GPE Promotes the Proliferation and Migration of Mouse Embryonic Neural Stem Cells and Their Progeny In Vitro



Abstract
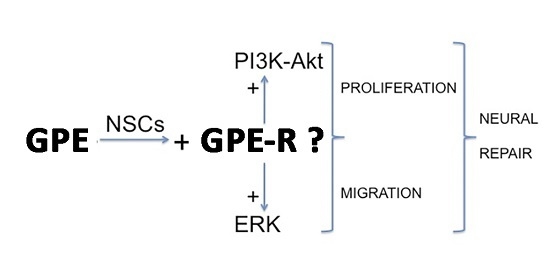
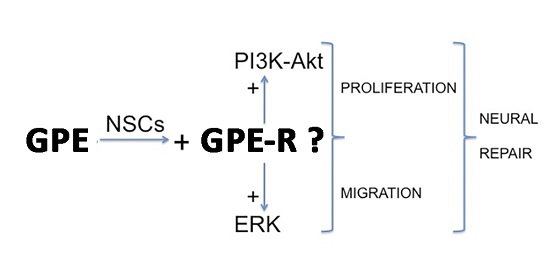
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
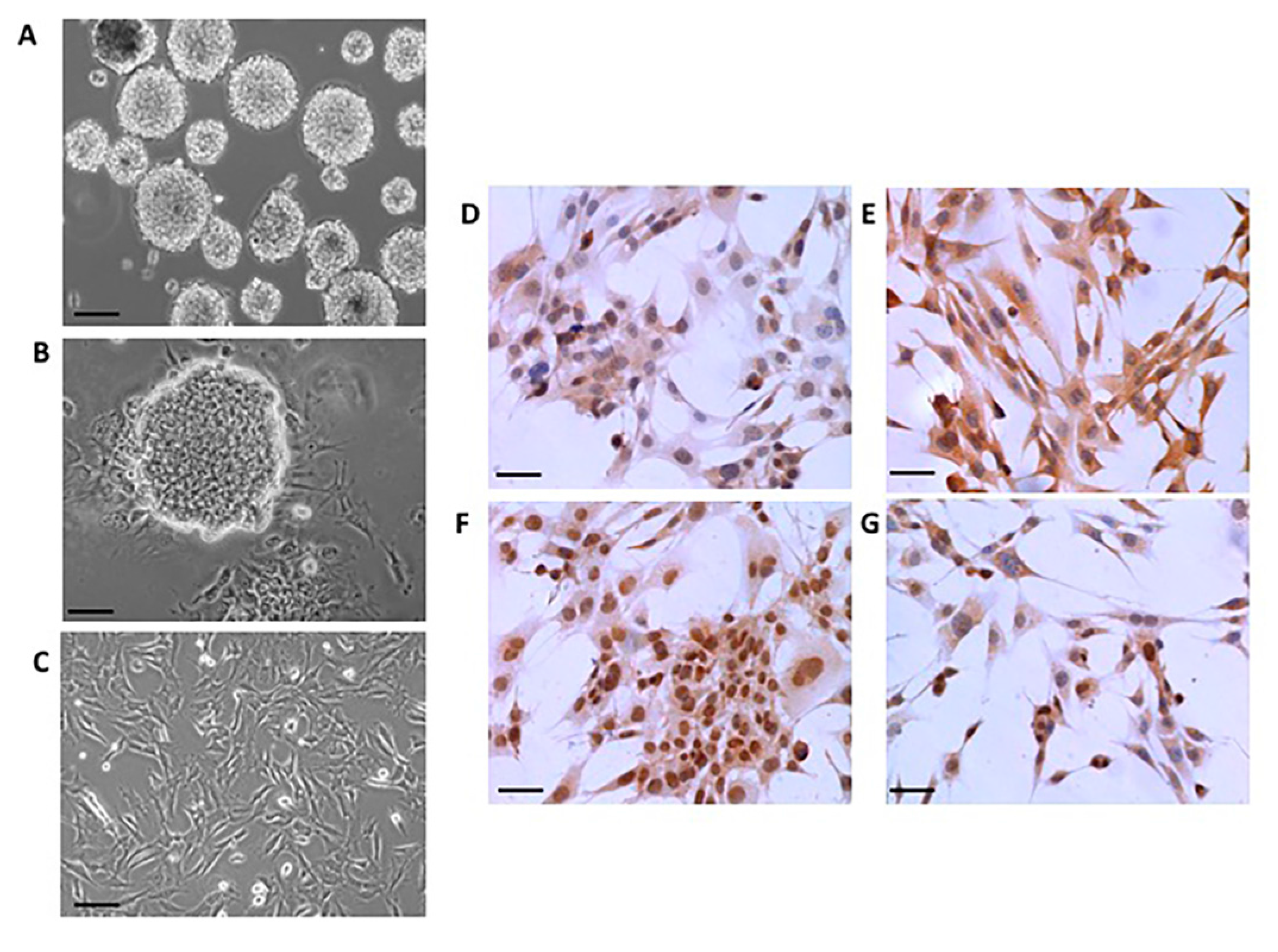
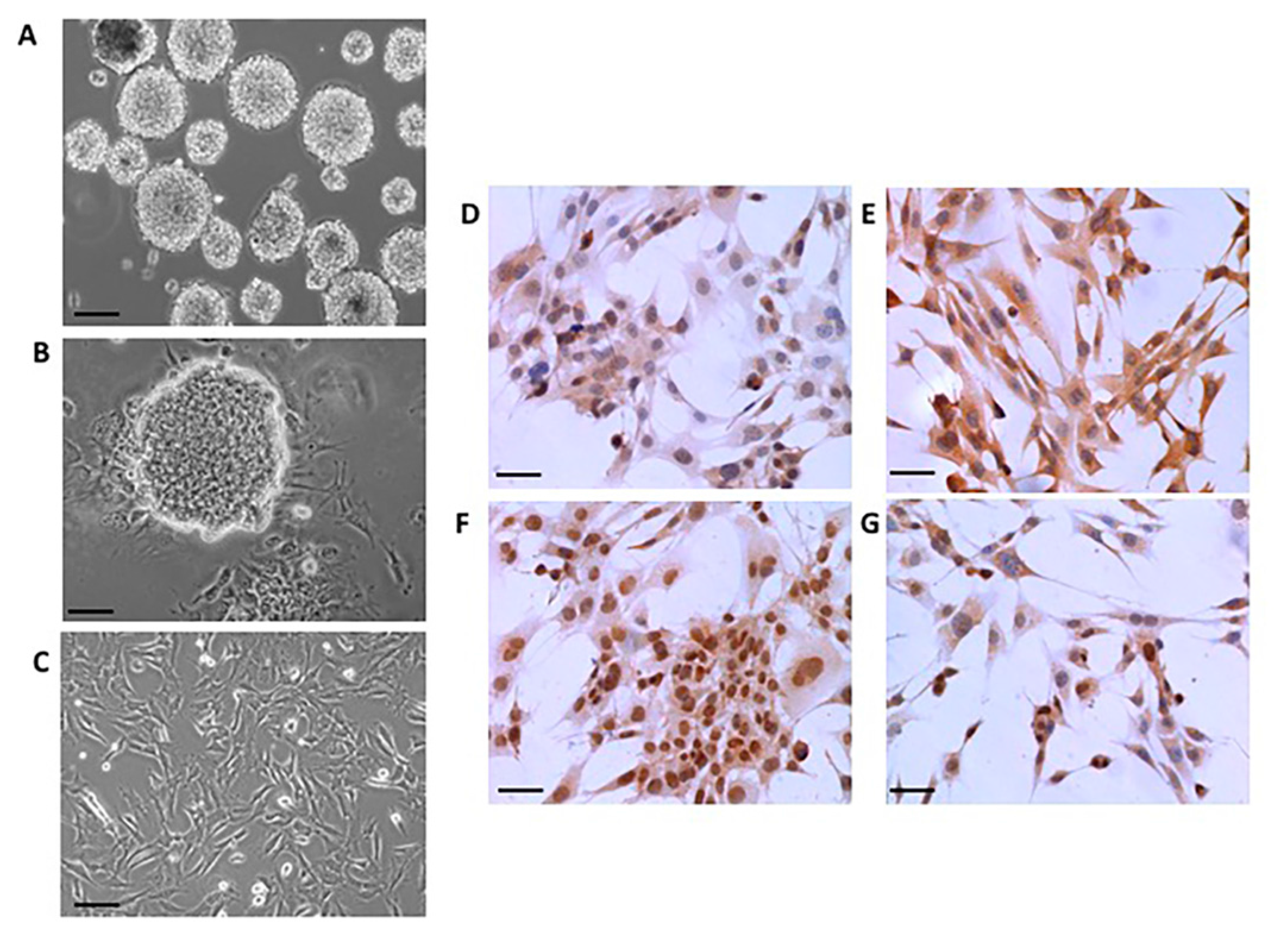
2.1. Neural Differentiation In Vitro
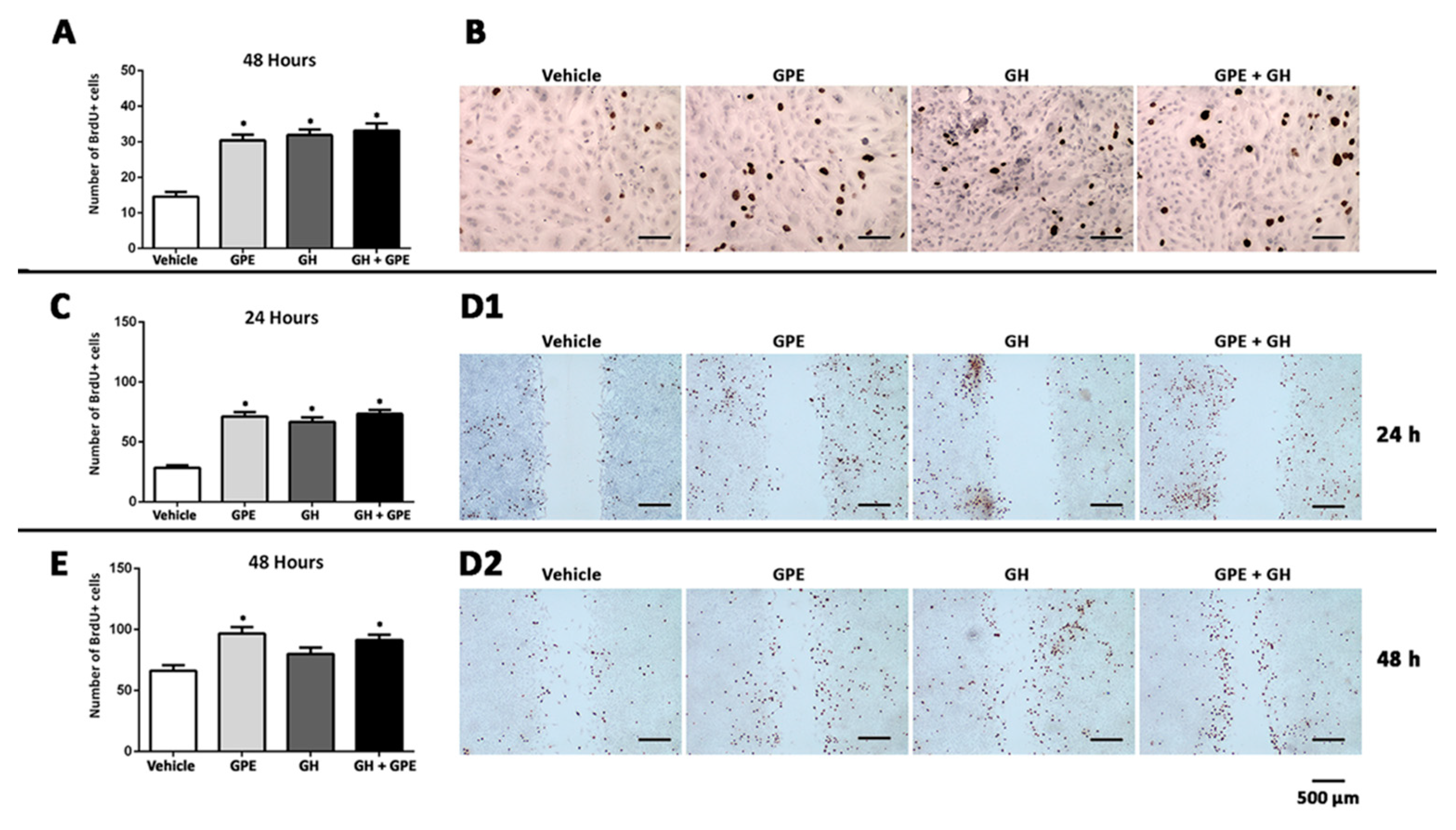
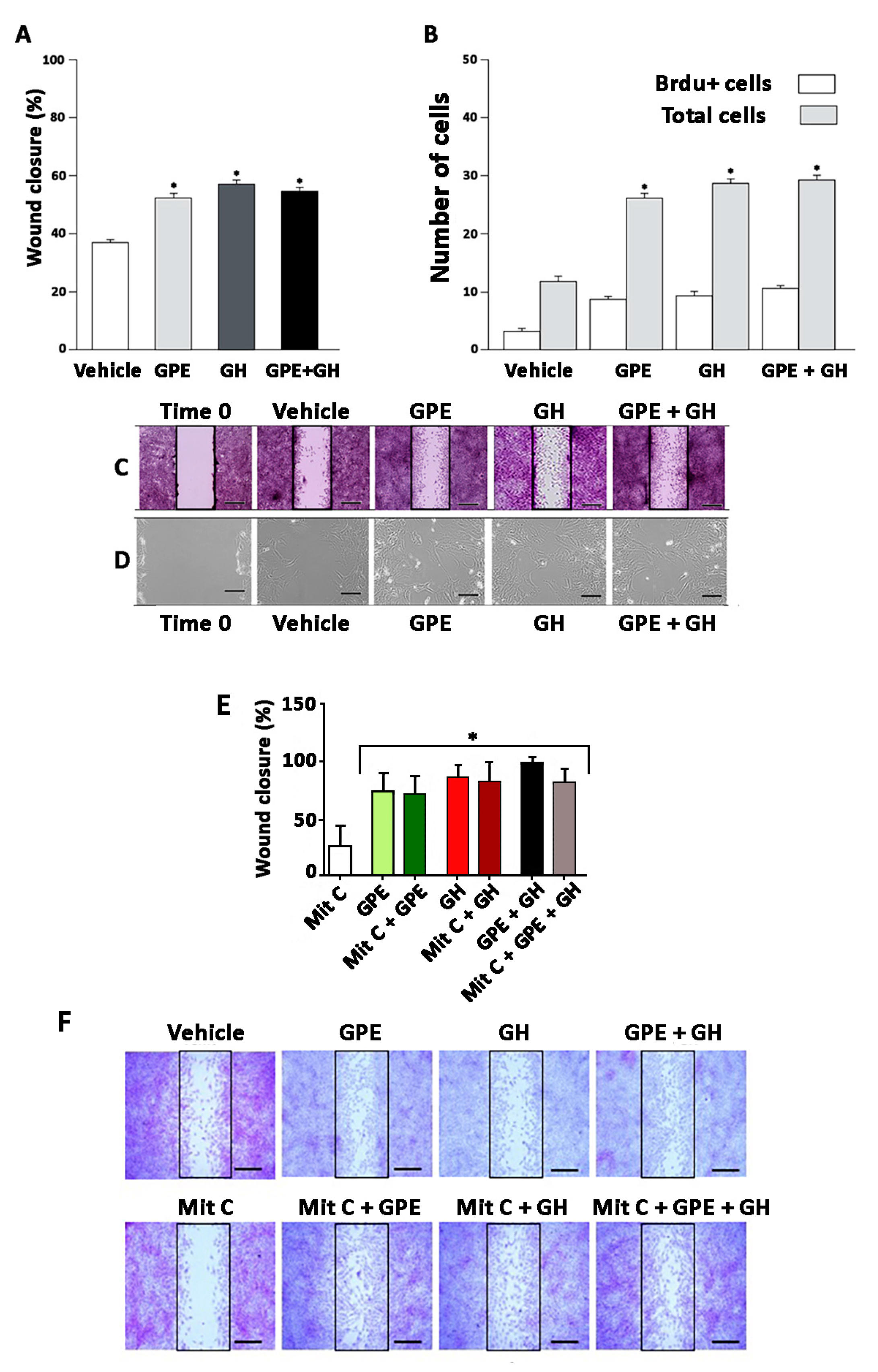
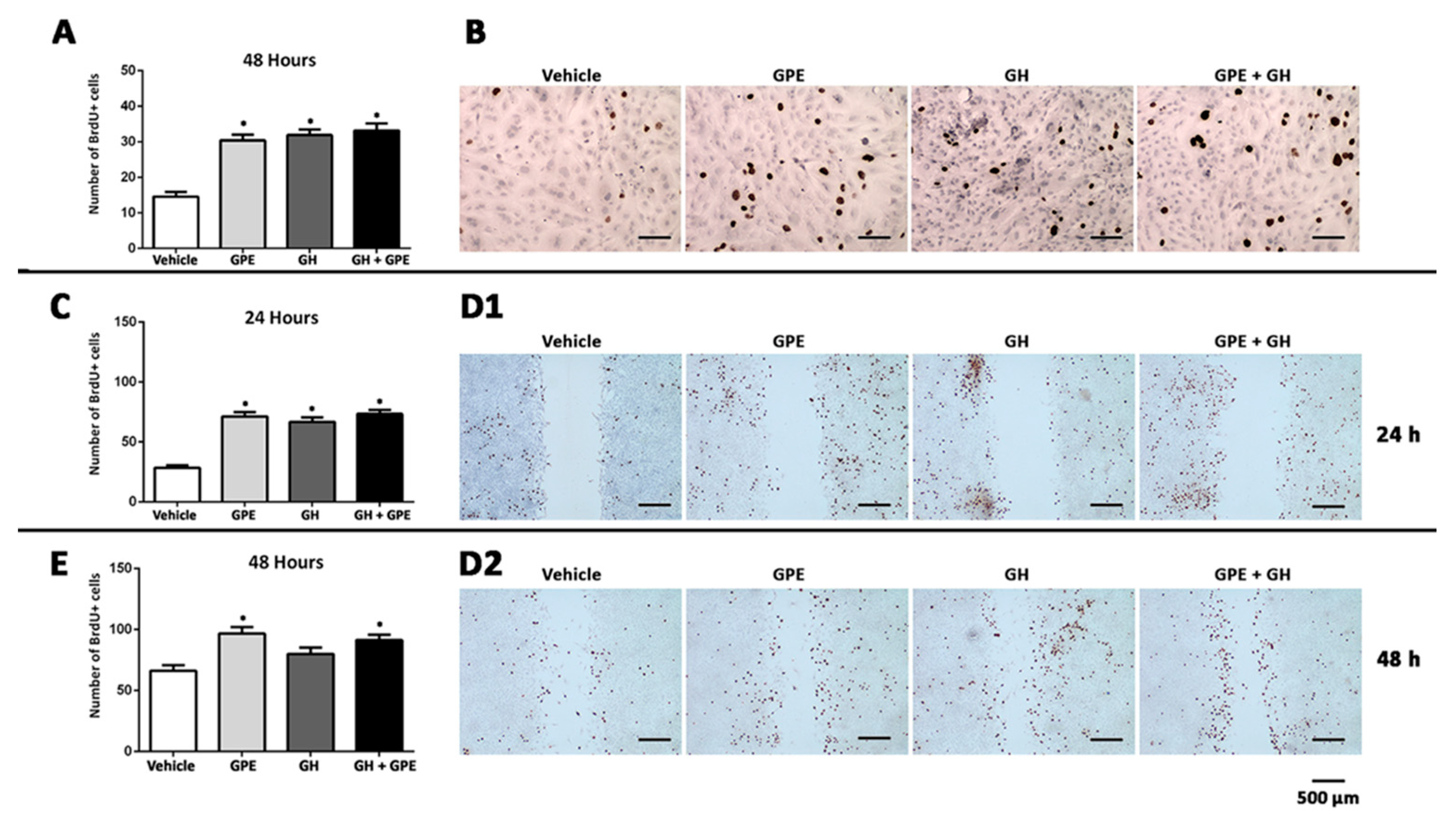
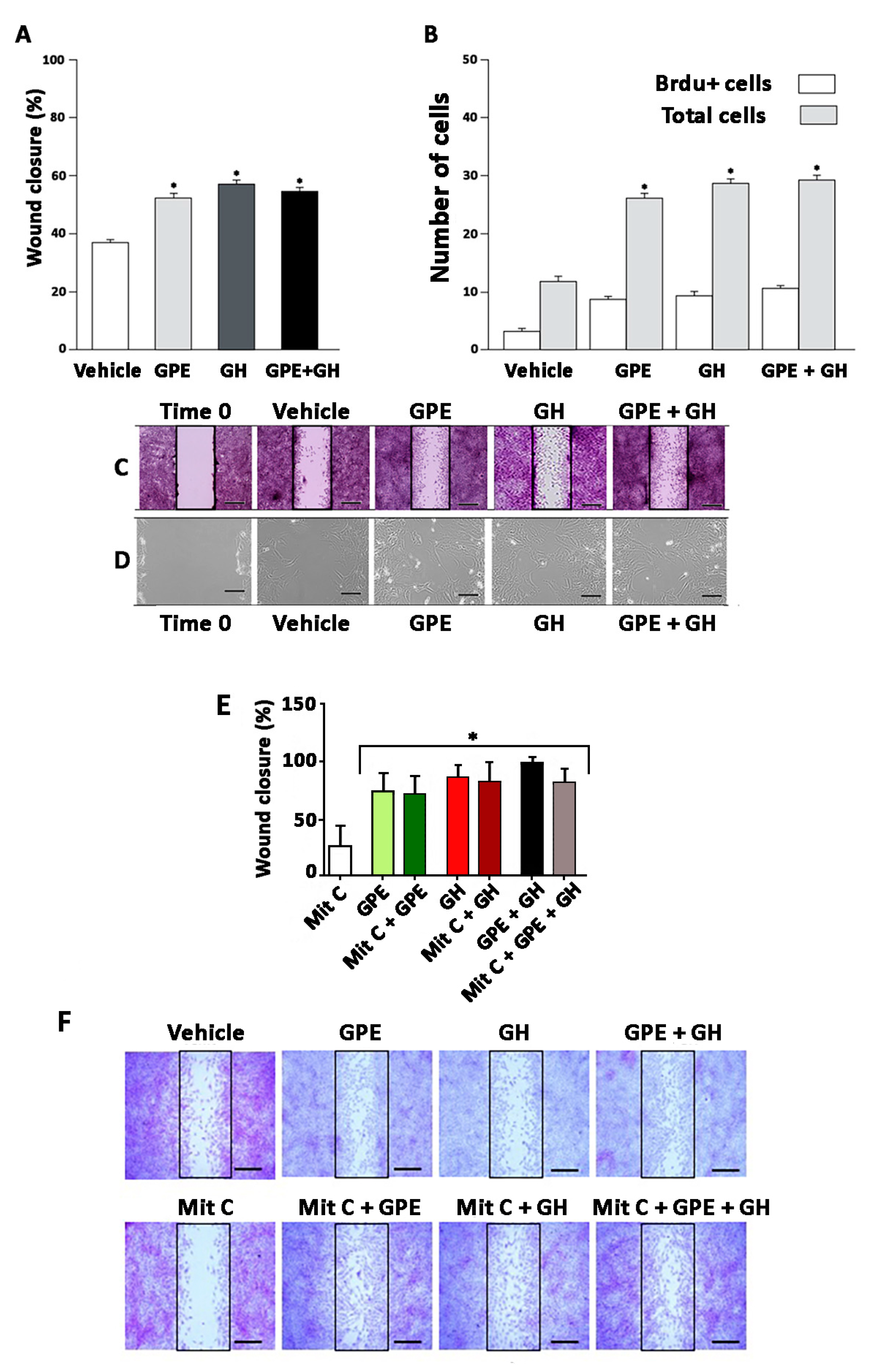
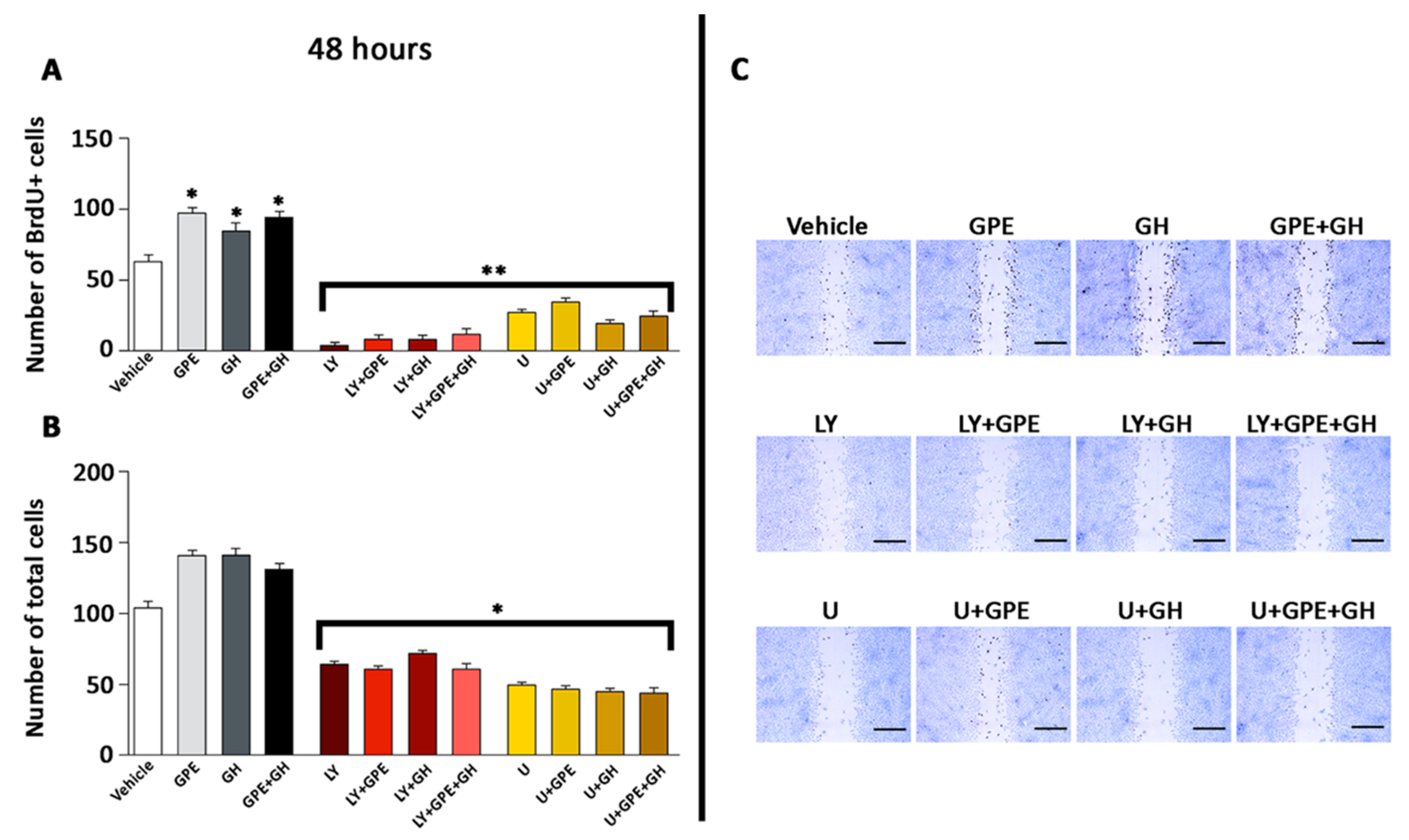
2.2. Cell Proliferation Induced by Gly-Pro-Glu (GPE) or Growth Hormone (GH)
2.3. Cell Migration Induced by GPE or GH
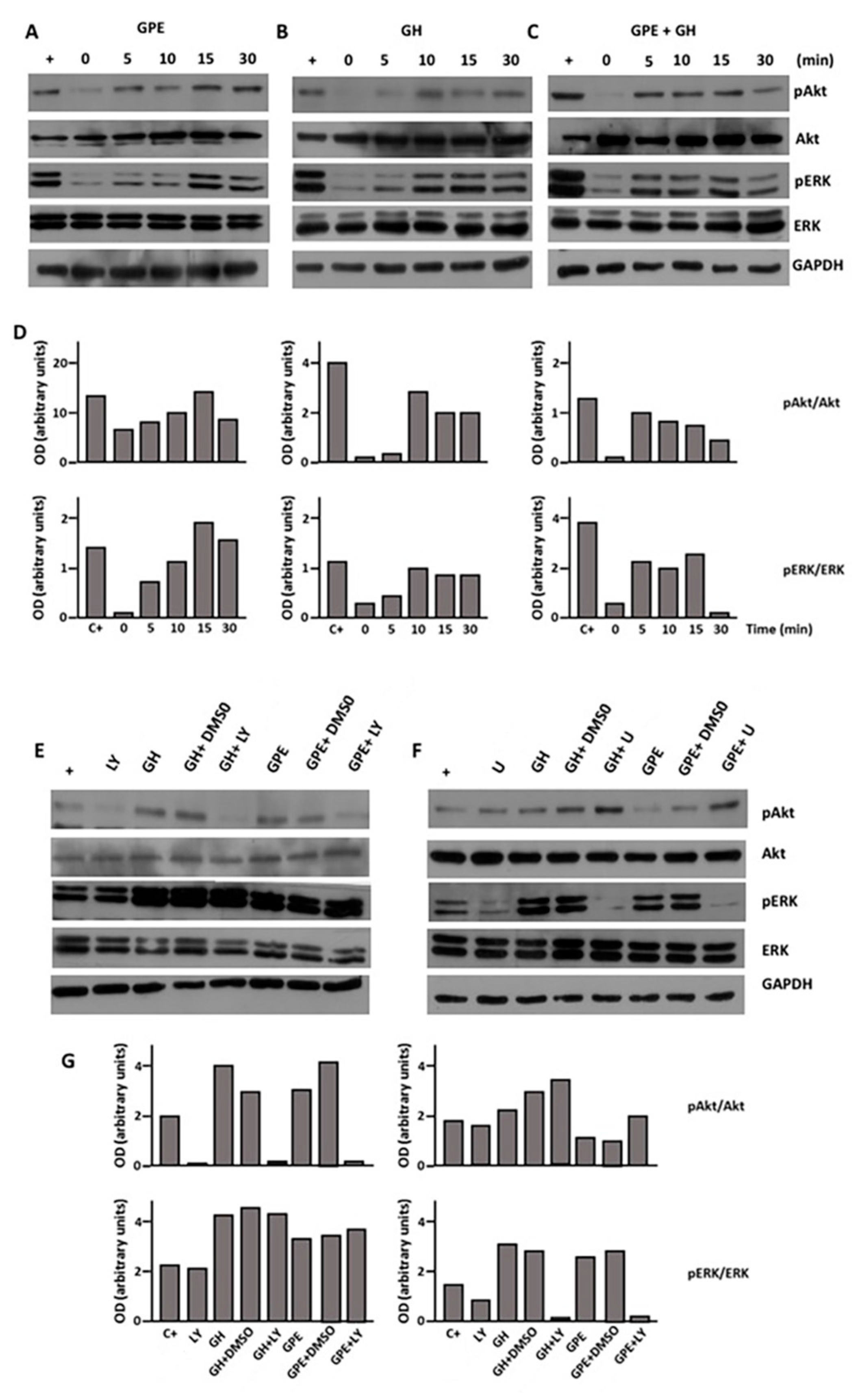
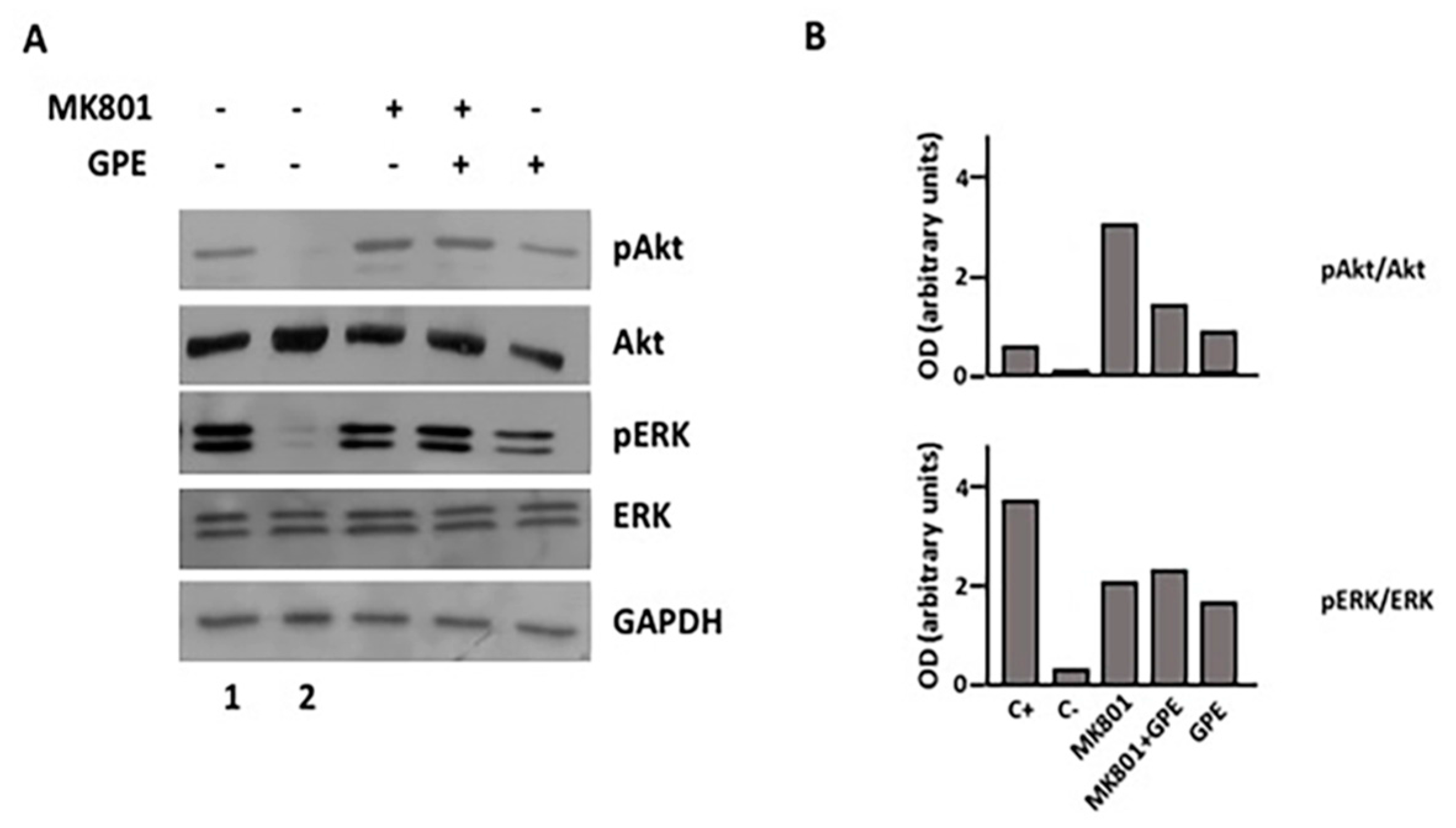
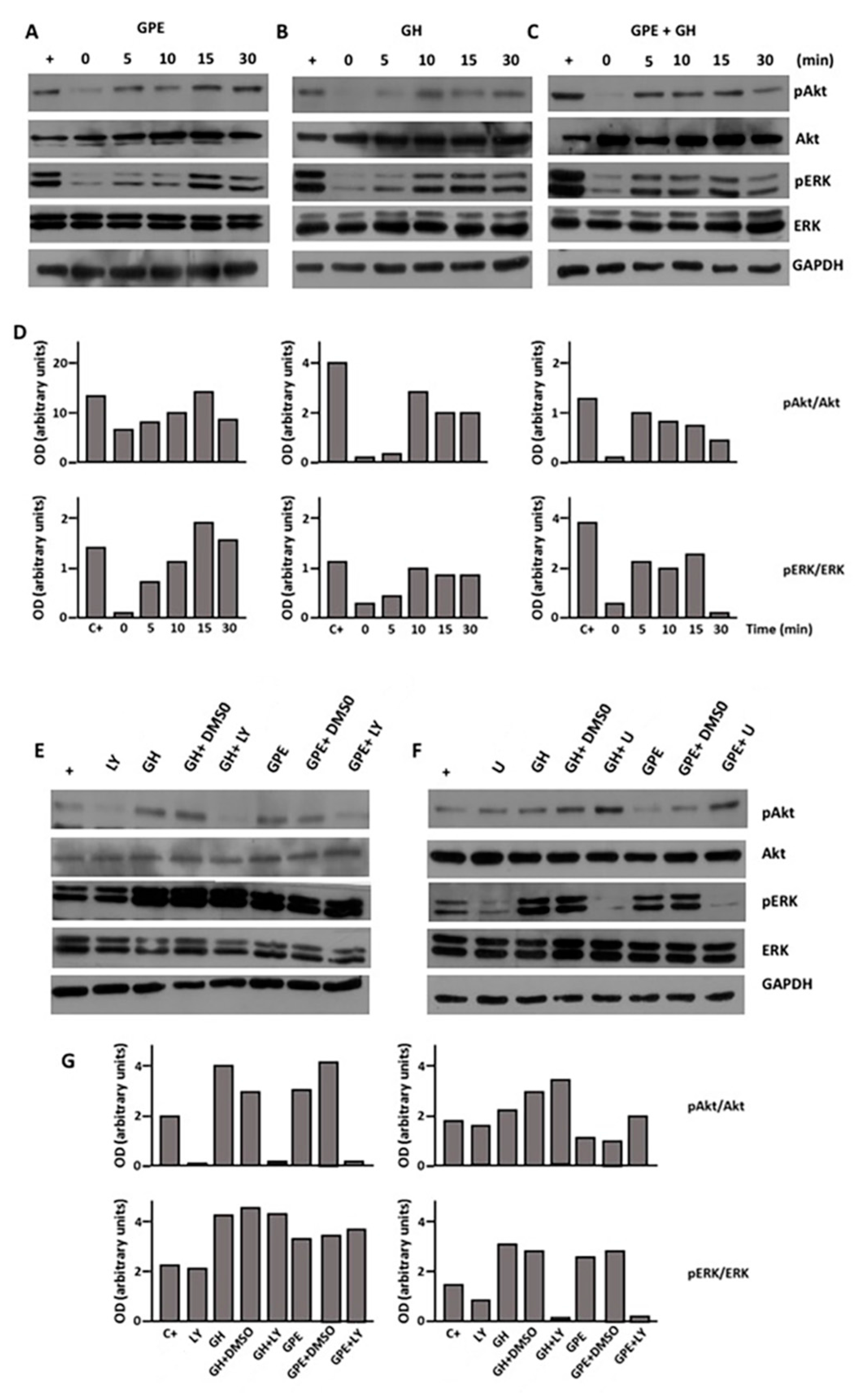
2.4. GPE Signaling Pathways
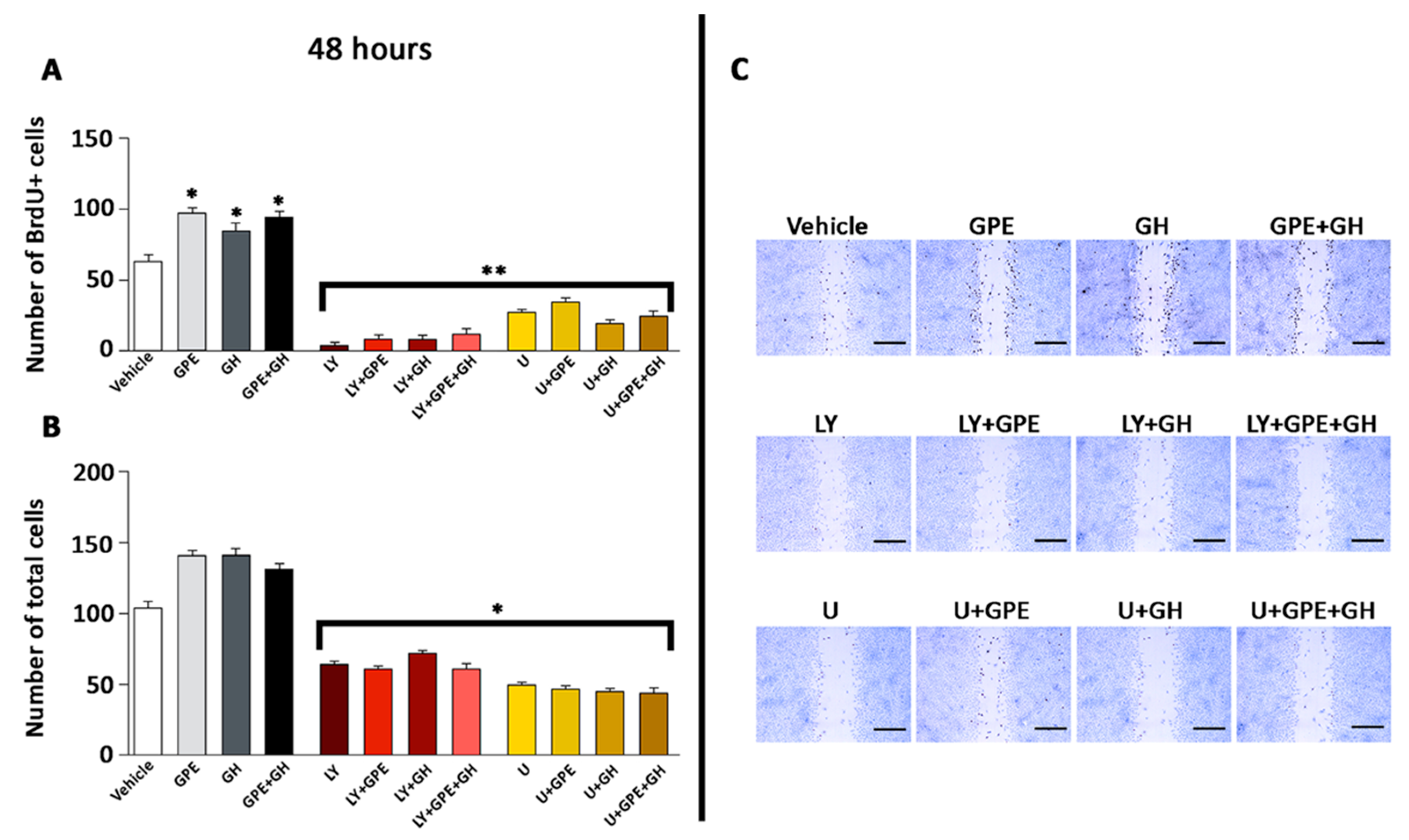
2.5. The Role of ERK and Akt on Cell Proliferation and Migration
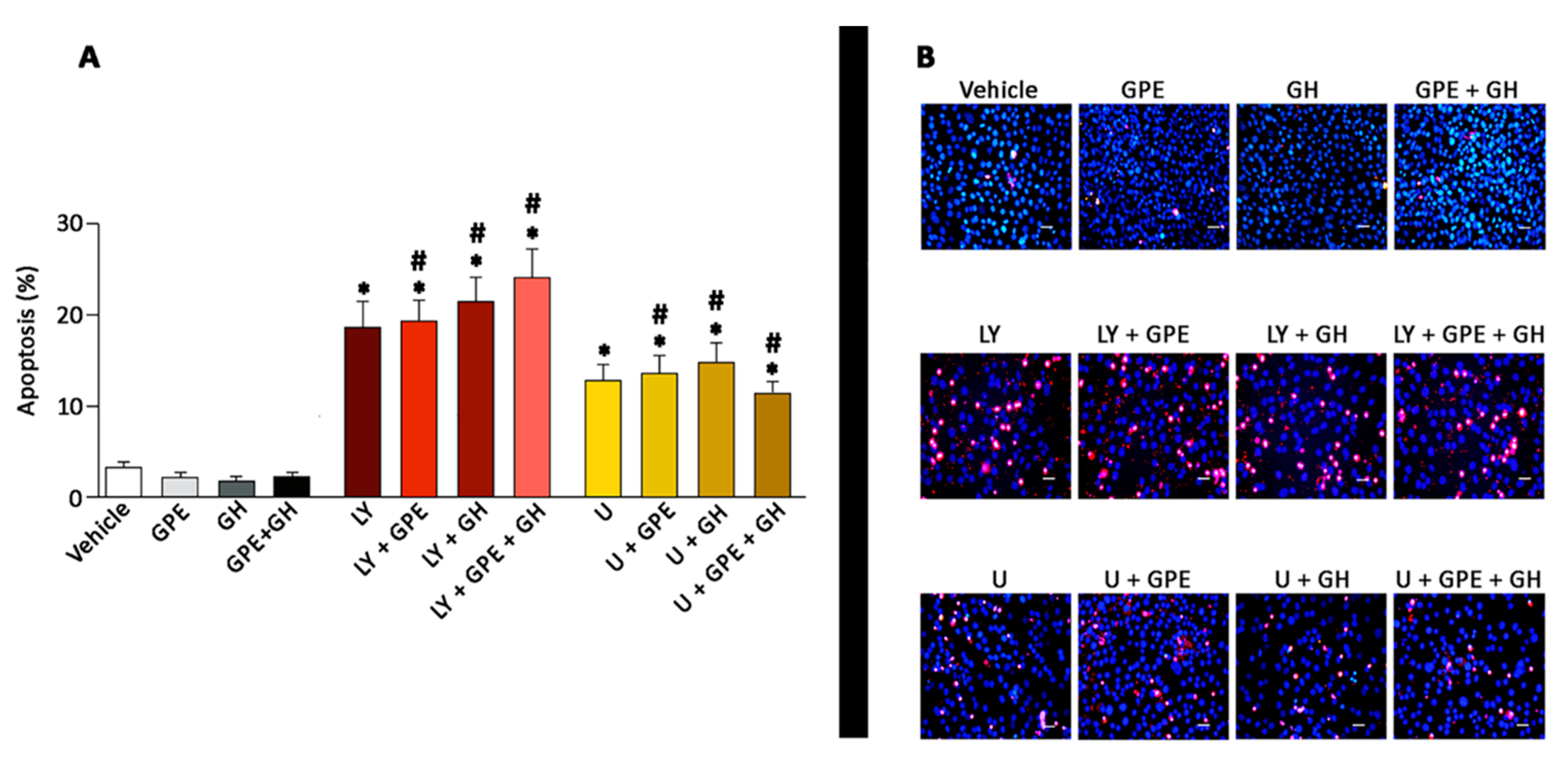
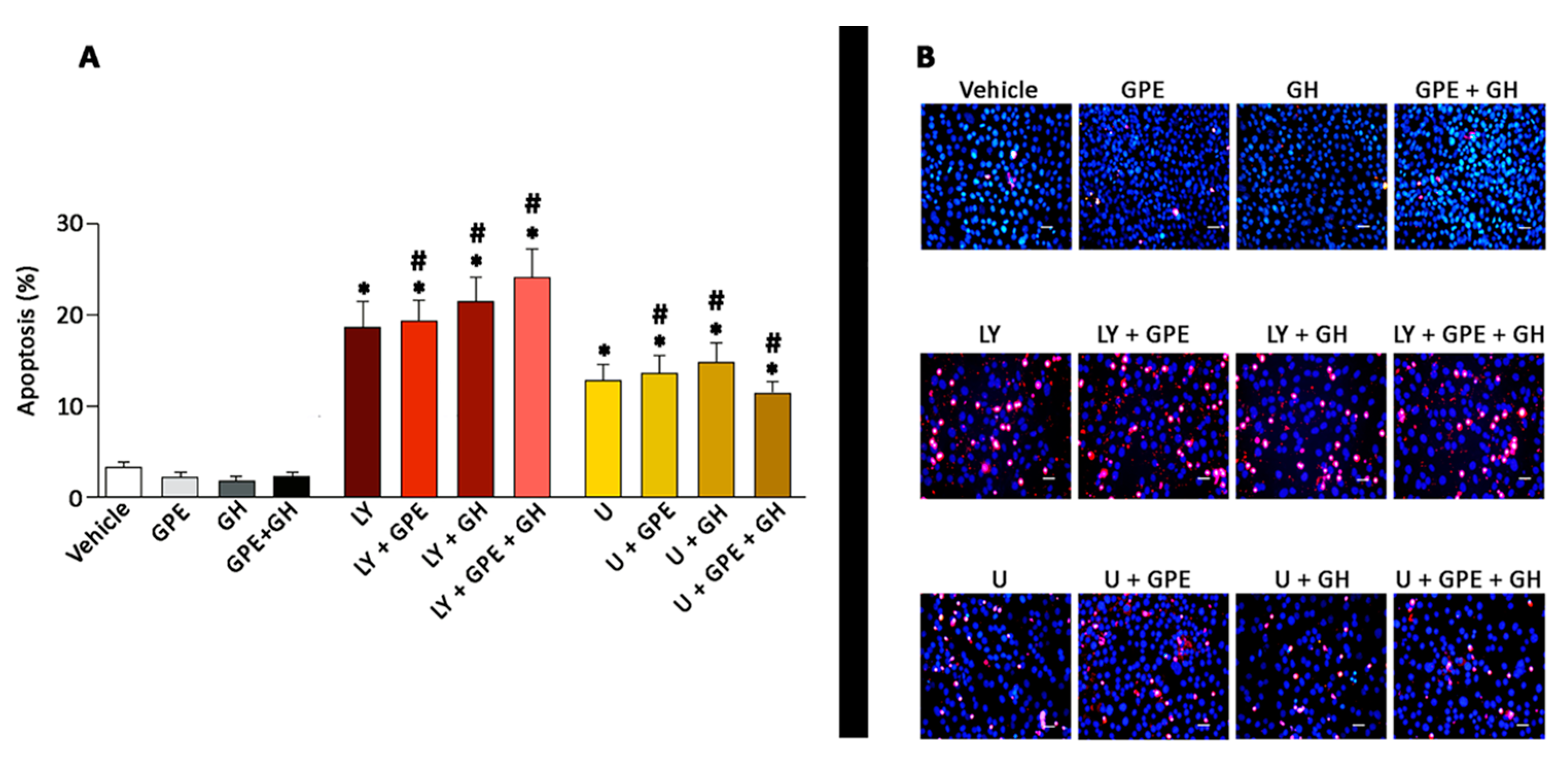
2.6. The Role of ERK and Akt on Cell Survival
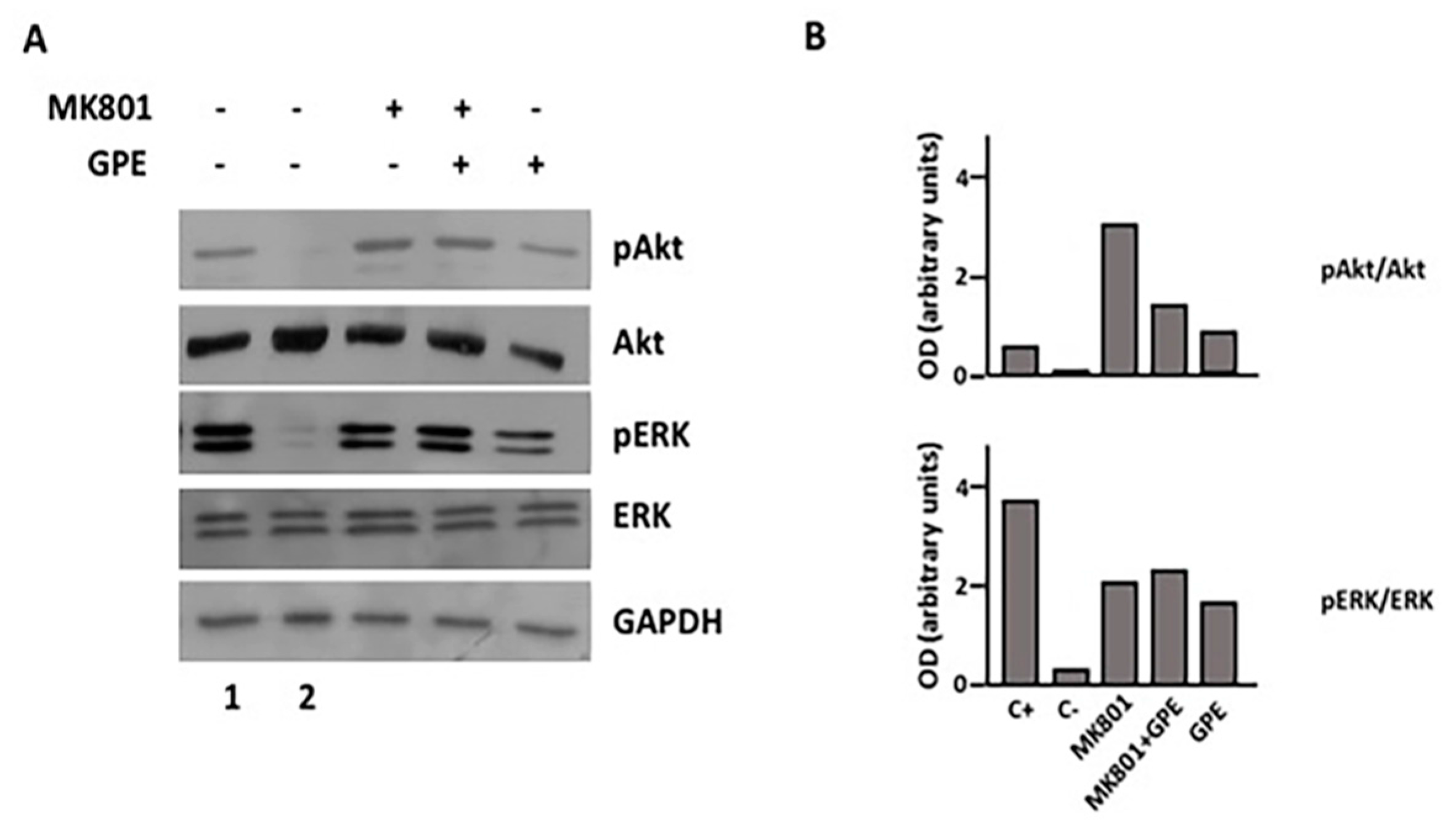
2.7. Blockade of the NMDA Receptor and GPE Signaling Pathways
3. Discussion
4. Materials and Methods
4.1. Cell Cultures and Treatments
4.2. Immunocytochemistry
4.3. Cell Proliferation Assays
4.4. Wound-Healing Assays
4.5. Western Blot
4.6. Determination of Cell Apoptosis
4.7. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Butler, A.A.; Roith, D.L. Control of growth by the somatropic axis: Growth hormone and the insulin-like growth factors have related and independent roles. Annu. Rev. Physiol. 2001, 63, 141–164. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.; Weinstein, D.; Bentov, I. Similarities and differences between insulin and IGF-I: Structures, receptors, and signalling pathways. Arch. Physiol. Biochem. 2008, 114, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Beck, K.D.; Powell-Braxton, L.; Widmer, H.R.; Valverde, J.; Hefti, F. Igf1 gene disruption results in reduced brain size, CNS hypomyelination, and loss of hippocampal granule and striatal parvalbumin-containing neurons. Neuron 1995, 14, 717–730. [Google Scholar] [CrossRef]
- Torres-Aleman, I. Insulin-like growth factors as mediators of functional plasticity in the adult brain. Horm. Metab. Res. 1999, 31, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Torres-Aleman, I. Role of insulin-like growth factors in neuronal plasticity and neuroprotection. Adv. Exp. Med. Biol. 2005, 567, 243–258. [Google Scholar] [PubMed]
- Llorens-Martín, M.; Torres-Alemán, I.; Trejo, J.L. Mechanisms mediating brain plasticity: IGF1 and adult hippocampal neurogenesis. Neuroscientist 2009, 15, 134–148. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Estévez, V.; Defterali, Ç.; Vicario-Abejón, C. IGF-I: A key growth factor that regulates neurogenesis and synaptogenesis from embryonic to adult stages of the brain. Front. Neurosci. 2016, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Murphy, L.J. Enzymatic conversion of IGF-I to des (1–3) IGF-I in rat serum and tissues: A further potential site of growth hormone regulation of IGF-I action. J. Endocrinol. 1995, 146, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Murphy, L.J. N-terminal truncated insulin-like growth factor-I in human urine. J. Clin. Endocrinol. Metab. 1995, 80, 1179–1183. [Google Scholar] [PubMed]
- Yamamoto, H.; Maake, C.; Murphy, L.J. Enhanced proteolytic activity directed against the N-terminal of IGF-I in diabetic rats. J. Endocrinol. 1999, 162, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Sara, V.R.; Carlsson-Skwirut, C.; Bergman, T.; Jörnvall, H.; Roberts, P.J.; Crawford, M.; Håkansson, L.N.; Civalero, I.; Nordberg, A. Identification of Gly-Pro-Glu (GPE), the aminoterminal tripeptide of insulin-like growth factor 1 which is truncated in brain, as a novel neuroactive peptide. Biochem. Biophys. Res. Commun. 1989, 165, 766–771. [Google Scholar] [CrossRef]
- Nilsson-Hakansson, L.; Civalero, I.; Zhang, X.; Carlsson-Skwirut, C.; Sara, V.R.; Nordberg, A. Effects of IGF-1, truncated IGF-1 and the tripeptide Gly-Pro-Glu on acetylcholine release from parietal cortex of rat brain. Neuroreport 1993, 4, 1111–1114. [Google Scholar] [PubMed]
- Guan, J.; Gluckman, P.D. IGF-1 derived small neuropeptides and analogues: A novel strategy for the development of pharmaceuticals for neurological conditions. Br. J. Pharmacol. 2009, 157, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Guan, J. Insulin-like growth factor-1 (IGF-1) derived neuropeptides, a novel strategy for the development of pharmaceuticals for managing ischemic brain injury. CNS Neurosci. Ther. 2011, 17, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Saura, J.; Curatolo, L.; Williams, C.E.; Gatti, S.; Benatti, L.; Peeters, C.; Guan, J.; Dragunow, M.; Post, C.; Faull, R.L.; et al. Neuroprotective effects of Gly-Pro-Glu, the N-terminal tripeptide of IGF-1, in the hippocampus in vitro. Neuroreport 1999, 10, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Waldvogel, H.J.; Faull, R.L.; Gluckman, P.D.; Williams, C.E. The effects of the N-terminal tripeptide of insulin-like growth factor-1, glycine-proline-glutamate in different regions following hypoxic-ischemic brain injury in adult rats. Neuroscience 1999, 89, 649–659. [Google Scholar] [CrossRef]
- Sizonenko, S.V.; Sirimanne, E.S.; Williams, C.E.; Gluckman, P.D. Neuroprotective effects of the N-terminal tripeptide of IGF-1, glycine-proline-glutamate, in the immature rat brain after hypoxic-ischemic injury. Brain Res. 2001, 922, 42–50. [Google Scholar] [CrossRef]
- Guan, J.; Thomas, G.B.; Lin, H.; Mathai, S.; Bachelor, D.C.; George, S.; Gluckman, P.D. Neuroprotective effects of the N-terminal tripeptide of insulin-like growth factor-1, glycine-proline-glutamate (GPE) following intravenous infusion in hypoxic-ischemic adult rats. Neuropharmacology 2004, 47, 892–903. [Google Scholar] [CrossRef] [PubMed]
- Alexi, T.; Hughes, P.E.; van Roon-Mom, W.M.; Faull, R.L.; Williams, C.E.; Clark, R.G.; Gluckman, P.D. The IGF-I amino-terminal tripeptide glycine-proline-glutamate (GPE) is neuroprotective to striatum in the quinolinic acid lesion animal model of Huntington’s disease. Exp. Neurol. 1999, 159, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Krishnamurthi, R.; Waldvogel, H.J.; Faull, R.L.; Clark, R.; Gluckman, P. N-terminal tripeptide of IGF-1 (GPE) prevents the loss of TH positive neurons after 6-OHDA induced nigral lesion in rats. Brain Res. 2000, 859, 286–292. [Google Scholar] [CrossRef]
- Aguado-Llera, D.; Martín-Martínez, M.; García-López, M.T.; Arilla-Ferreiro, E.; Barrios, V. Gly-Pro-Glu protects β-amyloid-induced somatostatin depletion in the rat cortex. Neuroreport 2004, 15, 1979–1982. [Google Scholar] [CrossRef] [PubMed]
- Park, S.E.; Lawson, M.; Dantzer, R.; Kelley, K.W.; McCusker, R.H. Insulin-like growth factor-I peptides act centrally to decrease depression-like behavior of mice treated intraperitoneally with lipopolysaccharide. J. Neuroinflamm. 2011, 8, 179. [Google Scholar] [CrossRef] [PubMed]
- Devesa, P.; Reimunde, P.; Gallego, R.; Devesa, J.; Arce, V.M. Growth hormone (GH) treatment may cooperate with locally-produced GH in increasing the proliferative response of hippocampal progenitors to kainite-induced injury. Brain Inj. 2011, 25, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Devesa, P.; Agasse, F.; Xapelli, S.; Almengló, C.; Devesa, J.; Malva, J.O.; Arce, V.M. Growth hormone pathways signaling for cell proliferation and survival in hippocampal neural precursors from postnatal mice. BMC Neurosci. 2014, 15, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Devesa, J.; Almengló, C.; Devesa, P. Multiple effects of growth hormone in the body: Is it really the hormone for growth? Clin. Med. Insights Endocrinol. Diabetes 2016, 9, 47–71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cui, W. Sox2, a key factor in the regulation of pluripotency and neural differentiation. World J. Stem. Cells 2014, 6, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Prot. 2007, 2, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Varoglu, M.; Sherman, D.H. Molecular characterization and analysis of the biosynthetic gene cluster for the antitumor antibiotic mitomycin C from Streptomyces lavendulae NRRL 2564. Chem. Biol. 1999, 6, 251–263. [Google Scholar] [CrossRef]
- Shapira, S.; Mathai, S.; Zhang, R.; Guan, J. Delayed peripheral administration of the N-terminal tripeptide of IGF-1 (GPE) reduces brain damage following microsphere induced embolic damage in young adult and aged rats. Neurosci. Lett. 2009, 454, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Arce, V.M.; Devesa, P.; Devesa, J. Role of growth hormone (GH) in the treatment on neural diseases: From neuroprotection to neural repair. Neurosci. Res. 2013, 76, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Ramos, J.W. The regulation of extracellular signal-regulated kinase (ERK) in mammalian cells. Int. J. Biochem. Cell Biol. 2008, 40, 2707–2719. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J. RAS and downstream RAF-MEK and PI3K-AKT signaling in neuronal development, function and dysfunction. Biol. Chem. 2016, 397, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhou, K.; Fu, Z.; Yu, D.; Huang, H.; Zang, X.; Mo, X. Brain development and Akt signaling: The crossroads of signaling pathway and neurodevelopmental diseases. J. Mol. Neurosci. 2017, 61, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Oosterholt, S.P.; Horrigan, J.; Jones, N.; Glass, L.; Pasqua, O.D. Population pharmacokinetics of NNZ-2566 in healthy subjects. Eur. J. Pharm. Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Harris, P.; Brimble, M.; Lei, Y.; Lu, J.; Yang, Y.; Gunn, A.J. The role for IGF-1-derived small neuropeptides as a therapeutic target for neurological disorders. Expert Opin. Ther. Targets 2015, 19, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Waldbillig, R.J.; Puro, D.G. Truncation of IGF-I yields two mitogens for retinal Müller glial cells. Brain Res. 1995, 686, 87–92. [Google Scholar] [CrossRef]
- De Diego, S.A.A.; Gutiérrez-Rodríguez, M.; de Vega, M.J.P.; González-Muñiz, R.; Herranz, R.; Martín-Martínez, M.; Cenarruzabeitia, E.; Frechilla, D.; del Río, J.; Jimeno, M.L.; et al. The neuroprotective activity of GPE tripeptide analogues does not correlate with glutamate receptor binding affinity. Bioorg. Med. Chem. Lett. 2006, 16, 3396–3400. [Google Scholar] [CrossRef] [PubMed]
- Vaaga, C.E.; Tovar, K.R.; Westbrook, G.L. The IGF-derived tripeptide Gly-Pro-Glu is a weak NMDA receptor agonist. J. Neurophysiol. 2014, 112, 1241–1245. [Google Scholar] [CrossRef] [PubMed]
- Bading, H.; Greenberg, M.E. Stimulation of protein tyrosine phosphorylation by NMDA receptor activation. Science 1991, 253, 912–914. [Google Scholar] [CrossRef] [PubMed]
- Krapivinsky, G.; Krapivinsky, L.; Manasian, Y.; Ivanov, A.; Tyzio, R.; Pellegrino, C.; Ben-Ari, Y.; Clapham, D.E.; Medina, I. The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron 2003, 40, 775–784. [Google Scholar] [CrossRef]
- Sutton, G.; Chandler, L.J. Activity-dependent NMDA receptor-mediated activation of protein kinase B/Akt in cortical neuronal cultures. J. Neurochem. 2002, 82, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.H.; Lu, X.C.; Shear, D.A.; Waghray, A.; Yao, C.; Tortella, F.C.; Dave, J.R. NNZ-2566 treatment inhibits neuroinflammation and pro-inflammatory cytokine expression induced by experimental penetrating ballistic-like brain injury in rats. J. Neuroinflamm. 2009, 6, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Pathipati, P.; Gorba, T.; Sceepens, A.; Goffin, V.; Sun, Y.; Fraser, M. Growth hormone and prolactin regulate human neural stem cell regenerative activity. Neuroscience 2011, 190, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Bickerdike, M.J.; Thomas, G.B.; Batchelor, D.C.; Sirimanne, E.S.; Leong, W.; Lin, H.; Sieg, F.; Wen, J.; Brimble, M.A.; Harris, P.W.; et al. NNZ-2566: A Gly-Pro-Glu analogue with neuroprotective efficacy in a rat model of acute focal stroke. J. Cereb. Blood Flow Metab. 2009, 29, 1924–1932. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.C.; Chen, R.W.; Yao, C.; Wei, H.; Yang, X.; Liao, Z.; Dave, J.R.; Tortella, F.C. NNZ-2566, a glypromate analog, improves functional recovery and attenuates apoptosis and inflammation in a rat model of penetrating ballistic-type brain injury. J. Neurotrauma 2009, 26, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Costoya, J.A.; Finidori, J.; Moutoussamy, S.; Señaris, R.; Devesa, J.; Arce, V.M. Activation of growth hormone receptor delivers an antiapoptotic signal: Evidence for a role of Akt in this pathway. Endocrinology 1999, 140, 5937–5943. [Google Scholar] [CrossRef] [PubMed]
- Latifi-Pupovci, H.; Kuçi, Z.; Wehner, S.; Bönig, H.; Lieberz, R.; Klingebiel, T.; Bader, P.; Kuç, S. In vitro migration and proliferation (“wound healing”) potential of mesenchymal stromal cells generated from human CD271+ bone marrow mononuclear cells. J. Transl. Med. 2015, 13, 315–324. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almengló, C.; Devesa, P.; Devesa, J.; Arce, V.M. GPE Promotes the Proliferation and Migration of Mouse Embryonic Neural Stem Cells and Their Progeny In Vitro. Int. J. Mol. Sci. 2017, 18, 1280. https://doi.org/10.3390/ijms18061280
Almengló C, Devesa P, Devesa J, Arce VM. GPE Promotes the Proliferation and Migration of Mouse Embryonic Neural Stem Cells and Their Progeny In Vitro. International Journal of Molecular Sciences. 2017; 18(6):1280. https://doi.org/10.3390/ijms18061280
Chicago/Turabian StyleAlmengló, Cristina, Pablo Devesa, Jesús Devesa, and Víctor M. Arce. 2017. "GPE Promotes the Proliferation and Migration of Mouse Embryonic Neural Stem Cells and Their Progeny In Vitro" International Journal of Molecular Sciences 18, no. 6: 1280. https://doi.org/10.3390/ijms18061280
APA StyleAlmengló, C., Devesa, P., Devesa, J., & Arce, V. M. (2017). GPE Promotes the Proliferation and Migration of Mouse Embryonic Neural Stem Cells and Their Progeny In Vitro. International Journal of Molecular Sciences, 18(6), 1280. https://doi.org/10.3390/ijms18061280