CRISPR/Cas9-Mediated Correction of the FANCD1 Gene in Primary Patient Cells


 ,
,
Abstract

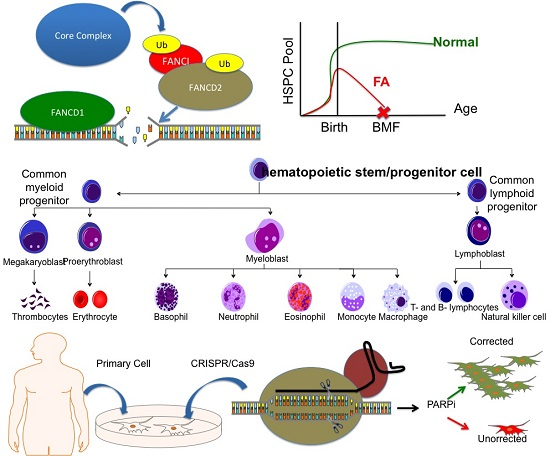
1. Introduction
2. Results
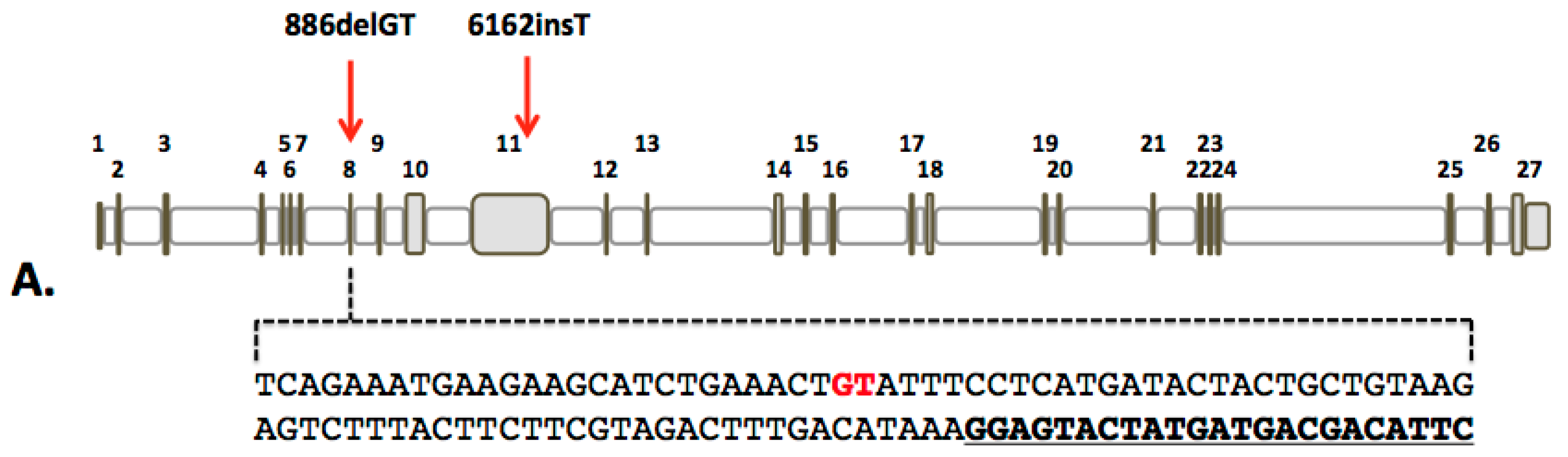
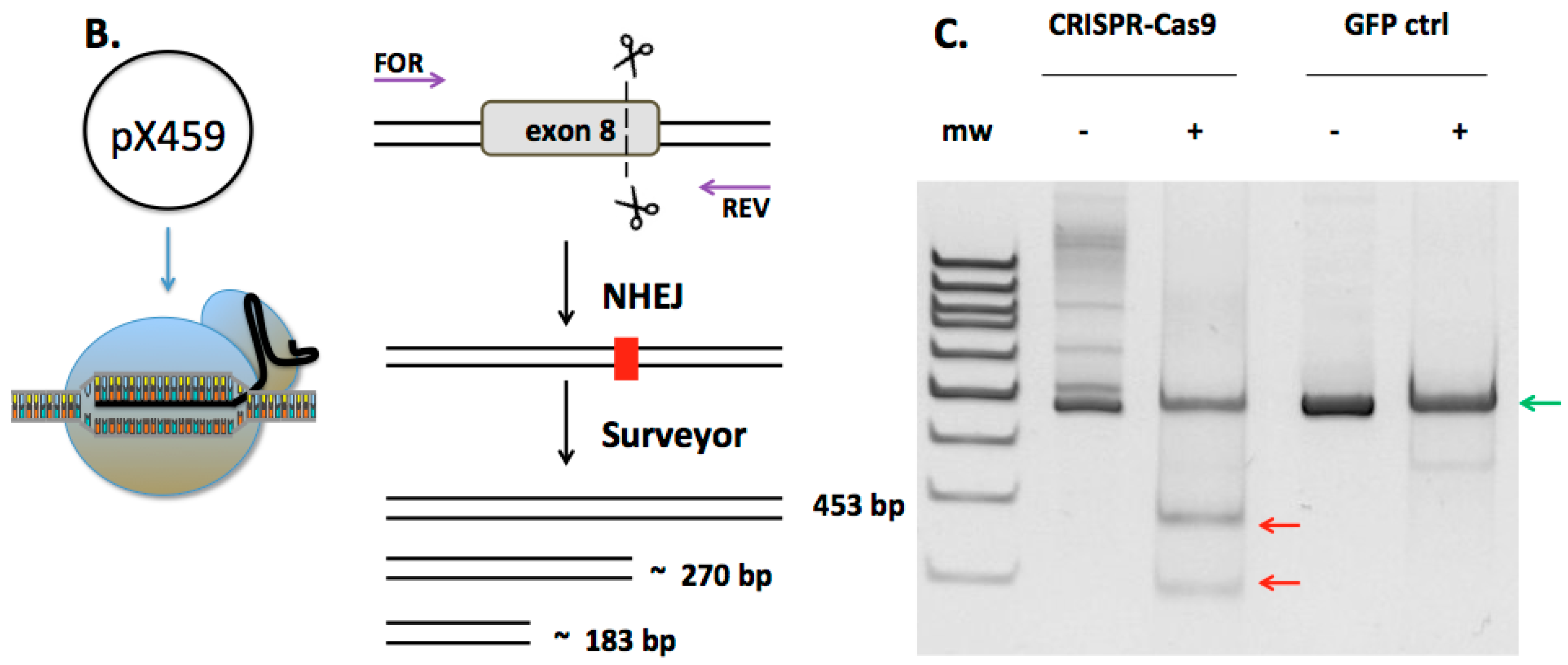
2.1. Design and Activity of CRISPR/Cas9 Gene-Editing Reagents for FANCD1 Gene Correction
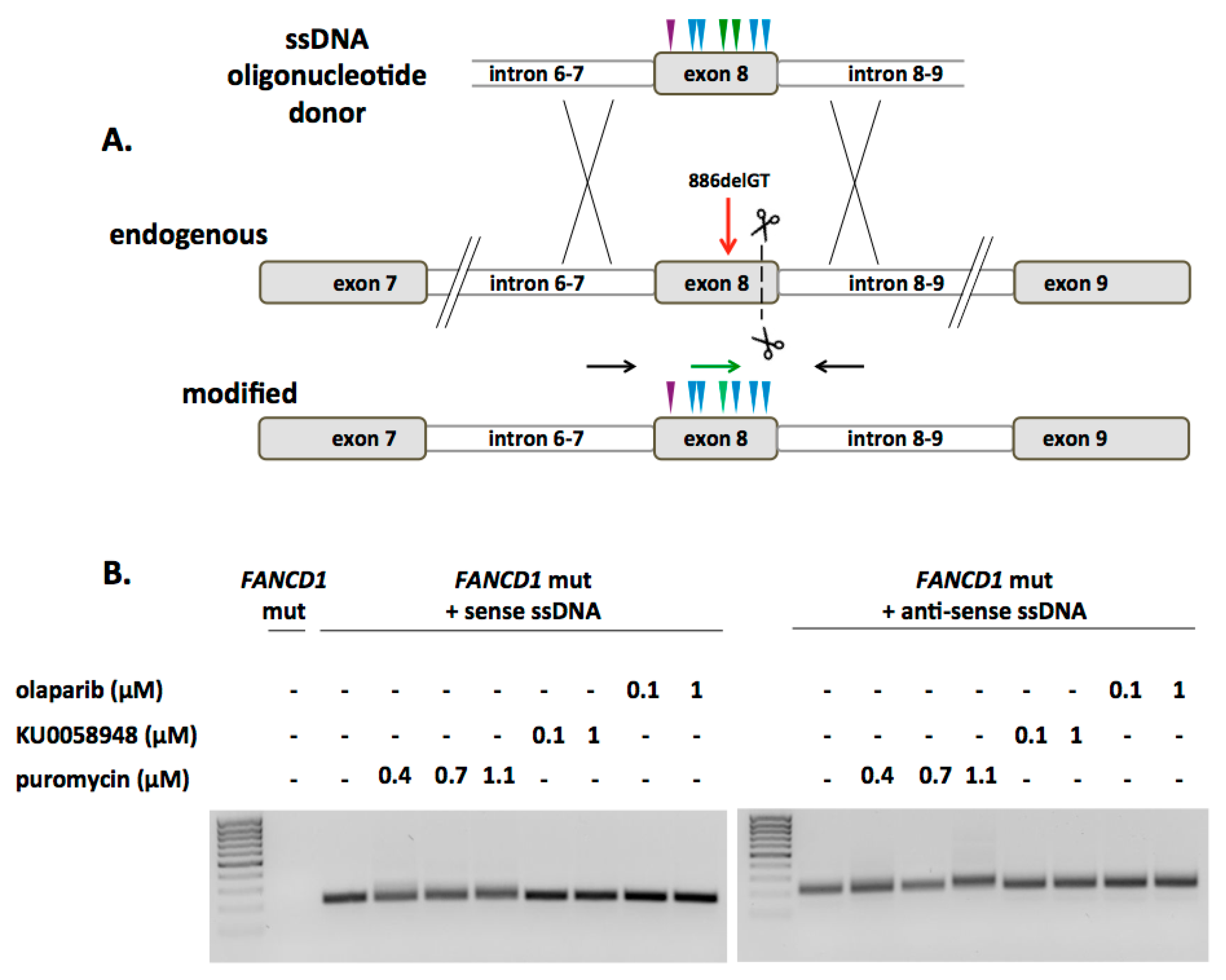
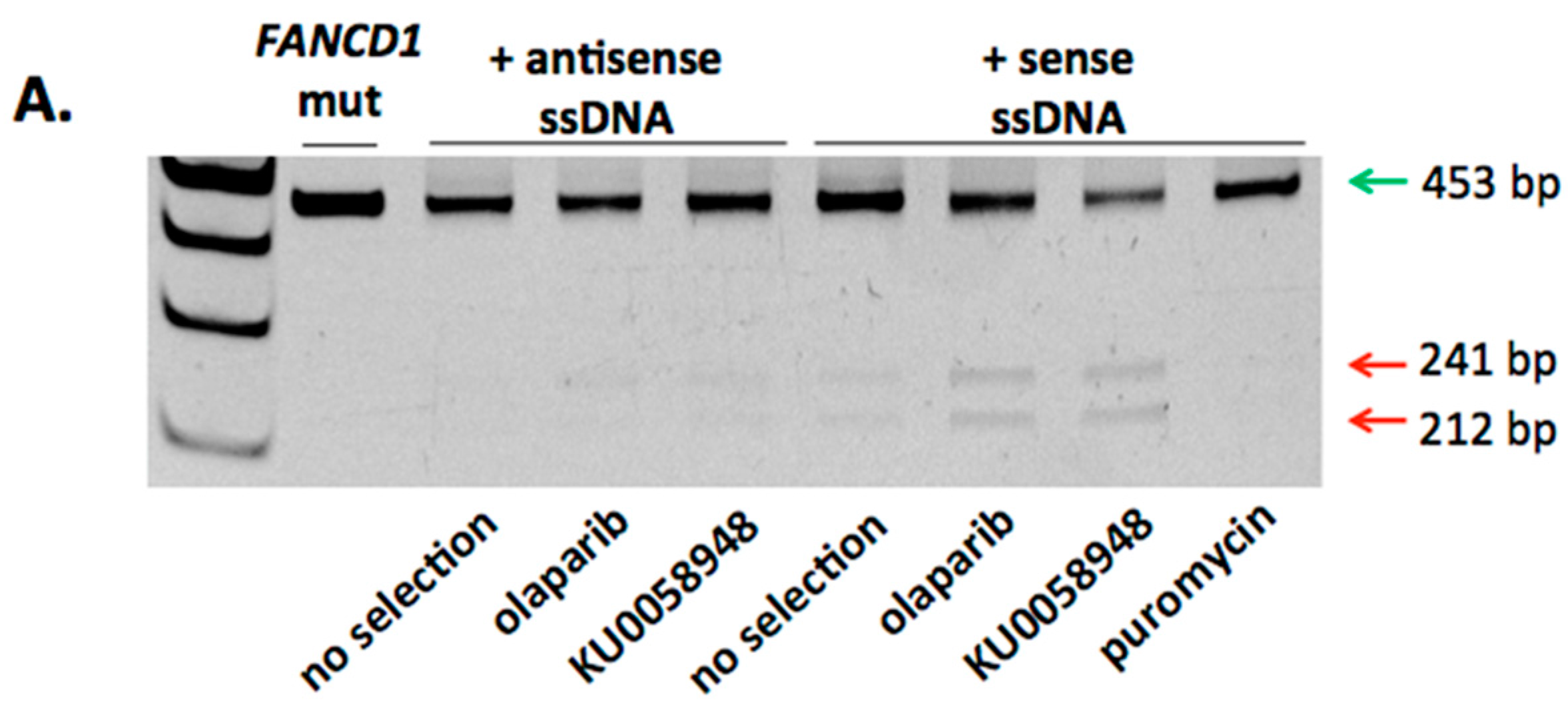
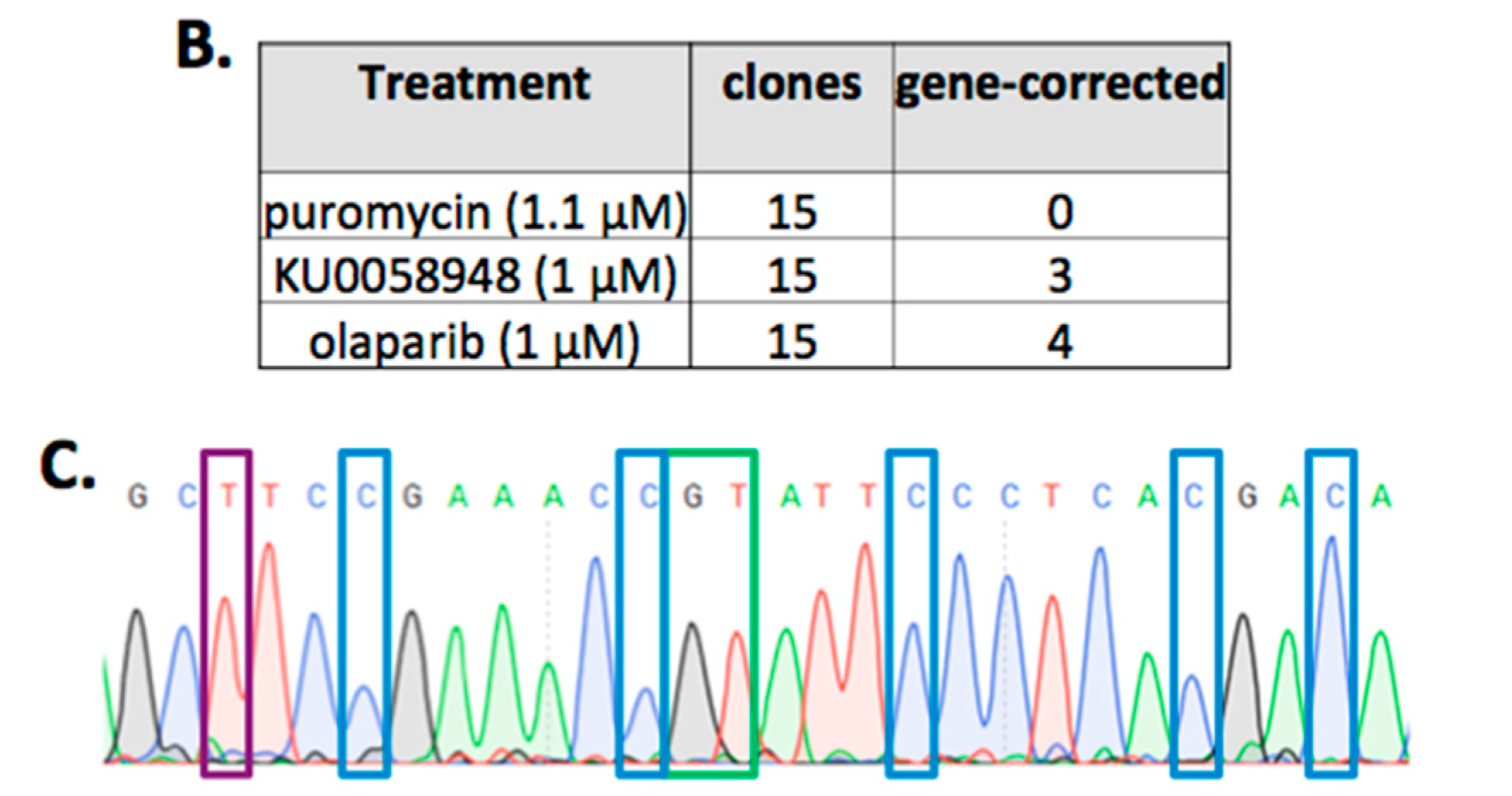
2.2. FANCD1 Gene Correction in Primary Patient Fibroblasts
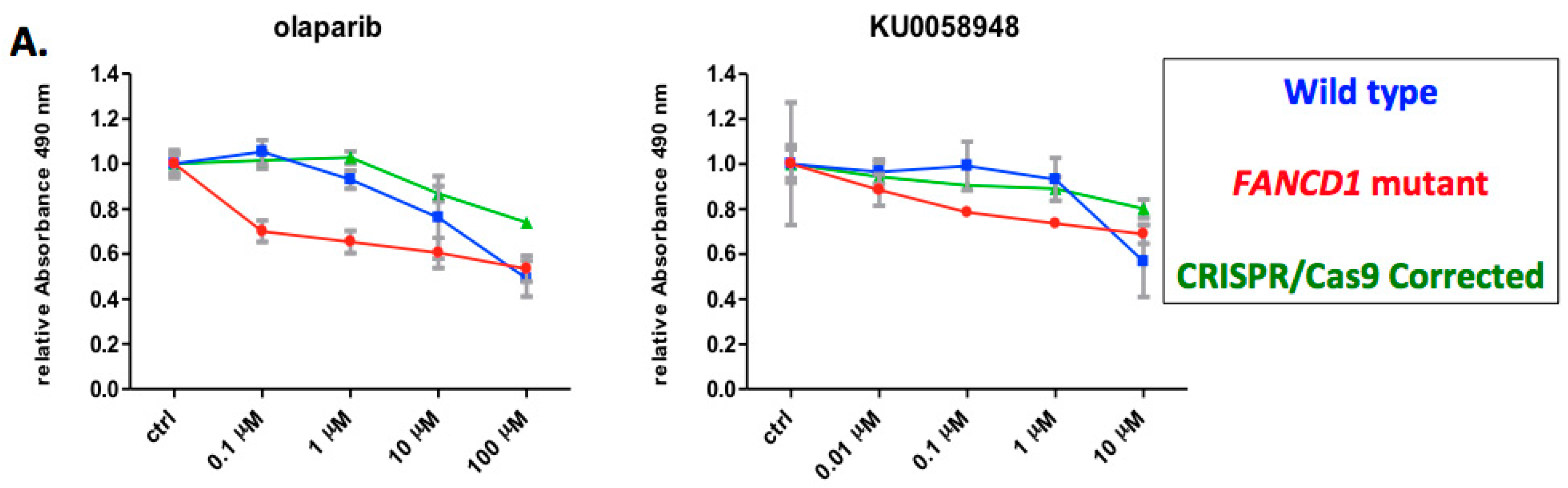
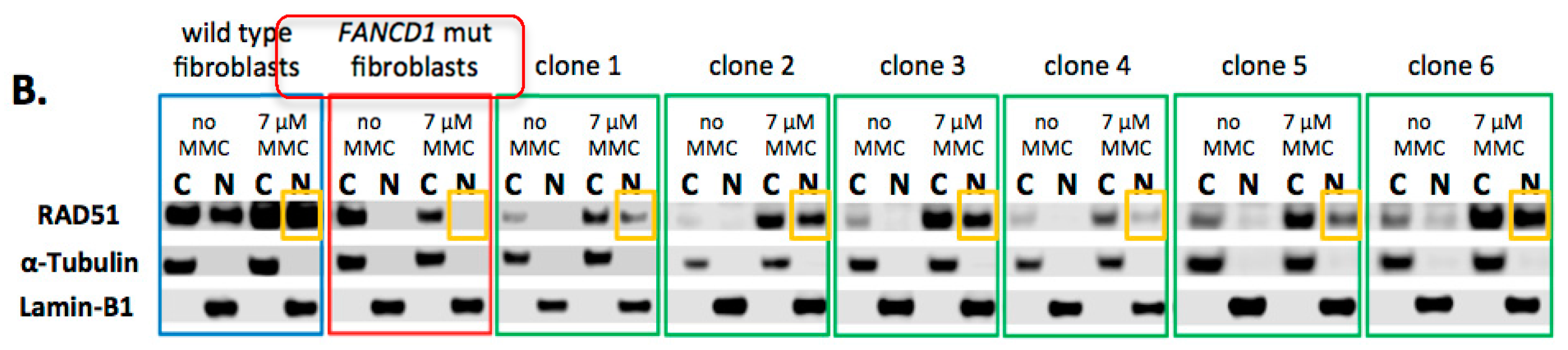
2.3. Phenotypic Rescue of Gene Corrected Primary Cells
2.4. CRISPR/Cas9 off Target Analysis
3. Discussion
4. Materials and Methods
4.1. Patient Samples
4.2. Culture Conditions
4.3. CRISPR/Cas9 Design
4.4. Gene Editing in Primary Cells
4.5. Selection
4.6. Off-Target Analysis
| FANCD1 surveyor F | AAACTTTATCACAGGGTATGTGCTT |
| FANCD1 surveyor R | CAGCATCATCTGACTTTCCAA |
| FANCD1 OT 1 Surveyor F | CCAGACCAGAAACCGAAAAA |
| FANCD1 OT 1 Surveyor R | TGGCAGTTTGTCCATTTGAA |
| FANCD1 OT 2 Surveyor F | CATCCTGAAAAATGATGGGATT |
| FANCD1 OT 2 Surveyor R | ATCTTCCTCCCTTCCTCCTG |
| FANCD1 OT 3 Surveyor F | CCCCCAACTACATTCGAAAA |
| FANCD1 OT 3 Surveyor R | AATTTGGTGGGTTCTACTTGTTT |
| FANCD1 OT 4 Surveyor F | TGAACGTCAGAAGGGCTAGAA |
| FANCD1 OT 4 Surveyor R | GACGTCAAGGTTGCAGTGAA |
| FANCD1 OT 5 Surveyor F | GGCCAGTGGTTCTCAACTTT |
| FANCD1 OT 5 Surveyor R | TGTTCCCATGAGTTTTGTGG |
| FANCD1 OT 6 Surveyor F | ACAAACTGCCGAACAAGAGG |
| FANCD1 OT 6 Surveyor R | AGGCTGAGTGGTACTCCATTG |
| FANCD1 OT 7 Surveyor F | TTGTGAATGAGGTGAGATGAGG |
| FANCD1 OT 7 Surveyor R | GATCTTGGCTCACTGCAACC |
| FANCD1 OT 8 Surveyor F | CATATTGTCTGGGTGCCACA |
| FANCD1 OT 8 Surveyor R | TCACCACAACCCCATAAAGC |
4.7. FANCD1 Functional Assay
4.8. Traffic Light Reporter Assay
4.9. Positive and Negative Strand Oligonucleotide Donor Assessment
4.10. Graphics
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| FA | Fanconi anemia |
| CRISPR/Cas9 | clustered regularly interspaced short palindromic repeats/Cas9 |
| BMF | bone marrow failure |
| ZFN | zinc-finger nucleases |
| TALEN | transcription activator-like effector nucleases |
| PARP | poly(ADP-ribose) polymerase |
| PARPi | PARP inhibition |
| gRNA | guide RNA |
| NHEJ | non-homologous endjoining |
| ODN | oligonucleotide donor |
| SNP | single-nucleotide polymorphisms |
| HDR | Homology directed repair |
| MMC | mitomycin-C |
| TERT | telomerase reverse transcriptase |
| HSPC | hematopoietic stem and progenitor cells |
| iPSC | Induced pluripotent stem cells |
References
- Bagby, G.C., Jr. Genetic basis of Fanconi anemia. Curr. Opin. Hematol. 2003, 10, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Joenje, H.; Patel, K.J. The emerging genetic and molecular basis of Fanconi anaemia. Nat. Rev. 2001, 2, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Cattoglio, C.; Facchini, G.; Sartori, D.; Antonelli, A.; Miccio, A.; Cassani, B.; Schmidt, M.; von Kalle, C.; Howe, S.; Thrasher, A.J.; et al. Hot spots of retroviral integration in human CD34+ hematopoietic cells. Blood 2007, 110, 1770–1778. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, R.; Eckenberg, R.; Paruzynski, A.; Bartholomae, C.C.; Nowrouzi, A.; Arens, A.; Howe, S.J.; Recchia, A.; Cattoglio, C.; Wang, W.; et al. Comprehensive genomic access to vector integration in clinical gene therapy. Nat. Med. 2009, 15, 1431–1436. [Google Scholar] [CrossRef] [PubMed]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Bartholomae, C.C.; Ranzani, M.; Benedicenti, F.; Sergi, L.S.; Ambrosi, A.; Ponzoni, M.; et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J. Clin. Investig. 2009, 119, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Stephen, S.L.; Montini, E.; Sivanandam, V.G.; Al-Dhalimy, M.; Kestler, H.A.; Finegold, M.; Grompe, M.; Kochanek, S. Chromosomal integration of adenoviral vector DNA in vivo. J. Virol. 2010, 84, 9987–9994. [Google Scholar] [CrossRef] [PubMed]
- Yant, S.R.; Wu, X.; Huang, Y.; Garrison, B.; Burgess, S.M.; Kay, M.A. High-resolution genome-wide mapping of transposon integration in mammals. Mol. Cell. Biol. 2005, 25, 2085–2094. [Google Scholar] [CrossRef] [PubMed]
- Porteus, M.H.; Carroll, D. Gene targeting using zinc finger nucleases. Nat. Biotechnol. 2005, 23, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Mussolino, C.; Alzubi, J.; Fine, E.J.; Morbitzer, R.; Cradick, T.J.; Lahaye, T.; Bao, G.; Cathomen, T. Talens facilitate targeted genome editing in human cells with high specificity and low cytotoxicity. Nucleic Acids Res. 2014, 42, 6762–6773. [Google Scholar] [CrossRef] [PubMed]
- Cermak, T.; Doyle, E.L.; Christian, M.; Wang, L.; Zhang, Y.; Schmidt, C.; Baller, J.A.; Somia, N.V.; Bogdanove, A.J.; Voytas, D.F. Efficient design and assembly of custom talen and other tal effector-based constructs for DNA targeting. Nucleic Acids Res. 2011, 39, e82. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Rio, P.; Banos, R.; Lombardo, A.; Quintana-Bustamante, O.; Alvarez, L.; Garate, Z.; Genovese, P.; Almarza, E.; Valeri, A.; Diez, B.; et al. Targeted gene therapy and cell reprogramming in Fanconi anemia. EMBO Mol. Med. 2014, 6, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Osborn, M.; Gabriel, R.; Webber, B.R.; DeFeo, A.P.; McElroy, A.N.; Jarjour, J.; Starker, C.G.; Wagner, J.E.; Joung, J.K.; Voytas, D.F.; et al. Fanconi anemia gene editing by the crispr/cas9 system. Human Gene Ther. 2014, 26, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Osborn, M.; Lonetree, C.L.; Webber, B.R.; Patel, D.; Dunmire, S.; McElroy, A.N.; DeFeo, A.P.; MacMillan, M.L.; Wagner, J.; Balzar, B.R.; et al. CRISPR/Cas9 targeted gene editing and cellular engineering in Fanconi anemia. Stem Cells Dev. 2016, 25, 1591–1603. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhang, J.; Yu, H.; Fei, P. Advances in the understanding of Fanconi anemia complementation group D2 protein (Fancd2) in human cancer. Cancer Cell. Microenviron. 2015, 2, e986. [Google Scholar] [PubMed]
- Longerich, S.; Li, J.; Xiong, Y.; Sung, P.; Kupfer, G.M. Stress and DNA repair biology of the Fanconi anemia pathway. Blood 2014, 124, 2812–2819. [Google Scholar] [CrossRef] [PubMed]
- Kottemann, M.C.; Smogorzewska, A. Fanconi anaemia and the repair of watson and crick DNA crosslinks. Nature 2013, 493, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.; Davies, S.M.; Harris, R.E.; Spunt, S.L.; Smolarek, T.; Zimmerman, S.; McMasters, R.; Wagner, L.; Mueller, R.; Auerbach, A.D.; et al. The clinical phenotype of children with Fanconi anemia caused by biallelic Fancd1/BRCA2 mutations. Pediatr. Blood Cancer 2012, 58, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Howlett, N.G.; Taniguchi, T.; Olson, S.; Cox, B.; Waisfisz, Q.; de Die-Smulders, C.; Persky, N.; Grompe, M.; Joenje, H.; Pals, G.; et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science 2002, 297, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Tutt, A.N.; Ashworth, A. Synthetic lethality and cancer therapy: Lessons learned from the development of parp inhibitors. Annu. Rev. Med. 2015, 66, 455–470. [Google Scholar] [CrossRef] [PubMed]
- Guschin, D.Y.; Waite, A.J.; Katibah, G.E.; Miller, J.C.; Holmes, M.C.; Rebar, E.J. A rapid and general assay for monitoring endogenous gene modification. In Engineered Zinc Finger Proteins; Mackay, J.P., Segal, D.J., Eds.; Springer: New York, NY, USA, 2010; Volume 649, pp. 247–256. [Google Scholar]
- Certo, M.T.; Ryu, B.Y.; Annis, J.E.; Garibov, M.; Jarjour, J.; Rawlings, D.J.; Scharenberg, A.M. Tracking genome engineering outcome at individual DNA breakpoints. Nat. Methods 2011, 8, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Renaud, J.B.; Boix, C.; Charpentier, M.; De Cian, A.; Cochennec, J.; Duvernois-Berthet, E.; Perrouault, L.; Tesson, L.; Edouard, J.; Thinard, R.; et al. Improved genome editing efficiency and flexibility using modified oligonucleotides with Talen and CRISPR-Cas9 nucleases. Cell Rep. 2016, 14, 2263–2272. [Google Scholar] [CrossRef] [PubMed]
- Godthelp, B.C.; Artwert, F.; Joenje, H.; Zdzienicka, M.Z. Impaired DNA damage-induced nuclear Rad51 Foci formation uniquely characterizes Fanconi anemia group D1. Oncogene 2002, 21, 5002–5005. [Google Scholar] [CrossRef] [PubMed]
- Digweed, M.; Rothe, S.; Demuth, I.; Scholz, R.; Schindler, D.; Stumm, M.; Grompe, M.; Jordan, A.; Sperling, K. Attenuation of the formation of DNA-repair foci containing Rad51 in Fanconi anaemia. Carcinogenesis 2002, 23, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Moreno, A.; Carrington, J.T.; Albergante, L.; Al Mamun, M.; Haagensen, E.J.; Komseli, E.S.; Gorgoulis, V.G.; Newman, T.J.; Blow, J.J. Unreplicated DNA remaining from unperturbed s phases passes through mitosis for resolution in daughter cells. Proc. Natl. Acad. Sci. USA 2016, 113, E5757–E5764. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, B.; Shimamura, A.; Moreau, L.; Baldinger, S.; Hag-alshiekh, M.; Bostrom, B.; Sencer, S.; D’Andrea, A.D. Association of biallelic BRCA2/FANCD1 mutations with spontaneous chromosomal instability and solid tumors of childhood. Blood 2004, 103, 2554–2559. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.; Maizels, N. Homology-directed repair of DNA nicks via pathways distinct from canonical double-strand break repair. Proc. Natl. Acad. Sci. USA 2014, 111, E924–E932. [Google Scholar] [CrossRef] [PubMed]
- Bialk, P.; Rivera-Torres, N.; Strouse, B.; Kmiec, E.B. Regulation of gene editing activity directed by single-stranded oligonucleotides and CRISPR/Cas9 systems. PLoS ONE 2015, 10, e0129308. [Google Scholar] [CrossRef] [PubMed]
- Cerrato, A.; Morra, F.; Celetti, A. Use of poly adp-ribose polymerase [PARP] inhibitors in cancer cells bearing ddr defects: The rationale for their inclusion in the clinic. J. Exp. Clin. Cancer Res. 2016, 35, 179. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Curtin, N.J. The role of parp in DNA repair and its therapeutic exploitation. Br. J. Cancer 2011, 105, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, A.; Ear, U.S.; Koller, B.H.; Weichselbaum, R.R.; Bishop, D.K. The breast cancer susceptibility gene BRCA1 is required for subnuclear assembly of Rad51 and survival following treatment with the DNA cross-linking agent cisplatin. J. Biol. Chem. 2000, 275, 23899–23903. [Google Scholar] [CrossRef] [PubMed]
- Esposito, M.T.; Zhao, L.; Fung, T.K.; Rane, J.K.; Wilson, A.; Martin, N.; Gil, J.; Leung, A.Y.; Ashworth, A.; So, C.W. Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors. Nat. Med. 2015, 21, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechonol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Anur, P.; Friedman, D.N.; Sklar, C.; Oeffinger, K.; Castiel, M.; Kearney, J.; Singh, B.; Prockop, S.E.; Kernan, N.A.; Scaradavou, A.; et al. Late effects in patients with Fanconi anemia following allogeneic hematopoietic stem cell transplantation from alternative donors. Bone Marrow Transplant. 2016, 51, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.A.; Davies, S.M.; Leemhuis, T.; Myers, K.; Kernan, N.A.; Prockop, S.E.; Scaradavou, A.; O’Reilly, R.J.; Williams, D.A.; Lehmann, L.; et al. Radiation-free, alternative-donor HCT for Fanconi anemia patients: Results from a prospective multi-institutional study. Blood 2017, 129, 2308–2315. [Google Scholar] [CrossRef] [PubMed]
- MacMillan, M.L.; DeFor, T.E.; Young, J.A.; Dusenbery, K.E.; Blazar, B.R.; Slungaard, A.; Zierhut, H.; Weisdorf, D.J.; Wagner, J.E. Alternative donor hematopoietic cell transplantation for Fanconi anemia. Blood 2015, 125, 3798–3804. [Google Scholar] [CrossRef] [PubMed]
- Adair, J.E.; Sevilla, J.; de Heredia, C.D.; Becker, P.S.; Kiem, H.P.; Bueren, J. Lessons learned from two decades of clinical trial experience in gene therapy for Fanconi anemia. Curr. Gene Ther. 2017, 16, 338–348. [Google Scholar] [CrossRef]
- Adair, J.E.; Becker, P.S.; Chandrasekaran, D.; Choi, G.; Woolfrey, A.E.; Burroughs, L.; Kiem, H.P. Gene therapy for Fanconi anemia in seattle: Clinical experience and next steps. Blood 2016, 128, 3510. [Google Scholar]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. Lmo2-associated clonal t cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Cumming, R.C.; Liu, J.M.; Youssoufian, H.; Buchwald, M. Suppression of apoptosis in hematopoietic factor-dependent progenitor cell lines by expression of the FAC gene. Blood 1996, 88, 4558–4567. [Google Scholar] [PubMed]
- Muller, L.U.; Milsom, M.D.; Harris, C.E.; Vyas, R.; Brumme, K.M.; Parmar, K.; Moreau, L.A.; Schambach, A.; Park, I.H.; London, W.B.; et al. Overcoming reprogramming resistance of Fanconi anemia cells. Blood 2012, 119, 5449–5457. [Google Scholar] [CrossRef] [PubMed]
- Sugimura, R.; Jha, D.K.; Han, A.; Soria-Valles, C.; da Rocha, E.L.; Lu, Y.F.; Goettel, J.A.; Serrao, E.; Rowe, R.G.; Malleshaiah, M.; et al. Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature 2017, 545, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Soulier, J.; Leblanc, T.; Larghero, J.; Dastot, H.; Shimamura, A.; Guardiola, P.; Esperou, H.; Ferry, C.; Jubert, C.; Feugeas, J.P.; et al. Detection of somatic mosaicism and classification of Fanconi anemia patients by analysis of the FA/BRCA pathway. Blood 2005, 105, 1329–1336. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.; Hanenberg, H.; Lobitz, S.; Friedl, R.; Herterich, S.; Dietrich, R.; Gruhn, B.; Schindler, D.; Hoehn, H. Reverse mosaicism in Fanconi anemia: Natural gene therapy via molecular self-correction. Cytogenet. Genome Res. 2002, 98, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Gregory, J.J., Jr.; Wagner, J.E.; Verlander, P.C.; Levran, O.; Batish, S.D.; Eide, C.R.; Steffenhagen, A.; Hirsch, B.; Auerbach, A.D. Somatic mosaicism in Fanconi anemia: Evidence of genotypic reversion in lymphohematopoietic stem cells. Proc. Natl. Acad. Sci. USA 2001, 98, 2532–2537. [Google Scholar] [CrossRef] [PubMed]
- Lo Ten Foe, J.R.; Kwee, M.L.; Rooimans, M.A.; Oostra, A.B.; Veerman, A.J.; van Weel, M.; Pauli, R.M.; Shahidi, N.T.; Dokal, I.; Roberts, I.; et al. Somatic mosaicism in fanconi anemia: Molecular basis and clinical significance. Eur J. Hum. Genet. 1997, 5, 137–148. [Google Scholar] [PubMed]
- Elliott, B.; Richardson, C.; Winderbaum, J.; Nickoloff, J.A.; Jasin, M. Gene conversion tracts from double-strand break repair in mammalian cells. Mol. Cellular Biol. 1998, 18, 93–101. [Google Scholar] [CrossRef]
- Urnov, F.D.; Miller, J.C.; Lee, Y.L.; Beausejour, C.M.; Rock, J.M.; Augustus, S.; Jamieson, A.C.; Porteus, M.H.; Gregory, P.D.; Holmes, M.C. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 2005, 435, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Topkar, V.V.; Zheng, Z.; Joung, J.K. Broadening the targeting range of staphylococcus aureus CRISPR-Cas9 by modifying pam recognition. Nat. Biotechnol. 2015, 33, 1293–1298. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Topkar, V.V.; Nguyen, N.T.; Zheng, Z.; Gonzales, A.P.; Li, Z.; Peterson, R.T.; Yeh, J.J.; et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 2015, 523, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Haeussler, M.; Schonig, K.; Eckert, H.; Eschstruth, A.; Mianne, J.; Renaud, J.B.; Schneider-Maunoury, S.; Shkumatava, A.; Teboul, L.; Kent, J.; et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016, 17, 148. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Off Target Candidate | 5′-Target Sequence-3′ | Gene | Mismatches |
|---|---|---|---|
| CTTACAGCAGTAGTATCATGAGG | FANCD1 | X | |
| 1 | TTTGCAGGAGCAGTATCATGAAG | GATA3 | 4 |
| 2 | CTTACAGTACTAGCATCATGGGG | AFF2 | 3 |
| 3 | CTTACTGAAGTAGTCTCAGGAAG | PATL1 | 5 |
| 4 | CTTACAGAAGTATTATCACCGGG | MFAP1 | 5 |
| 5 | TTTACAGCAGCAGTAGAATGGAG | NUMB | 6 |
| 6 | CTTACTGCAGAAGTTTCATCCAG | TCERG1 | 6 |
| 7 | CGCACAGCAGTAGCATCCTGGAG | USP54 | 6 |
| 8 | CTTGCAGCAGGAGGATCGTGCAG | SPON1 | 6 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skvarova Kramarzova, K.; Osborn, M.J.; Webber, B.R.; DeFeo, A.P.; McElroy, A.N.; Kim, C.J.; Tolar, J. CRISPR/Cas9-Mediated Correction of the FANCD1 Gene in Primary Patient Cells. Int. J. Mol. Sci. 2017, 18, 1269. https://doi.org/10.3390/ijms18061269
Skvarova Kramarzova K, Osborn MJ, Webber BR, DeFeo AP, McElroy AN, Kim CJ, Tolar J. CRISPR/Cas9-Mediated Correction of the FANCD1 Gene in Primary Patient Cells. International Journal of Molecular Sciences. 2017; 18(6):1269. https://doi.org/10.3390/ijms18061269
Chicago/Turabian StyleSkvarova Kramarzova, Karolina, Mark J. Osborn, Beau R. Webber, Anthony P. DeFeo, Amber N. McElroy, Chong Jai Kim, and Jakub Tolar. 2017. "CRISPR/Cas9-Mediated Correction of the FANCD1 Gene in Primary Patient Cells" International Journal of Molecular Sciences 18, no. 6: 1269. https://doi.org/10.3390/ijms18061269
APA StyleSkvarova Kramarzova, K., Osborn, M. J., Webber, B. R., DeFeo, A. P., McElroy, A. N., Kim, C. J., & Tolar, J. (2017). CRISPR/Cas9-Mediated Correction of the FANCD1 Gene in Primary Patient Cells. International Journal of Molecular Sciences, 18(6), 1269. https://doi.org/10.3390/ijms18061269