Interactions between a Heparin Trisaccharide Library and FGF-1 Analyzed by NMR Methods




Abstract
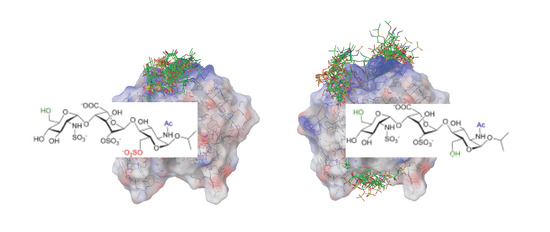
:
1. Introduction
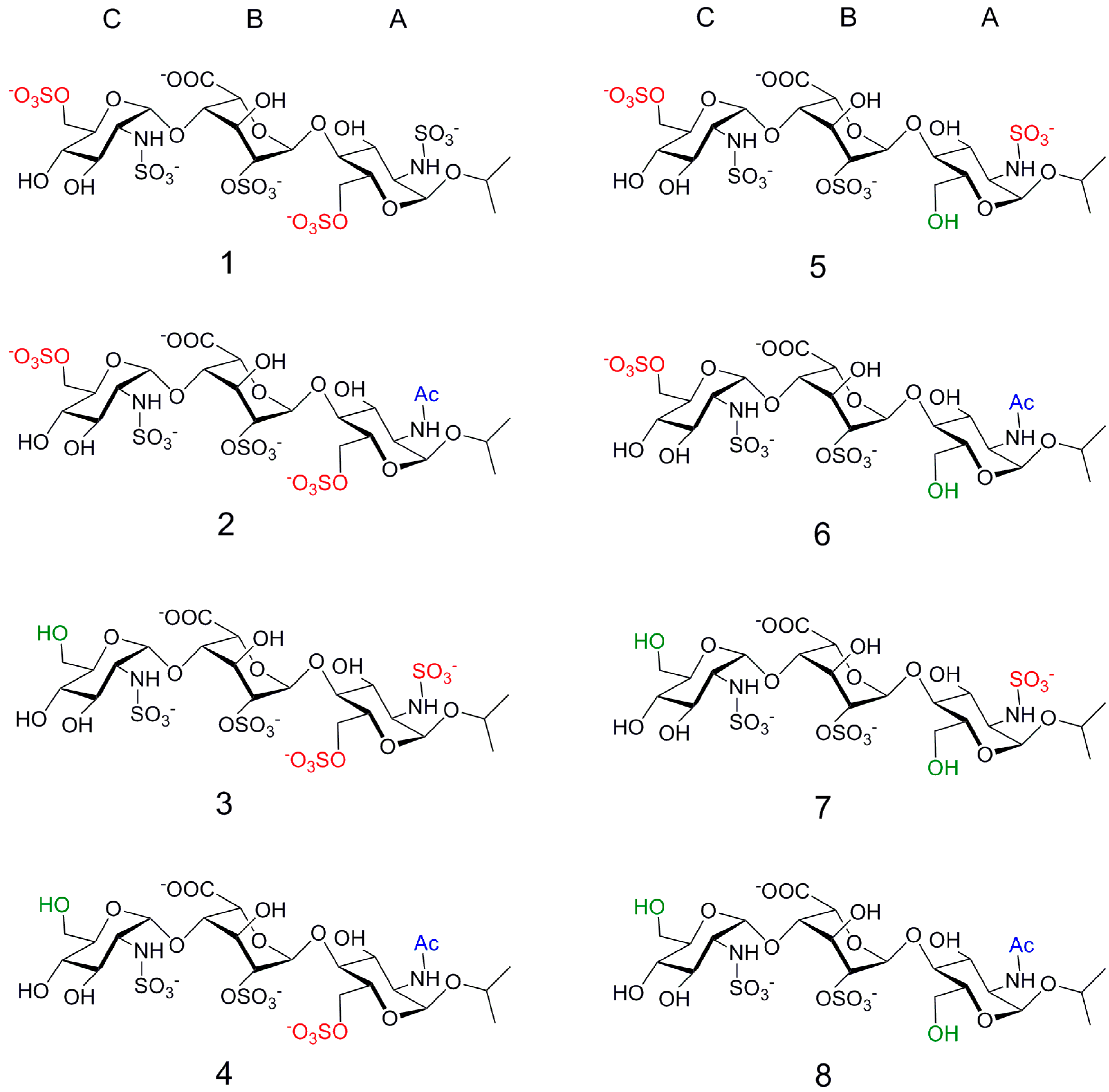
2. Results
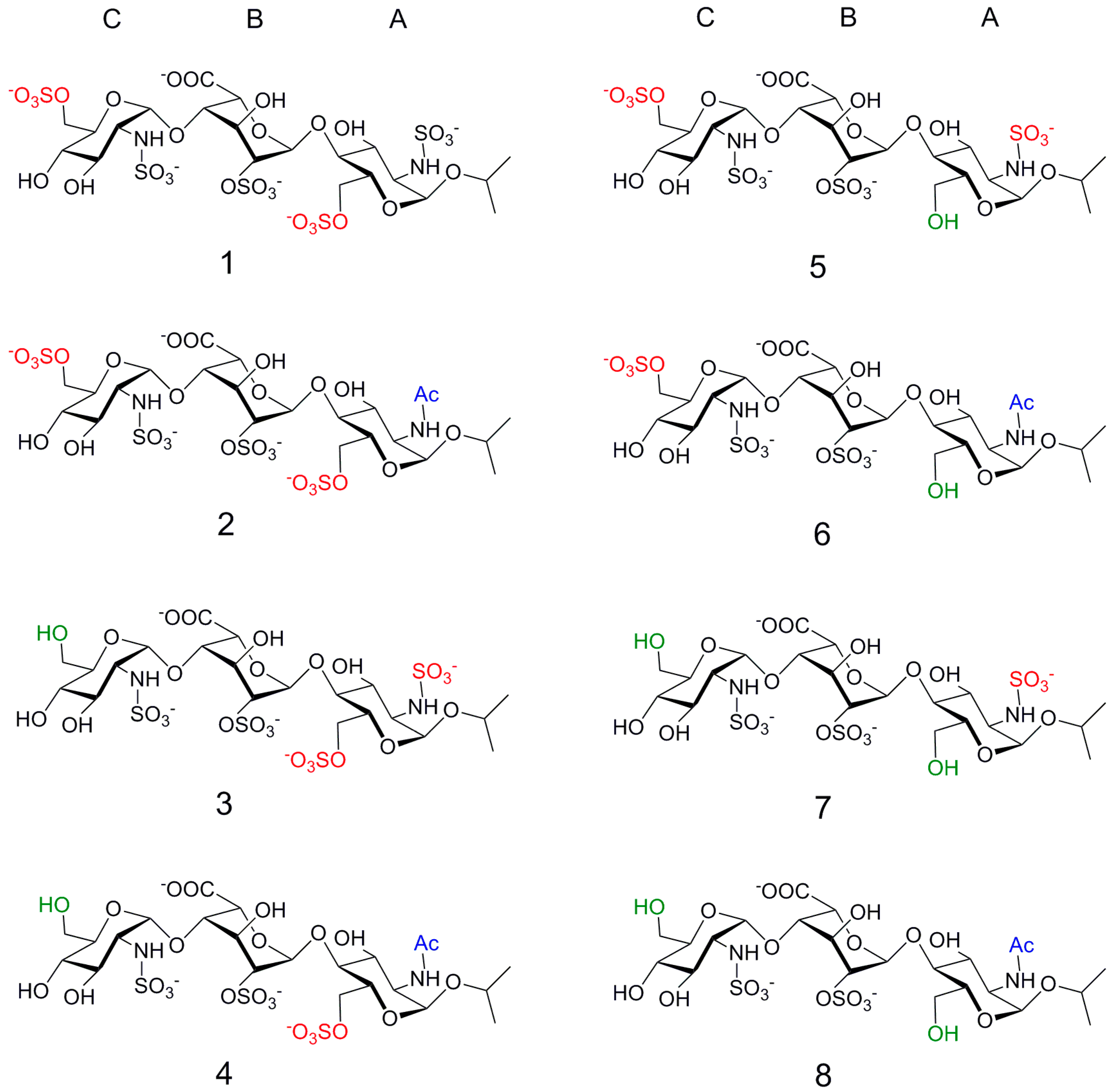
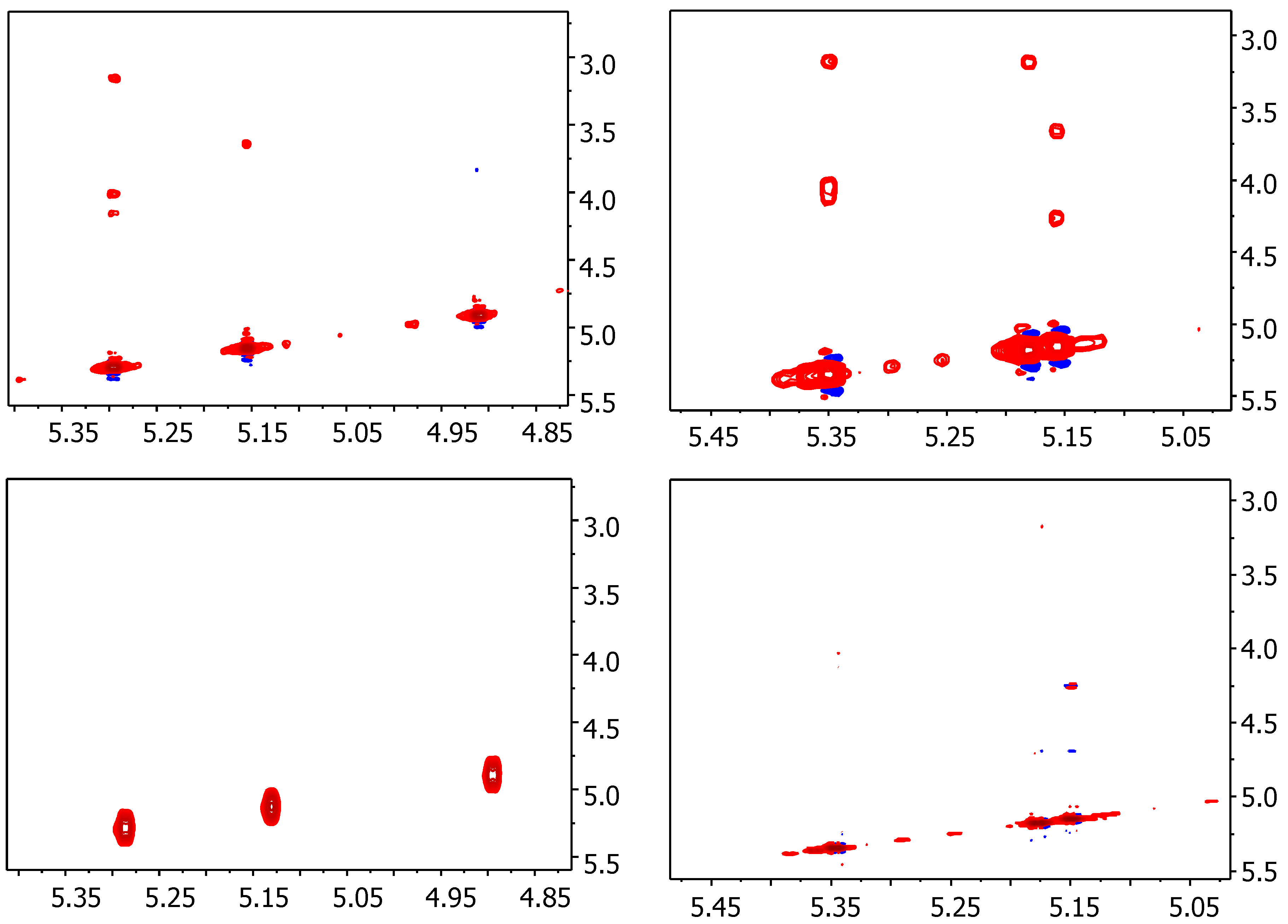
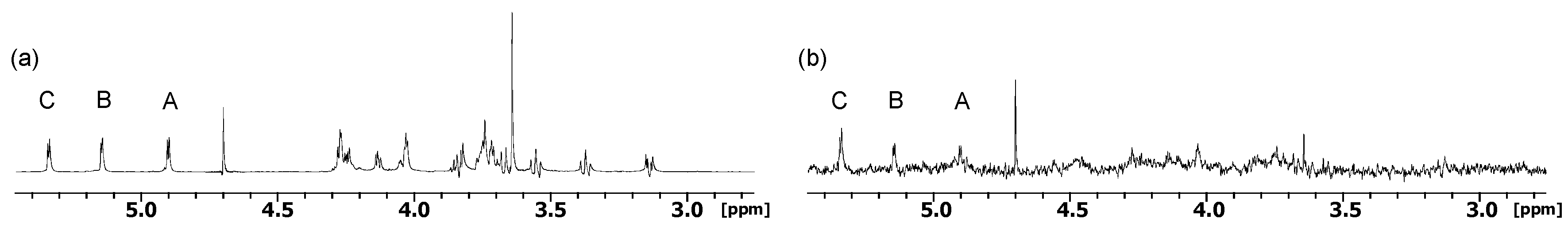
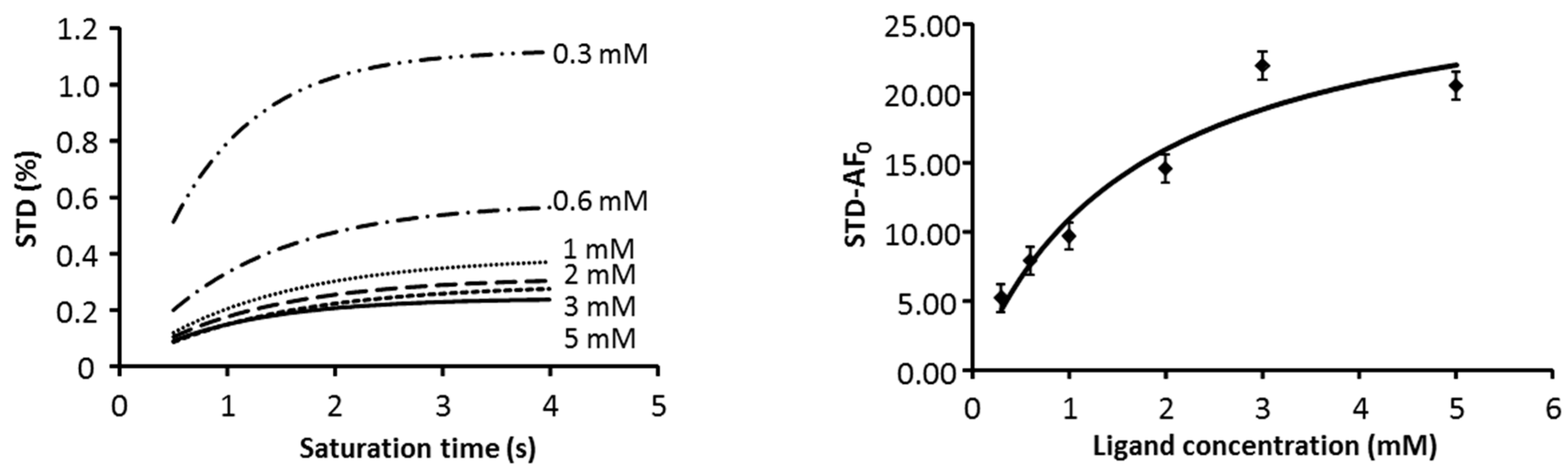
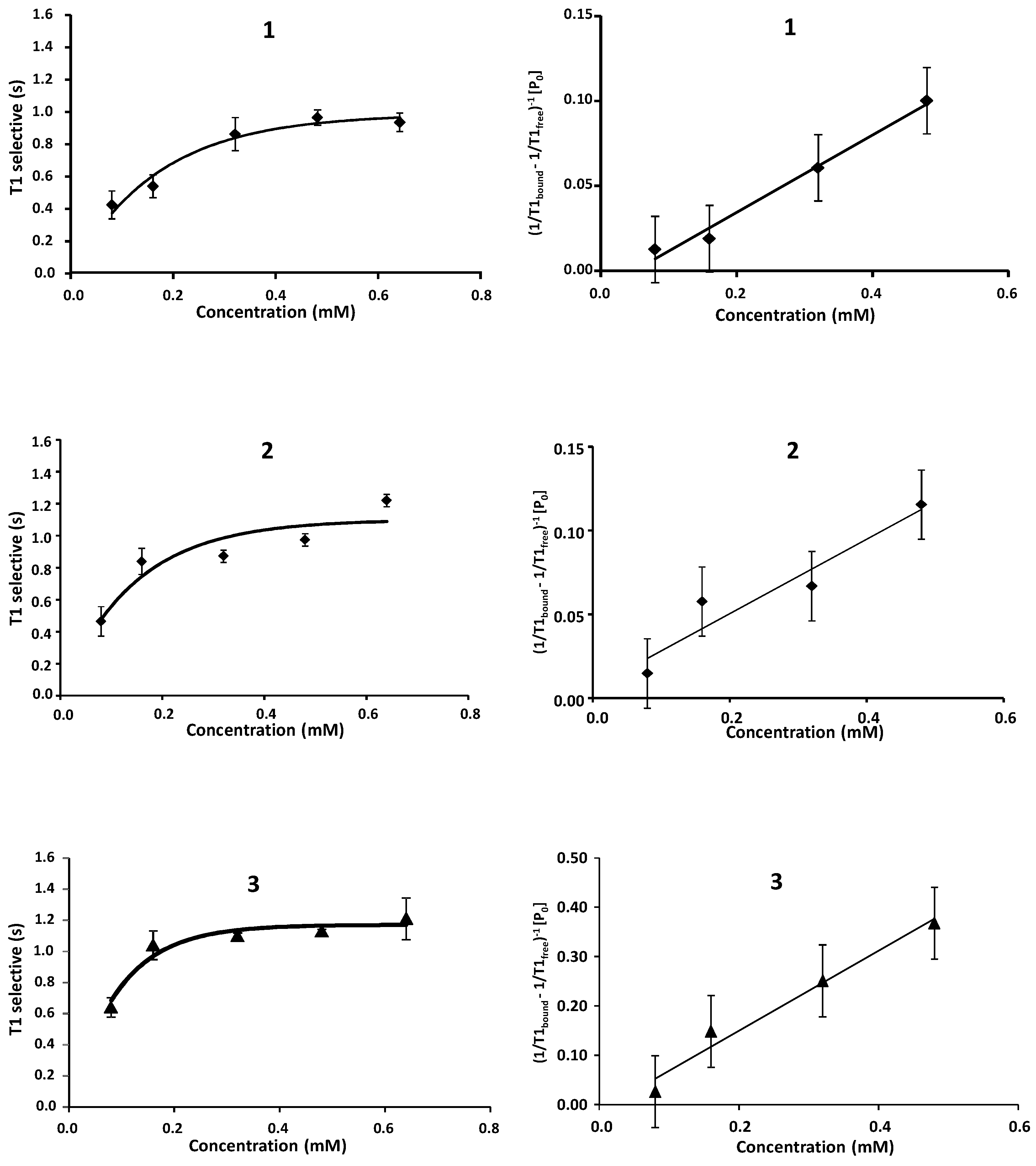
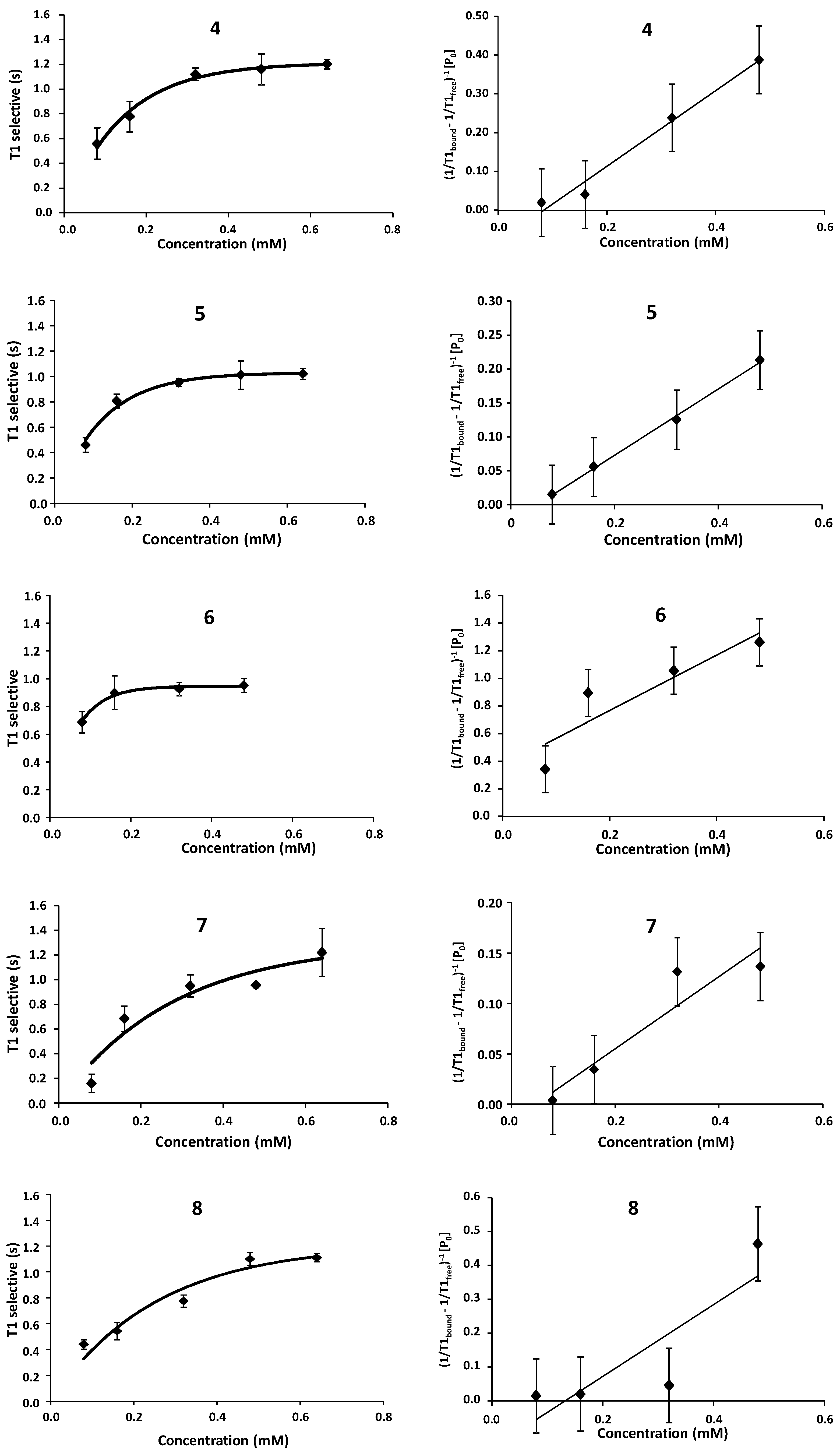
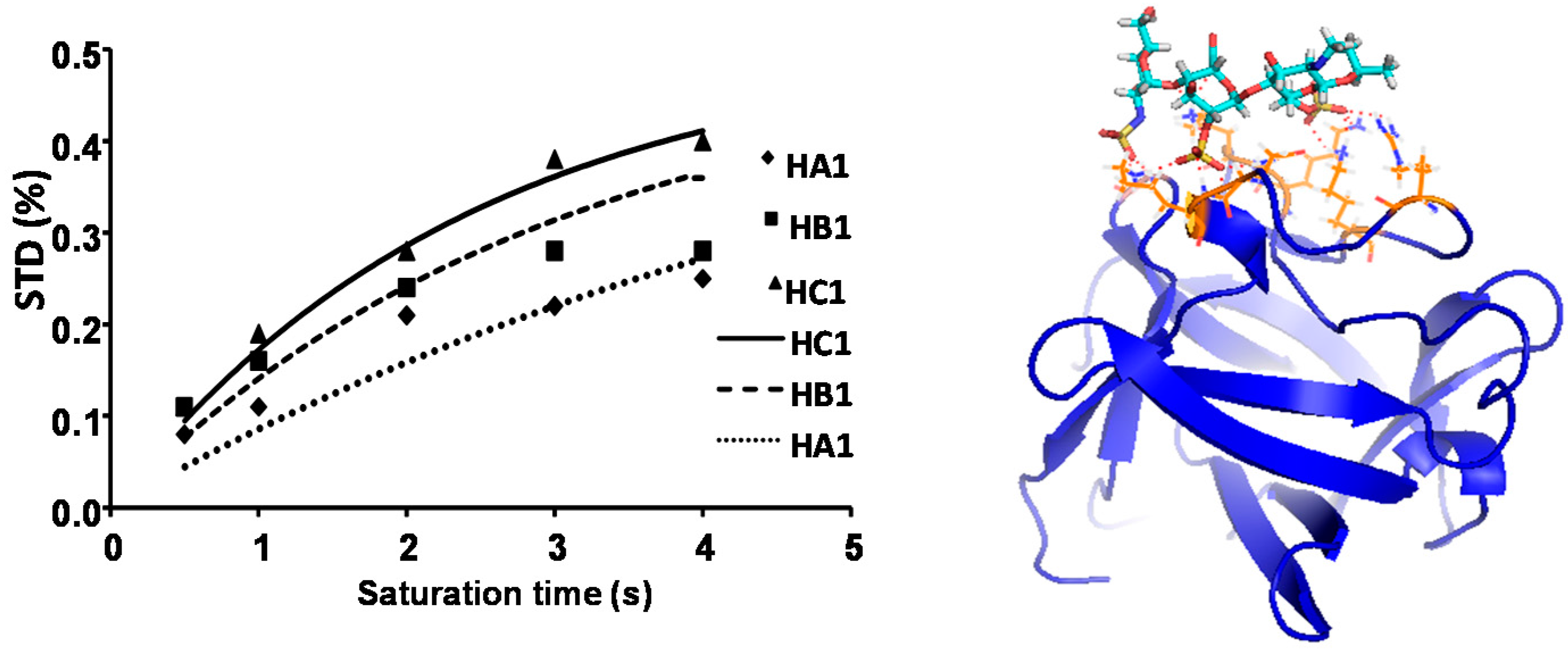
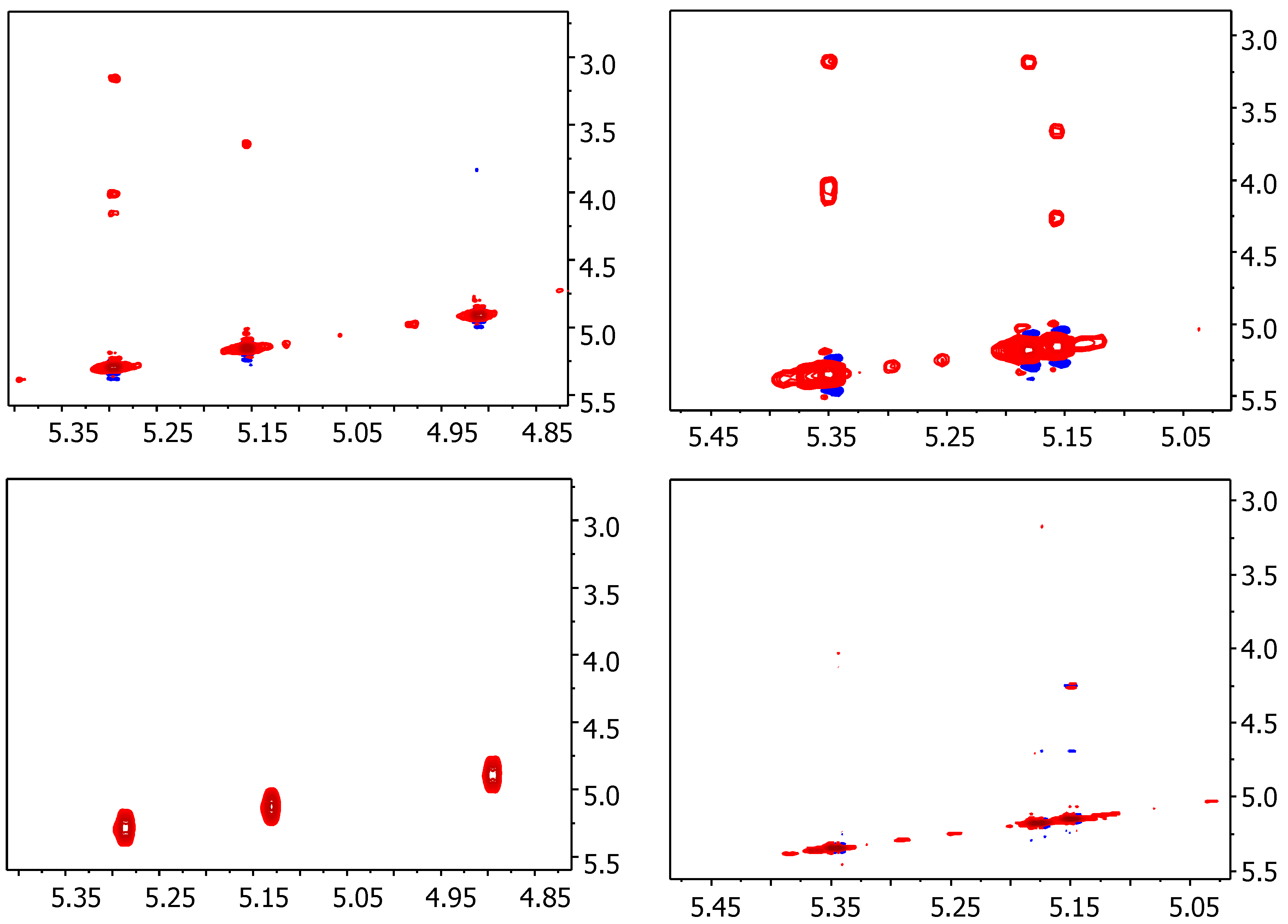
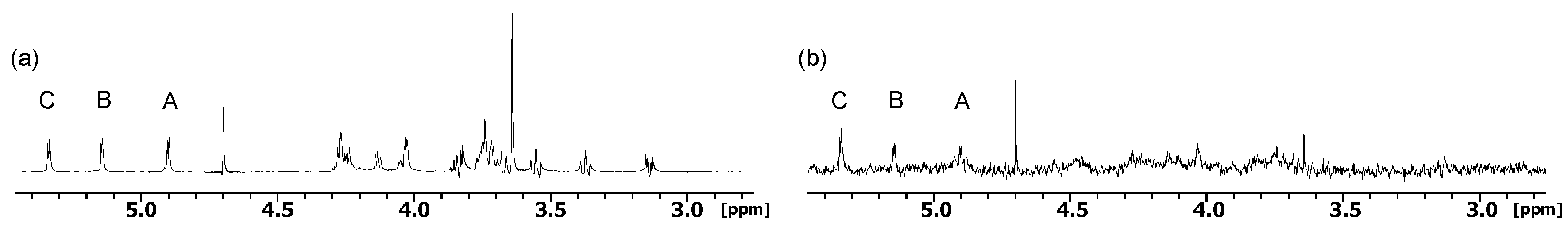
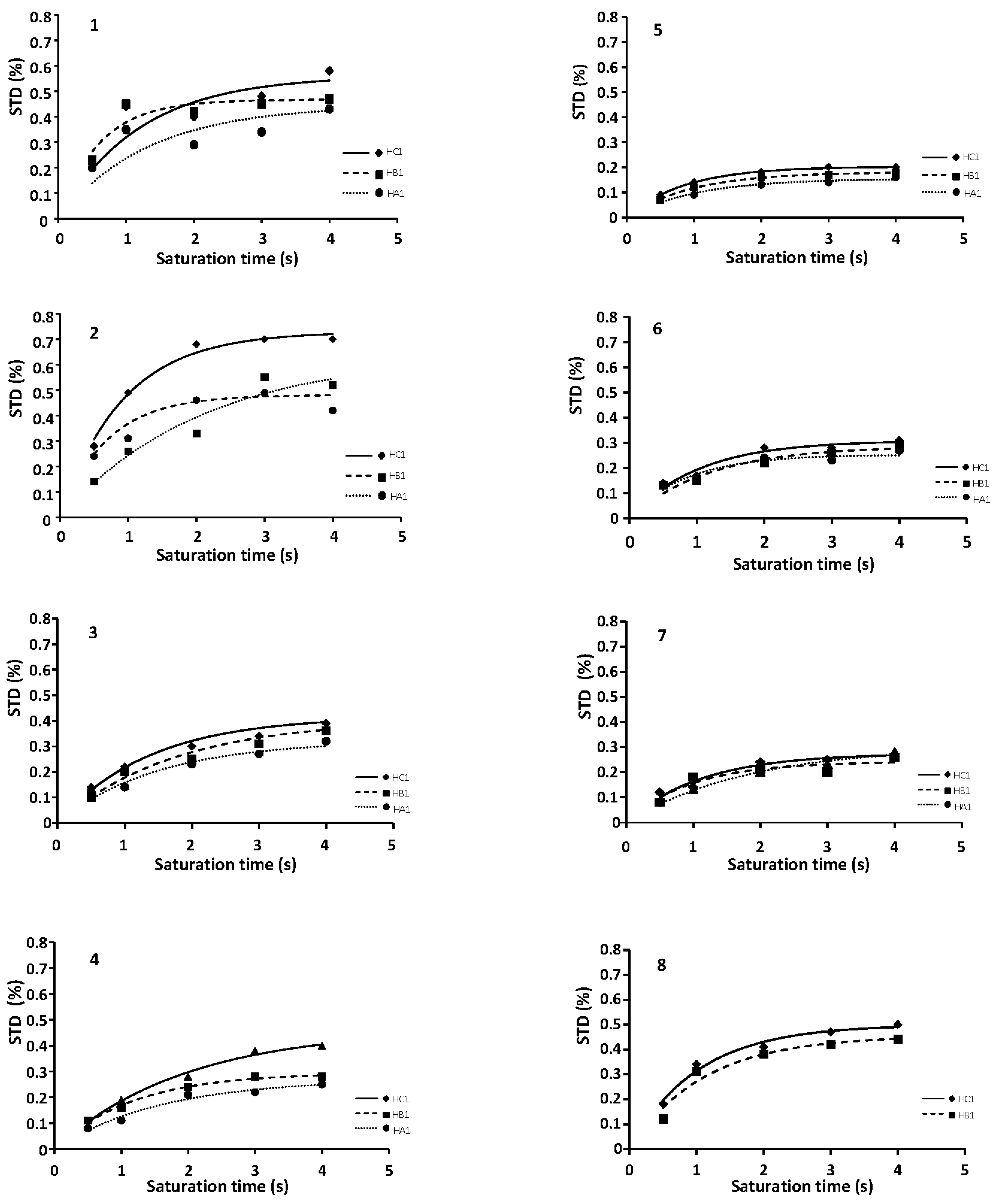
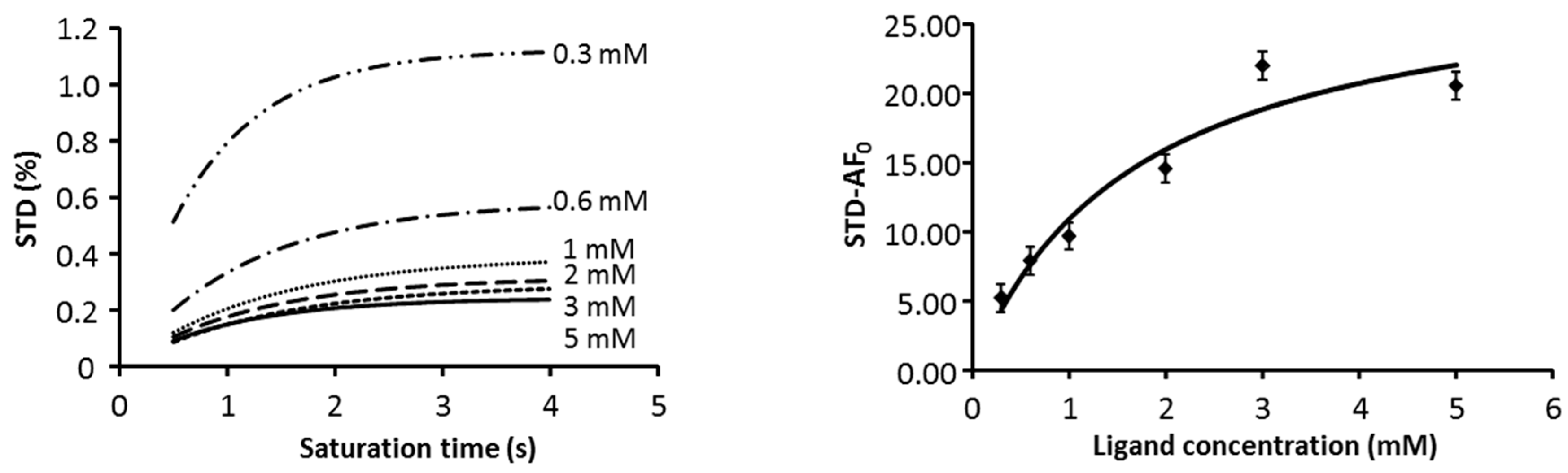
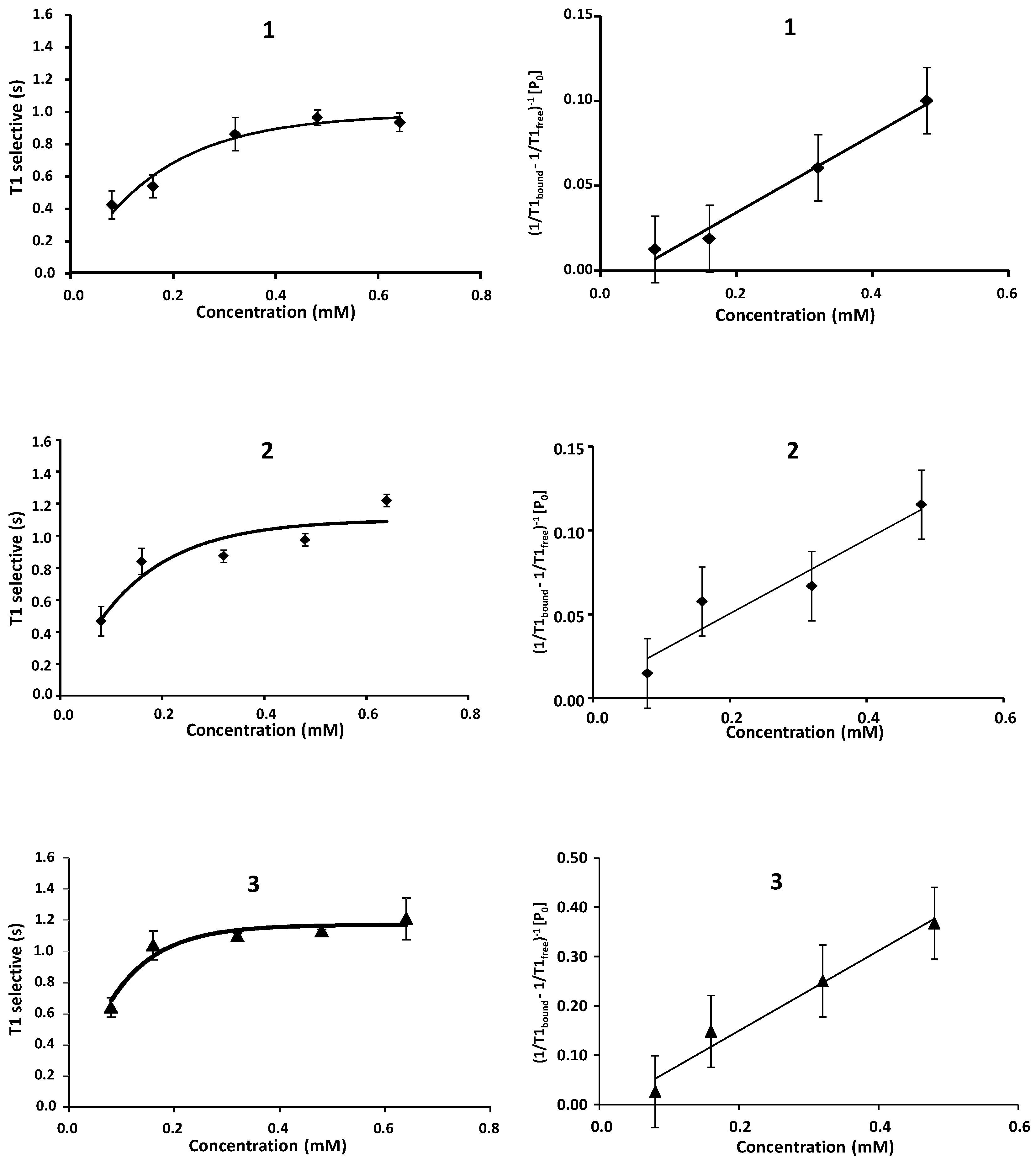
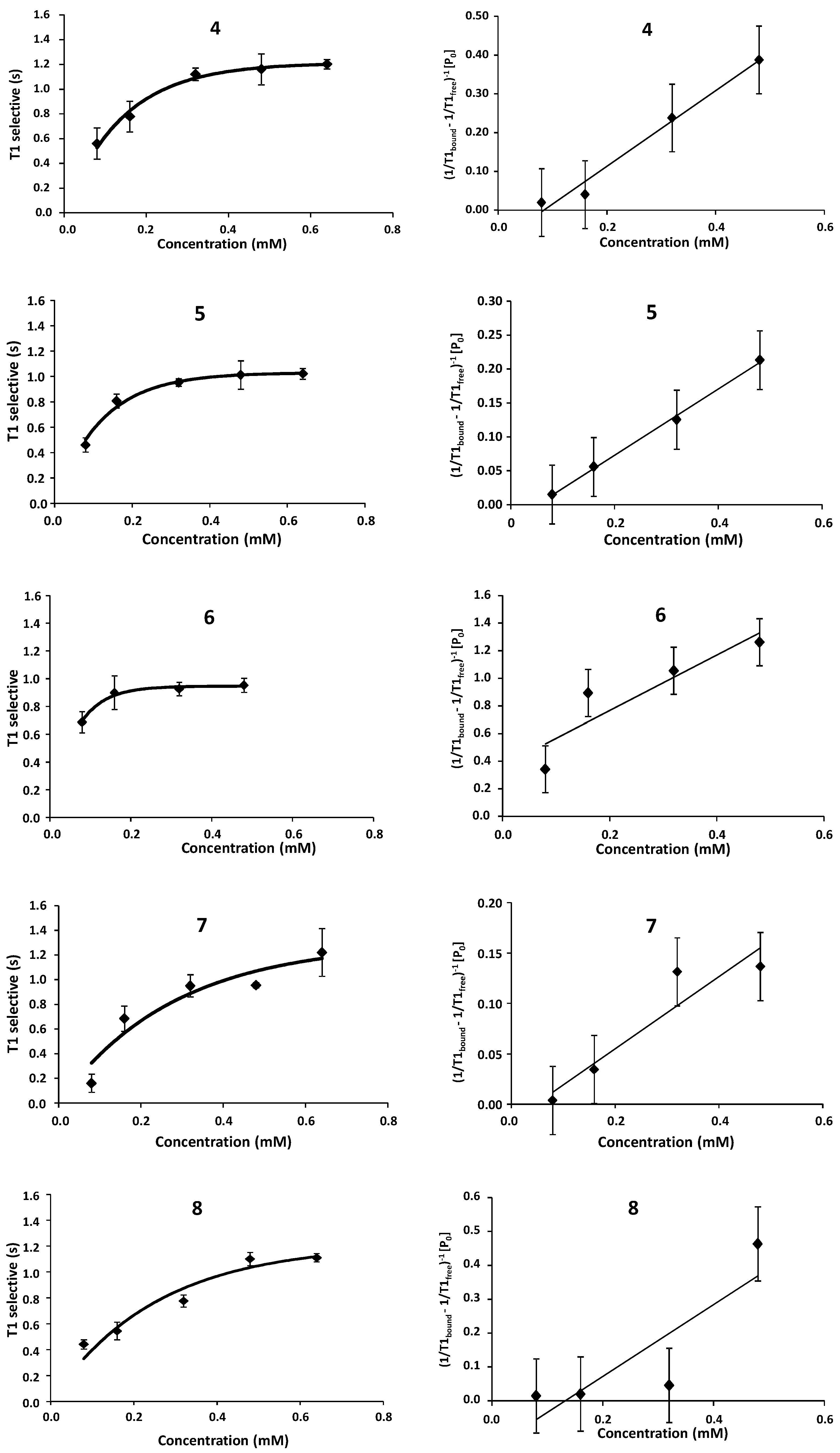
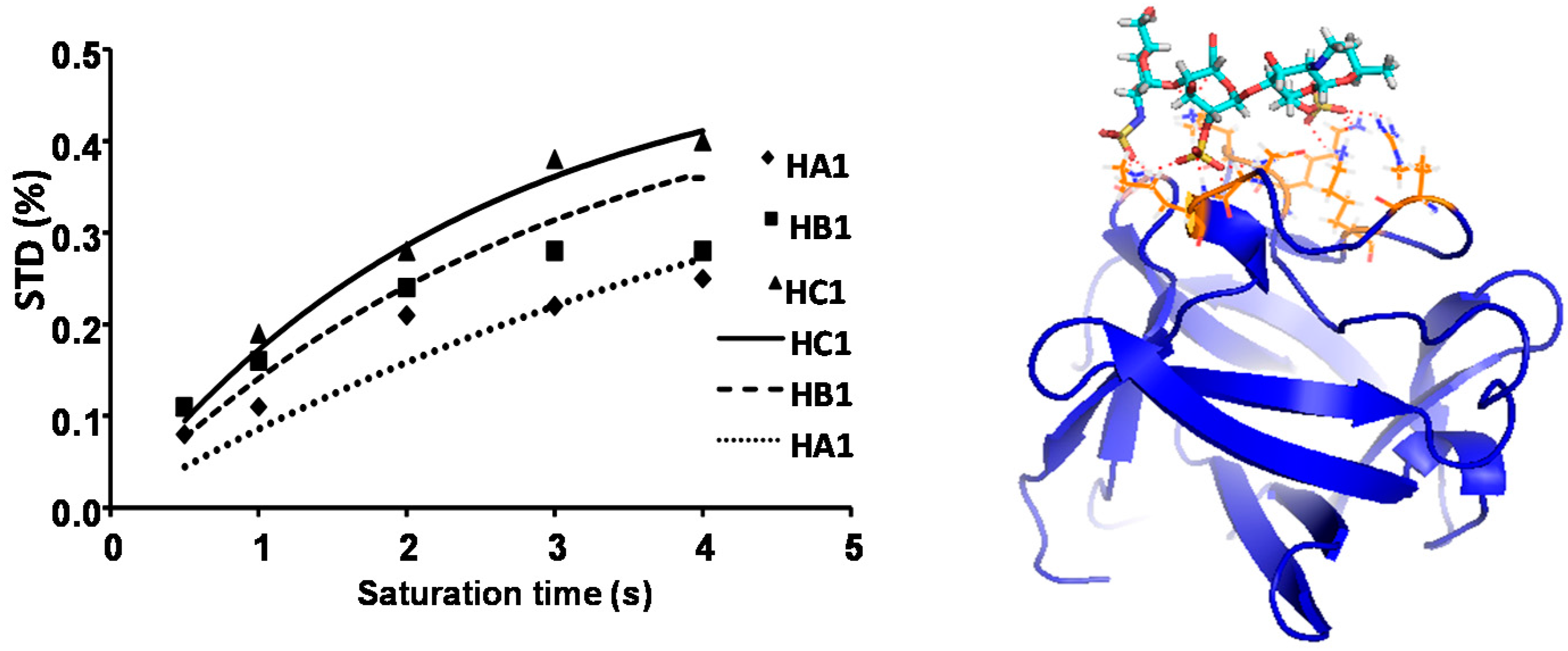
2.1. NMR
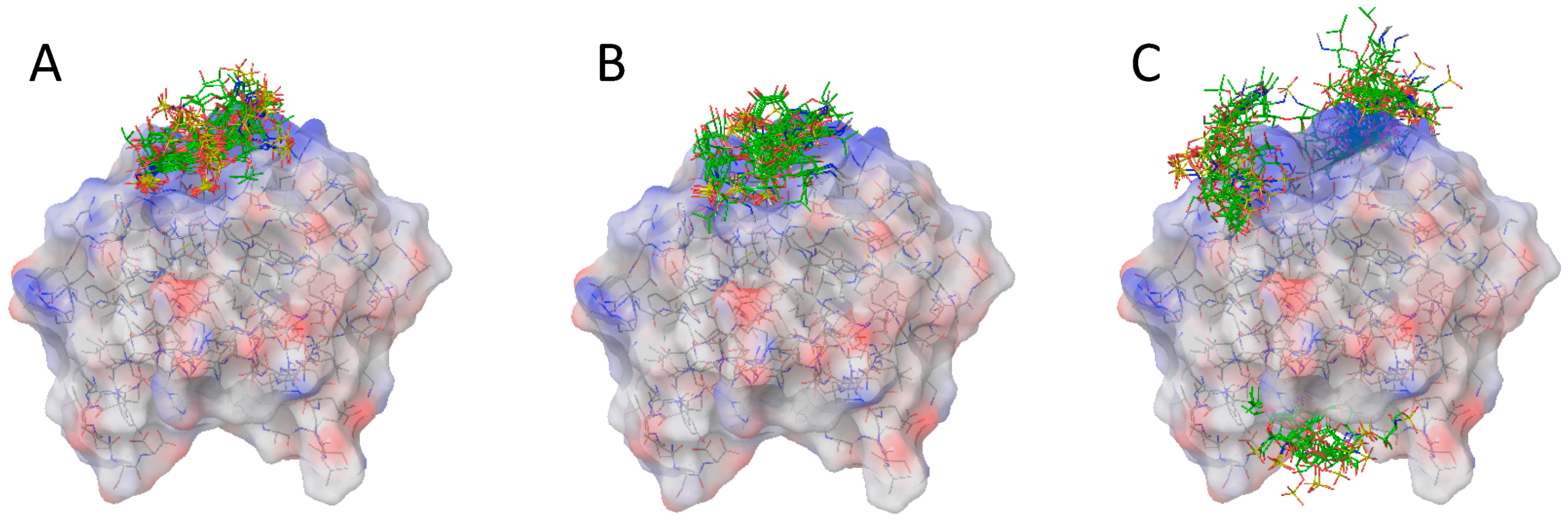
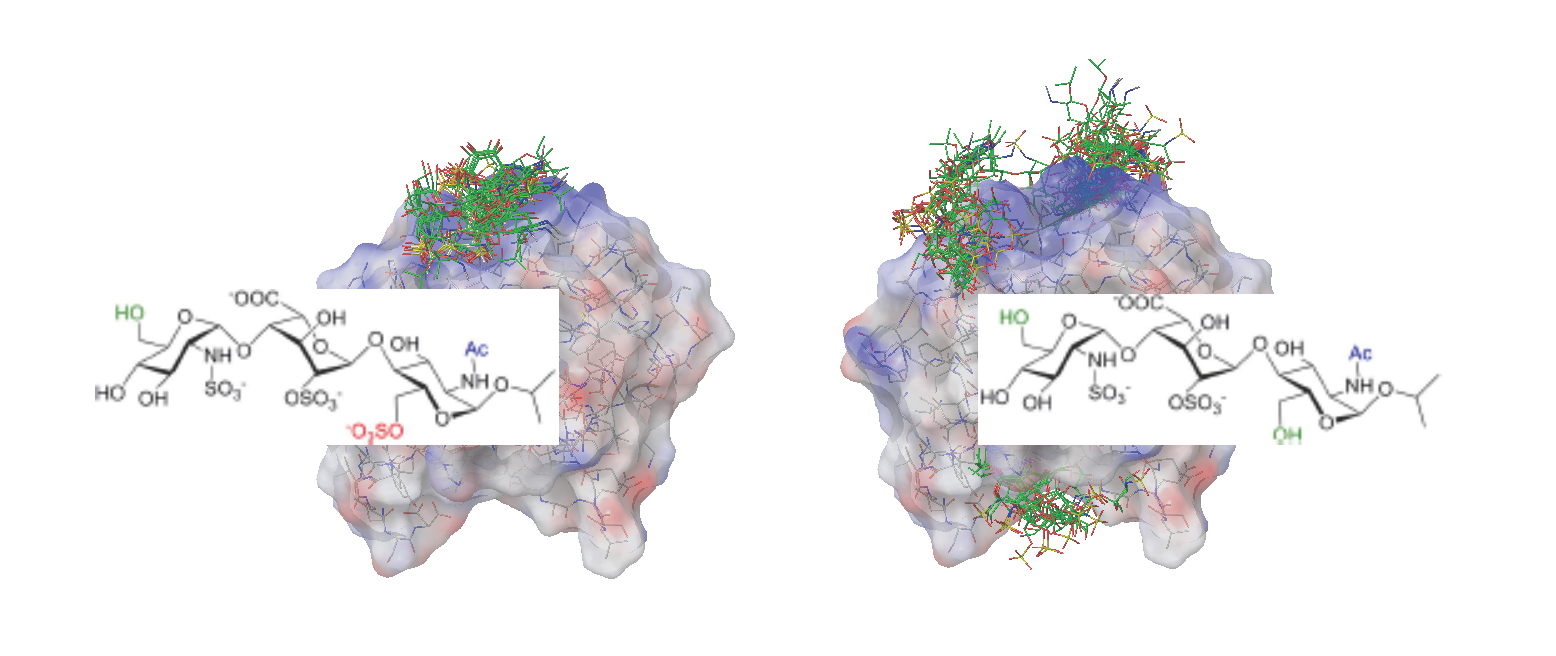
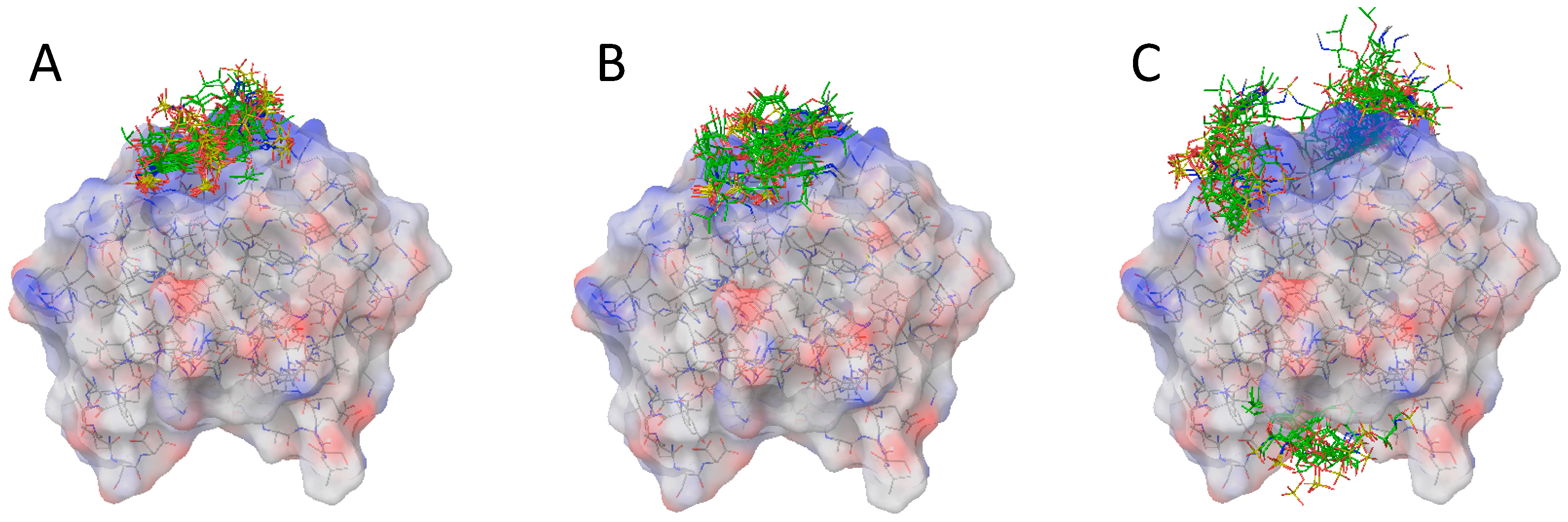
2.2. Docking
2.3. CORCEMA
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yayon, A.; Klagsbrun, M.; Esko, J.D.; Leder, P.; Ornitz, D.M. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell 1991, 64, 841–848. [Google Scholar] [CrossRef]
- Eswarakumar, V.P.; Lax, I.; Schlessinger, J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, J.; Plotnikov, A.N.; Ibrahimi, O.A.; Eliseenkova, A.V.; Yeh, B.K.; Yayon, A.; Linhardt, R.J.; Mohammadi, M. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell 2000, 6, 743–750. [Google Scholar] [CrossRef]
- Plotnikov, A.N.; Hubbard, S.R.; Schlessinger, J.; Mohammadi, M. Crystal structures of two FGF-FGFR complexes reveal the determinants of ligand-receptor specificity. Cell 2000, 101, 413–424. [Google Scholar] [CrossRef]
- Saxena, K.; Schieborr, U.; Anderka, O.; Duchardt-Ferner, E.; Elshorst, B.; Gande, S.L.; Janzon, J.; Kudlinzki, D.; Sreeramulu, S.; Dreyer, M.K.; et al. Influence of heparin mimetics on assembly of the FGF center dot FGFR4 signaling complex. J. Biol. Chem. 2010, 285, 26628–26640. [Google Scholar] [CrossRef] [PubMed]
- DiGabriele, A.D.; Lax, I.; Chen, D.I.; Svahn, C.M.; Jaye, M.; Schlessinger, J.; Hendrickson, W.A. Structure of a heparin-linked biologically active dimer of fibroblast growth factor. Nature 1998, 393, 812–817. [Google Scholar] [PubMed]
- Canales, A.; Lozano, R.; Lopez-Mendez, B.; Angulo, J.; Ojeda, R.; Nieto, P.M.; Martin-Lomas, M.; Gimenez-Gallego, G.; Jimenez-Barbero, J. Solution nmr structure of a human FGF-1 monomer, activated by a hexasaccharide heparin-analogue. FEBS J. 2006, 273, 4716–4727. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-García, J.C.; García-Jiménez, M.J.; Carrero, P.; Canales, Á.; Jiménez-Barbero, J.; Martín-Lomas, M.; Imberty, A.; de Paz, J.L.; Angulo, J.; Lortat-Jacob, H.; et al. Importance of the polarity of the glycosaminoglycan chain on the interaction with FGF-1. Glycobiology 2014, 24, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, J.; Powell, A.; Guimond, S. Heparan sulfate: Decoding a dynamic multifunctional cell regulator. Trends Cell Biol. 2001, 11, 75–82. [Google Scholar] [CrossRef]
- Jastrebova, N.; Vanwildemeersch, M.; Lindahl, U.; Spillmann, D. Heparan sulfate domain organization and sulfation modulate FGF-induced cell signaling. J. Biol. Chem. 2010, 285, 26842–26851. [Google Scholar] [CrossRef] [PubMed]
- Guimond, S.E.; Turnbull, J.E. Fibroblast growth factor receptor signalling is dictated by specific heparan sulphate saccharides. Curr. Biol. 1999, 9, 1343–1346. [Google Scholar] [CrossRef]
- De Paz, J.L.; Angulo, J.; Lassaletta, J.M.; Nieto, P.M.; Redondo-Horcajo, M.; Lozano, R.M.; Gimenez-Gallego, G.; Martin-Lomas, M. The activation of fibroblast growth factors by heparin: Synthesis, structure, and biological activity of heparin-like oligosaccharides. ChemBioChem 2001, 2, 673–685. [Google Scholar] [CrossRef]
- Munoz-Garcia, J.C.; Solera, C.; Carrero, P.; de Paz, J.L.; Angulo, J.; Nieto, P.M. 3D structure of a heparin mimetic analogue of a FGF-1 activator. A nmr and molecular modelling study. Org. Biomol. Chem. 2013, 11, 8269–8275. [Google Scholar] [CrossRef] [PubMed]
- Angulo, J.; Ojeda, R.; de Paz, J.L.; Lucas, R.; Nieto, P.M.; Lozano, R.M.; Redondo-Horcajo, M.; Gimenez-Gallego, G.; Martin-Lomas, M. The activation of fibroblast growth factors (FGFs) by glycosaminoglycans: Influence of the sulfation pattern on the biological activity of FGF-1. ChemBioChem 2004, 5, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; El Hadri, A.; Richard, S.; Denis, F.; Holte, K.; Duffner, J.; Yu, F.; Galcheva-Gargova, Z.; Capila, I.; Schultes, B.; et al. Synthesis and biological evaluation of a unique heparin mimetic hexasaccharide for structure–activity relationship studies. J. Med. Chem. 2014, 57, 4511–4520. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, M.; Agulles, T.; Bisio, A.; Hricovini, M.; Lay, L.; Naggi, A.; Poletti, L.; Sturiale, L.; Torri, G.; Casu, B. Minimal heparin/heparan sulfate sequences for binding to fibroblast growth factor-1. Biochem. Biophys. Res. Commun. 2002, 292, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.-P.; Zhong, Y.-Q.; Chen, Z.-G.; Chen, C.-Y.; Shi, Z.; Zulueta, M.M.L.; Ku, C.-C.; Lee, P.-Y.; Wang, C.-C.; Hung, S.-C. Divergent synthesis of 48 heparan sulfate-based disaccharides and probing the specific sugar–fibroblast growth factor-1 interaction. J. Am. Chem. Soc. 2012, 134, 20722–20727. [Google Scholar] [CrossRef] [PubMed]
- Seeberger, P.H. Automated oligosaccharide synthesis. Chem. Soc. Rev. 2008, 37, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Mikhailov, D.; Linhardt, R.J.; Mayo, K.H. NMR solution conformation of heparin-derived hexasaccharide. Biochem. J. 1997, 328, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-García, J.C.; Corzana, F.; de Paz, J.L.; Angulo, J.; Nieto, P.M. Conformations of the iduronate ring in short heparin fragments described by time averaged distance restrained molecular dynamics. Glycobiology 2013, 23, 1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Garcia, J.C.; Lopez-Prados, J.; Angulo, J.; Diaz-Contreras, I.; Reichardt, N.; de Paz, J.L.; Martin-Lomas, M.; Nieto, P.M. Effect of the substituents of the neighboring ring in the conformational equilibrium of iduronate in heparin-like trisaccharides. Chem. Eur. J. 2012, 18, 16319–16331. [Google Scholar] [CrossRef] [PubMed]
- Mulloy, B.; Forster, M.J.; Jones, C.; Davies, D.B. NMR and molecular-modeling studies of the solution conformation of heparin. Biochem. J. 1993, 293, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Maccarana, M.; Casu, B.; Lindahl, U. Minimal sequence in heparin/heparan sulfate required for binding of basic fibroblast growth factor. J. Biol. Chem. 1993, 268, 23898–23905. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-García, J.C.; Chabrol, E.; Vivès, R.R.; Thomas, A.; de Paz, J.L.; Rojo, J.; Imberty, A.; Fieschi, F.; Nieto, P.M.; Angulo, J. Langerin–heparin interaction: Two binding sites for small and large ligands as revealed by a combination of nmr spectroscopy and cross-linking mapping experiments. J. Am. Chem. Soc. 2015, 137, 4100–4110. [Google Scholar] [CrossRef] [PubMed]
- Ojeda, R.; Angulo, J.; Nieto, P.M.; Martin-Lomas, M. The activation of fibroblast growth factors by heparin: Synthesis and structural study of rationally modified heparin-like oligosaccharides. Can. J. Chem.-Rev. Can. Chim. 2002, 80, 917–936. [Google Scholar] [CrossRef]
- Lucas, R.; Angulo, J.; Nieto, P.M.; Martin-Lomas, M. Synthesis and structural study of two new heparin-like hexasaccharides. Org. Biomol. Chem. 2003, 1, 2253–2266. [Google Scholar] [CrossRef] [PubMed]
- De Paz, J.L.; Martin-Lomas, M. Synthesis and biological evaluation of a heparin-like hexasaccharide with the structural motifs for binding to fgf and fgfr. Eur. J. Org. Chem. 2005, 1849–1858. [Google Scholar] [CrossRef]
- Maza, S.; Kayser, M.M.; Macchione, G.; Lopez-Prados, J.; Angulo, J.; de Paz, J.L.; Nieto, P.M. Synthesis of chondroitin/dermatan sulfate-like oligosaccharides and evaluation of their protein affinity by fluorescence polarization. Org. Biomol. Chem. 2013, 11, 3510–3525. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.; Meyer, B. Characterization of ligand binding by saturation transfer difference nmr spectroscopy. Angew. Chem. Int. Ed. 1999, 38, 1784–1788. [Google Scholar] [CrossRef]
- Angulo, J.; Nieto, P.M. STD-NMR: Application to transient interactions between biomolecules-a quantitative approach. Eur. Biophys. J. 2011, 40, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Ni, F. Recent developments in transferred noe methods. Prog. Nucl. Magn. Reson. Spectrosc. 1994, 26, 517–606. [Google Scholar] [CrossRef]
- Angulo, J.; Enriquez-Navas, P.M.; Nieto, P.M. Ligand-receptor binding affinities from saturation transfer difference (STD) NMR spectroscopy: The binding isotherm of std initial growth rates. Chem. Eur. J. 2010, 16, 7803–7812. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Herr, A.B.; Nilsson, M.; Westman, J.; Svahn, C.M.; Waksman, G. FGF binding and FGF receptor activation by synthetic heparan-derived di- and trisaccharides. Science 1995, 268, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Jemth, P.; Kreuger, J.; Kusche-Gullberg, M.; Sturiale, L.; Gimenez-Gallego, G.; Lindahl, U. Biosynthetic oligosaccharide libraries for identification of protein-binding heparan sulfate motifs—Exploring the structural diversity by screening for fibroblast growth factor (FGF) 1 and FGF2 binding. J. Biol. Chem. 2002, 277, 30567–30573. [Google Scholar] [CrossRef] [PubMed]
- Fielding, L. Nmr methods for the determination of protein-ligand dissociation constants. Prog. Nucl. Magn. Reson. Spectrosc. 2007, 51, 219–242. [Google Scholar] [CrossRef]
- Schrödinger Release 2015–4: Maestro 10.4, G. Macromodel 11.0 Schrödinger, LLC: New York, NY, USA, 2015.
- Jayalakshmi, V.; Krishna, N.R. Complete relaxation and conformational exchange matrix (corcema) analysis of intermolecular saturation transfer effects in reversibly forming ligand-receptor complexes. J. Magn. Reson. 2002, 155, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Enríquez-Navas, P.M.; Guzzi, C.; Muñoz-García, J.C.; Nieto, P.M.; Angulo, J. Structures of glycans bound to receptors from saturation transfer difference (STD) NMR spectroscopy: Quantitative analysis by using CORCEMA-ST. Methods Mol. Biol. 2015, 1273, 475–487. [Google Scholar] [PubMed]
- Rama Krishna, N.; Jayalakshmi, V. Complete relaxation and conformational exchange matrix analysis of std-nmr spectra of ligand-receptor complexes. Prog. Nucl. Magn. Reson. Spectrosc. 2006, 49, 1–25. [Google Scholar] [CrossRef]
- Mayer, M.; James, T.L. NMR-based characterization of phenothiazines as a rna binding scaffold. J. Am. Chem. Soc. 2004, 126, 4453–4460. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.W.; Moore, J.; Abdul-Manan, N. NMR experiments for lead generation in drug discovery. Prog. Nucl. Magn. Reson. Spectrosc. 2004, 44, 225–256. [Google Scholar] [CrossRef]
- Yuan, Y.; Bleile, D.W.; Wen, X.; Sanders, D.A.R.; Itoh, K.; Liu, H.W.; Pinto, B.M. Investigation of binding of UDP-Galf and UDP-[3-F]Galf to udp-galactopyranose mutase by STD-NMR spectroscopy, molecular dynamics, and CORCEMA-ST calculations. J. Am. Chem. Soc. 2008, 130, 3157–3168. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | STD0 | Relative STD0 | ||||
|---|---|---|---|---|---|---|
| H1-A | H1-B | H1-C | H1-A | H1-B | H1-C | |
| 1 | 0.28 | 0.34 | 0.42 | 66 | 80 | 100 |
| 2 | 0.11 | 0.36 | 0.24 | 31 | 100 | 68 |
| 3 | 0.17 | 0.23 | 0.20 | 72 | 100 | 88 |
| 4 | 0.17 | 0.25 | 0.24 | 67 | 100 | 94 |
| 5 | 0.07 | 0.06 | 0.06 | 100 | 82 | 87 |
| 6 | 0.08 | 0.12 | 0.12 | 62 | 100 | 94 |
| 7 | 0.18 | 0.08 | 0.11 | 100 | 48 | 64 |
| 8 | - | 0.18 | 0.19 | - | 99 | 100 |
| Compound | IC50 (μM) |
|---|---|
| 1 | 49.2 ± 0.3 |
| 2 | 26.1 ± 0.7 |
| 3 | 15.4 ± 0.4 |
| 4 | 84.7 ± 0.4 |
| 5 | 50.6 ± 0.1 |
| 6 | 180.9 ± 5.5 |
| 7 | 46.7 ± 0.9 |
| 8 | 132.4 ± 1.8 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Jiménez, M.J.; Gil-Caballero, S.; Canales, Á.; Jiménez-Barbero, J.; De Paz, J.L.; Nieto, P.M. Interactions between a Heparin Trisaccharide Library and FGF-1 Analyzed by NMR Methods. Int. J. Mol. Sci. 2017, 18, 1293. https://doi.org/10.3390/ijms18061293
García-Jiménez MJ, Gil-Caballero S, Canales Á, Jiménez-Barbero J, De Paz JL, Nieto PM. Interactions between a Heparin Trisaccharide Library and FGF-1 Analyzed by NMR Methods. International Journal of Molecular Sciences. 2017; 18(6):1293. https://doi.org/10.3390/ijms18061293
Chicago/Turabian StyleGarcía-Jiménez, María José, Sergio Gil-Caballero, Ángeles Canales, Jesús Jiménez-Barbero, José L. De Paz, and Pedro M. Nieto. 2017. "Interactions between a Heparin Trisaccharide Library and FGF-1 Analyzed by NMR Methods" International Journal of Molecular Sciences 18, no. 6: 1293. https://doi.org/10.3390/ijms18061293
APA StyleGarcía-Jiménez, M. J., Gil-Caballero, S., Canales, Á., Jiménez-Barbero, J., De Paz, J. L., & Nieto, P. M. (2017). Interactions between a Heparin Trisaccharide Library and FGF-1 Analyzed by NMR Methods. International Journal of Molecular Sciences, 18(6), 1293. https://doi.org/10.3390/ijms18061293