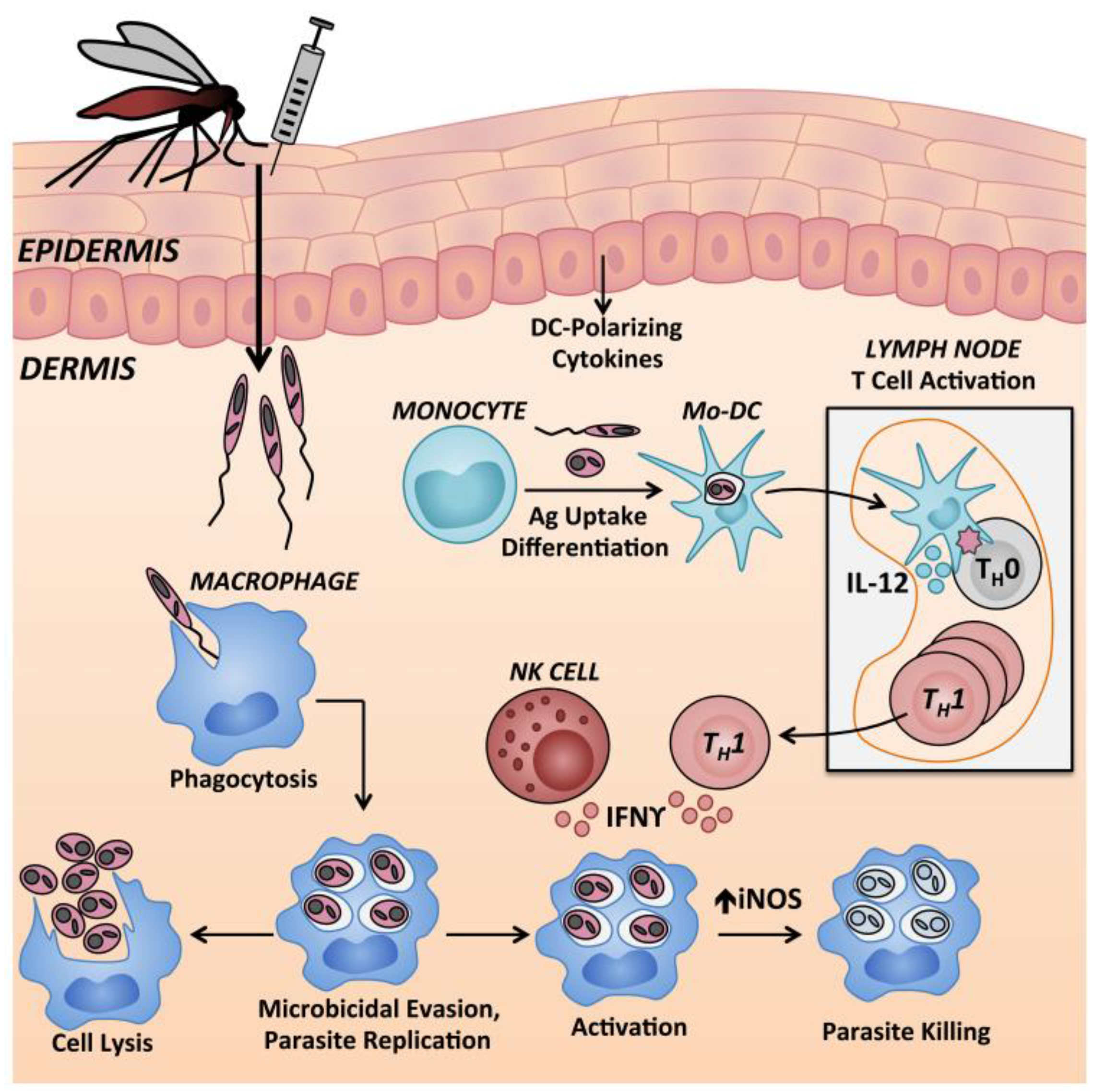
9.1. Neutrophils
9.2. Langerhans Cells
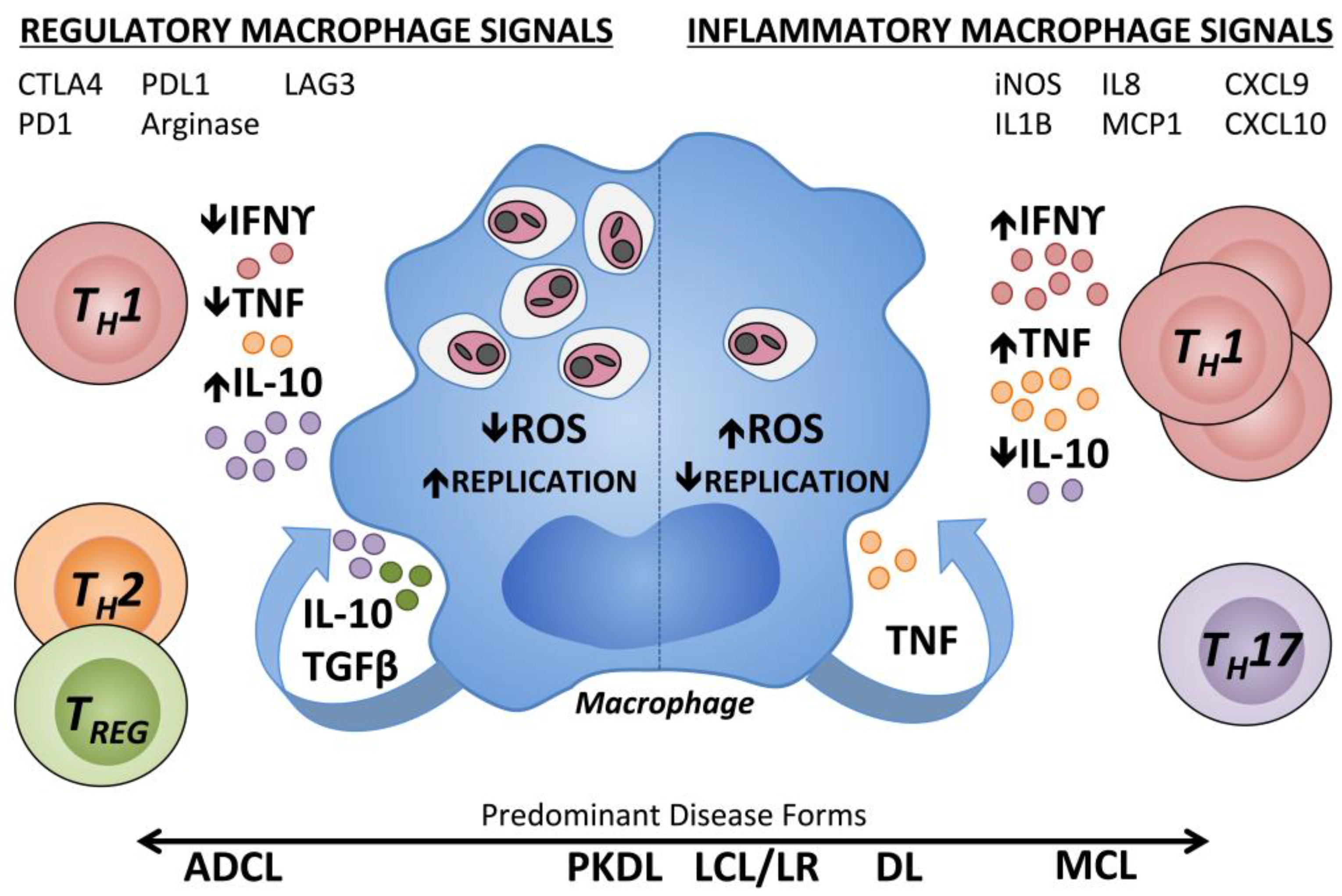
9.3. Monocytes and Macrophages
9.4. Natural Killer Cells
9.5. Keratinocytes
10. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| LCL | Localized cutaneous leishmaniasis (also called cutaneous leishmaniasis or CL) |
| ADCL | Anergic diffuse cutaneous leishmaniasis (also called diffuse cutaneous leishmaniasis or DCL) |
| DL | Disseminated leishmaniasis (also called disseminated cutaneous leishmaniasis) |
| LC | Langerhans’ cell |
| LR | Leishmaniasis recidivans |
| MDM | Monocyte derived macrophage |
| MCL | Mucocutaneous leishmaniasis (also called mucosal leishmaniasis or ML) |
| PBMC | Peripheral blood mononuclear cells |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
References
- WHO. Weekly Epidemiological Record. Available online: http://www.who.int/wer/2016/wer9122.pdf?ua=1 (accessed on 31 August 2016).
- Grinnage-Pulley, T.; Scott, B.; Petersen, C.A. A mother’s gift: Congenital transmission of trypanosoma and leishmania species. PLoS Pathog. 2016, 12, e1005302. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.D.; Sousa, A.Q. Clinical spectrum of leishmaniasis. Clin. Infect. Dis. 1996, 22, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Malaviya, P.; Picado, A.; Singh, S.P.; Hasker, E.; Singh, R.P.; Boelaert, M.; Sundar, S. Visceral leishmaniasis in Muzaffarpur district, Bihar, India from 1990 to 2008. PLoS ONE 2011, 6, e14751. [Google Scholar]
- Al-Salem, W.S.; Pigott, D.M.; Subramaniam, K.; Haines, L.R.; Kelly-Hope, L.; Molyneux, D.H.; Hay, S.I.; Acosta-Serrano, A. Cutaneous leishmaniasis and conflict in Syria. Emerg. Infect. Dis. 2016, 22, 931–933. [Google Scholar] [CrossRef] [PubMed]
- Pavli, A.; Maltezou, H.C. Leishmaniasis, an emerging infection in travelers. Int. J. Infect. Dis. 2010, 14, e1032–e1039. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, G., Jr.; Tesh, R.B.; McMahon-Pratt, D. A review of the geographic distribution and epidemiology of leishmaniasis in the new world. Am. J. Trop. Med. Hyg. 1989, 41, 687–725. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Bumb, R.A.; Ansari, N.A.; Mehta, R.D.; Salotra, P. Cutaneous leishmaniasis caused by leishmania tropica in Bikaner, India: Parasite identification and characterization using molecular and immunologic tools. Am. J. Trop. Med. Hyg. 2007, 76, 896–901. [Google Scholar] [PubMed]
- Van Griensven, J.; Gadisa, E.; Aseffa, A.; Hailu, A.; Beshah, A.M.; Diro, E. Treatment of cutaneous leishmaniasis caused by Leishmania aethiopica: A systematic review. PLoS Negl. Trop. Dis. 2016, 10, e0004495. [Google Scholar] [CrossRef] [PubMed]
- Oliveira Neto, M.P.; Grimaldi, G., Jr.; Momen, H.; Pacheco, R.S.; Marzochi, M.C.; McMahon Pratt, D. Active cutaneous leishmaniasis in Brazil, induced by Leishmania donovani chagasi. Memorias do Instituto Oswaldo Cruz 1986, 81, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Karunaweera, N.D.; Pratlong, F.; Siriwardane, H.V.; Ihalamulla, R.L.; Dedet, J.P. Sri lankan cutaneous leishmaniasis is caused by Leishmania donovani zymodeme mon-37. Trans. R. Soc. Trop. Med. Hyg. 2003, 97, 380–381. [Google Scholar] [CrossRef]
- Schriefer, A.; Wilson, M.E.; Carvalho, E.M. Recent developments leading toward a paradigm switch in the diagnostic and therapeutic approach to human leishmaniasis. Curr. Opin. Infect. Dis. 2008, 21, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Constant, S.L.; Lee, K.S.; Bottomly, K. Site of antigen delivery can influence T cell priming: Pulmonary environment promotes preferential Th2-type differentiation. Eur. J. Immunol. 2000, 30, 840–847. [Google Scholar] [CrossRef]
- Costa, J.M.; Marsden, P.D.; Llanos-Cuentas, E.A.; Netto, E.M.; Carvalho, E.M.; Barral, A.; Rosa, A.C.; Cuba, C.C.; Magalhaes, A.V.; Barreto, A.C. Disseminated cutaneous leishmaniasis in a field clinic in Bahia, Brazil: A report of eight cases. J. Trop. Med. Hyg. 1986, 89, 319–323. [Google Scholar] [PubMed]
- Osorio, L.E.; Castillo, C.M.; Ochoa, M.T. Mucosal leishmaniasis due to Leishmania (Viannia) panamensis in Colombia: Clinical characteristics. Am. J. Trop. Med. Hyg. 1998, 59, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Santrich, C.; Segura, I.; Arias, A.L.; Saravia, N.G. Mucosal disease caused by Leishmania braziliensis guyanensis. Am. J. Trop. Med. Hyg. 1990, 42, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.M.; Franke, E.D.; Cachay, M.I.; Tejada, A.; Cruz, M.E.; Kreutzer, R.D.; Barker, D.C.; McCann, S.H.; Watts, D.M. Geographic distribution and clinical description of leishmaniasis cases in Peru. Am. J. Trop. Med. Hyg. 1998, 59, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Barral, A.; Pedral-Sampaio, D.; Grimaldi Junior, G.; Momen, H.; McMahon-Pratt, D.; Ribeiro de Jesus, A.; Almeida, R.; Badaro, R.; Barral-Netto, M.; Carvalho, E.M.; et al. Leishmaniasis in Bahia, Brazil: Evidence that Leishmania amazonensis produces a wide spectrum of clinical disease. Am. J. Trop. Med. Hyg. 1991, 44, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Velasco, O.; Savarino, S.J.; Walton, B.C.; Gam, A.A.; Neva, F.A. Diffuse cutaneous leishmaniasis in Mexico. Am. J. Trop. Med. Hyg. 1989, 41, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Convit, J.; Pinardi, M.E.; Rondon, A.J. Diffuse cutaneous leishmaniasis: A disease due to an immunological defect of the host. Trans. R. Soc. Trop. Med. Hyg. 1972, 66, 603–610. [Google Scholar] [CrossRef]
- Akuffo, H.; Maasho, K.; Blostedt, M.; Hojeberg, B.; Britton, S.; Bakhiet, M. Leishmania aethiopica derived from diffuse leishmaniasis patients preferentially induce mRNA for interleukin-10 while those from localized leishmaniasis patients induce interferon-γ. J. Infect. Dis. 1997, 175, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Develoux, M.; Diallo, S.; Dieng, Y.; Mane, I.; Huerre, M.; Pratlong, F.; Dedet, J.P.; Ndiaye, B. Diffuse cutaneous leishmaniasis due to leishmania major in Senegal. Trans. R. Soc. Trop. Med. Hyg. 1996, 90, 396–397. [Google Scholar] [CrossRef]
- Bryceson, A.D. Diffuse cutaneous leishmaniasis in Ethiopia. I. The clinical and histological features of the disease. Trans. R. Soc. Trop. Med. Hyg. 1969, 63, 708–737. [Google Scholar] [CrossRef]
- Carvalho, E.M.; Barral, A.; Costa, J.M.; Bittencourt, A.; Marsden, P. Clinical and immunopathological aspects of disseminated cutaneous leishmaniasis. Acta Trop. 1994, 56, 315–325. [Google Scholar] [CrossRef]
- Hashiguchi, Y.; Gomez, E.L.; Kato, H.; Martini, L.R.; Velez, L.N.; Uezato, H. Diffuse and disseminated cutaneous leishmaniasis: Clinical cases experienced in Ecuador and a brief review. Trop. Med. Health 2016, 44, 2. [Google Scholar] [CrossRef] [PubMed]
- Couppie, P.; Clyti, E.; Sainte-Marie, D.; Dedet, J.P.; Carme, B.; Pradinaud, R. Disseminated cutaneous leishmaniasis due to Leishmania guyanensis: Case of a patient with 425 lesions. Am. J. Trop. Med. Hyg. 2004, 71, 558–560. [Google Scholar] [PubMed]
- Turetz, M.L.; Machado, P.R.; Ko, A.I.; Alves, F.; Bittencourt, A.; Almeida, R.P.; Mobashery, N.; Johnson, W.D., Jr.; Carvalho, E.M. Disseminated leishmaniasis: A new and emerging form of leishmaniasis observed in Northeastern Brazil. J. Infect. Dis. 2002, 186, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Zijlstra, E.E.; Khalil, E.A.; Kager, P.A.; El-Hassan, A.M. Post-kala-azar dermal leishmaniasis in the Sudan: Clinical presentation and differential diagnosis. Br. J. Dermatol. 2000, 143, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Zijlstra, E.E.; Musa, A.M.; Khalil, E.A.; El-Hassan, I.M.; El-Hassan, A.M. Post-kala-azar dermal leishmaniasis. Lancet Infect. Dis. 2003, 3, 87–98. [Google Scholar] [CrossRef]
- Musa, A.M.; Khalil, E.A.; Raheem, M.A.; Zijlstra, E.E.; Ibrahim, M.E.; Elhassan, I.M.; Mukhtar, M.M.; El Hassan, A.M. The natural history of sudanese post-kala-azar dermal leishmaniasis: Clinical, immunological and prognostic features. Ann. Trop. Med. Parasitol. 2002, 96, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Peacock, C.S.; Seeger, K.; Harris, D.; Murphy, L.; Ruiz, J.C.; Quail, M.A.; Peters, N.; Adlem, E.; Tivey, A.; Aslett, M.; et al. Comparative genomic analysis of three leishmania species that cause diverse human disease. Nat. Genet. 2007, 39, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.B.; Hilley, J.D.; Dickens, N.J.; Wilkes, J.; Bates, P.A.; Depledge, D.P.; Harris, D.; Her, Y.; Herzyk, P.; Imamura, H.; et al. Chromosome and gene copy number variation allow major structural change between species and strains of leishmania. Genome Res. 2011, 21, 2129–2142. [Google Scholar] [CrossRef] [PubMed]
- Clayton, C.; Shapira, M. Post-transcriptional regulation of gene expression in trypanosomes and leishmanias. Mol. Biochem. Parasitol. 2007, 156, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Abdeladhim, M.; Kamhawi, S.; Valenzuela, J.G. What’s behind a sand fly bite? The profound effect of sand fly saliva on host hemostasis, inflammation and immunity. Infect. Genet. Evol. 2014, 28, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Castellucci, L.; Jamieson, S.E.; Almeida, L.; Oliveira, J.; Guimaraes, L.H.; Lessa, M.; Fakiola, M.; Jesus, A.R.; Nancy Miller, E.; Carvalho, E.M.; et al. Wound healing genes and susceptibility to cutaneous leishmaniasis in Brazil. Infect. Genet. Evol. 2012, 12, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Castellucci, L.C.; Almeida, L.F.; Jamieson, S.E.; Fakiola, M.; Carvalho, E.M.; Blackwell, J.M. Host genetic factors in american cutaneous leishmaniasis: A critical appraisal of studies conducted in an endemic area of Brazil. Memorias do Instituto Oswaldo Cruz 2014, 109, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Ribas-Silva, R.C.; Ribas, A.D.; Dos Santos, M.C.; da Silva, W.V.; Lonardoni, M.V.; Borelli, S.D.; Silveira, T.G. Association between HLA genes and american cutaneous leishmaniasis in endemic regions of southern Brazil. BMC Infect. Dis. 2013, 13, 198. [Google Scholar] [CrossRef] [PubMed]
- Samaranayake, N.; Fernando, S.D.; Neththikumara, N.F.; Rodrigo, C.; Karunaweera, N.D.; Dissanayake, V.H. Association of HLA class I and II genes with cutaneous leishmaniasis: A case control study from Sri Lanka and a systematic review. BMC Infect. Dis. 2016, 16, 292. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.E.; Carneiro, M.B.; Dos Santos, L.M.; Vieira, L.Q. Indigenous microbiota and leishmaniasis. Parasite Immunol. 2016, 38, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Sakthianandeswaren, A.; Foote, S.J.; Handman, E. The role of host genetics in leishmaniasis. Trends Parasitol. 2009, 25, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.; Kropf, P.; Choi, B.S.; Dillon, R.; Podinovskaia, M.; Bates, P.; Muller, I. Proteophosophoglycans regurgitated by leishmania-infected sand flies target the l-arginine metabolism of host macrophages to promote parasite survival. PLoS Pathog. 2009, 5, e1000555. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.E.; Corware, K.; Muller, I.; Bates, P.A. Leishmania infantum proteophosphoglycans regurgitated by the bite of its natural sand fly vector, lutzomyia longipalpis, promote parasite establishment in mouse skin and skin-distant tissues. Microbes Infect. Inst. Pasteur 2010, 12, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Vannier-Santos, M.A.; Martiny, A.; de Souza, W. Cell biology of Leishmania spp.: Invading and evading. Curr. Pharm. Des. 2002, 8, 297–318. [Google Scholar] [CrossRef] [PubMed]
- Ehrchen, J.M.; Roebrock, K.; Foell, D.; Nippe, N.; von Stebut, E.; Weiss, J.M.; Munck, N.A.; Viemann, D.; Varga, G.; Muller-Tidow, C.; et al. Keratinocytes determine Th1 immunity during early experimental leishmaniasis. PLoS Pathog. 2010, 6, e1000871. [Google Scholar] [CrossRef] [PubMed]
- Leon, B.; Lopez-Bravo, M.; Ardavin, C. Monocyte-derived dendritic cells formed at the infection site control the induction of protective T helper 1 responses against leishmania. Immunity 2007, 26, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Sacks, D.; Noben-Trauth, N. The immunology of susceptibility and resistance to leishmania major in mice. Nat. Rev. Immunol. 2002, 2, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro-Gomes, F.L.; Sacks, D. The influence of early neutrophil-leishmania interactions on the host immune response to infection. Front. Cell. Infect. Microbiol. 2012, 2, 59. [Google Scholar] [CrossRef] [PubMed]
- Lessa, H.A.; Lessa, M.M.; Guimaraes, L.H.; Lima, C.M.; Arruda, S.; Machado, P.R.; Carvalho, E.M. A proposed new clinical staging system for patients with mucosal leishmaniasis. Trans. R. Soc. Trop. Med. Hyg. 2012, 106, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, E.; Hatz, C.; Blum, J. New world cutaneous leishmaniasis in travellers. Lancet Infect. Dis. 2006, 6, 342–349. [Google Scholar] [CrossRef]
- Hochedez, P.; Caumes, E. Common skin infections in travelers. J. Travel Med. 2008, 15, 252–262. [Google Scholar] [CrossRef] [PubMed]
- El Hajj, L.; Thellier, M.; Carriere, J.; Bricaire, F.; Danis, M.; Caumes, E. Localized cutaneous leishmaniasis imported into Paris: A review of 39 cases. Int. J. Dermatol. 2004, 43, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Barral, A.; Guerreiro, J.; Bomfim, G.; Correia, D.; Barral-Netto, M.; Carvalho, E.M. Lymphadenopathy as the first sign of human cutaneous infection by Leishmania braziliensis. Am. J. Trop. Med. Hyg. 1995, 53, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Ziaei, H.; Sadeghian, G.; Hejazi, S.H. Distribution frequency of pathogenic bacteria isolated from cutaneus leishmaniasis lesions. Korean J. Parasitol. 2008, 46, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Unger, A.; O’Neal, S.; Machado, P.R.; Guimaraes, L.H.; Morgan, D.J.; Schriefer, A.; Bacellar, O.; Glesby, M.J.; Carvalho, E.M. Association of treatment of american cutaneous leishmaniasis prior to ulcer development with high rate of failure in northeastern Brazil. Am. J. Trop. Med. Hyg. 2009, 80, 574–579. [Google Scholar] [PubMed]
- Schubach, A.; Haddad, F.; Oliveira-Neto, M.P.; Degrave, W.; Pirmez, C.; Grimaldi, G., Jr.; Fernandes, O. Detection of leishmania DNA by polymerase chain reaction in scars of treated human patients. J. Infect. Dis. 1998, 178, 911–914. [Google Scholar] [CrossRef] [PubMed]
- Schubach, A.; Marzochi, M.C.; Cuzzi-Maya, T.; Oliveira, A.V.; Araujo, M.L.; Oliveira, A.L.; Pacheco, R.S.; Momen, H.; Conceicao-Silva, F.; Coutinho, S.G.; et al. Cutaneous scars in american tegumentary leishmaniasis patients: A site of Leishmania (Viannia) braziliensis persistence and viability eleven years after antimonial therapy and clinical cure. Am. J. Trop. Med. Hyg. 1998, 58, 824–827. [Google Scholar] [CrossRef] [PubMed]
- Barral, A.; Teixeira, M.; Reis, P.; Vinhas, V.; Costa, J.; Lessa, H.; Bittencourt, A.L.; Reed, S.; Carvalho, E.M.; Barral-Netto, M. Transforming growth factor-β in human cutaneous leishmaniasis. Am. J. Pathol. 1995, 147, 947–954. [Google Scholar] [PubMed]
- Bourreau, E.; Prevot, G.; Gardon, J.; Pradinaud, R.; Launois, P. High intralesional interleukin-10 messenger RNA expression in localized cutaneous leishmaniasis is associated with unresponsiveness to treatment. J. Infect. Dis. 2001, 184, 1628–1630. [Google Scholar] [CrossRef] [PubMed]
- Salhi, A.; Rodrigues, V., Jr.; Santoro, F.; Dessein, H.; Romano, A.; Castellano, L.R.; Sertorio, M.; Rafati, S.; Chevillard, C.; Prata, A.; et al. Immunological and genetic evidence for a crucial role of IL-10 in cutaneous lesions in humans infected with Leishmania braziliensis. J. Immunol. 2008, 180, 6139–6148. [Google Scholar] [CrossRef] [PubMed]
- Castellano, L.R.; Filho, D.C.; Argiro, L.; Dessein, H.; Prata, A.; Dessein, A.; Rodrigues, V. Th1/Th2 immune responses are associated with active cutaneous leishmaniasis and clinical cure is associated with strong interferon-γ production. Hum. Immunol. 2009, 70, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, W.N.; Ribeiro, L.E.; Schrieffer, A.; Machado, P.; Carvalho, E.M.; Bacellar, O. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of human tegumentary leishmaniasis. Cytokine 2014, 66, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Novais, F.O.; Carvalho, L.P.; Passos, S.; Roos, D.S.; Carvalho, E.M.; Scott, P.; Beiting, D.P. Genomic profiling of human Leishmania braziliensis lesions identifies transcriptional modules associated with cutaneous immunopathology. J. Investig. Dermatol. 2015, 135, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Bacellar, O.; Faria, D.; Nascimento, M.; Cardoso, T.M.; Gollob, K.J.; Dutra, W.O.; Scott, P.; Carvalho, E.M. Interleukin 17 production among patients with american cutaneous leishmaniasis. J. Infect. Dis. 2009, 200, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Boaventura, V.S.; Santos, C.S.; Cardoso, C.R.; de Andrade, J.; Dos Santos, W.L.; Clarencio, J.; Silva, J.S.; Borges, V.M.; Barral-Netto, M.; Brodskyn, C.I.; et al. Human mucosal leishmaniasis: Neutrophils infiltrate areas of tissue damage that express high levels of Th17-related cytokines. Eur. J. Immunol. 2010, 40, 2830–2836. [Google Scholar] [CrossRef] [PubMed]
- Abebe, T.; Hailu, A.; Woldeyes, M.; Mekonen, W.; Bilcha, K.; Cloke, T.; Fry, L.; Seich Al Basatena, N.K.; Corware, K.; Modolell, M.; et al. Local increase of arginase activity in lesions of patients with cutaneous leishmaniasis in Ethiopia. PLoS Negl. Trop. Dis. 2012, 6, e1684. [Google Scholar] [CrossRef] [PubMed]
- Iniesta, V.; Carcelen, J.; Molano, L.; Peixoto, P.M.V.; Redondo, E.; Parra, P.; Mangas, M.; Monroy, I.; Campo, M.L.; Nieto, C.G.; et al. Arginase I induction during Leishmania major infection mediates the development of disease. Infect. Immun. 2005, 73, 6085–6090. [Google Scholar] [CrossRef] [PubMed]
- Giudice, A.; Vendrame, C.; Bezerra, C.; Carvalho, L.P.; Delavechia, T.; Carvalho, E.M.; Bacellar, O. Macrophages participate in host protection and the disease pathology associated with Leishmania braziliensis infection. BMC Infect. Dis. 2012, 12, 75. [Google Scholar] [CrossRef] [PubMed]
- Follador, I.; Araujo, C.; Bacellar, O.; Araujo, C.B.; Carvalho, L.P.; Almeida, R.P.; Carvalho, E.M. Epidemiologic and immunologic findings for the subclinical form of Leishmania braziliensis infection. Clin. Infect. Dis. 2002, 34, E54–E58. [Google Scholar] [CrossRef] [PubMed]
- Novoa, R.; Bacellar, O.; Nascimento, M.; Cardoso, T.M.; Ramasawmy, R.; Oliveira, W.N.; Schriefer, A.; Carvalho, E.M. IL-17 and regulatory cytokines (IL-10 and IL-27) in L. Braziliensis infection. Parasite Immunol. 2011, 33, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Schnorr, D.; Muniz, A.C.; Passos, S.; Guimaraes, L.H.; Lago, E.L.; Bacellar, O.; Glesby, M.J.; Carvalho, E.M. IFN-γ production to leishmania antigen supplements the leishmania skin test in identifying exposure to L. Braziliensis infection. PLoS Negl. Trop. Dis. 2012, 6, e1947. [Google Scholar] [CrossRef] [PubMed]
- Bittar, R.C.; Nogueira, R.S.; Vieira-Goncalves, R.; Pinho-Ribeiro, V.; Mattos, M.S.; Oliveira-Neto, M.P.; Coutinho, S.G.; Da-Cruz, A.M. T-cell responses associated with resistance to leishmania infection in individuals from endemic areas for Leishmania (Viannia) braziliensis. Memorias do Instituto Oswaldo Cruz 2007, 102, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, M.W.; Fukutani, K.F.; Andrade, B.B.; Curvelo, R.P.; Cristal, J.R.; Carvalho, A.M.; Barral, A.; Van Weyenbergh, J.; Barral-Netto, M.; de Oliveira, C.I. Gene expression profile of high IFN-γ producers stimulated with Leishmania braziliensis identifies genes associated with cutaneous leishmaniasis. PLoS Negl. Trop. Dis. 2016, 10, e0005116. [Google Scholar] [CrossRef] [PubMed]
- Pompeu, M.M.; Brodskyn, C.; Teixeira, M.J.; Clarencio, J.; Van Weyenberg, J.; Coelho, I.C.; Cardoso, S.A.; Barral, A.; Barral-Netto, M. Differences in γ interferon production in vitro predict the pace of the in vivo response to Leishmania amazonensis in healthy volunteers. Infect. Immun. 2001, 69, 7453–7460. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, L.R.; Dutra, W.O.; Almeida, R.P.; Bacellar, O.; Carvalho, E.M.; Gollob, K.J. Activated inflammatory T cells correlate with lesion size in human cutaneous leishmaniasis. Immunol. Lett. 2005, 101, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, F.; Bafica, A.; Rosato, A.B.; Favali, C.B.; Costa, J.M.; Cafe, V.; Barral-Netto, M.; Barral, A. Lesion size correlates with leishmania antigen-stimulated TNF-levels in human cutaneous leishmaniasis. Am. J. Trop. Med. Hyg. 2011, 85, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Melby, P.C.; Andrade-Narvaez, F.J.; Darnell, B.J.; Valencia-Pacheco, G.; Tryon, V.V.; Palomo-Cetina, A. Increased expression of proinflammatory cytokines in chronic lesions of human cutaneous leishmaniasis. Infect. Immun. 1994, 62, 837–842. [Google Scholar] [PubMed]
- Hejazi, S.; Hoseini, S.; Javanmard, S.; Zarkesh, S.; Khamesipour, A. Interleukin-10 and transforming growth factor-β in early and late lesions of patients with Leishmania major induced cutaneous leishmaniasis. Iran. J. Parasitol. 2012, 7, 16–23. [Google Scholar] [PubMed]
- Bourreau, E.; Ronet, C.; Darcissac, E.; Lise, M.C.; Sainte Marie, D.; Clity, E.; Tacchini-Cottier, F.; Couppie, P.; Launois, P. Intralesional regulatory T-cell suppressive function during human acute and chronic cutaneous leishmaniasis due to Leishmania guyanensis. Infect. Immun. 2009, 77, 1465–1474. [Google Scholar] [CrossRef] [PubMed]
- Gaze, S.T.; Dutra, W.O.; Lessa, M.; Lessa, H.; Guimaraes, L.H.; Jesus, A.R.; Carvalho, L.P.; Machado, P.; Carvalho, E.M.; Gollob, K.J. Mucosal leishmaniasis patients display an activated inflammatory T-cell phenotype associated with a nonbalanced monocyte population. Scand. J. Immunol. 2006, 63, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.M.; Dillon, L.A.; Carvalho, L.P.; Passos, S.; Novais, F.O.; Hughitt, V.K.; Beiting, D.P.; Carvalho, E.M.; Scott, P.; El-Sayed, N.M.; et al. Meta-transcriptome profiling of the human Leishmania braziliensis cutaneous lesion. PLoS Negl. Trop. Dis. 2016, 10, e0004992. [Google Scholar] [CrossRef] [PubMed]
- Bomfim, G.; Andrade, B.B.; Santos, S.; Clarencio, J.; Barral-Netto, M.; Barral, A. Cellular analysis of cutaneous leishmaniasis lymphadenopathy: Insights into the early phases of human disease. Am. J. Trop. Med. Hyg. 2007, 77, 854–859. [Google Scholar] [PubMed]
- Rodriguez-Pinto, D.; Saravia, N.G.; McMahon-Pratt, D. CD4 T cell activation by B cells in human Leishmania (Viannia) infection. BMC Infect. Dis. 2014, 14, 108. [Google Scholar] [CrossRef] [PubMed]
- Junqueira Pedras, M.; Orsini, M.; Castro, M.; Passos, V.M.; Rabello, A. Antibody subclass profile against Leishmania braziliensis and Leishmania amazonensis in the diagnosis and follow-up of mucosal leishmaniasis. Diagn. Microbiol. Infect. Dis. 2003, 47, 477–485. [Google Scholar] [CrossRef]
- Faria, D.R.; Souza, P.E.; Duraes, F.V.; Carvalho, E.M.; Gollob, K.J.; Machado, P.R.; Dutra, W.O. Recruitment of CD8+ T cells expressing granzyme a is associated with lesion progression in human cutaneous leishmaniasis. Parasite Immunol. 2009, 31, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, T.M.; Machado, A.; Costa, D.L.; Carvalho, L.P.; Queiroz, A.; Machado, P.; Scott, P.; Carvalho, E.M.; Bacellar, O. Protective and pathological functions of CD8+ T cells in Leishmania braziliensis infection. Infect. Immun. 2015, 83, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Santos, C.; Brodskyn, C.I. The role of CD4 and CD8 T cells in human cutaneous leishmaniasis. Front. Public Health 2014, 2, 165. [Google Scholar] [CrossRef] [PubMed]
- Marsden, P.D. Mucosal leishmaniasis (“espundia” Escomel, 1911). Trans. R. Soc. Trop. Med. Hyg. 1986, 80, 859–876. [Google Scholar] [CrossRef]
- Jones, T.C.; Johnson, W.D., Jr.; Barretto, A.C.; Lago, E.; Badaro, R.; Cerf, B.; Reed, S.G.; Netto, E.M.; Tada, M.S.; Franca, T.F.; et al. Epidemiology of american cutaneous leishmaniasis due to Leishmania braziliensis braziliensis. J. Infect. Dis. 1987, 156, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Machado-Coelho, G.L.; Caiaffa, W.T.; Genaro, O.; Magalhaes, P.A.; Mayrink, W. Risk factors for mucosal manifestation of American cutaneous leishmaniasis. Trans. R. Soc. Trop. Med. Hyg. 2005, 99, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Marsden, P.D. Mucocutaneous leishmaniasis. BMJ 1990, 301, 656–657. [Google Scholar] [CrossRef] [PubMed]
- Saravia, N.G.; Segura, I.; Holguin, A.F.; Santrich, C.; Valderrama, L.; Ocampo, C. Epidemiologic, genetic, and clinical associations among phenotypically distinct populations of Leishmania (Viannia) in Colombia. Am. J. Trop. Med. Hyg. 1998, 59, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Amato, V.S.; Tuon, F.F.; Bacha, H.A.; Neto, V.A.; Nicodemo, A.C. Mucosal leishmaniasis. Current scenario and prospects for treatment. Acta Trop. 2008, 105, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Franke, E.D.; Wignall, F.S.; Cruz, M.E.; Rosales, E.; Tovar, A.A.; Lucas, C.M.; Llanos-Cuentas, A.; Berman, J.D. Efficacy and toxicity of sodium stibogluconate for mucosal leishmaniasis. Ann. Intern. Med. 1990, 113, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Lessa, H.A.; Machado, P.; Lima, F.; Cruz, A.A.; Bacellar, O.; Guerreiro, J.; Carvalho, E.M. Successful treatment of refractory mucosal leishmaniasis with pentoxifylline plus antimony. Am. J. Trop. Med. Hyg. 2001, 65, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Amato, V.S.; Tuon, F.F.; Siqueira, A.M.; Nicodemo, A.C.; Neto, V.A. Treatment of mucosal leishmaniasis in Latin America: Systematic review. Am. J. Trop. Med. Hyg. 2007, 77, 266–274. [Google Scholar] [PubMed]
- Ridley, D.S.; De Magalhaes, A.V.; Marsden, P.D. Histological analysis and the pathogenesis of mucocutaneous leishmaniasis. J. Pathol. 1989, 159, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Bacellar, O.; Lessa, H.; Schriefer, A.; Machado, P.; Ribeiro de Jesus, A.; Dutra, W.O.; Gollob, K.J.; Carvalho, E.M. Up-regulation of Th1-type responses in mucosal leishmaniasis patients. Infect. Immun. 2002, 70, 6734–6740. [Google Scholar] [CrossRef] [PubMed]
- Faria, D.R.; Gollob, K.J.; Barbosa, J., Jr.; Schriefer, A.; Machado, P.R.; Lessa, H.; Carvalho, L.P.; Romano-Silva, M.A.; de Jesus, A.R.; Carvalho, E.M.; et al. Decreased in situ expression of interleukin-10 receptor is correlated with the exacerbated inflammatory and cytotoxic responses observed in mucosal leishmaniasis. Infect. Immun. 2005, 73, 7853–7859. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Lindoso, J.A. Current diagnosis and treatment of cutaneous and mucocutaneous leishmaniasis. Expert Rev. Anti-Infect. Ther. 2010, 8, 419–433. [Google Scholar] [CrossRef] [PubMed]
- Machado, P.R.; Rosa, M.E.; Costa, D.; Mignac, M.; Silva, J.S.; Schriefer, A.; Teixeira, M.M.; Bacellar, O.; Carvalho, E.M. Reappraisal of the immunopathogenesis of disseminated leishmaniasis: In situ and systemic immune response. Trans. R. Soc. Trop. Med. Hyg. 2011, 105, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Schriefer, A.L.F.; Goes Neto, A.; Guimaraes, L.H.; Carvalho, L.P.; Almeida, R.P.; Machado, P.R.; Lessa, H.A.; Jesus, A.R.; Riley, L.W.; Carvalho, E.M. Multiclonal Leishmania braziliensis population structure and its clinical implication in a region of endemicity for american tegumentary leishmaniasis. Infect. Immun. 2004, 72, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, A.; Sousa, R.; Heine, C.; Cardoso, M.; Guimaraes, L.H.; Machado, P.R.; Carvalho, E.M.; Riley, L.W.; Wilson, M.E.; Schriefer, A. Association between an emerging disseminated form of leishmaniasis and Leishmania (Viannia) braziliensis strain polymorphisms. J. Clin. Microbiol. 2012, 50, 4028–4034. [Google Scholar] [CrossRef] [PubMed]
- Schriefer, A.; Guimaraes, L.H.; Machado, P.R.; Lessa, M.; Lessa, H.A.; Lago, E.; Ritt, G.; Goes-Neto, A.; Schriefer, A.L.; Riley, L.W.; et al. Geographic clustering of leishmaniasis in northeastern Brazil. Emerg. Infect. Dis. 2009, 15, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Leopoldo, P.T.; Machado, P.R.; Almeida, R.P.; Schriefer, A.; Giudice, A.; de Jesus, A.R.; Ho, J.L.; Guimaraes, L.H.; Bacellar, O.; Carvalho, E.M. Differential effects of antigens from l. Braziliensis isolates from disseminated and cutaneous leishmaniasis on in vitro cytokine production. BMC Infect. Dis. 2006, 6, 75. [Google Scholar] [CrossRef] [PubMed]
- Castellucci, L.; Menezes, E.; Oliveira, J.; Magalhaes, A.; Guimaraes, L.H.; Lessa, M.; Ribeiro, S.; Reale, J.; Noronha, E.F.; Wilson, M.E.; et al. IL6-174 G/C promoter polymorphism influences susceptibility to mucosal but not localized cutaneous leishmaniasis in Brazil. J. Infect. Dis. 2006, 194, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Castellucci, L.; Jamieson, S.E.; Miller, E.N.; Menezes, E.; Oliveira, J.; Magalhaes, A.; Guimaraes, L.H.; Lessa, M.; de Jesus, A.R.; Carvalho, E.M.; et al. CXCR1 and SLC11A1 polymorphisms affect susceptibility to cutaneous leishmaniasis in Brazil: A case-control and family-based study. BMC Med. Genet. 2010, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Castellucci, L.; Jamieson, S.E.; Miller, E.N.; de Almeida, L.F.; Oliveira, J.; Magalhaes, A.; Guimaraes, L.H.; Lessa, M.; Lago, E.; de Jesus, A.R.; et al. FLI1 polymorphism affects susceptibility to cutaneous leishmaniasis in Brazil. Genes Immun. 2011, 12, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Bomfim, G.; Nascimento, C.; Costa, J.; Carvalho, E.M.; Barral-Netto, M.; Barral, A. Variation of cytokine patterns related to therapeutic response in diffuse cutaneous leishmaniasis. Exp. Parasitol. 1996, 84, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Silveira, F.T.; Lainson, R.; Corbett, C.E. Clinical and immunopathological spectrum of american cutaneous leishmaniasis with special reference to the disease in amazonian Brazil: A review. Memorias do Instituto Oswaldo Cruz 2004, 99, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Convit, J.; Kerdel-Vegas, F.; Gordon, B. Disseminated anergic cutaneous leishmaniasis. Br. J. Dermatol. 1962, 74, 132–135. [Google Scholar] [CrossRef] [PubMed]
- Petersen, E.A.; Neva, F.A.; Oster, C.N.; Bogaert Diaz, H. Specific inhibition of lymphocyte-proliferation responses by adherent suppressor cells in diffuse cutaneous leishmaniasis. N. Engl. J. Med. 1982, 306, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Barral, A.; Costa, J.M.; Bittencourt, A.L.; Barral-Netto, M.; Carvalho, E.M. Polar and subpolar diffuse cutaneous leishmaniasis in Brazil: Clinical and immunopathologic aspects. Int. J. Dermatol. 1995, 34, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Diaz, N.L.; Arvelaez, F.A.; Zerpa, O.; Tapia, F.J. Inducible nitric oxide synthase and cytokine pattern in lesions of patients with american cutaneous leishmaniasis. Clin. Exp. Dermatol. 2006, 31, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Zerpa, O.; Ulrich, M.; Blanco, B.; Polegre, M.; Avila, A.; Matos, N.; Mendoza, I.; Pratlong, F.; Ravel, C.; Convit, J. Diffuse cutaneous leishmaniasis responds to miltefosine but then relapses. Br. J. Dermatol. 2007, 156, 1328–1335. [Google Scholar] [CrossRef] [PubMed]
- Calvopina, M.; Gomez, E.A.; Sindermann, H.; Cooper, P.J.; Hashiguchi, Y. Relapse of new world diffuse cutaneous leishmaniasis caused by Leishmania (Leishmania) mexicana after miltefosine treatment. Am. J. Trop. Med. Hyg. 2006, 75, 1074–1077. [Google Scholar] [PubMed]
- Salaiza-Suazo, N.; Volkow, P.; Tamayo, R.; Moll, H.; Gillitzer, R.; Perez-Torres, A.; Perez-Montfort, R.; Dominguez, J.D.; Velasco-Castrejon, O.; Crippa, M.; et al. Treatment of two patients with diffuse cutaneous leishmaniasis caused by Leishmania mexicana modifies the immunohistological profile but not the disease outcome. Trop. Med. Int. Health 1999, 4, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Caneda-Guzman, I.C.; Salaiza-Suazo, N.; Fernandez-Figueroa, E.A.; Carrada-Figueroa, G.; Aguirre-Garcia, M.; Becker, I. NK cell activity differs between patients with localized and diffuse cutaneous leishmaniasis infected with Leishmania mexicana: A comparative study of TLRs and cytokines. PLoS ONE 2014, 9, e112410. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Asoyan, A.; Rabenstein, H.; Nakano, N.; Obst, R. Role of antigen persistence and dose for CD4+ T-cell exhaustion and recovery. Proc. Natl. Acad. Sci. USA 2010, 107, 20453–20458. [Google Scholar] [CrossRef] [PubMed]
- Kahan, S.M.; Wherry, E.J.; Zajac, A.J. T cell exhaustion during persistent viral infections. Virology 2015, 479–480, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Franke, E.D.; Lucas, C.M.; Tovar, A.A.; Kruger, J.H.; De Rivera, M.V.; Wignall, F.S. Diffuse cutaneous leishmaniasis acquired in Peru. Am. J. Trop. Med. Hyg. 1990, 43, 260–262. [Google Scholar] [CrossRef] [PubMed]
- Convit, J.; Castellanos, P.L.; Ulrich, M.; Castes, M.; Rondon, A.; Pinardi, M.E.; Rodriquez, N.; Bloom, B.R.; Formica, S.; Valecillos, L.; et al. Immunotherapy of localized, intermediate, and diffuse forms of american cutaneous leishmaniasis. J. Infect. Dis. 1989, 160, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.I.; Dorta, M.L.; Pereira, A.J.; Bastos, R.P.; Oliveira, M.A.; Pinto, S.A.; Galdino, H., Jr.; Mayrink, W.; Barcelos, W.; Toledo, V.P.; et al. Increase of NK cells and proinflammatory monocytes are associated with the clinical improvement of diffuse cutaneous leishmaniasis after immunochemotherapy with BCG/leishmania antigens. Am. J. Trop. Med. Hyg. 2009, 81, 378–383. [Google Scholar] [PubMed]
- Badaro, R.; Johnson, W.D., Jr. The role of interferon-γ in the treatment of visceral and diffuse cutaneous leishmaniasis. J. Infect. Dis. 1993, 167 (Suppl. 1), S13–S17. [Google Scholar] [CrossRef] [PubMed]
- Marovich, M.A.; Lira, R.; Shepard, M.; Fuchs, G.H.; Kruetzer, R.; Nutman, T.B.; Neva, F.A. Leishmaniasis recidivans recurrence after 43 years: A clinical and immunologic report after successful treatment. Clin. Infect. Dis. 2001, 33, 1076–1079. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, I.; Fekri, A.R.; Aflatoonian, M.R.; Khamesipour, A.; Mahboudi, F.; Dowlati, Y.; Nadim, A.; Modabber, F. Leishmaniasis recidivans among school children in bam, south-east Iran, 1994–2006. Int. J. Dermatol. 2010, 49, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Momeni, A.Z.; Aminjavaheri, M. Treatment of recurrent cutaneous leishmaniasis. Int. J. Dermatol. 1995, 34, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Berlin, C. Leishmaniasis recidiva cutis; leishmanid. Arch. Dermatol. Syphilol. 1940, 41, 874–886. [Google Scholar] [CrossRef]
- Oliveira-Neto, M.P.; Mattos, M.; Souza, C.S.; Fernandes, O.; Pirmez, C. Leishmaniasis recidiva cutis in new world cutaneous leishmaniasis. Int. J. Dermatol. 1998, 37, 846–849. [Google Scholar] [CrossRef] [PubMed]
- Strick, R.A.; Borok, M.; Gasiorowski, H.C. Recurrent cutaneous leishmaniasis. J. Am. Acad. Dermatol. 1983, 9, 437–443. [Google Scholar] [CrossRef]
- Wortmann, G.W.; Aronson, N.E.; Miller, R.S.; Blazes, D.; Oster, C.N. Cutaneous leishmaniasis following local trauma: A clinical pearl. Clin. Infect. Dis. 2000, 31, 199–201. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.F.; Lira, R.; Kamhawi, S.; Belkaid, Y.; Wynn, T.A.; Sacks, D. Il-10 and TGF-β control the establishment of persistent and transmissible infections produced by leishmania tropica in C57BL/6 mice. J. Immunol. 2008, 180, 4090–4097. [Google Scholar] [CrossRef] [PubMed]
- Girgla, H.S.; Marsden, R.A.; Singh, G.M.; Ryan, T.J. Post-kala-azar dermal leishmaniasis. Br. J. Dermatol. 1977, 97, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, V.; Mukherjee, A. Post-kala-azar dermal leishmaniasis. Int. J. Dermatol. 1995, 34, 85–91. [Google Scholar] [CrossRef] [PubMed]
- El Hassan, A.M.; Ghalib, H.W.; Zijlstra, E.E.; Eltoum, I.A.; Satti, M.; Ali, M.S.; Ali, H.M. Post kala-azar dermal leishmaniasis in the Sudan: Clinical features, pathology and treatment. Trans. R. Soc. Trop. Med. Hyg. 1992, 86, 245–248. [Google Scholar] [CrossRef]
- Zijlstra, E.E.; El-Hassan, A.M. Leishmanin and tuberculin sensitivity in leishmaniasis in the Sudan, with special reference to kala-azar. Trans. R. Soc. Trop. Med. Hyg. 1993, 87, 425–427. [Google Scholar] [CrossRef]
- Ritmeijer, K.; Veeken, H.; Melaku, Y.; Leal, G.; Amsalu, R.; Seaman, J.; Davidson, R.N. Ethiopian visceral leishmaniasis: Generic and proprietary sodium stibogluconate are equivalent; HIV co-infected patients have a poor outcome. Trans. R. Soc. Trop. Med. Hyg. 2001, 95, 668–672. [Google Scholar] [CrossRef]
- Ramesh, V.; Kaushal, H.; Mishra, A.K.; Singh, R.; Salotra, P. Clinico-epidemiological analysis of post kala-azar dermal leishmaniasis (PKDL) cases in india over last two decades: A hospital based retrospective study. BMC Public Health 2015, 15, 1092. [Google Scholar] [CrossRef] [PubMed]
- Ismail, A.; Kharazmi, A.; Permin, H.; El Hassan, A.M. Detection and characterization of leishmania in tissues of patients with post kala-azar dermal leishmaniasis using a specific monoclonal antibody. Trans. R. Soc. Trop. Med. Hyg. 1997, 91, 283–285. [Google Scholar] [CrossRef]
- Gasim, S.; Elhassan, A.M.; Khalil, E.A.; Ismail, A.; Kadaru, A.M.; Kharazmi, A.; Theander, T.G. High levels of plasma IL-10 and expression of IL-10 by keratinocytes during visceral leishmaniasis predict subsequent development of post-kala-azar dermal leishmaniasis. Clin. Exp. Immunol. 1998, 111, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Mishra, J.; Singh, R.; Singh, S. Animal reservoirs of visceral leishmaniasis in India. J. Parasitol. 2013, 99, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.; Roychoudhury, J.; Palit, P.; Ali, N. Cationic liposomal sodium stibogluconate (SSG), a potent therapeutic tool for treatment of infection by SSG-sensitive and -resistant Leishmania donovani. Antimicrob. Agents Chemother. 2015, 59, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Sen Gupta, P.C.; Mukherjee, A.M. Recurrence of kala-azar associated with post-kala-azar dermal leishmaniasis. J. Indian Med. Assoc. 1968, 50, 1–5. [Google Scholar] [PubMed]
- Nandy, A.; Addy, M.; Maji, A.K.; Guha, S.K.; Banerjee, D.; Chaudhuri, D. Recurrence of kala-azar after PKDL: Role of co-factors. Trop. Med. Int. Health 1998, 3, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Haldar, J.P.; Ghose, S.; Saha, K.C.; Ghose, A.C. Cell-mediated immune response in Indian kala-azar and post-kala-azar dermal leishmaniasis. Infect. Immun. 1983, 42, 702–707. [Google Scholar] [PubMed]
- Ghosh, M.K.; Nandy, A.; Addy, M.; Maitra, T.K.; Ghose, A.C. Subpopulations of T lymphocytes in the peripheral blood, dermal lesions and lymph nodes of post kala-azar dermal leishmaniasis patients. Scand. J. Immunol. 1995, 41, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Ghalib, H.W.; Piuvezam, M.R.; Skeiky, Y.A.; Siddig, M.; Hashim, F.A.; El-Hassan, A.M.; Russo, D.M.; Reed, S.G. Interleukin 10 production correlates with pathology in human Leishmania donovani infections. J. Clin. Investig. 1993, 92, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Ismail, A.; Khalil, E.A.; Musa, A.M.; El Hassan, I.M.; Ibrahim, M.E.; Theander, T.G.; El Hassan, A.M. The pathogenesis of post kala-azar dermal leishmaniasis from the field to the molecule: Does ultraviolet light (UVB) radiation play a role? Med. Hypotheses 2006, 66, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, K.; Wessner, B.; Laggner, U.; Ploder, M.; Tamandl, D.; Friedl, J.; Zugel, U.; Steinmeyer, A.; Pollak, A.; Roth, E.; et al. Vitamin D3 down-regulates monocyte TLR expression and triggers hyporesponsiveness to pathogen-associated molecular patterns. Eur. J. Immunol. 2006, 36, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Leung, D.Y.; Richers, B.N.; Liu, Y.; Remigio, L.K.; Riches, D.W.; Goleva, E. Vitamin D inhibits monocyte/macrophage proinflammatory cytokine production by targeting MAPK phosphatase-1. J. Immunol. 2012, 188, 2127–2135. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, D.; Mukherjee, S.; Roy, S.; Dalton, J.E.; Kundu, S.; Sarkar, A.; Das, N.K.; Kaye, P.M.; Chatterjee, M. M2 polarization of monocytes-macrophages is a hallmark of Indian post kala-azar dermal leishmaniasis. PLoS Negl. Trop. Dis. 2015, 9, e0004145. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Whitcomb, J.P.; Deagostino, M.; Ballentine, M.; Fu, J.; Tenniswood, M.; Welsh, J.; Cantorna, M.; McDowell, M.A. The role of vitamin D and vitamin D receptor in immunity to Leishmania major infection. J. Parasitol. Res. 2012, 2012, 134645. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.W.; Ramasamy, G.; McCall, L.I.; Haydock, A.; Ranasinghe, S.; Abeygunasekara, P.; Sirimanna, G.; Wickremasinghe, R.; Myler, P.; Matlashewski, G. Genetic analysis of leishmania donovani tropism using a naturally attenuated cutaneous strain. PLoS Pathog. 2014, 10, e1004244. [Google Scholar] [CrossRef] [PubMed]
- Nuwayri-Salti, N.; Matta, M.; Shbaklo, Z.; Lakkis, M.; Kabbani, Z.E. Behavior in a mouse model of isolates of leishmania donovani sensu lato cultured from the blood of patients with chronic cutaneous lesions. Am. J. Trop. Med. Hyg. 1998, 58, 710–714. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.E.; Jeronimo, S.M.; Pearson, R.D. Immunopathogenesis of infection with the visceralizing Leishmania species. Microb. Pathog. 2005, 38, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, J.M.; Fakiola, M.; Ibrahim, M.E.; Jamieson, S.E.; Jeronimo, S.B.; Miller, E.N.; Mishra, A.; Mohamed, H.S.; Peacock, C.S.; Raju, M.; et al. Genetics and visceral leishmaniasis: Of mice and man. Parasite Immunol. 2009, 31, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Scott, P.; Novais, F.O. Cutaneous leishmaniasis: Immune responses in protection and pathogenesis. Nat. Rev. Immunol. 2016, 16, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Scott, P.; Natovitz, P.; Coffman, R.L.; Pearce, E.; Sher, A. CD4+ T cell subsets in experimental cutaneous leishmaniasis. Memorias do Instituto Oswaldo Cruz 1988, 83 (Suppl. 1), 256–259. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, F.P.; Sadick, M.D.; Holaday, B.J.; Coffman, R.L.; Locksley, R.M. Reciprocal expression of interferon γ or interleukin 4 during the resolution or progression of murine leishmaniasis. Evidence for expansion of distinct helper T cell subsets. J. Exp. Med. 1989, 169, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.J.; Filipe-Santos, O.; Eberl, G.; Aebischer, T.; Spath, G.F.; Bousso, P. CD4+ T cells rely on a cytokine gradient to control intracellular pathogens beyond sites of antigen presentation. Immunity 2012, 37, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Filipe-Santos, O.; Pescher, P.; Breart, B.; Lippuner, C.; Aebischer, T.; Glaichenhaus, N.; Spath, G.F.; Bousso, P. A dynamic map of antigen recognition by CD4 T cells at the site of Leishmania major infection. Cell Host Microbe 2009, 6, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Mendez, S.; Lira, R.; Kadambi, N.; Milon, G.; Sacks, D. A natural model of Leishmania major infection reveals a prolonged “silent” phase of parasite amplification in the skin before the onset of lesion formation and immunity. J. Immunol. 2000, 165, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.E.; Recker, T.J.; Rodriguez, N.E.; Young, B.M.; Burnell, K.K.; Streit, J.A.; Kline, J.N. The TGF-β response to leishmania chagasi in the absence of IL-12. Eur. J. Immunol. 2002, 32, 3556–3565. [Google Scholar] [CrossRef]
- Perez, H.; Malave, I.; Arredondo, B. The effects of protein malnutrition on the course of leishmania mexicana infection in C57BL/6 mice: Nutrition and susceptibility to leishmaniasis. Clin. Exp. Immunol. 1979, 38, 453–460. [Google Scholar] [PubMed]
- Rocha, F.J.; Schleicher, U.; Mattner, J.; Alber, G.; Bogdan, C. Cytokines, signaling pathways, and effector molecules required for the control of Leishmania (Viannia) braziliensis in mice. Infect. Immun. 2007, 75, 3823–3832. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira Cardoso, F.; de Souza Cda, S.; Mendes, V.G.; Abreu-Silva, A.L.; Goncalves da Costa, S.C.; Calabrese, K.S. Immunopathological studies of Leishmania amazonensis infection in resistant and in susceptible mice. J. Infect. Dis. 2010, 201, 1933–1940. [Google Scholar] [CrossRef] [PubMed]
- Lipoldova, M.; Demant, P. Genetic susceptibility to infectious disease: Lessons from mouse models of leishmaniasis. Nat. Rev. Genet. 2006, 7, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Loeuillet, C.; Banuls, A.L.; Hide, M. Study of leishmania pathogenesis in mice: Experimental considerations. Parasit. Vectors 2016, 9, 144. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, J.M. Genetic susceptibility to leishmanial infections: Studies in mice and man. Parasitology 1996, 112, S67–S74. [Google Scholar] [PubMed]
- Aebischer, T.; Moody, S.F.; Handman, E. Persistence of virulent Leishmania major in murine cutaneous leishmaniasis: A possible hazard for the host. Infect. Immun. 1993, 61, 220–226. [Google Scholar] [PubMed]
- Aebischer, T.; Morris, L.; Handman, E. Intravenous injection of irradiated leishmania major into susceptible BALB/c mice: Immunization or protective tolerance. Int. Immunol. 1994, 6, 1535–1543. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Hoffmann, K.F.; Mendez, S.; Kamhawi, S.; Udey, M.C.; Wynn, T.A.; Sacks, D.L. The role of interleukin (IL)-10 in the persistence of Leishmania major in the skin after healing and the therapeutic potential of anti-IL-10 receptor antibody for sterile cure. J. Exp. Med. 2001, 194, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Liew, F.Y.; Hale, C.; Howard, J.G. Immunologic regulation of experimental cutaneous leishmaniasis. V. Characterization of effector and specific suppressor T cells. J. Immunol. 1982, 128, 1917–1922. [Google Scholar] [PubMed]
- Muller, I. Role of T cell subsets during the recall of immunologic memory to Leishmania major. Eur. J. Immunol. 1992, 22, 3063–3069. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.; Russell, D.G. The interaction of leishmania species with macrophages. Adv. Parasitol. 1992, 31, 175–254. [Google Scholar] [PubMed]
- Moll, H.; Flohe, S.; Rollinghoff, M. Dendritic cells in leishmania major-immune mice harbor persistent parasites and mediate an antigen-specific T cell immune response. Eur. J. Immunol. 1995, 25, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, C.; Donhauser, N.; Doring, R.; Rollinghoff, M.; Diefenbach, A.; Rittig, M.G. Fibroblasts as host cells in latent leishmaniosis. J. Exp. Med. 2000, 191, 2121–2130. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Piccirillo, C.A.; Mendez, S.; Shevach, E.M.; Sacks, D.L. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature 2002, 420, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.E.; Ackermann, M.R.; Wille, U.; Hunter, C.A.; Scott, P. Early enhanced th1 response after Leishmania amazonensis infection of C57BL/6 interleukin-10-deficient mice does not lead to resolution of infection. Infect. Immun. 2002, 70, 2151–2158. [Google Scholar] [CrossRef] [PubMed]
- Padigel, U.M.; Alexander, J.; Farrell, J.P. The role of interleukin-10 in susceptibility of BALB/c mice to infection with Leishmania mexicana and Leishmania amazonensis. J. Immunol. 2003, 171, 3705–3710. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Sun, J.; Soong, L. Impaired expression of inflammatory cytokines and chemokines at early stages of infection with Leishmania amazonensis. Infect. Immun. 2003, 71, 4278–4288. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, F.P.; Schoenhaut, D.S.; Rerko, R.M.; Rosser, L.E.; Gately, M.K. Recombinant interleukin 12 cures mice infected with Leishmania major. J. Exp. Med. 1993, 177, 1505–1509. [Google Scholar] [CrossRef] [PubMed]
- Sypek, J.P.; Chung, C.L.; Mayor, S.E.; Subramanyam, J.M.; Goldman, S.J.; Sieburth, D.S.; Wolf, S.F.; Schaub, R.G. Resolution of cutaneous leishmaniasis: Interleukin 12 initiates a protective T helper type 1 immune response. J. Exp. Med. 1993, 177, 1797–1802. [Google Scholar] [CrossRef] [PubMed]
- Chatelain, R.; Varkila, K.; Coffman, R.L. IL-4 induces a Th2 response in leishmania major-infected mice. J. Immunol. 1992, 148, 1182–1187. [Google Scholar] [PubMed]
- Peters, N.C.; Egen, J.G.; Secundino, N.; Debrabant, A.; Kimblin, N.; Kamhawi, S.; Lawyer, P.; Fay, M.P.; Germain, R.N.; Sacks, D. In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science 2008, 321, 970–974. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro-Gomes, F.L.; Peters, N.C.; Debrabant, A.; Sacks, D.L. Efficient capture of infected neutrophils by dendritic cells in the skin inhibits the early anti-leishmania response. PLoS Pathog. 2012, 8, e1002536. [Google Scholar] [CrossRef] [PubMed]
- Conceicao, J.; Davis, R.; Carneiro, P.P.; Giudice, A.; Muniz, A.C.; Wilson, M.E.; Carvalho, E.M.; Bacellar, O. Characterization of neutrophil function in human cutaneous leishmaniasis caused by Leishmania braziliensis. PLoS Negl. Trop. Dis. 2016, 10, e0004715. [Google Scholar] [CrossRef] [PubMed]
- Van Zandbergen, G.; Klinger, M.; Mueller, A.; Dannenberg, S.; Gebert, A.; Solbach, W.; Laskay, T. Cutting edge: Neutrophil granulocyte serves as a vector for leishmania entry into macrophages. J. Immunol. 2004, 173, 6521–6525. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro-Gomes, F.L.; Romano, A.; Lee, S.; Roffe, E.; Peters, N.C.; Debrabant, A.; Sacks, D. Apoptotic cell clearance of Leishmania major-infected neutrophils by dendritic cells inhibits cCD8+ T-cell priming in vitro by MER tyrosine kinase-dependent signaling. Cell Death Dis. 2015, 6, e2018. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.; Fuchs, H.; Rappersberger, K.; Rollinghoff, M.; Moll, H. Parasitism of epidermal Langerhans cells in experimental cutaneous leishmaniasis with Leishmania major. J. Infect. Dis. 1993, 167, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Moll, H.; Fuchs, H.; Blank, C.; Rollinghoff, M. Langerhans cells transport leishmania major from the infected skin to the draining lymph node for presentation to antigen-specific T cells. Eur. J. Immunol. 1993, 23, 1595–1601. [Google Scholar] [CrossRef] [PubMed]
- Ritter, U.; Meissner, A.; Scheidig, C.; Korner, H. CD8 α- and langerin-negative dendritic cells, but not Langerhans cells, act as principal antigen-presenting cells in leishmaniasis. Eur. J. Immunol. 2004, 34, 1542–1550. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.G.; Hsu, A.; Mandell, M.A.; Roediger, B.; Hoeller, C.; Mrass, P.; Iparraguirre, A.; Cavanagh, L.L.; Triccas, J.A.; Beverley, S.M.; et al. Migratory dermal dendritic cells act as rapid sensors of protozoan parasites. PLoS Pathog. 2008, 4, e1000222. [Google Scholar] [CrossRef] [PubMed]
- Brewig, N.; Kissenpfennig, A.; Malissen, B.; Veit, A.; Bickert, T.; Fleischer, B.; Mostbock, S.; Ritter, U. Priming of CD8+ and CD4+ T cells in experimental leishmaniasis is initiated by different dendritic cell subtypes. J. Immunol. 2009, 182, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Kautz-Neu, K.; Noordegraaf, M.; Dinges, S.; Bennett, C.L.; John, D.; Clausen, B.E.; von Stebut, E. Langerhans cells are negative regulators of the anti-leishmania response. J. Exp. Med. 2011, 208, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, C.H.; Ueno, N.; Shao, J.Q.; Schroeder, K.R.; Moore, K.C.; Donelson, J.E.; Wilson, M.E. The effects of macrophage source on the mechanism of phagocytosis and intracellular survival of leishmania. Microbes Infect. Inst. Pasteur 2011, 13, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Ueno, N.; Wilson, M.E. Receptor-mediated phagocytosis of leishmania: Implications for intracellular survival. Trends Parasitol. 2012, 28, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Stafford, J.L.; Neumann, N.F.; Belosevic, M. Macrophage-mediated innate host defense against protozoan parasites. Crit. Rev. Microbiol. 2002, 28, 187–248. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Murray, P.J. Understanding local macrophage phenotypes in disease: Modulating macrophage function to treat cancer. Nat. Med. 2015, 21, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Minakami, R.; Sumimotoa, H. Phagocytosis-coupled activation of the superoxide-producing phagocyte oxidase, a member of the NADPH oxidase (NOX) family. Int. J. Hematol. 2006, 84, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Gantt, K.R.; Goldman, T.L.; McCormick, M.L.; Miller, M.A.; Jeronimo, S.M.; Nascimento, E.T.; Britigan, B.E.; Wilson, M.E. Oxidative responses of human and murine macrophages during phagocytosis of Leishmania chagasi. J. Immunol. 2001, 167, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Van Assche, T.; Deschacht, M.; da Luz, R.A.; Maes, L.; Cos, P. Leishmania-macrophage interactions: Insights into the redox biology. Free Radic. Biol. Med. 2011, 51, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Liew, F.Y.; Millott, S.; Parkinson, C.; Palmer, R.M.; Moncada, S. Macrophage killing of leishmania parasite in vivo is mediated by nitric oxide from l-arginine. J. Immunol. 1990, 144, 4794–4797. [Google Scholar] [PubMed]
- Bogdan, C.; Moll, H.; Solbach, W.; Rollinghoff, M. Tumor necrosis factor-α in combination with interferon-γ, but not with interleukin 4 activates murine macrophages for elimination of Leishmania major amastigotes. Eur. J. Immunol. 1990, 20, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Almeida, T.F.; Palma, L.C.; Mendez, L.C.; Noronha-Dutra, A.A.; Veras, P.S. Leishmania amazonensis fails to induce the release of reactive oxygen intermediates by CBA macrophages. Parasite Immunol. 2012, 34, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Matheoud, D.; Moradin, N.; Bellemare-Pelletier, A.; Shio, M.T.; Hong, W.J.; Olivier, M.; Gagnon, E.; Desjardins, M.; Descoteaux, A. Leishmania evades host immunity by inhibiting antigen cross-presentation through direct cleavage of the SNARE VAMP8. Cell Host Microbe 2013, 14, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Channon, J.Y.; Roberts, M.B.; Blackwell, J.M. A study of the differential respiratory burst activity elicited by promastigotes and amastigotes of Leishmania donovani in murine resident peritoneal macrophages. Immunology 1984, 53, 345–355. [Google Scholar] [PubMed]
- Nandan, D.; Reiner, N.E. Leishmania donovani engages in regulatory interference by targeting macrophage protein tyrosine phosphatase SHP-1. Clin. Immunol. 2005, 114, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.Q.; Charles, I.G.; Smith, A.; Ure, J.; Feng, G.J.; Huang, F.P.; Xu, D.; Muller, W.; Moncada, S.; Liew, F.Y. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature 1995, 375, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Mukbel, R.M.; Patten, C., Jr.; Gibson, K.; Ghosh, M.; Petersen, C.; Jones, D.E. Macrophage killing of Leishmania amazonensis amastigotes requires both nitric oxide and superoxide. Am. J. Trop. Med. Hyg. 2007, 76, 669–675. [Google Scholar] [PubMed]
- Novais, F.O.; Nguyen, B.T.; Beiting, D.P.; Carvalho, L.P.; Glennie, N.D.; Passos, S.; Carvalho, E.M.; Scott, P. Human classical monocytes control the intracellular stage of Leishmania braziliensis by reactive oxygen species. J. Infect. Dis. 2014, 209, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, J.B.; Misukonis, M.A.; Shami, P.J.; Mason, S.N.; Sauls, D.L.; Dittman, W.A.; Wood, E.R.; Smith, G.K.; McDonald, B.; Bachus, K.E.; et al. Human mononuclear phagocyte inducible nitric oxide synthase (iNOS): Analysis of iNOS mRNA, iNOS protein, biopterin, and nitric oxide production by blood monocytes and peritoneal macrophages. Blood 1995, 86, 1184–1195. [Google Scholar] [PubMed]
- Bogdan, C. Natural killer cells in experimental and human leishmaniasis. Front. Cell. Infect. Microbiol. 2012, 2, 69. [Google Scholar] [CrossRef] [PubMed]
- Scharton, T.M.; Scott, P. Natural killer cells are a source of interferon γ that drives differentiation of CD4+ T cell subsets and induces early resistance to Leishmania major in mice. J. Exp. Med. 1993, 178, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Muller, K.; van Zandbergen, G.; Hansen, B.; Laufs, H.; Jahnke, N.; Solbach, W.; Laskay, T. Chemokines, natural killer cells and granulocytes in the early course of Leishmania major infection in mice. Med. Microbiol. Immunol. 2001, 190, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Bajenoff, M.; Breart, B.; Huang, A.Y.; Qi, H.; Cazareth, J.; Braud, V.M.; Germain, R.N.; Glaichenhaus, N. Natural killer cell behavior in lymph nodes revealed by static and real-time imaging. J. Exp. Med. 2006, 203, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Sanabria, M.X.; Vargas-Inchaustegui, D.A.; Xin, L.; Soong, L. Role of natural killer cells in modulating dendritic cell responses to leishmania amazonensis infection. Infect. Immun. 2008, 76, 5100–5109. [Google Scholar] [CrossRef] [PubMed]
- Mailliard, R.B.; Son, Y.I.; Redlinger, R.; Coates, P.T.; Giermasz, A.; Morel, P.A.; Storkus, W.J.; Kalinski, P. Dendritic cells mediate NK cell help for Th1 and CTL responses: Two-signal requirement for the induction of NK cell helper function. J. Immunol. 2003, 171, 2366–2373. [Google Scholar] [CrossRef] [PubMed]
- Prajeeth, C.K.; Haeberlein, S.; Sebald, H.; Schleicher, U.; Bogdan, C. Leishmania-infected macrophages are targets of NK cell-derived cytokines but not of NK cell cytotoxicity. Infect. Immun. 2011, 79, 2699–2708. [Google Scholar] [CrossRef] [PubMed]
- Wakil, A.E.; Wang, Z.E.; Ryan, J.C.; Fowell, D.J.; Locksley, R.M. Interferon γ derived from CD4+ T cells is sufficient to mediate T helper cell type 1 development. J. Exp. Med. 1998, 188, 1651–1656. [Google Scholar] [CrossRef] [PubMed]
- Laouar, Y.; Sutterwala, F.S.; Gorelik, L.; Flavell, R.A. Transforming growth factor-β controls T helper type 1 cell development through regulation of natural killer cell interferon-γ. Nat. Immunol. 2005, 6, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Takashima, A.; Bergstresser, P.R. Cytokine-mediated communication by keratinocytes and Langerhans cells with dendritic epidermal T cells. Semin. Immunol. 1996, 8, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Roebrock, K.; Sunderkotter, C.; Munck, N.A.; Wolf, M.; Nippe, N.; Barczyk, K.; Varga, G.; Vogl, T.; Roth, J.; Ehrchen, J. Epidermal expression of I-TAC (CXCL11) instructs adaptive Th2-type immunity. FASEB J. 2014, 28, 1724–1734. [Google Scholar] [CrossRef] [PubMed]
- Scorza, B.M.; Wacker, M.A.; Messingham, K.; Fairley, J.; Kim, P.; Klingelhutz, A.; Wilson, M.E. Differential activation of human keratinocytes by Leishmania spp. causing localized or disseminated disease. J. Investig. Dermatol. 2017, accepted/in press. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Syndrome | Causative Species 1 | Clinical Manifestation |
|---|---|---|
| Localized Cutaneous Leishmaniasis (LCL) | L. (L.) major [3] L. (L.) mexicana [7] L. (L.) amazonensis [7] L. (V.) braziliensis [7] L. (L.) tropica [8] L. (L.) aethiopica [9] L. (V.) panamanensis [7] L. (L.) infantum [10] L. (L.) donovani [11] | Single or limited number of lesions; ulcers formed can be wet or dry with raised crateriform border. Moderate parasite loads in biopsies of the ulcer border; positive DTH 2 response [12] |
| Mucosal Leishmaniasis [13] | L. (V.) braziliensis [14] L. (V.) panamanensis [15] L. (V.) guyanensis [16] L. (L.) amazonensis [14,17] | Highly inflammatory lesions involving mucosal membranes; can be disfiguring. Rare parasite forms present in biopsies; strong DTH response [12] |
| Anergic Diffuse Cutaneous Leishmaniasis (ADCL) | L. (L.) amazonensis [18] L. (L.) mexicana [19] L. (V.) pifanoi [20] L. (L.) aethiopica [21] L. (L.) major [22] | Multiple, disseminated, non-ulcerative nodular lesions; many parasites in lesions; absent DTH response (anergy) [20,23] |
| Disseminated Leishmaniasis (DL) | L. (V.) braziliensis [24] L. (V.) panamanensis [25] L. (V.) guyanensis [25,26] L. (L.) amazonensis [24] | Numerous papular/acneiform lesions in ≥2 non-contiguous areas of the body, commonly involving mucosal membranes. Few parasites in lesions; strong DTH response [27] |
| Post-Kala Azar Dermal Leishmaniasis (PKDL) | L. (L.) donovani [1] | Hypopigmented macular, maculopapular, or nodular rash. Interferon γ (IFNγ) response to Leishmania antigens. Parasites are present in lesions [28,29,30] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scorza, B.M.; Carvalho, E.M.; Wilson, M.E. Cutaneous Manifestations of Human and Murine Leishmaniasis. Int. J. Mol. Sci. 2017, 18, 1296. https://doi.org/10.3390/ijms18061296
Scorza BM, Carvalho EM, Wilson ME. Cutaneous Manifestations of Human and Murine Leishmaniasis. International Journal of Molecular Sciences. 2017; 18(6):1296. https://doi.org/10.3390/ijms18061296
Chicago/Turabian StyleScorza, Breanna M., Edgar M. Carvalho, and Mary E. Wilson. 2017. "Cutaneous Manifestations of Human and Murine Leishmaniasis" International Journal of Molecular Sciences 18, no. 6: 1296. https://doi.org/10.3390/ijms18061296
APA StyleScorza, B. M., Carvalho, E. M., & Wilson, M. E. (2017). Cutaneous Manifestations of Human and Murine Leishmaniasis. International Journal of Molecular Sciences, 18(6), 1296. https://doi.org/10.3390/ijms18061296