Autocrine Human Growth Hormone Promotes Invasive and Cancer Stem Cell-Like Behavior of Hepatocellular Carcinoma Cells by STAT3 Dependent Inhibition of CLAUDIN-1 Expression

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
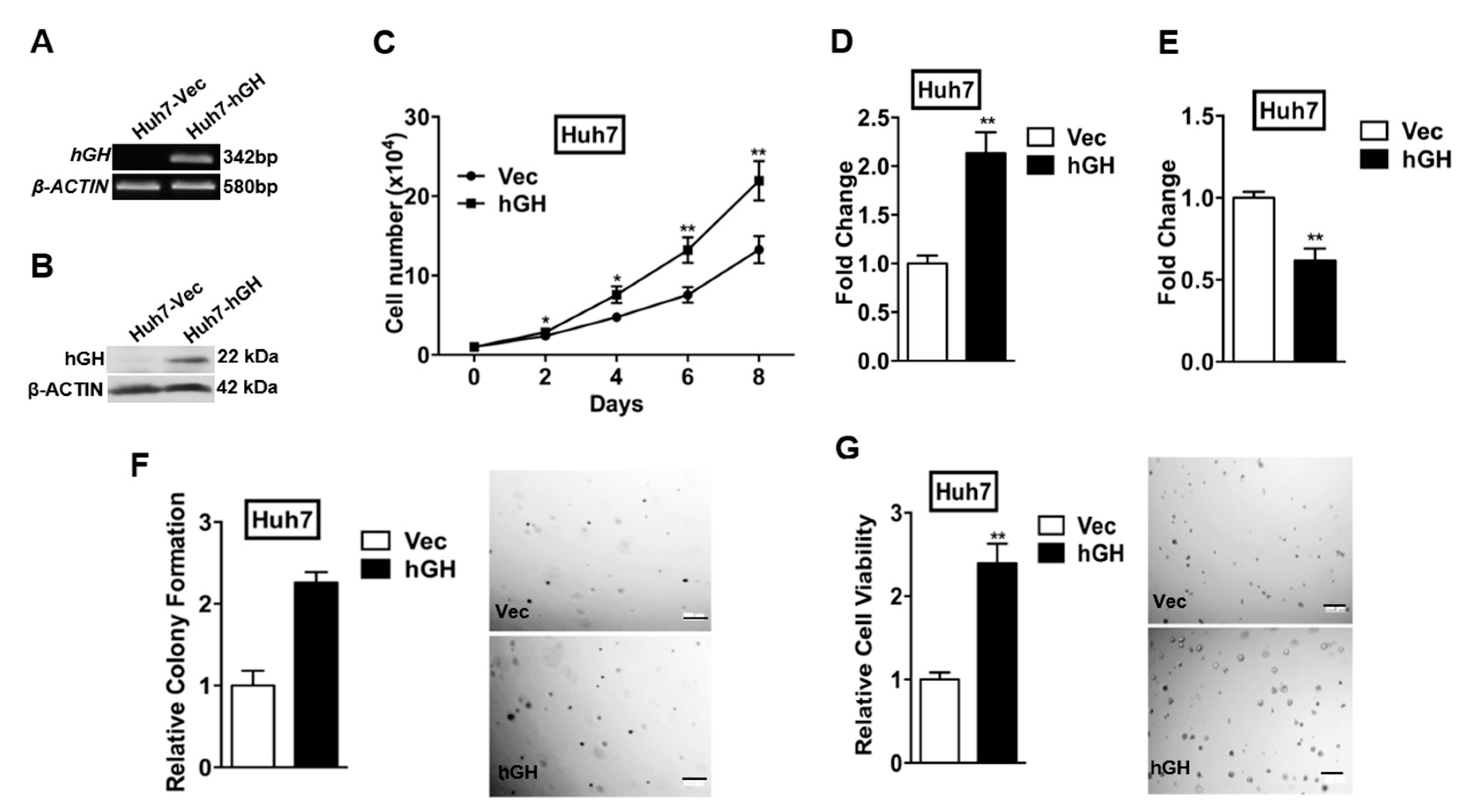
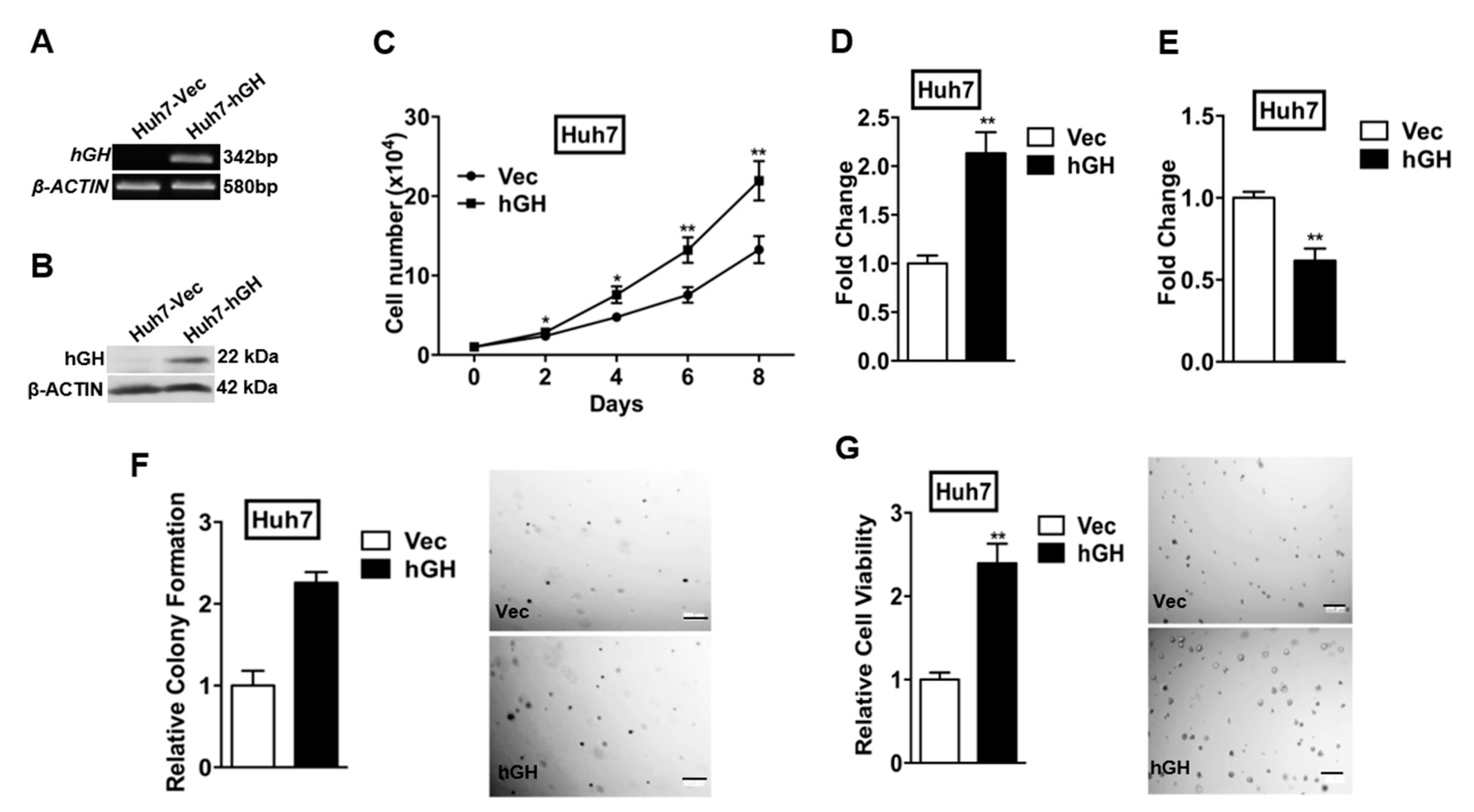
2.1. Forced Expression of Human Growth Hormone (hGH) Promotes Monolayer, Anchorage-Independent and Three-Dimensional (3D) Matrigel Growth of Human Hepatocellular Carcinoma (HCC) Cells, and Protects Human HCC Cells from Apoptosis
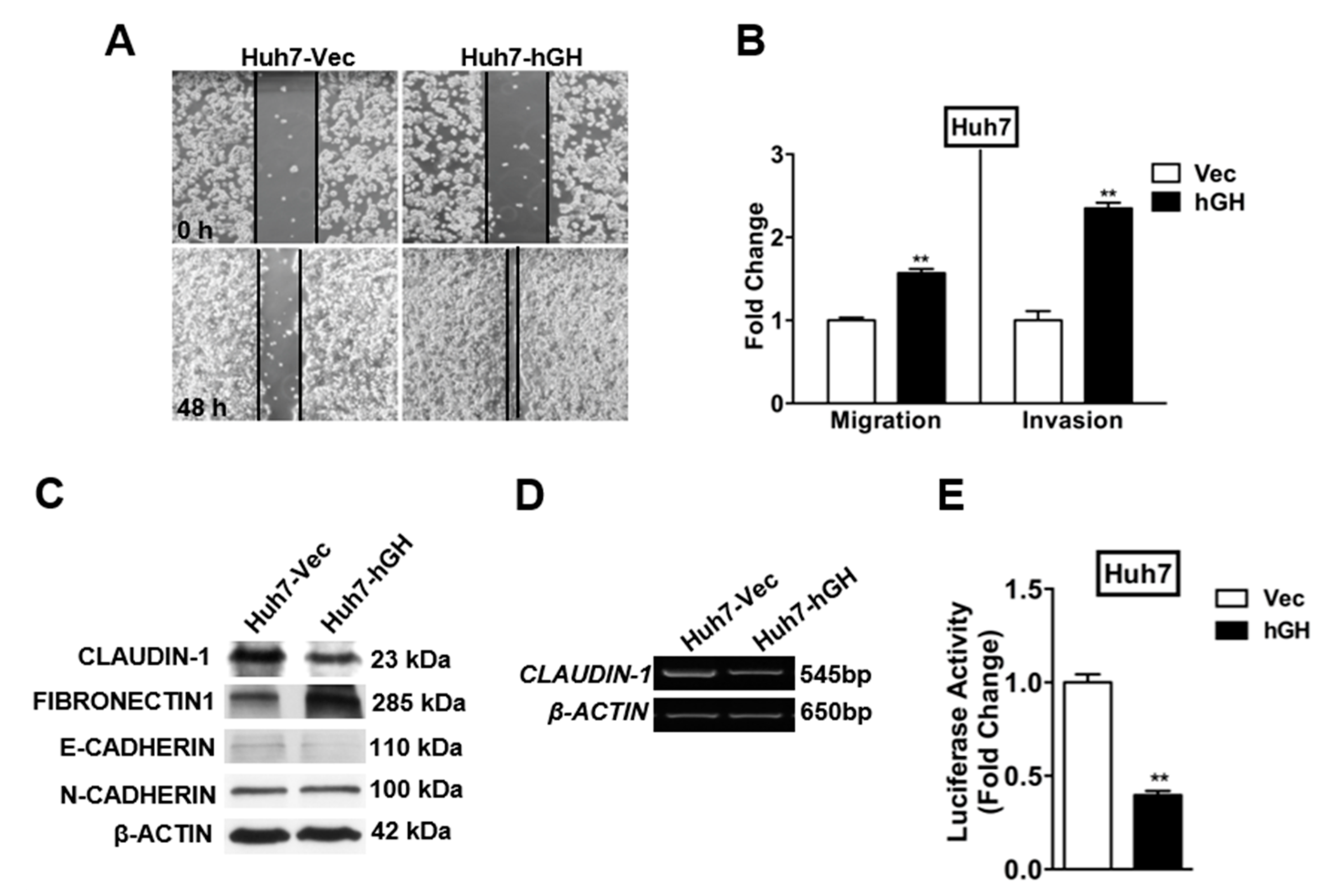
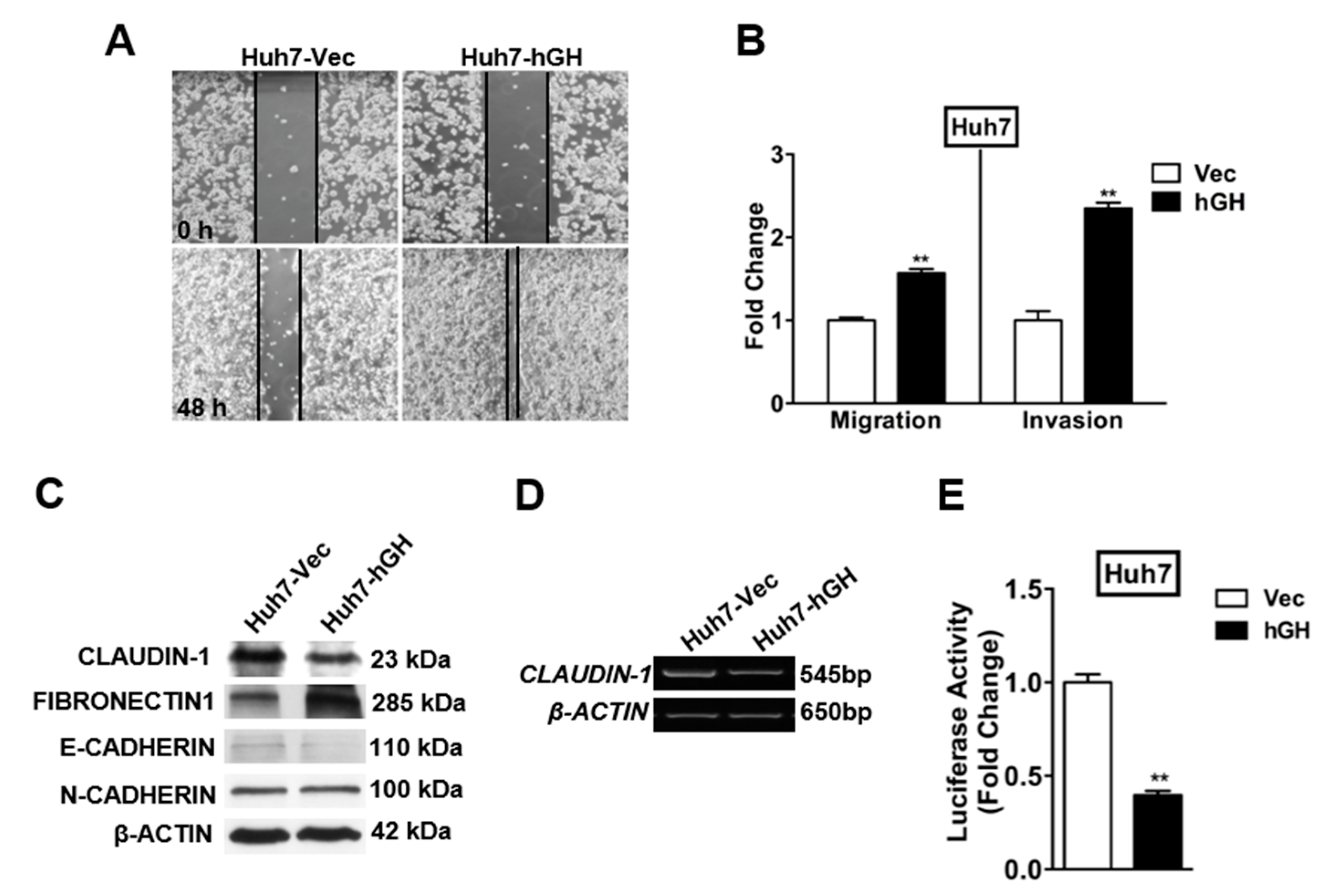
2.2. Forced Expression of hGH Promotes Migration and Invasion of HCC Cells
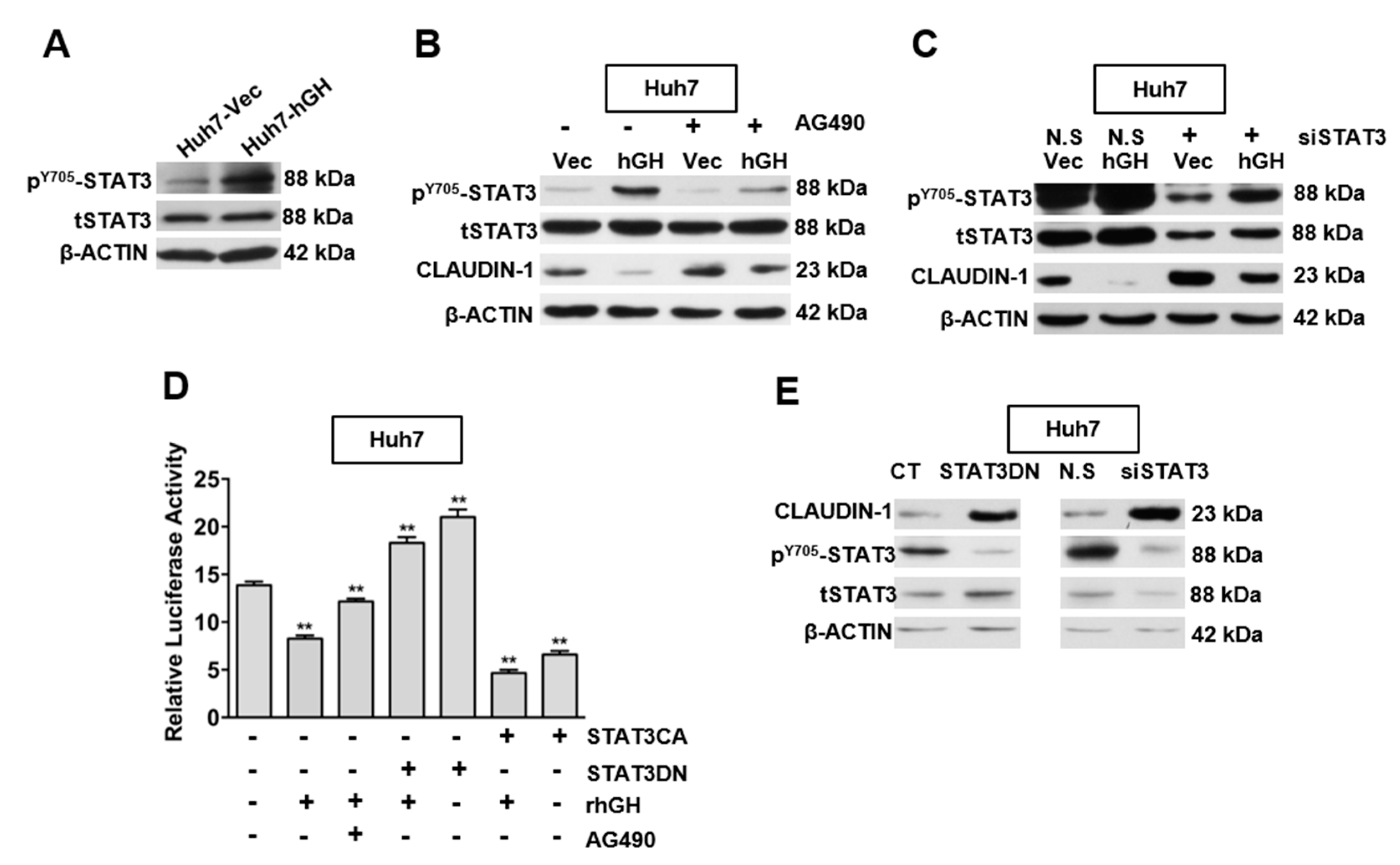
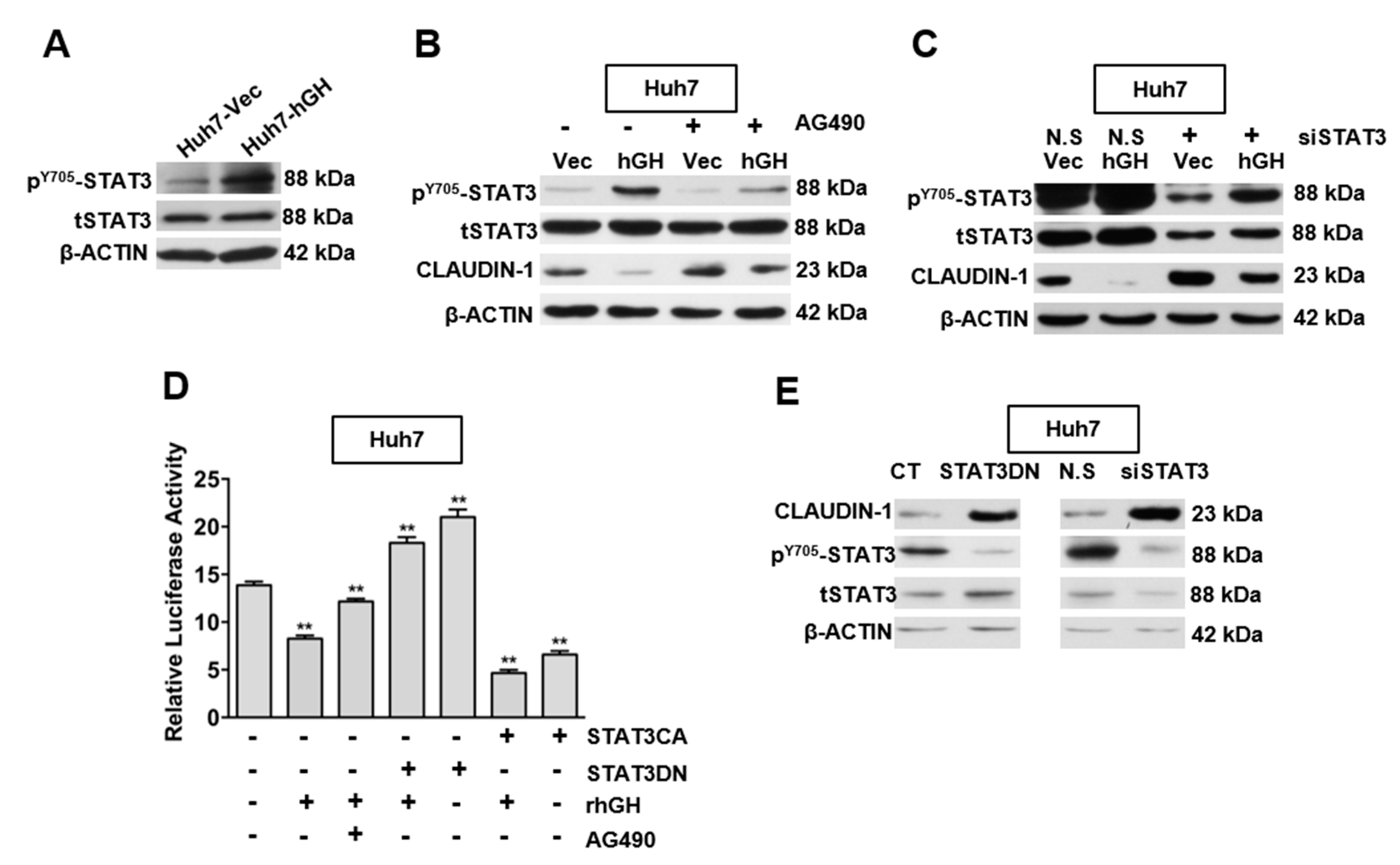
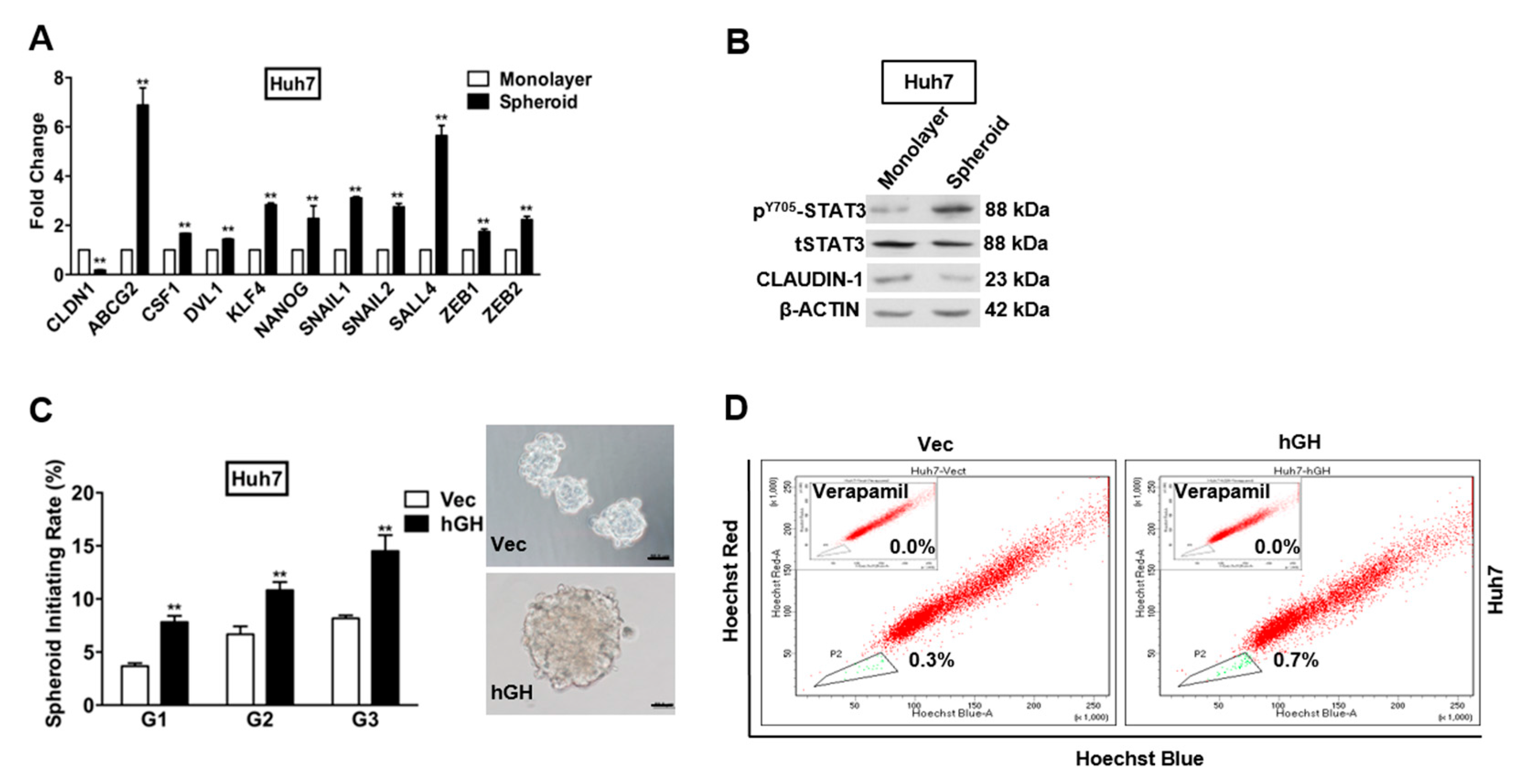
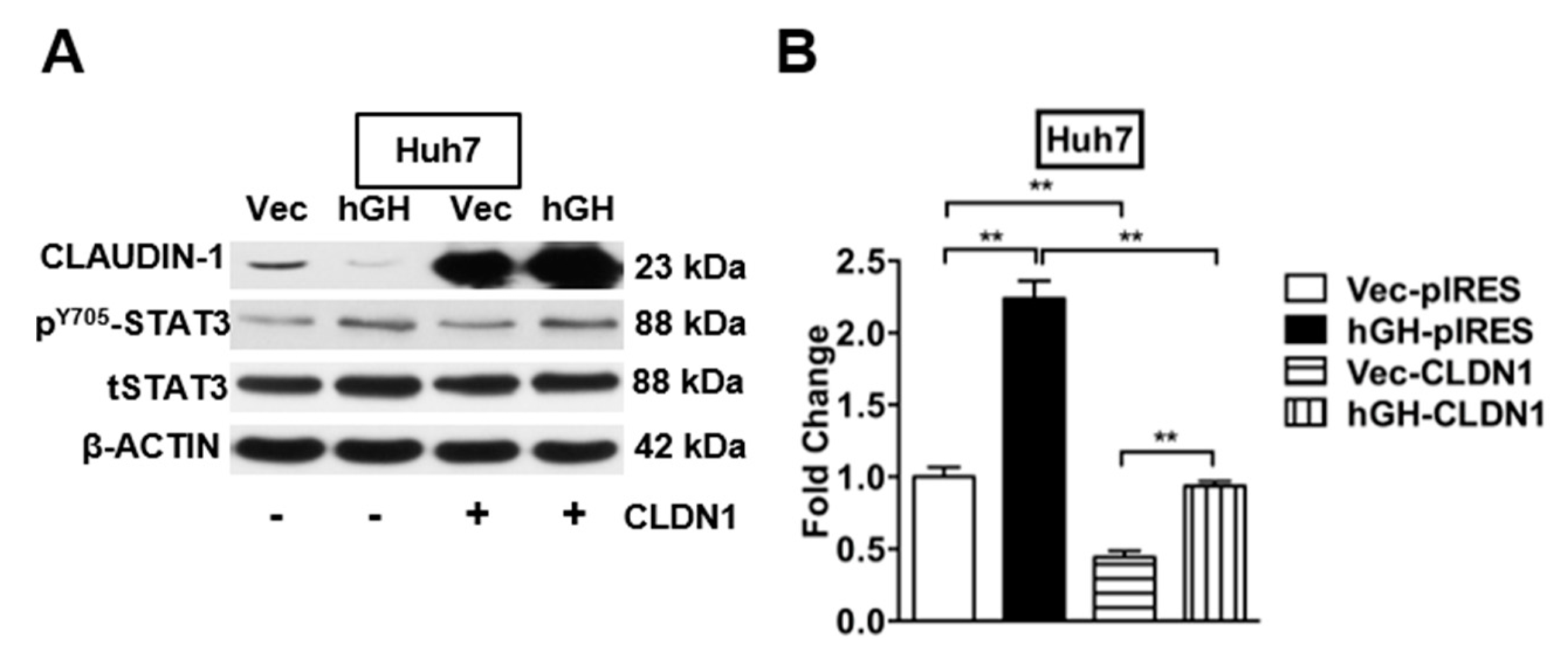
2.3. hGH Inhibits CLAUDIN-1 Expression Through Activation of Signal Transducer and Activator of Transcription 3 (STAT3)
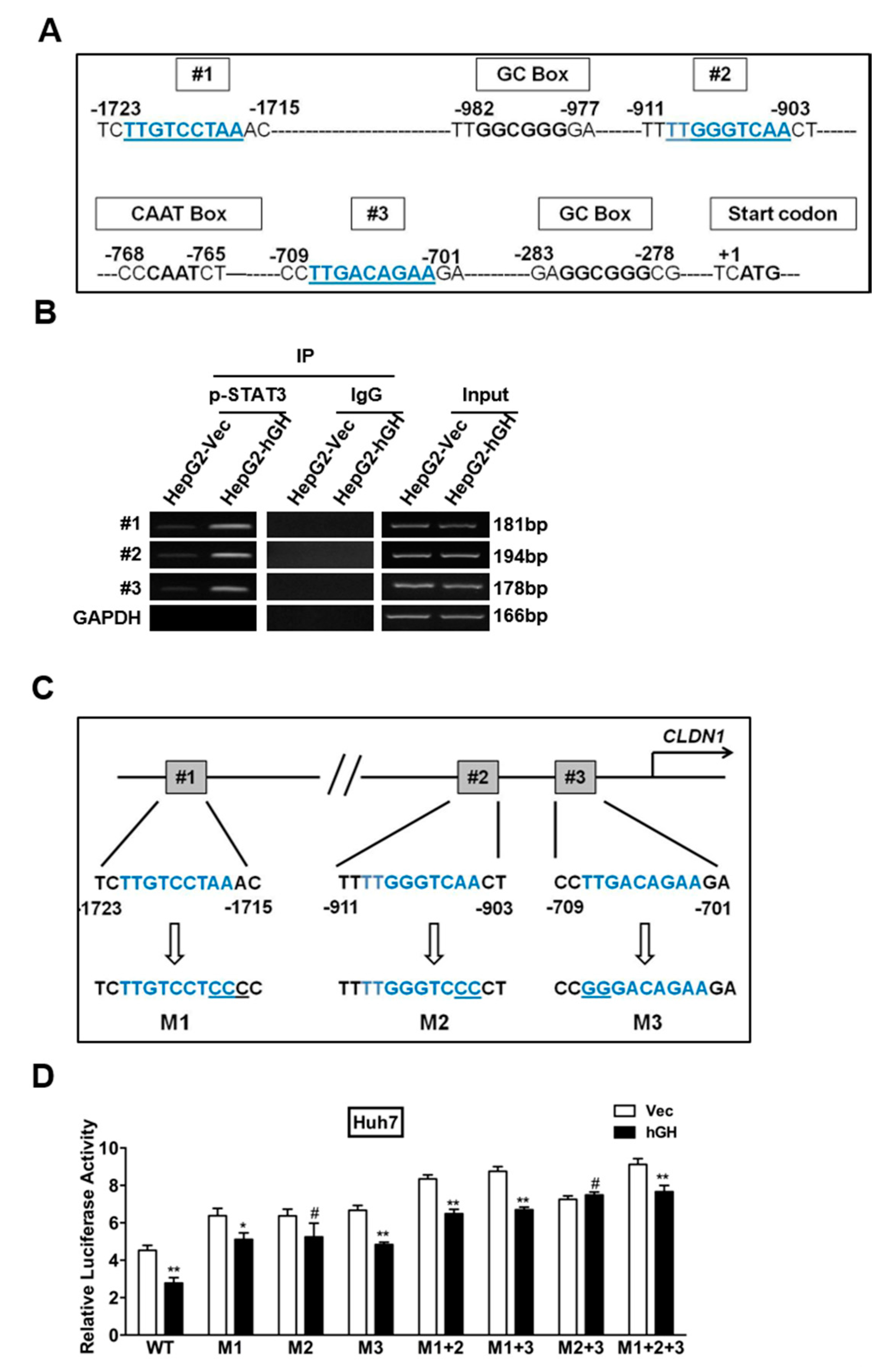
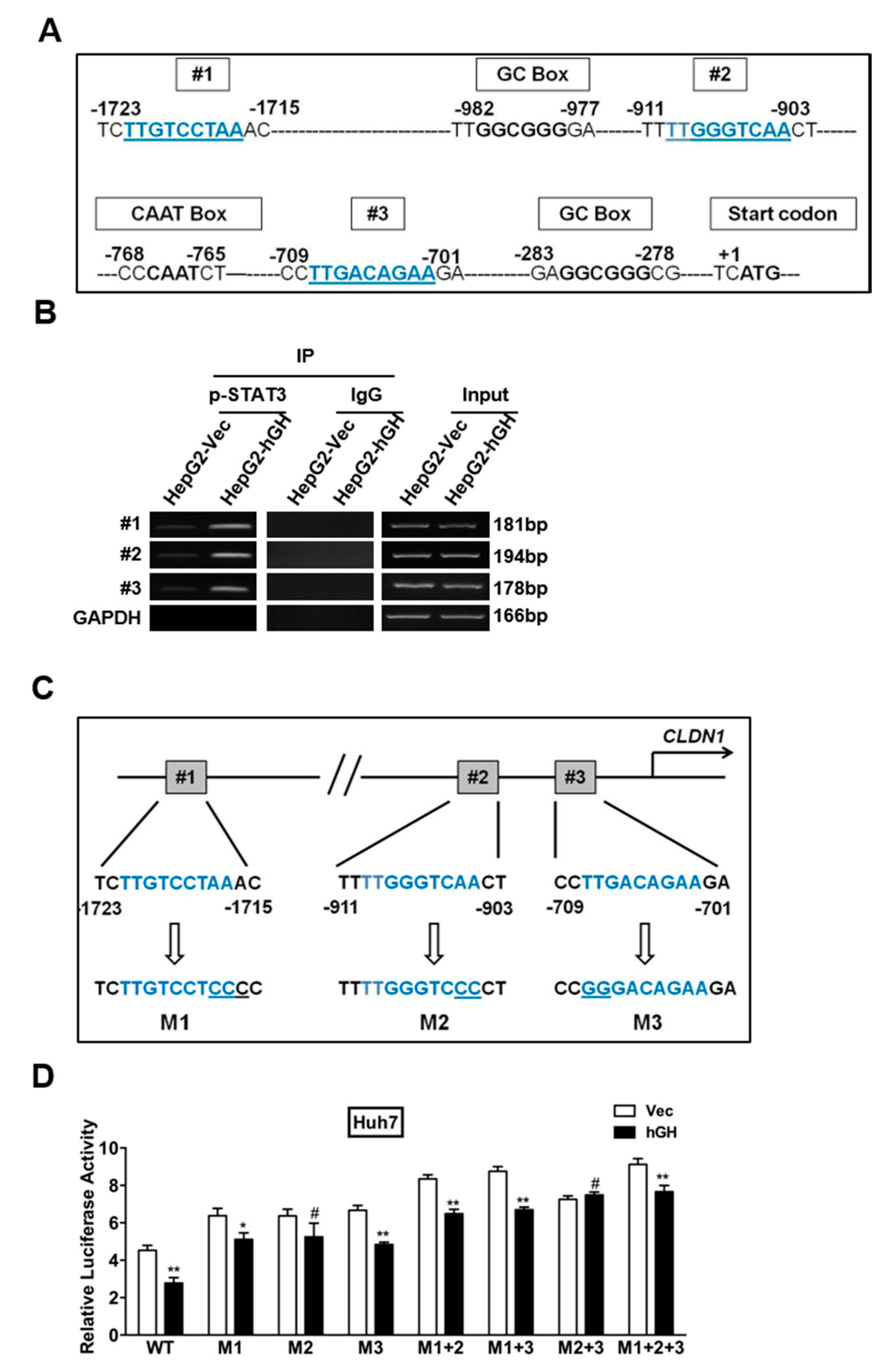
2.4. Identification of a STAT3-Targeted Region within Human CLAUDIN-1 Promoter
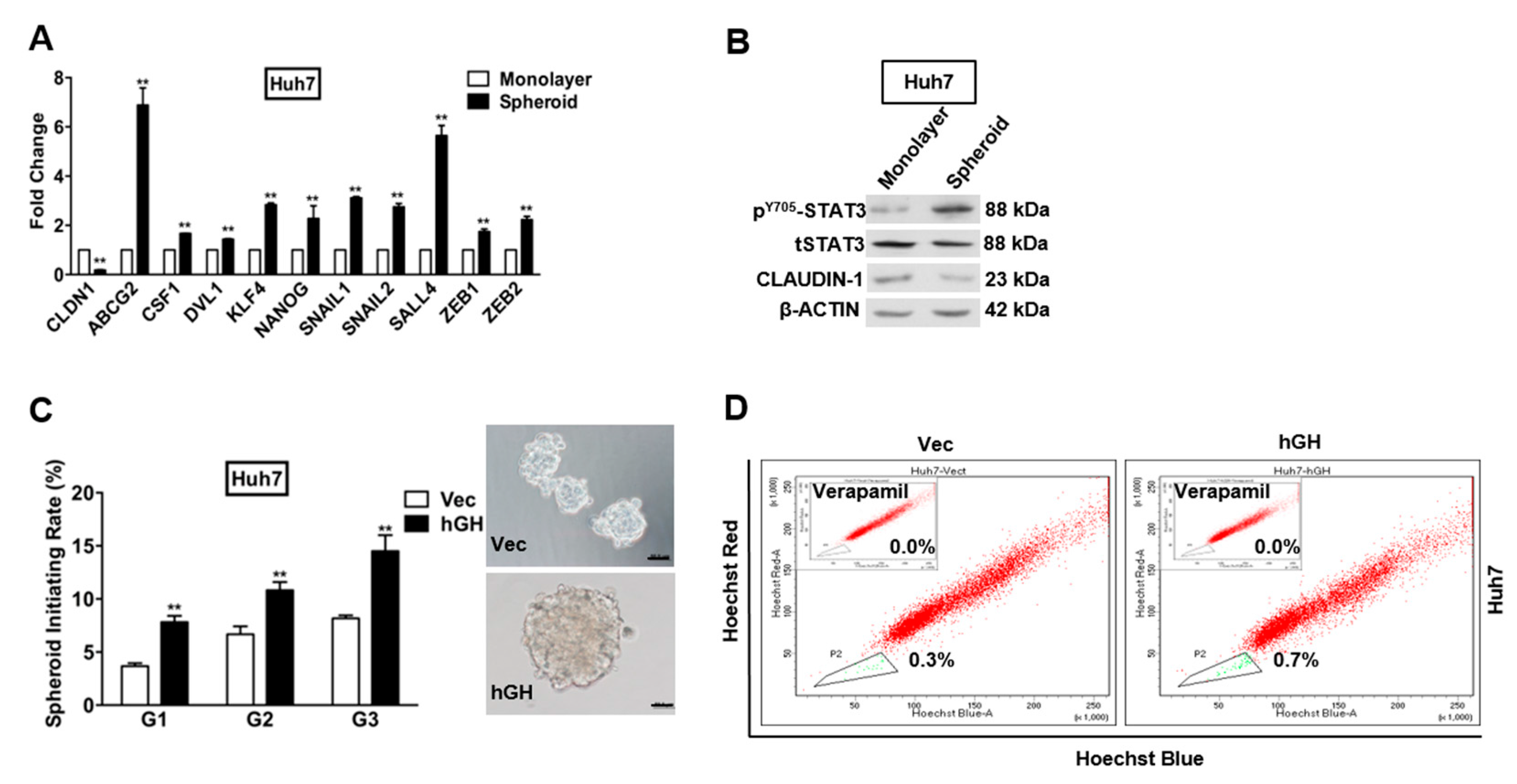
2.5. Forced Expression of hGH Promotes Cancer Stem Cell (CSC)-Like Behavior of HCC Cells
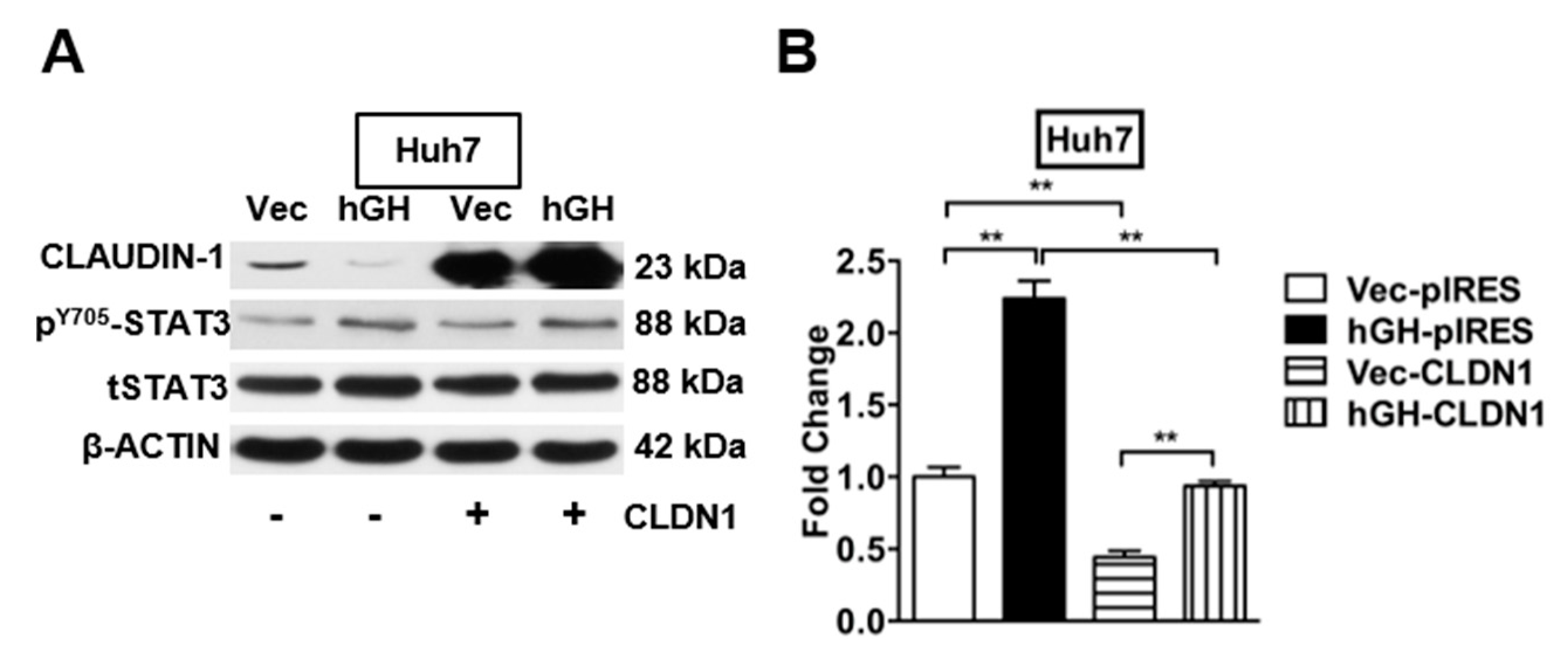
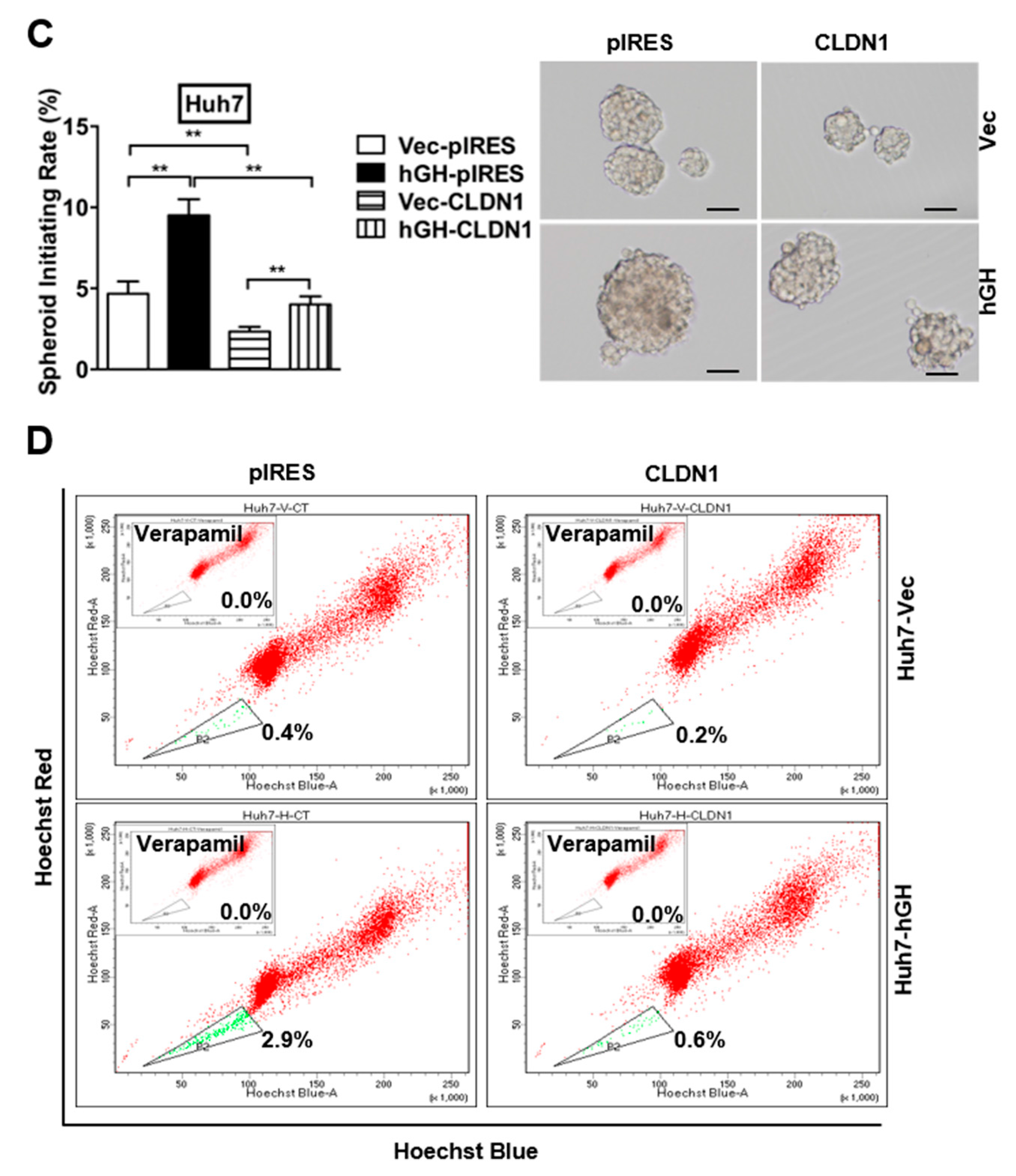
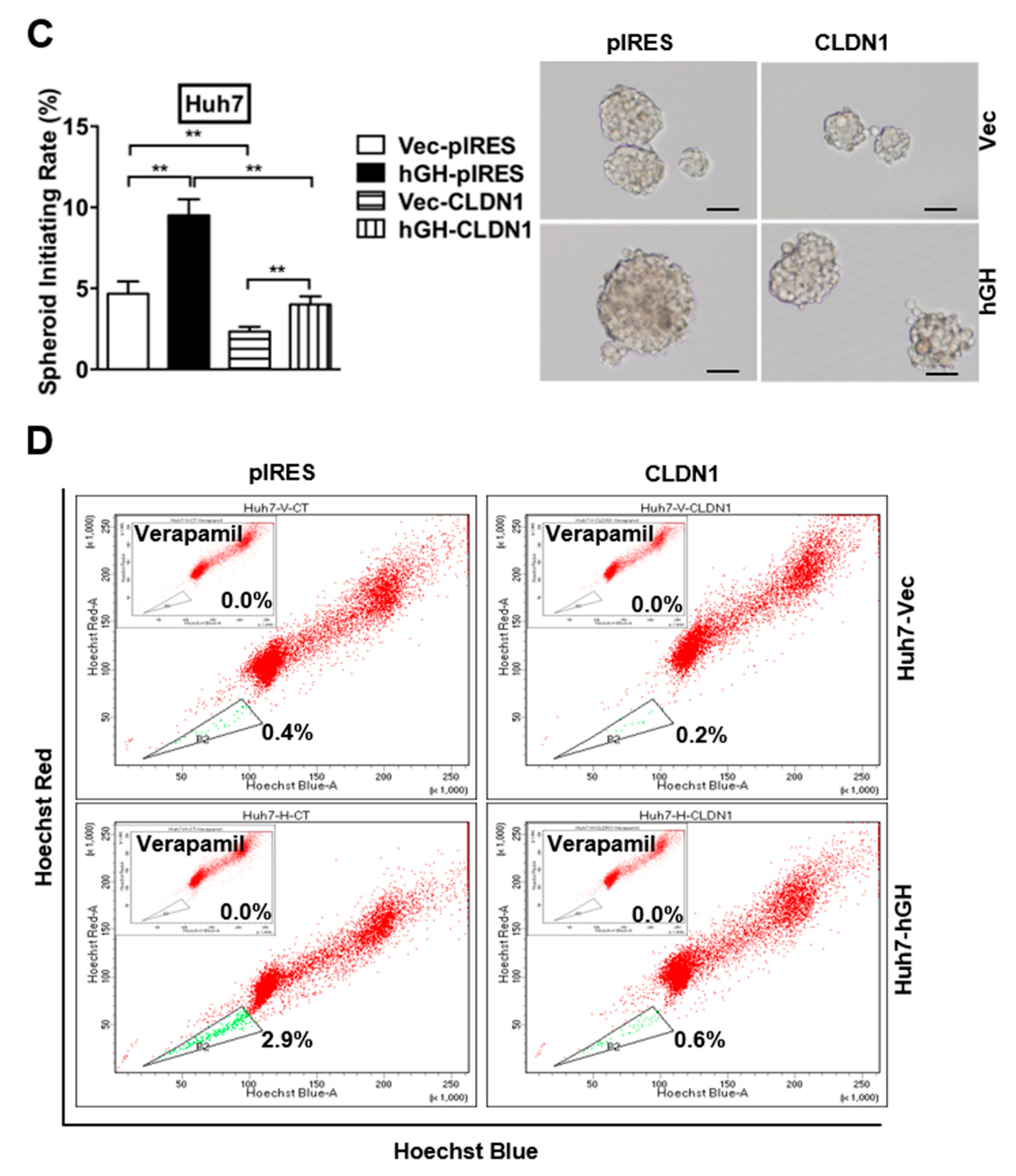
2.6. The Autocrine hGH-Stimulated Invasive and CSC-Like Behaviors in HCC Cells Are Abrogated by Forced Expression of CLAUDIN-1
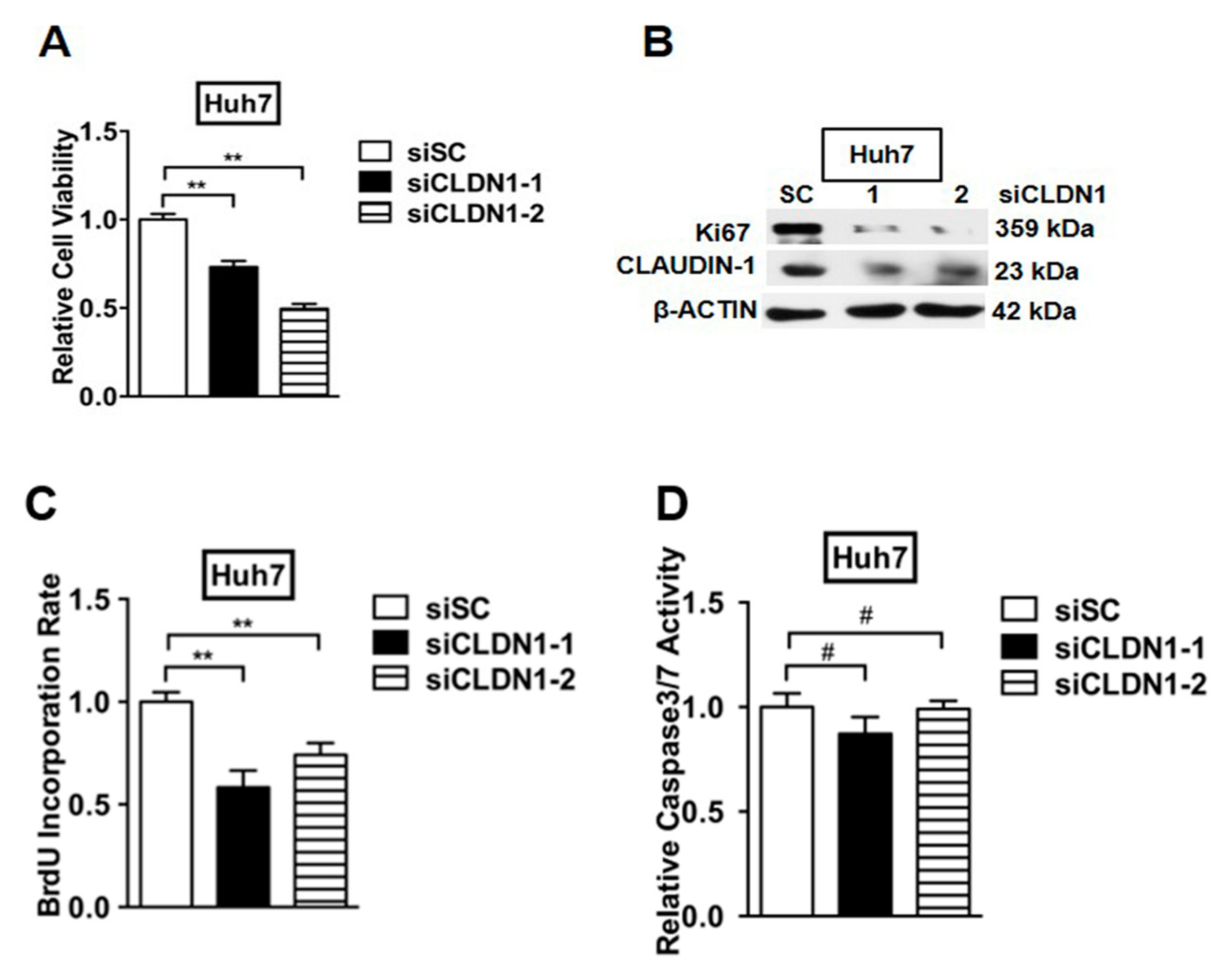
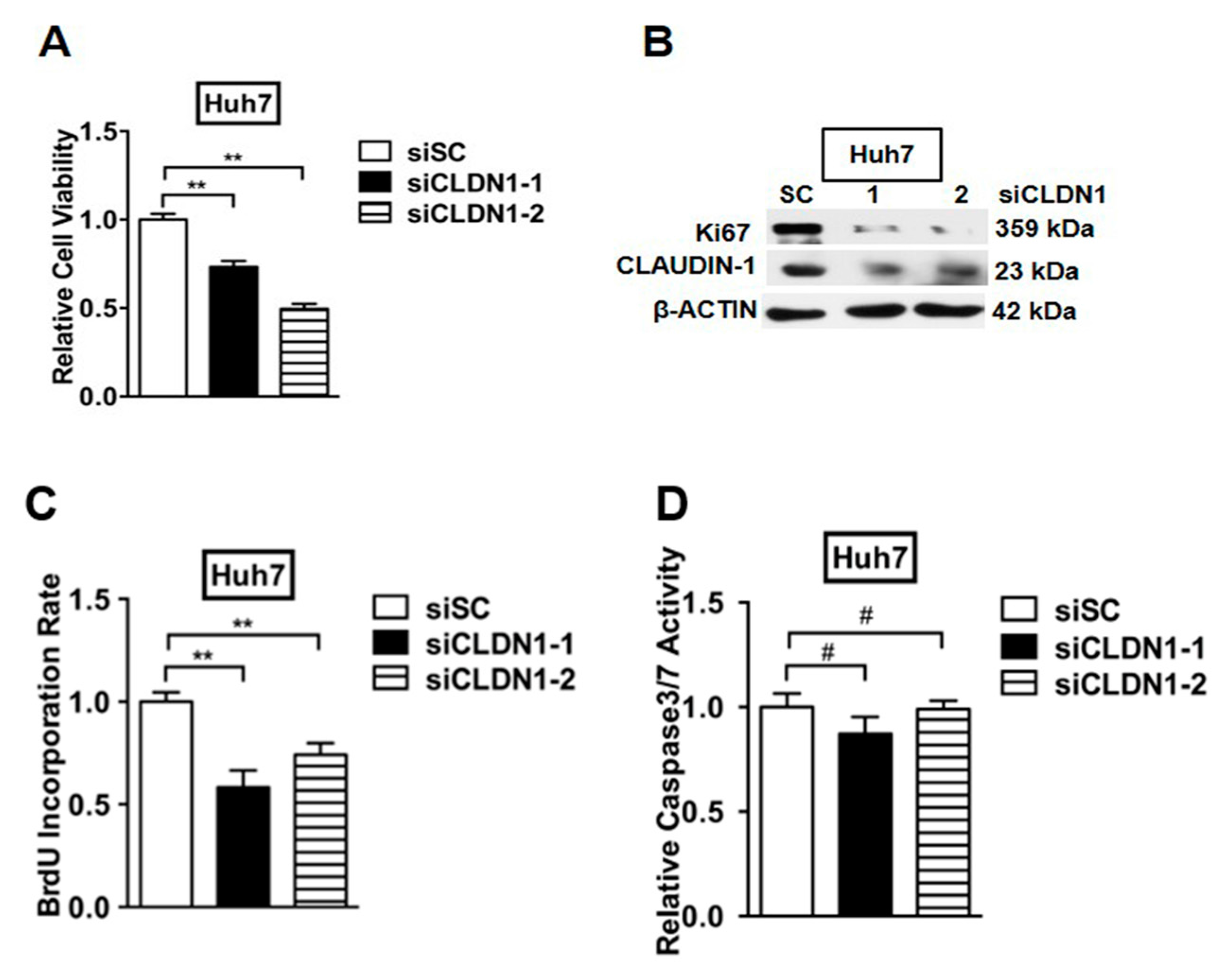
2.7. Depletion of CLAUDIN-1 Enhances Invasive and CSC-Like Behaviors of HCC Cells
2.8. Depletion of CLAUDIN-1 Attenuates Cell Proliferation without Affecting Cell Apoptosis
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Transfection
4.2. In Vitro Cell Function Assays
4.3. Spheroid Formation Assay
4.4. Side Population
4.5. Polymerase Chain Reaction (PCR) and Western Blot Analysis
4.6. Chromatin Immunoprecipitation
4.7. STAT3-Binding Site Mutated CLAUDIN-1 Promoter-Driven Luciferase Reporter Constructs
4.8. Construction of CLAUDIN-1 Expression Plasmids
4.9. Luciferase Reporter Assay
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| hGH | Human growth hormone |
| HCC | Hepatocellular carcinoma |
| EMT | Epithelial to mesenchymal transition |
| CSCs | cancer stem cells |
References
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef] [PubMed]
- Tung-Ping Poon, R.; Fan, S.T.; Wong, J. Risk factors, prevention, and management of postoperative recurrence after resection of hepatocellular carcinoma. Ann. Surg. 2000, 232, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-L.; Xing, B.-C.; Han, H.-B.; Zhao, W.; Hu, M.-H.; Xu, Z.-L.; Li, J.-Y.; Xie, Y.; Gu, J.; Wang, Y.; et al. The properties of tumor-initiating cells from a hepatocellular carcinoma patient’s primary and recurrent tumor. Carcinogenesis 2010, 31, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.M.; Haraguchi, N.; Ishii, H.; Ohkuma, M.; Okano, M.; Mimori, K.; Eguchi, H.; Yamamoto, H.; Nagano, H.; Sekimoto, M.; et al. Increased CD13 expression reduces reactive oxygen species, promoting survival of liver cancer stem cells via an epithelial-mesenchymal transition-like phenomenon. Ann. Surg. Oncol. 2012, 19, S539–S548. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.K.; Emerald, B.S.; Mertani, H.C.; Lobie, P.E. The oncogenic potential of growth hormone. Growth Horm. IGF Res. 2006, 16, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Webb, S.M.; Casanueva, F.; Wass, J.A. Oncological complications of excess GH in acromegaly. Pituitary 2002, 5, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Aguirre, J.; Balasubramanian, P.; Guevara-Aguirre, M.; Wei, M.; Madia, F.; Cheng, C.W.; Hwang, D.; Martin-Montalvo, A.; Saavedra, J.; Ingles, S.; et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci. Transl. Med. 2011, 3, 70ra13. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.T.; Zhou, J.; Adesanya, O.O.; Wang, J.; LeRoith, D.; Bondy, C.A. Growth hormone treatment induces mammary gland hyperplasia in aging primates. Nat. Med. 1997, 3, 1141–1144. [Google Scholar] [CrossRef] [PubMed]
- Harvey, S. Extrapituitary growth hormone. Endocrine 2010, 38, 335–359. [Google Scholar] [CrossRef] [PubMed]
- Waters, M.J.; Conway-Campbell, B.L. The oncogenic potential of autocrine human growth hormone in breast cancer. Proc. Natl. Acad. Sci. USA 2004, 101, 14992–14993. [Google Scholar] [CrossRef] [PubMed]
- Pandey, V.; Perry, J.K.; Mohankumar, K.M.; Kong, X.J.; Liu, S.M.; Wu, Z.S.; Mitchell, M.D.; Zhu, T.; Lobie, P.E. Autocrine human growth hormone stimulates oncogenicity of endometrial carcinoma cells. Endocrinology 2008, 149, 3909–3919. [Google Scholar] [CrossRef] [PubMed]
- Mukhina, S.; Mertani, H.C.; Guo, K.; Lee, K.O.; Gluckman, P.D.; Lobie, P.E. Phenotypic conversion of human mammary carcinoma cells by autocrine human growth hormone. Proc. Natl. Acad. Sci. USA 2004, 101, 15166–15171. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Qian, P.; Zhang, X.; Zhang, M.; Wang, H.; Wu, M.; Kong, X.; Tan, S.; Ding, K.; Perry, J.K.; et al. Autocrine/paracrine human growth hormone-stimulated microRNA 96-182-183 cluster promotes epithelial-mesenchymal transition and invasion in breast cancer. J. Biol. Chem. 2015, 290, 13812–13829. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Zhang, X.; Wu, Z.S.; Wang, J.J.; Lau, A.Y.; Zhu, T.; Lobie, P.E. Autocrine human growth hormone stimulates the tumor initiating capacity and metastasis of estrogen receptor-negative mammary carcinoma cells. Cancer Lett. 2015, 365, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.Z.; Kong, X.J.; Banerjee, A.; Muniraj, N.; Pandey, V.; Steiner, M.; Perry, J.K.; Zhu, T.; Liu, D.X.; Lobie, P.E. STAT3α is oncogenic for endometrial carcinoma cells and mediates the oncogenic effects of autocrine human growth hormone. Endocrinology 2010, 151, 4133–4145. [Google Scholar] [CrossRef] [PubMed]
- Snibson, K.J.; Bhathal, P.S.; Hardy, C.L.; Brandon, M.R.; Adams, T.E. High, persistent hepatocellular proliferation and apoptosis precede hepatocarcinogenesis in growth hormone transgenic mice. Liver 1999, 19, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Snibson, K.J.; Bhathal, P.S.; Adams, T.E. Overexpressed growth hormone (GH) synergistically promotes carcinogen-initiated liver tumour growth by promoting cellular proliferation in emerging hepatocellular neoplasms in female and male GH-transgenic mice. Liver 2001, 21, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Bugni, J.M.; Poole, T.M.; Drinkwater, N.R. The little mutation suppresses DEN-induced hepatocarcinogenesis in mice and abrogates genetic and hormonal modulation of susceptibility. Carcinogenesis 2001, 22, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Caballero, T.; Mertani, H.M.; Lambert, A.; Gallego, R.; Fraga, M.; Pintos, E.; Forteza, J.; Chevallier, M.; Lobie, P.E.; Vonderhaar, B.K.; et al. Increased expression of growth hormone and prolactin receptors in hepatocellular carcinomas. Endocrine 2000, 12, 265–271. [Google Scholar] [CrossRef]
- Kong, X.; Wu, W.; Yuan, Y.; Pandey, V.; Wu, Z.; Lu, X.; Zhang, W.; Chen, Y.; Wu, M.; Zhang, M.; et al. Human growth hormone and human prolactin function as autocrine/paracrine promoters of progression of hepatocellular carcinoma. Oncotarget 2016, 7, 29465–29479. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.B.; Sharma, A.; Dhawan, P. Claudin family of proteins and cancer: An overview. J. Oncol. 2010, 2010, 541957. [Google Scholar] [CrossRef] [PubMed]
- Turksen, K.; Troy, T.C. Junctions gone bad: Claudins and loss of the barrier in cancer. Biochim. Biophys. Acta 2011, 1816, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Resnick, M.B.; Konkin, T.; Routhier, J.; Sabo, E.; Pricolo, V.E. CLAUDIN-1 is a strong prognostic indicator in stage II colonic cancer: A tissue microarray study. Mod. Pathol. 2005, 18, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, P.; Singh, A.B.; Deane, N.G.; No, Y.; Shiou, S.R.; Schmidt, C.; Neff, J.; Washington, M.K.; Beauchamp, R.D. CLAUDIN-1 regulates cellular transformation and metastatic behavior in colon cancer. J. Clin. Investig. 2005, 115, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.B.; Sharma, A.; Smith, J.J.; Krishnan, M.; Chen, X.; Eschrich, S.; Washington, M.K.; Yeatman, T.J.; Beauchamp, R.D.; Dhawan, P. CLAUDIN-1 up-regulates the repressor ZEB-1 to inhibit E-cadherin expression in colon cancer cells. Gastroenterology 2011, 141, 2140–2153. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.L.; Ito, K.; Ko, T.K.; Liu, Q.; Salto–Tellez, M.; Yeoh, K.G.; Fukamachi, H.; Ito, Y. CLAUDIN-1 has tumor suppressive activity and is a direct target of RUNX3 in gastric epithelial cells. Gastroenterology 2010, 138, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Suzuki, S.; Sakaguchi, T.; Nakamura, T.; Baba, S.; Reinecker, H.C.; Nakamura, S.; Konno, H. Loss of CLAUDIN-1 expression correlates with malignancy of hepatocellular carcinoma. J. Surg. Res. 2007, 139, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Starling-Emerald, B.; Zhang, X.; Lee, K.O.; Gluckman, P.D.; Mertani, H.C.; Lobie, P.E. Oncogenic transformation of human mammary epithelial cells by autocrine human growth hormone. Cancer Res. 2005, 65, 317–324. [Google Scholar] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.Y. From PTK-STAT signaling to caspase expression and apoptosis induction. Cell. Death Differ. 1999, 6, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Zhang, W.; Kone, B.C. Signal transducers and activators of transcription 3 (STAT3) inhibits transcription of the inducible nitric oxide synthase gene by interacting with nuclear factor κB. Biochem. J. 2002, 367, 97–105. [Google Scholar] [CrossRef] [PubMed]
- De la Iglesia, N.; Konopka, G.; Lim, K.L.; Nutt, C.L.; Bromberg, J.F.; Frank, D.A.; Mischel, P.S.; Louis, D.N.; Bonni, A. Deregulation of a STAT3-interleukin 8 signaling pathway promotes human glioblastoma cell proliferation and invasiveness. J. Neurosci. 2008, 28, 5870–5878. [Google Scholar] [CrossRef] [PubMed]
- Dontu, G.; Abdallah, W.M.; Foley, J.M.; Jackson, K.W.; Clarke, M.F.; Kawamura, M.J.; Wicha, M.S. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003, 17, 1253–1270. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Ji, J.; Budhu, A.; Forgues, M.; Yang, W.; Wang, H.Y.; Jia, H.; Ye, Q.; Qin, L.X.; Wauthier, E.; et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology 2009, 136, 1012–1024. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, N.; Ishii, H.; Mimori, K.; Tanaka, F.; Ohkuma, M.; Kim, H.M.; Akita, H.; Takiuchi, D.; Hatano, H.; Nagano, H.; et al. CD13 is a therapeutic target in human liver cancer stem cells. J. Clin. Investig. 2010, 120, 3326–3339. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Kita, K.; Zheng, Y.W.; Yokosuka, O.; Saisho, H.; Iwama, A.; Nakauchi, H.; Taniguchi, H. Side population purified from hepatocellular carcinoma cells harbors cancer stem cell-like properties. Hepatology 2006, 44, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Wu, J.; Jiang, C. Dysregulation of signaling pathways and putative biomarkers in liver cancer stem cells (Review). Oncol. Rep. 2013, 29, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Cheung, S.T.; Leung, K.L.; Ip, Y.C.; Chen, X.; Fong, D.Y.; Ng, I.O.; Fan, S.T.; So, S. CLAUDIN-10 expression level is associated with recurrence of primary hepatocellular carcinoma. Clin. Cancer Res. 2005, 11, 551–556. [Google Scholar] [PubMed]
- Baumann, G.; Amburn, K.; Shaw, M.A. The circulating growth hormone (GH)-binding protein complex: A major constituent of plasma GH in man. Endocrinology 1988, 122, 976–984. [Google Scholar] [CrossRef]
- Scheuer, A.; Grün, R.; Lehmann, F. Peptide hormones in liver cirrhosis and hepatocellular carcinoma. Oncodev. Biol. Med. 1980, 2, 1–10. [Google Scholar]
- Carter-Su, C.; Schwartz, J.; Smit, L.S. Molecular mechanism of growth hormone action. Annu. Rev. Physiol. 1996, 58, 187–207. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, Y.; Berry, P.A.; Jiang, J.; Lobie, P.E.; Langenheim, J.F.; Chen, W.Y.; Frank, S.J. Growth hormone signaling in human T47D breast cancer cells: Potential role for a growth hormone receptor-prolactin receptor complex. Mol. Endocrinol. 2011, 25, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Menashe, I.; Maeder, D.; Garcia-Closas, M.; Figueroa, J.D.; Bhattacharjee, S.; Rotunno, M.; Kraft, P.; Hunter, D.J.; Chanock, S.J.; Rosenberg, P.S.; et al. Pathway analysis of breast cancer genome-wide association study highlights three pathways and one canonical signaling cascade. Cancer Res. 2010, 70, 4453–4459. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Morella, K.K.; Ripperger, J.; Lai, C.F.; Gearing, D.P.; Fey, G.H.; Campos, S.P.; Baumann, H. Receptors for interleukin-3 (IL-3) and growth hormone mediate an IL-6-type transcriptional induction in the presence of JAK2 or STAT3. Blood 1995, 86, 1671–1679. [Google Scholar] [PubMed]
- Han, Y.; Leaman, D.W.; Watling, D.; Rogers, N.C.; Groner, B.; Kerr, I.M.; Wood, W.I.; Stark, G.R. Participation of JAK and STAT proteins in growth hormone-induced signaling. J. Biol. Chem. 1996, 271, 5947–5952. [Google Scholar] [CrossRef] [PubMed]
- Taub, R. Liver regeneration: From myth to mechanism. Nat. Rev. Mol. Cell. Biol. 2004, 5, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Gritsko, T.; Williams, A.; Turkson, J.; Kaneko, S.; Bowman, T.; Huang, M.; Nam, S.; Eweis, I.; Diaz, N.; Sullivan, D.; et al. Persistent activation of STAT3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin. Cancer Res. 2006, 12, 11–19. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Karin, M. NF-κB and STAT3—Key players in liver inflammation and cancer. Cell. Res. 2011, 21, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.H.; Xu, G.L.; Jia, W.D.; Li, J.S.; Ma, J.L.; Ren, W.H.; Ge, Y.S.; Yu, J.H.; Liu, W.B.; Wang, W. Activation of STAT3 signal pathway correlates with twist and E-cadherin expression in hepatocellular carcinoma and their clinical significance. J. Surg. Res. 2012, 174, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Hoevel, T.; Macek, R.; Swisshelm, K.; Kubbies, M. Reexpression of the TJ protein CLDN1 induces apoptosis in breast tumor spheroids. Int. J. Cancer 2004, 108, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.H.; Kim, M.J.; Park, M.J.; Park, I.C.; Hwang, S.G.; An, S.; Choi, Y.H.; Yoon, G.; Lee, S.J. Claudin-1 acts through c-Abl-protein kinase Cδ (PKCδ) signaling and has a causal role in the acquisition of invasive capacity in human liver cells. J. Biol. Chem. 2010, 285, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.; Yoon, C.H.; Kim, R.K.; Lim, E.J.; Oh, Y.S.; Hwang, S.G.; An, S.; Yoon, G.; Gye, M.C.; Yi, J.M.; et al. Claudin-1 induces epithelial-mesenchymal transition through activation of the c-Abl-ERK signaling pathway in human liver cells. Oncogene 2013, 32, 4873–4882. [Google Scholar] [CrossRef] [PubMed]
- Farazi, P.A.; DePinho, R.A. Hepatocellular carcinoma pathogenesis: From genes to environment. Nat. Rev. Cancer 2006, 6, 674–687. [Google Scholar] [CrossRef] [PubMed]
- Farazi, P.A.; Glickman, J.; Jiang, S.; Yu, A.; Rudolph, K.L.; DePinho, R.A. Differential impact of telomere dysfunction on initiation and progression of hepatocellular carcinoma. Cancer Res. 2003, 63, 5021–5027. [Google Scholar] [PubMed]
- Lee, T.K.; Castilho, A.; Cheung, V.C.; Tang, K.H.; Ma, S.; Ng, I.O. CD24+ liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell 2011, 9, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, J.U.; Thorgeirsson, S.S. Stem cells in hepatocarcinogenesis: Evidence from genomic data. Semin. Liver Dis. 2010, 30, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Ramalho-Santos, M.; Yoon, S.; Matsuzaki, Y.; Mulligan, R.C.; Melton, D.A. “Stemness”: Transcriptional profiling of embryonic and adult stem cells. Science 2002, 298, 597–600. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, S.; Honeth, G.; Ginestier, C.; Shinomiya, I.; Marlow, R.; Buchupalli, B.; Gazinska, P.; Brown, J.; Catchpole, S.; Liu, S.; et al. Growth hormone is secreted by normal breast epithelium upon progesterone stimulation and increases proliferation of stem/progenitor cells. Stem Cell Rep. 2014, 2, 780–793. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Kitisin, K.; Jogunoori, W.; Li, C.; Deng, C.X.; Mueller, S.C.; Ressom, H.W.; Rashid, A.; He, A.R.; Mendelson, J.S.; et al. Progenitor/stem cells give rise to liver cancer due to aberrant TGF-β and IL-6 signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 2445–2450. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Morel, A.P.; Lievre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef] [PubMed]
- Song, L.B.; Li, J.; Liao, W.T.; Feng, Y.; Yu, C.P.; Hu, L.J.; Kong, Q.L.; Xu, L.H.; Zhang, X.; Liu, W.L.; et al. The polycomb group protein Bmi-1 represses the tumor suppressor PTEN and induces epithelial-mesenchymal transition in human nasopharyngeal epithelial cells. J. Clin. Investig. 2009, 119, 3626–3636. [Google Scholar] [CrossRef] [PubMed]
- Dang, H.; Ding, W.; Emerson, D.; Rountree, C.B. Snail1 induces epithelial-to-mesenchymal transition and tumor initiating stem cell characteristics. BMC Cancer 2011, 11, 396. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Cheng, W.; Lai, D.; Huang, Y.; Guo, L. Characterization of primary ovarian cancer cells in different culture systems. Oncol. Rep. 2010, 23, 1277–1284. [Google Scholar] [PubMed]
- Qin, W.; Ren, Q.; Liu, T.; Huang, Y.; Wang, J. MicroRNA-155 is a novel suppressor of ovarian cancer-initiating cells that targets CLDN1. FEBS Lett. 2013, 587, 1434–1439. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Estrada, O.M.; Culleres, A.; Soriano, F.X.; Peinado, H.; Bolos, V.; Martinez, F.O.; Reina, M.; Cano, A.; Fabre, M.; Vilaro, S. The transcription factors Slug and Snail act as repressors of Claudin-1 expression in epithelial cells. Biochem. J. 2006, 394, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Lopez, D.; Niu, G.; Huber, P.; Carter, W.B. Tumor-induced upregulation of Twist, Snail, and Slug represses the activity of the human VE-cadherin promoter. Arch. Biochem. Biophys. 2009, 482, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Ikenouchi, J.; Matsuda, M.; Furuse, M.; Tsukita, S. Regulation of tight junctions during the epithelium-mesenchyme transition: Direct repression of the gene expression of claudins/occludin by Snail. J. Cell. Sci. 2003, 116, 1959–1967. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E., Jr. STATs and gene regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Kone, B.C. The STAT3 DNA-binding domain mediates interaction with NF-κB p65 and inducible nitric oxide synthase transrepression in mesangial cells. J. Am. Soc. Nephrol. 2004, 15, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y. Cancer Science Institute of Singapore and Department of Pharmacology, National University of Singapore, Singapore. NF-κB Elements in the 5′ Proximal Region of CLAUDIN-1 Promoter. Personal communication, 2017. [Google Scholar]
- Yadav, A.; Kumar, B.; Datta, J.; Teknos, T.N.; Kumar, P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol. Cancer Res. 2011, 9, 1658–1667. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W.; Hsu, S.C.; Xia, W.; Cao, X.; Shih, J.Y.; Wei, Y.; Abbruzzese, J.L.; Hortobagyi, G.N.; Hung, M.C. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007, 67, 9066–9076. [Google Scholar] [CrossRef] [PubMed]
- Brunet-Dunand, S.E.; Vouyovitch, C.; Araneda, S.; Pandey, V.; Vidal, L.J.; Print, C.; Mertani, H.C.; Lobie, P.E.; Perry, J.K. Autocrine human growth hormone promotes tumor angiogenesis in mammary carcinoma. Endocrinology 2009, 150, 1341–1352. [Google Scholar] [CrossRef] [PubMed]
- Goodell, M.A.; Brose, K.; Paradis, G.; Conner, A.S.; Mulligan, R.C. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J. Exp. Med. 1996, 183, 1797–1806. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-J.; You, M.-L.; Chong, Q.-Y.; Pandey, V.; Zhuang, Q.-S.; Liu, D.-X.; Ma, L.; Zhu, T.; Lobie, P.E. Autocrine Human Growth Hormone Promotes Invasive and Cancer Stem Cell-Like Behavior of Hepatocellular Carcinoma Cells by STAT3 Dependent Inhibition of CLAUDIN-1 Expression. Int. J. Mol. Sci. 2017, 18, 1274. https://doi.org/10.3390/ijms18061274
Chen Y-J, You M-L, Chong Q-Y, Pandey V, Zhuang Q-S, Liu D-X, Ma L, Zhu T, Lobie PE. Autocrine Human Growth Hormone Promotes Invasive and Cancer Stem Cell-Like Behavior of Hepatocellular Carcinoma Cells by STAT3 Dependent Inhibition of CLAUDIN-1 Expression. International Journal of Molecular Sciences. 2017; 18(6):1274. https://doi.org/10.3390/ijms18061274
Chicago/Turabian StyleChen, Yi-Jun, Ming-Liang You, Qing-Yun Chong, Vijay Pandey, Qiu-Shi Zhuang, Dong-Xu Liu, Lan Ma, Tao Zhu, and Peter E. Lobie. 2017. "Autocrine Human Growth Hormone Promotes Invasive and Cancer Stem Cell-Like Behavior of Hepatocellular Carcinoma Cells by STAT3 Dependent Inhibition of CLAUDIN-1 Expression" International Journal of Molecular Sciences 18, no. 6: 1274. https://doi.org/10.3390/ijms18061274
APA StyleChen, Y.-J., You, M.-L., Chong, Q.-Y., Pandey, V., Zhuang, Q.-S., Liu, D.-X., Ma, L., Zhu, T., & Lobie, P. E. (2017). Autocrine Human Growth Hormone Promotes Invasive and Cancer Stem Cell-Like Behavior of Hepatocellular Carcinoma Cells by STAT3 Dependent Inhibition of CLAUDIN-1 Expression. International Journal of Molecular Sciences, 18(6), 1274. https://doi.org/10.3390/ijms18061274