
Personalized Medicine: New Perspectives for the Diagnosis and the Treatment of Renal Diseases




Abstract
:
{kind=link}
{kind=link}
1. Introduction
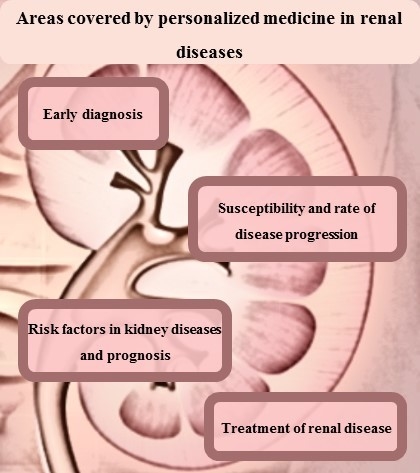
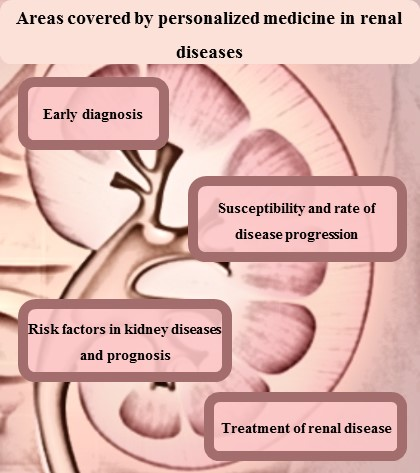
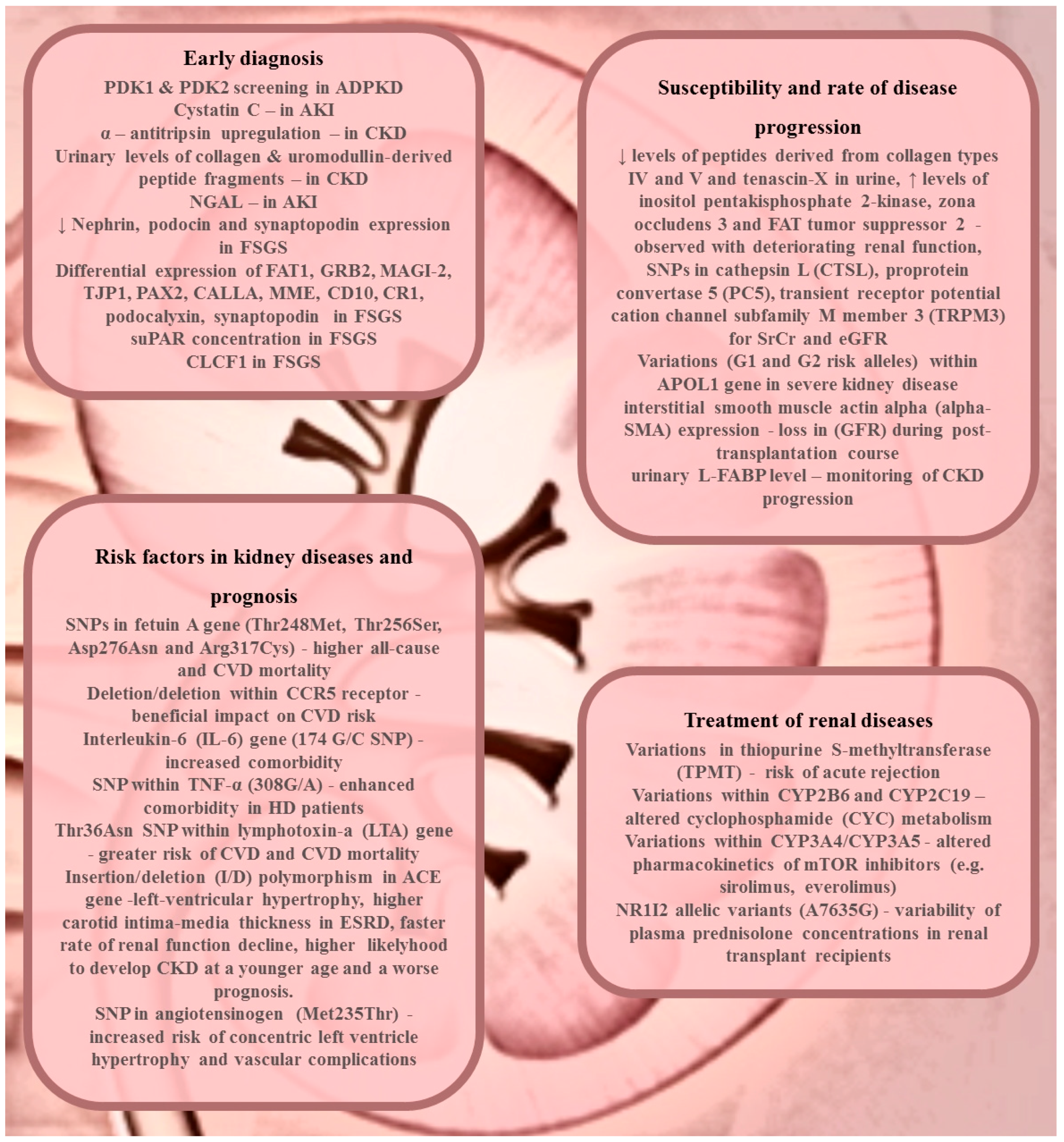
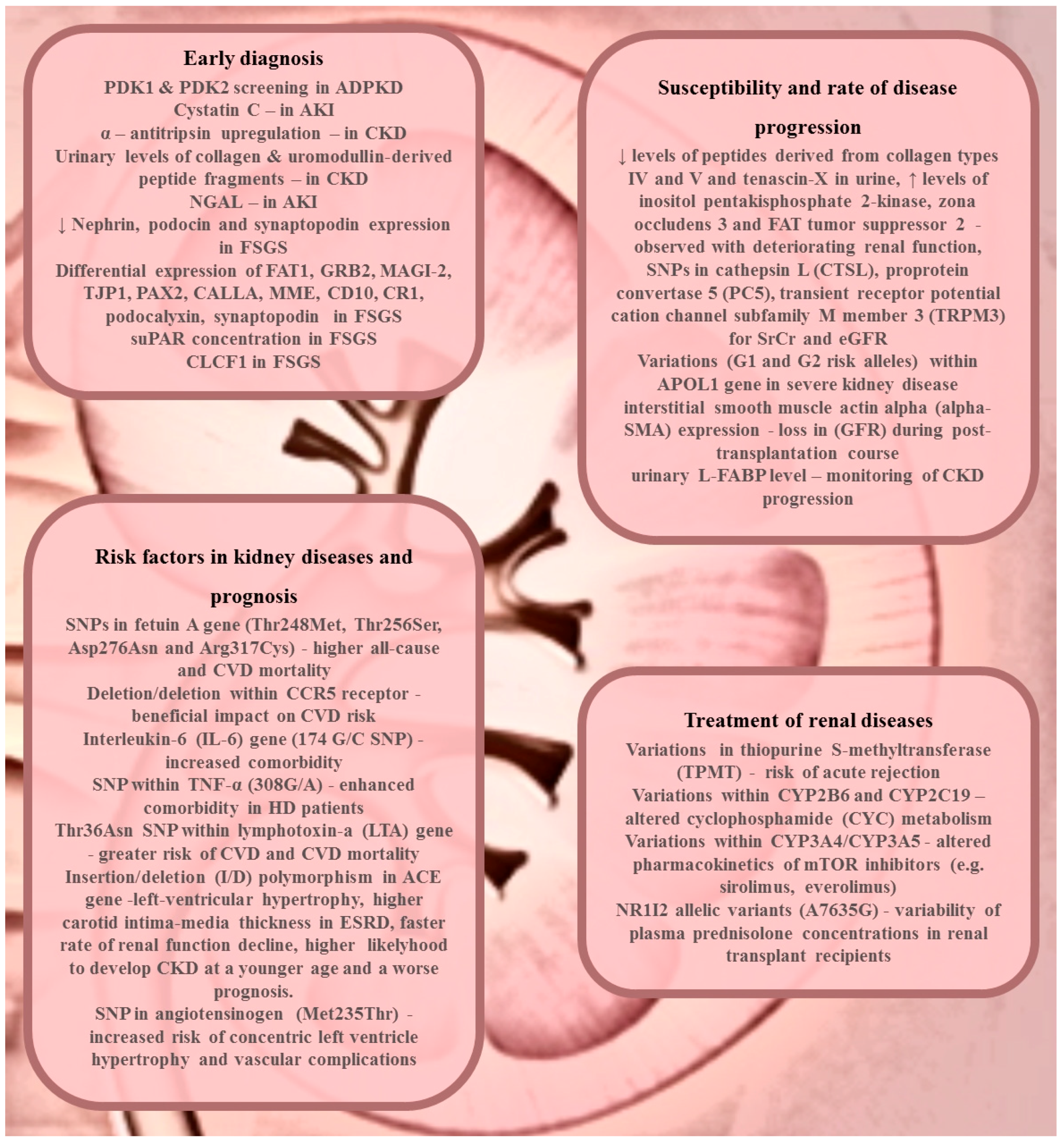
2. Renal Disease Diagnosis or Early Detection
3. Susceptibility and Rate of Disease Progression
4. Treatment of Renal Diseases
5. Risk Factors in Kidney Diseases and Prognosis
6. Financial Aspect
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Padullés, A.; Rama, I.; Llaudó, I.; Lloberas, N. Developments in renal pharmacogenomics and applications in chronic kidney disease. Pharm. Pers. Med. 2014, 7, 251–266. [Google Scholar]
- Coresh, J.; Selvin, E.; Stevens, L.A. Prevalence of chronic kidney disease in the United States. J. Am. Med. Assoc. 2007, 298, 2038–2047. [Google Scholar] [CrossRef] [PubMed]
- Hallan, S.I.; Coresh, J.; Astor, B.C. International comparison of the relationship of chronic kidney disease prevalence and ESRD risk. J. Am. Soc. Nephrol. 2006, 17, 2275–2284. [Google Scholar] [CrossRef] [PubMed]
- Schleidgen, S.; Klingler, C.; Bertram, T.; Rogowski, W.H.; Marckmann, G. What is personalized medicine: Sharpening a vague term based on a systematic literature review. BMC Med. Ethics 2013, 14, 55. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.K. Textbook of Personalised Medicine; Springer: Dordrecht, The Netherlands, 2009. [Google Scholar]
- Collins, F.S.; Lander, E.S.; Rogers, J.; Waterston, R.H. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar]
- Agyeman, A.A.; Ofori-Asenso, R. Perspective: Does personalized medicine hold the future for medicine? J. Pharm. Bioallied. Sci. 2015, 7, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, R.S.; Herman, J.G.; McCaffrey, T.; Raj, D.S.C. Beyond genetics: Epigenetic code in chronic kidney disease. Kidney Int. 2011. [Google Scholar] [CrossRef] [PubMed]
- Callinan, P.A.; Feinberg, A.P. The emerging science of epigenomics. Hum. Mol. Genet. 2006, 15, R95–R101. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Vogelstein, B.; Velculescu, V.E.; Papadopoulos, N.; Kinzler, K.W. The antisense transcriptomes of human cells. Science 2008, 322, 1855–1857. [Google Scholar] [CrossRef] [PubMed]
- Garfield, D.A.; Wray, G.A. The Evolution of Gene Regulatory Interactions. BioScience 2010, 60, 15–23. [Google Scholar] [CrossRef]
- Foroutan, B. Personalized Medicine: A Review with Regard to Biomarkers. J. Bioequiv. Availab. 2015, 7, 244–256. [Google Scholar] [CrossRef]
- Gibbs, R.A.; Belmont, J.W.; Boudreau, A.; Leal, S.M.; Hardenbol, P.; Pasternak, S.; Wheeler, D.A.; Willis, T.D.; Yu, F.; Yang, H.; et al. A haplotype map of the human genome. Nature 2005, 437, 1299–1320. [Google Scholar]
- Personal Genome Project. Volunteers from the General Public Working Together with Researchers to Advance Personal Genomics 2013. Available online: http://www.personalgenomes.org (accessed on 5 May 2017).
- NHGRI. Genome-Wide Association Studies 2013. Available online: http://www.genome.gov/20019523 (accessed on 3 May 2017).
- ENCODE Project Consortium. The ENCODE (ENCyclopedia of DNA Elements) Project. Science 2004, 306, 636–640. [Google Scholar]
- Siva, N. 1000 Genomes project. Nat. Biotechnol. 2008, 26, 256. [Google Scholar] [PubMed]
- Kaiser, J. A plan to capture human diversity in 1000 genomes. Science 2008, 319, 395. [Google Scholar] [CrossRef] [PubMed]
- The 1000 Genomes Project Consortium. An integrated map of genetic variation from 1092 human genomes. Nature 2012, 491, 56–65. [Google Scholar]
- Business Wire. The University of Cambridge, Cancer Research Technology, Cancer Research UK and Perlegen Sciences Collaborate to Analyze Thousands of DNA Samples from Breast Cancer Patients 2005. Available online: http://www.businesswire.com/news/home/20050215006276/en/University-Cambridge-Cancer-Research-Technology-Cancer-Research (accessed on 9 August 2013).
- Farlex Perlegen Sciences to Analyze Genetics of Common Diseases in Postmenopausal Women; Collaboration with Women’s Health Initiative Funded by the National Institutes of Health 2005. Available online: http://www.businesswire.com/news/home/20050630005199/en/Perlegen-Sciences-Analyze-Genetics-Common-Diseases-Postmenopausal#VW2J50ZJUdU (accessed on 2 May 2013).
- Gerdes, M.J.; Sood, A.; Sevinsky, C.; Pris, A.D.; Zavodszky, M.I.; Ginty, F. Emerging understanding of multiscale tumor heterogeneity. Front. Oncol. 2014, 4, 366. [Google Scholar] [CrossRef] [PubMed]
- Khella, H.W.Z.; Bakhet, M.; Lichner, Z.; Romaschin, A.D.; Jewett, M.A.S.; Yousef, G.M. MicroRNAs in kidney disease: An emerging understanding. Am. J. Kidney Dis. 2013, 61, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Brennan, E.P.; Morine, M.J.; Walsh, D.W.; Roxburgh, S.A.; Lindenmeyer, M.T.; Brazil, D.P.; Gaora, P.Ó.; Roche, H.M.; Sadlier, D.M.; Cohen, C.D.; et al. Next-generation sequencing identifies TGF-β1-associated gene expression profiles in renal epithelial cells reiterated in human diabetic nephropathy. Biochim. Biophys. Acta 2012, 1822, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Albert, G.I.; Schell, C.; Kirschner, K.M.; Schäfer, S.; Naumann, R.; Müller, A.; Kretz, O.; Kuropka, B.; Girbig, M.; Hübner, N.; et al. The GYF domain protein CD2BP2 is critical for embryogenesis and podocyte function. J. Mol. Cell Biol. 2015, 7, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Yoshigoe, K.; Qin, X.; Liu, J.S.; Yang, J.Y.; Niemierko, A.; Deng, Y.; Liu, Y.; Dunker, A.K.; Chen, Z.; et al. Identification of genes and pathways involved in kidney renal clear cell carcinoma. BMC Bioinform. 2014, 15, S2. [Google Scholar] [CrossRef] [PubMed]
- Akkina, S.; Becker, B.N. MicroRNAs in kidney function and disease. Translat. Res. 2011, 157, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Kasinath, B.S.; Feliers, D. The complex world of kidney microRNAs. Kidney Int. 2011, 80, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Qin, S.; Ho, J. Systems biology approach to identify transcriptome reprogramming and candidate microRNA targets during the progression of polycystic kidney disease. BMC Syst. Biol. 2011, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.L.; DiStefano, J.K. The role of non-coding RNAs in diabetic nephropathy: Potential applications as biomarkers for disease development and progression. Diabetes Res. Clin. Pract. 2013, 99, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Nassirpour, R.; Mathur, S.; Gosink, M.M.; Li, Y.; Shoieb, A.M.; Wood, J.; O’Neil, S.P.; Homer, B.L.; Whiteley, L.O. Identification of tubular injury microRNA biomarkers in urine: Comparison of next-generation sequencing and qPCR-based profiling platforms. BMC Genom. 2014, 15, 485. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.G.; Pollock, C.A. Biomarkers in kidney fibrosis: Are they useful? Kidney Int. Suppl. 2014, 4, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Trujillano, D.; Bullich, G.; Ossowski, S.; Ballarín, J.; Torra, R.; Estivill, X.; Ars, E. Diagnosis of autosomal dominant polycystic kidney disease using efficient PKD1 and PKD2 targeted next-generation sequencing. Mol. Genet. Genom. Med. 2014, 2, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Porath, B.; Gainullin, V.G.; Cornec-Le Gall, E.; Dillinger, E.K.; Heyer, C.M.; Hopp, K.; Edwards, M.E.; Madsen, C.D.; Mauritz, S.R.; Banks, C.J.; et al. Mutations in GANAB, Encoding the Glucosidase IIα Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease. Am. J. Hum. Genet. 2016, 98, 1193–1207. [Google Scholar] [CrossRef] [PubMed]
- Helal, I.; Reed, B.; McFann, K.; Yan, X.D.; Fick-Brosnahan, G.M.; Cadnapaphornchai, M.; Schrier, R.W. Glomerular hyperfiltration and renal progression in children with autosomal dominant polycystic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 2439–2443. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Chong, J.; Ong, A.C.M. Autosomal dominant polycystic kidney disease: Recent advances in clinical management. F1000Research 2016, 5, 2029. [Google Scholar] [CrossRef] [PubMed]
- Good, D.M.; Zürbig, P.; Argilés, A.; Bauer, H.W.; Behrens, G.; Coon, J.J.; Dakna, M.; Decramer, S.; Delles, C.; Dominiczak, A.F.; et al. Naturally occurring human urinary peptides for use in diagnosis of chronic kidney disease. Mol. Cell. Proteom. 2010, 9, 2424–2437. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; Greene, T.; Beck, G.J.; Caggiula, A.W.; Kusek, J.W.; Hunsicker, L.G.; Klahr, S. Dietary protein restriction and the progression of chronic renal disease: What have all of the results of the MDRD study shown? Modification of Diet in Renal Disease Study group. J. Am. Soc. Nephrol. 1999, 10, 2426–2439. [Google Scholar] [PubMed]
- Vriesendorp, R.; Donker, A.J.; de Zeeuw, D.; de Jong, P.E.; van der Hem, G.K.; Brentjens, J.R. Effects of non-steroidal anti-inflammatory drugs on proteinuria. Am. J. Med. 1986, 81, 84–94. [Google Scholar] [CrossRef]
- Lv, J.; Xu, D.; Perkovic, V.; Ma, X.; Johnson, D.W.; Woodward, M.; Levin, A.; Zhang, H.; Wang, H. Corticosteroid therapy in IgA nephropathy. J. Am. Soc. Nephrol. 2012, 23, 1108–1116. [Google Scholar] [CrossRef] [PubMed]
- Lambers Heerspink, H.J.; de Zeeuw, D. Novel drugs and intervention strategies for the treatment of chronic kidney disease. Br. J. Clin. Pharmacol. 2013, 76, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Herget-Rosenthal, S.; Marggraf, G.; Husing, J.; Goring, F.; Pietruck, F.; Janssen, O.; Philipp, T.; Kribben, A. Early detection of acute renal failure by serum cystatin C. Kidney Int. 2004, 66, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Pollock, A.S.; Mahimkar, R.; Olson, J.L.; Lovett, D.H. Matrix metalloproteinase 2 and basement membrane integrity: A unifying mechanism for progressive renal injury. FASEB J. 2006, 20, 1898–1900. [Google Scholar] [CrossRef] [PubMed]
- Varghese, S.A.; Powell, T.B.; Budisavljevic, M.N.; Oates, J.C.; Raymond, J.R.; Almeida, J.S.; Arthur, J.M. Urine biomarkers predict the cause of glomerular disease. J. Am. Soc. Nephrol. 2007, 18, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.; Dent, C.L.; Ma, Q.; Dastrala, S.; Grenier, F.; Workman, R.; Syed, H.; Ali, S.; Barasch, J.; Devarajan, P. Urine NGAL predicts severity of acute kidney injury after cardiac surgery: A prospective study. Clin. J. Am. Soc. Nephrol. 2008, 3, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Dent, C.L.; Ma, Q.; Dastrala, S.; Bennett, M.; Mitsnefes, M.M.; Barasch, J.; Devarajan, P. Plasma neutrophil gelatinase-associated lipocalin predicts acute kidney injury, morbidity and mortality after pediatric cardiac surgery: A prospective uncontrolled cohort study. Crit. Care 2007, 11, R127. [Google Scholar] [CrossRef] [PubMed]
- Mishra, J.; Ma, Q.; Prada, A.; Mitsnefes, M.; Zahedi, K.; Yang, J.; Barasch, J.; Devarajan, P. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J. Am. Soc. Nephrol. 2003, 14, 2534–2543. [Google Scholar] [CrossRef] [PubMed]
- Mishra, J.; Dent, C.; Tarabishi, R.; Mitsnefes, M.M.; Ma, Q.; Kelly, C.; Ruff, S.M.; Zahedi, K.; Shao, M.; Bean, J.; et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet 2005, 365, 1231–1238. [Google Scholar] [CrossRef]
- McIlroy, D.R.; Wagener, G.; Lee, H.T. Neutrophil Gelatinase-Associated Lipocalin and Acute Kidney Injury after Cardiac Surgery: The Effect of Baseline Renal Function on Diagnostic Performance. Clin. J. Am. Soc. Nephrol. 2010, 5, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Hodgin, J.B.; Borczuk, A.C.; Nasr, S.H.; Markowitz, G.S.; Nair, V. A Molecular Profile of Focal Segmental Glomerulosclerosis from Formalin-Fixed, Paraffin-Embedded Tissue. Am. J. Pathol. 2010, 177, 1674–1686. [Google Scholar] [CrossRef] [PubMed]
- Schwab, K.; Witte, D.; Aronow, B.; Devarajan, P.; Potter, S.S.; Patterson, L.T. Microarray analysis of focal segmental glomerulosclerosis. Am. J. Nephrol. 2004, 24, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; El Hindi, S.; Li, J.; Fornoni, A.; Goes, N.; Sageshima, J.; Maiguel, D.; Karumanchi, S.A.; Yap, H.K.; Saleem, M.; et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat. Med. 2011, 17, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Maas, R.J.; Deegens, J.K.; Wetzels, J.F. Permeability factors in idiopathic nephrotic syndrome: Historical perspectives and lessons for the future. Nephrol. Dial. Transplant. 2014, 29, 2207–2216. [Google Scholar] [CrossRef] [PubMed]
- Maas, R.J.; Wetzels, J.F.; Deegens, J.K. Serum-soluble urokinase receptor concentration in primary FSGS. Kidney Int. 2012, 81, 1043–1044. [Google Scholar] [CrossRef] [PubMed]
- Cathelin, D.; Placier, S.; Ploug, M.; Verpont, M.C.; Vandermeersch, S.; Luque, Y.; Hertig, A.; Rondeau, E.; Mesnard, L. Administration of recombinant soluble urokinase receptor per se is not sufficient to induce podocyte alterations and proteinuria in mice. J. Am. Soc. Nephrol. 2014, 25, 1662–1668. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Zhou, J.; Gauchat, J.-F.; Sharma, R.; McCarthy, E.T.; Srivastava, T.; Savin, V.J. Janus kinase 2/signal transducer and activator of transcription 3 inhibitors attenuate the effect of cardiotrophin-like cytokine factor 1 and human focal segmental glomerulosclerosis serum on glomerular filtration barrier. Transl. Res. 2015, 166, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.P.; Cossey, L.N.; Beck, L.H. THSD7A staining of membranous glomerulopathy in clinical practice reveals cases with dual autoantibody positivity. Modern Pathol. 2016, 29, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Hoxha, E.; Thiele, I.; Zahner, G.; Panzer, U.; Harendza, S.; Stahl, R.A. Phospholipase 2 receptor autoantibiodies and clinical outcome in patients with primary membranous nephropathy. J. Am. Soc. Nephrol. 2014, 25, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Otu, H.H.; Can, H.; Spentzos, D.; Nelson, R.G.; Hanson, R.L.; Looker, H.C.; Knowler, W.C.; Monroy, M.; Libermann, T.A.; Karumanchi, S.A.; et al. Prediction of diabetic nephropathy using urine proteomic profiling 10 years prior to development of nephropathy. Diabetes Care 2007, 30, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.L.; Perkins, B.; Boratyn, G.M.; Ficociello, L.H.; Wilkey, D.W.; Barati, M.T.; Bertram, C.C.; Page, G.P.; Rovin, B.H.; Warram, J.H.; et al. Urinary peptidome may predict renal function decline in type 1 diabetes and microalbuminuria. J. Am. Soc. Nephrol. 2009, 20, 2065–2074. [Google Scholar] [CrossRef] [PubMed]
- Kottgen, A.; Kao, W.H.; Hwang, S.J.; Boerwinkle, E.; Yang, Q.; Levy, D.; Benjamin, E.J.; Larson, M.G.; Astor, B.C.; Coresh, J.; et al. Genome-wide association study for renal traits in the Framingham Heart and Atherosclerosis Risk in Communities Studies. BMC Med. Genet. 2008, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Arar, N.H.; Voruganti, V.S.; Nath, S.D.; Thameem, F.; Bauer, R.; Cole, S.A.; Blangero, J.; MacCluer, J.W.; Comuzzie, A.G.; Abboud, H.E. A genome-wide search for linkage to chronic kidney disease in a community-based sample: The SAFHS. Nephrol. Dial. Transplant. 2008, 23, 3184–3191. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Mok, A.; Miskie, B.; Hegele, R.A. Single-nucleotide polymorphisms of the proprotein convertase subtilisin/kexin type 5 (PCSK5) gene. J. Hum. Genet. 2001, 46, 730–732. [Google Scholar] [CrossRef] [PubMed]
- Kao, L.; Klag, M.J.; Meoni, L.A.; Reich, D.; Berthier-Schaad, Y.; Li, M.; Coresh, J.; Patterson, N.; Tandon, A.; Powe, N.R.; et al. A genome-wide admixture scan identifies MYH9 as a candidate locus associated with non-diabetic end stage renal disease in African Americans. Nat. Genet. 2008, 40, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.C.; Coresh, J.; Fornage, M.; Astor, B.C.; Grams, M.; Franceschini, N.; Boerwinkle, E.; Parekh, R.S.; Kao, W.H. APOL1 Variants Associate with Increased Risk of CKD among African Americans. JASN 2013, 24, 1484–1491. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Friedman, D.J.; Ross, M.D.; Lecordier, L.; Uzureau, P.; Freedman, B.I.; Bowden, D.W.; Langefeld, C.D.; Oleksyk, T.K.; Uscinski Knob, A.L.; et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 2010, 329, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Kopp, J.B.; Nelson, G.W.; Sampath, K.; Johnson, R.C.; Genovese, G.; An, P.; Friedman, D.; Briggs, W.; Dart, R.; Korbet, S.; et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J. Am. Soc. Nephrol. 2011, 22, 2129–2137. [Google Scholar] [CrossRef] [PubMed]
- Papeta, N.; Kiryluk, K.; Patel, A.; Sterken, R.; Kacak, N.; Snyder, H.J.; Imus, P.H.; Mhatre, A.N.; Lawani, A.K.; Julian, B.A.; et al. APOL1 variants increase risk for FSGS and HIVAN but not IgA nephropathy. J. Am. Soc. Nephrol. 2011, 22, 1991–1996. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.J.; Kozlitina, J.; Genovese, G.; Jog, P.; Pollak, M.R. Population-based risk assessment of APOL1 on renal disease. J. Am. Soc. Nephrol. 2011, 22, 2098–2105. [Google Scholar] [CrossRef] [PubMed]
- Ksiazek, P.; Bednarek-Skublewska, A.; Buraczynska, M. The C677T methylenetetrahydrofolate reductase gene mutation and nephropathy in type 2 diabetes mellitus. Med. Sci. Monit. 2004, 10, BR47–BR51. [Google Scholar] [PubMed]
- Badid, C.; Desmouliere, A.; Babici, D.; Hadj-Aissa, A.; McGregor, B.; Lefrancois, N.; Touraine, J.L.; Laville, M. Interstitial expression of α-SMA: An early marker of chronic renal allograft dysfunction. Nephrol. Dial. Transplant. 2002, 17, 1993–1998. [Google Scholar] [CrossRef] [PubMed]
- Kamijo, A.; Sugaya, T.; Hikawa, A.; Yamanouchi, M.; Hirata, Y.; Ishimitsu, T.; Numabe, A.; Takagi, M.; Hayakawa, H.; Tabei, F.; et al. Clinical evaluation of urinary excretion of liver-type fatty acid-binding protein as a marker for the monitoring of chronic kidney disease: A multicenter trial. J. Lab. Clin. Med. 2005, 145, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Matsui, K.; Kamijo-Ikemorif, A.; Sugaya, T.; Yasuda, T.; Kimura, K. Renal liver-type fatty acid binding protein (L-FABP) attenuates acute kidney injury in aristolochic acid nephrotoxicity. Am. J. Pathol. 2011, 178, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Veerkamp, J.H.; Peeters, R.A.; Maatman, R.G. Structural and functional features of different types of cytoplasmic fatty acid-binding proteins. Biochim. Biophys. Acta 1991, 4, 1–24. [Google Scholar] [CrossRef]
- Veerkamp, J.H.; van Kuppevelt, T.H.; Maatman, R.G.; Prinsen, C.F. Structural and functional aspects of cytosolic fatty acid-binding proteins. Prostaglandins Leukot. Essent. Fat. Acids 1993, 49, 887–906. [Google Scholar] [CrossRef]
- Wang, G.; Gong, Y.; Anderson, J.; Sun, D.; Minuk, G.; Roberts, M.S.; Burczynski, F.J. Antioxidative function of L-FABP in L-FABP stably transfected Chang liver cells. Hepatology 2005, 42, 871–879. [Google Scholar] [CrossRef] [PubMed]
- De Souza, C.M.; Braosi, A.P.; Luczyszyn, S.M.; Avila, A.R.; de Brito, R.B., Jr.; Ignácio, S.A.; Probst, C.M.; Riella, M.C.; Sotomaior, V.S.; Mira, M.T.; Pecoits-Filho, R.; Trevilatto, P.C. Association between vitamin D receptor gene polymorphisms and susceptibility to chronic kidney disease and periodontitis. Blood Purif. 2007, 25, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Mini, E.; Nobili, S. Pharmacogenetics: Implementing personalized medicine. Clin. Cases Miner. Bone Metab. 2009, 6, 17–24. [Google Scholar] [PubMed]
- Zaza, G.; Granata, S.; Sallustio, F.; Grandaliano, G.; Schena, F.P. Pharmacogenomics: A new paradigm to personalize treatments in nephrology patients. Clin. Exp. Immunol. 2010, 159, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Plosker, G.; Foster, R.H. Tacrolimus: A further update of its pharmacology and therapeutic use in the management of organ transplantation. Drugs 2000, 59, 323–389. [Google Scholar] [CrossRef] [PubMed]
- Fulton, B.; Markham, A. Mycophenolate mofetil: A review of its pharmacodynamic and pharmacokinetic properties and clinical efficacy in renal transplantation. Drugs 1996, 51, 278–298. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.E. The use of other drugs to allow a lower dosage of cyclosporine to be used. Therapeutic and pharmacoeconomic considerations. Clin. Pharmacokinet. 1997, 32, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Kurzawski, M.; Dziewanowski, K.; Lener, A.; Drozdzik, M. TPMT but not ITPA gene polymorphism influences the risk of azathioprine intolerance in renal transplant recipients. Eur. J. Clin. Pharmacol. 2009, 65, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Kurzawski, M.; Dziewanowski, K.; Gawrońska-Szklarz, B.; Domański, L.; Drozdzik, M. The impact of thiopurine S-methyltransferase polymorphism on azathioprine-induced myelotoxicity in renal transplant recipients. Ther. Drug Monit. 2005, 27, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Yates, C.R.; Krynetski, E.Y.; Loennechen, T.; Fessing, M.Y.; Tai, H.L.; Pui, C.H.; Relling, M.V.; Evans, W.E. Molecular diagnosis of thiopurine S-methyltransferase deficiency: Genetic basis for azathioprine and mercaptopurine intolerance. Ann. Intern. Med. 1997, 126, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.E.; Hon, Y.Y.; Bomgaars, L.; Coutre, S.; Holdsworth, M.; Janco, R.; Kalwinsky, D.; Keller, F.; Khatib, Z.; Margolin, J.; Murray, J. Preponderance of thiopurine S-methyltransferase deficiency and heterozygosity among patients intolerant to mercaptopurine or azathioprine. J. Clin. Oncol. 2001, 19, 2293–2301. [Google Scholar] [CrossRef] [PubMed]
- Dervieux, T.; Médard, Y.; Baudouin, V.; Maisin, A.; Zhang, D.; Broly, F.; Loirat, C.; Jacqz-Aigrain, E. Thiopurine methyltransferase activity and its relationship to the occurrence of rejection episodes in paediatric renal transplant recipients treated with azathioprine. Br. J. Clin. Pharmacol. 1999, 48, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Timm, R.; Kaiser, R.; Lotsch, J.; Heider, U.; Sezer, O.; Weisz, K.; Montemurro, M.; Roots, I.; Cascorbi, I. Association of cyclophosphamide pharmacokinetics to polymorphic cytochrome. Pharm. J. 2005, 5, 365–373. [Google Scholar]
- Solus, J.F.; Arietta, B.J.; Harris, J.R.; Sexton, D.P.; Steward, J.Q.; McMunn, C.; Ihrie, P.; Mehall, J.M.; Edwards, T.L.; Dawson, E.P. Genetic variation in eleven phase I drug metabolism genes in an ethnically diverse population. Pharmacogenomics 2004, 5, 895–931. [Google Scholar] [CrossRef] [PubMed]
- Takada, K.; Arefayene, M.; Desta, Z.; Yarboro, C.H.; Boumdpas, D.T.; Balow, J.E.; Flockhart, D.A.; Illei, G.G. Cytochrome P450 pharmacogenetics as a predictor of toxicity and clinical response to pulse cyclophosphamide in lupus nephritis. Arthritis Rheum. 2004, 50, 2202–2210. [Google Scholar] [CrossRef] [PubMed]
- Le Meur, Y.; Djebli, N.; Szelag, J.C.; Hoizey, G.; Toupance, O.; Rérolle, J.P.; Marquet, P. CYP3A5*3 influences sirolimus oral clearance in de novo and stable renal transplant recipients. Clin. Pharmacol. Ther. 2006, 80, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Stenton, S.B.; Partovi, N.; Ensom, M.H. Sirolimus: The evidence for clinical pharmacokinetic monitoring. Clin. Pharmacokinet. 2005, 44, 769–786. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.T.; Andrews, L.M.; van Gelder, T.; Shi, Y.Y.; van Schaik, R.H.; Wang, L.L.; Hesselink, D.A. Pharmacogenetic aspects of the use of tacrolimus in renal transplantation: Recent developments and ethnic considerations. Expert Opin. Drug Metab. Toxicol. 2016, 12, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Hebert, M.F.; Isoherranen, N.; Davis, C.L.; Marsh, C.; Shen, D.D.; Thummel, K.E. Effect of CYP3A5 polymorphism on tacrolimus metabolic clearance in vitro. Drug Metab. Dispos. 2006, 34, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, P.A.; Oetting, W.S.; Brearley, A.M.; Leduc, R.; Guan, W.; Schladt, D.; Matas, A.J.; Lamba, V.; Julian, B.A.; Mannon, R.B.; Israni, A.; DeKAF Investigators. Novel polymorphisms associated with tacrolimus trough concentrations: Results from a multicenter kidney transplant consortium. Transplantation 2011, 91, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Tavira, B.; Díaz-Corte, C.; Coronel, D.; Ortega, F.; Coto, E. Pharmacogenetics of tacrolimus: From bench to bedside? Nefrologia 2014, 34, 11–17. [Google Scholar] [PubMed]
- Kuehl, P.; Zhang, J.; Lin, Y.; Lamba, J.; Assem, M.; Schuetz, J.; Watkins, P.B.; Daly, A.; Wrighton, S.A.; Hall, S.D.; et al. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat. Genet. 2001, 27, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Hustert, E.; Haberl, M.; Burk, O.; Wolbold, R.; He, Y.Q.; Klein, K.; Nuessler, A.C.; Neuhaus, P.; Klattig, J.; Eiselt, R.; et al. The genetic determinants of the CYP3A5 polymorphism. Pharmacogenetics 2001, 11, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Quteineh, L.; Verstuyft, C.; Furlan, V.; Durrbach, A.; Letierce, A.; Ferlicot, S.; Taburet, A.M.; Charpentier, B.; Becquemont, L. Influence of CYP3A5 genetic polymorphism on tacrolimus daily dose requirements and acute rejection in renal graft recipients. Basic Clin. Pharmacol. Toxicol. 2008, 103, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Satoh, S.; Saito, M.; Inoue, T.; Kagaya, H.; Miura, M.; Inoue, K.; Komatsuda, A.; Tsuchiya, N.; Suzuki, T.; Habuchi, T. CYP3A5 *1 allele associated with tacrolimus trough concentrations but not subclinical acute rejection or chronic allograft nephropathy in Japanese renal transplant recipients. Eur. J. Clin. Pharmacol. 2009, 65, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Dirks, N.L.; Huth, B.; Yates, C.R.; Meibohm, B. Pharmacokinetics of immunosuppressants: A perspective on ethnic differences. Int. J. Clin. Pharmacol. Ther. 2004, 42, 701–718. [Google Scholar] [CrossRef] [PubMed]
- Neylan, J.F. Racial differences in renal transplantation after immunosuppression with tacrolimus versus cyclosporine. FK506 Kidney Transplant Study Group. Transplantation 1998, 65, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Kuypers, D.R.; de Jonge, H.; Naesens, M.; Lerut, E.; Verbeke, K.; Vanrenterghem, Y. CYP3A5 and CYP3A4 but not MDR1 single-nucleotide polymorphisms determine long-term tacrolimus disposition and drug-related nephrotoxicity in renal recipients. Clin. Pharmacol. Ther. 2007, 82, 711–725. [Google Scholar] [CrossRef] [PubMed]
- Amirimani, B.; Ning, B.; Deitz, A.C.; Weber, B.L.; Kadlubar, F.F.; Rebbeck, T.R. Increased transcriptional activity of the CYP3A4*1B promoter variant. Environ. Mol. Mutagen. 2003, 42, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Thervet, E.; Loriot, M.A.; Barbier, S.; Buchler, M.; Ficheux, M.; Choukroun, G.; Toupance, O.; Touchard, G.; Alberti, C.; Le Pogamp, P.; et al. Optimization of initial tacrolimus dose using pharmacogenetic testing. Clin. Pharmacol. Ther. 2010, 87, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Halloran, P.F. Immunosuppressive drugs for kidney transplantation. N. Engl. J. Med. 2004, 351, 2715–2729. [Google Scholar] [CrossRef] [PubMed]
- Sousa, A.R.; Lane, S.J.; Cidlowski, J.A.; Staynov, D.Z.; Lee, T.H. Glucocorticoid resistance in asthma is associated with elevated in vivo expression of the glucocorticoid receptor β-isoform. J. Allergy Clin. Immunol. 2000, 105, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Miura, M.; Satoh, S.; Inoue, K.; Kagaya, H.; Saito, M.; Inoue, T.; Habuchi, T.; Suzuki, T. Influence of CYP3A5, ABCB1 and NR1I2 polymorphisms on prednisolone pharmacokinetics in renal transplant recipients. Steroids 2008, 73, 1052–1059. [Google Scholar] [CrossRef] [PubMed]
- Sarwal, M.; Chua, M.; Kambham, N.; Hsieh, S.; Satterwhite, T.; Masek, M.; Salvatierra, O. Molecular heterogeneity in acute renal allograft rejection identified by DNA microarray profiling. N. Engl. J. Med. 2003, 349, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Aranda, J.M., Jr.; Scornik, J.C.; Normann, S.J.; Lottenberg, R.; Schofield, R.S.; Pauly, D.F.; Miles, M.; Hill, J.A.; Sleasman, J.W.; Skoda-Smith, S. Anti-CD20 monoclonal antibody (rituximab) therapy for acute cardiac humoral rejection: A case report. Transplantation 2002, 73, 907–910. [Google Scholar] [CrossRef] [PubMed]
- Zarkhin, V.; Kambham, N.; Li, L.; Kwok, S.; Hsieh, S.-C.; Salvatierra, O.; Sarwal, M. Characterization of intra-graft B cells during renal allograft rejection. Kidney Int. 2008, 74, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Stenvinkel, P.; Wang, K.; Qureshi, A.R.; Axelsson, J.; Pecoits-Filho, R.; Gao, P.; Barany, P.; Lindholm, B.; Jogestrand, T.; Heimbürger, O.; et al. Low fetuin-A levels are associated with cardiovascular death: Impact of variations in the gene encoding fetuin. Kidney Int. 2005, 67, 2383–2392. [Google Scholar] [CrossRef] [PubMed]
- Braunersreuther, V.; Mach, F.; Steffens, S. The specific role of chemokines in atherosclerosis. Thromb. Haemost. 2007, 97, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Muntinghe, F.L.; Verduijn, M.; Zuurman, M.W.; Grootendorst, D.C.; Carrero, J.J.; Qureshi, A.R.; Luttropp, K.; Nordfors, L.; Lindholm, B.; Brandenburg, V.; et al. CCR5 deletion protects against inflammation-associated mortality in dialysis patients. J. Am. Soc. Nephrol. 2009, 20, 1641–1649. [Google Scholar] [CrossRef] [PubMed]
- Veillard, N.R.; Kwak, B.; Pelli, G.; Mulhaupt, F.; James, R.W.; Proudfoot, A.E.; Mach, F. Antagonism of RANTES receptors reduces atherosclerotic plaque formation in mice. Circ. Res. 2004, 94, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, V.S.; Guo, D.; Rao, M.; Jaber, B.L.; Tighiouart, H.; Freeman, R.L.; Huang, C.; King, A.J.; Pereira, B.J.; HEMO Study Group. Cytokine gene polymorphisms in hemodialysis patients: association with comorbidity, functionality, and serum albumin. Kidney Int. 2004, 65, 1449–1460. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Berthier-Schaad, Y.; Fallin, M.D.; Fink, N.E.; Tracy, R.P.; Klag, M.J.; Smith, M.W.; Coresh, J. IL-6 haplotypes, inflammation, and risk for cardiovascular disease in a multiethnic dialysis cohort. J. Am. Soc. Nephrol. 2006, 17, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Berthier-Schaad, Y.; Plantinga, L.; Fink, N.E.; Tracy, R.P.; Kao, W.H.; Klag, M.J.; Smith, M.W.; Coresh, J. Functional variants in the lymphotoxin-α gene predict cardiovascular disease in dialysis patients. J. Am. Soc. Nephrol. 2006, 17, 3158–3166. [Google Scholar] [CrossRef] [PubMed]
- Zaza, G.; Pontrelli, P.; Pertosa, G.; Granata, S.; Rossini, M.; Porreca, S.; Staal, F.J.; Gesualdo, L.; Grandaliano, G.; Schena, F.P. Dialysis-related systemic micro-inflammation is associated with specific genomic patterns. Nephrol. Dial. Transplant. 2008, 23, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Jaber, B.L.; Balakrishnan, V.S. Gene polymorphism association studies in dialysis: Cardiovascular disease. Semin. Dial. 2005, 18, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Gumprecht, J.; Zychma, M.J.; Grzeszczak, W.; Zukowska-Szczechowska, E. Angiotensin I-converting enzyme gene insertion/deletion and angiotensinogen M235T polymorphisms: Risk of chronic renal failure. End-Stage Renal Disease Study Group. Kidney Int. 2000, 58, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Perez-Oller, L.; Torra, R.; Badenas, C.; Mila, M.; Darnell, A. Influence of the ACE gene polymorphism in the progression of renal failure in autosomal dominant polycystic kidney disease. Am. J. Kidney Dis. 1999, 34, 273–278. [Google Scholar] [CrossRef]
- Wang, A.Y.; Chan, J.C.; Wang, M.; Poon, E.; Lui, S.F.; Li, P.K.; Sanderson, J. Cardiac hypertrophy and remodeling in relation to ACE and angiotensinogen genes genotypes in Chinese dialysis patients. Kidney Int. 2003, 63, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- Friedman, E.A.; Friedman, A.L. Payment for donor kidneys: Pros and cons. Review. Kidney Int. 2006, 69, 960–962. [Google Scholar] [CrossRef] [PubMed]
- Wanless, D. Securing our Future Health: Taking a Long-Term View Final Report; HM Treasury: London, UK, 2002; pp. 13–34. [Google Scholar]
- Programme Budgeting Tools and Data. National expenditure data. London, UK: Department of Health. Available online: http://www.dh.gov.uk/en/Managingyourorganisation/Financeandplanning/Programmebudgeting/DH_075743 (accessed on 2 December 2011).
- Honeycutt, A.A.; Segel, J.E.; Zhuo, X.; Hoerger, T.J.; Imai, K.; Williams, D. Medical Costs of CKD in the Medicare Population. JASN 2013, 24, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- U.S. Renal Data System (USRDS). USRDS 2010 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States; National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2010.
- U.S. Renal Data System (USRDS). USRDS 2011 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States; National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2011.
- Kerr, M.; Bray, B.; Medcalf, J.; O’Donoghue, D.J.; Matthews, B. Estimating the financial cost of chronic kidney disease to the NHS in England. Nephrol. Dial. Transplant. 2012, 27, iii73–iii80. [Google Scholar] [CrossRef] [PubMed]
- Cappuccio, F.P. Ethnicity and cardiovascular risk: Variations in people of African ancestry and South Asian origin. J. Hum. Hypertens. 1997, 11, 571–576. [Google Scholar] [CrossRef] [PubMed]
- NKF News 2016. Available online: https://www.kidney.org/news/newsroom/factsheets/African-Americans-and-CKD (accessed on 20 March 2017).
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gluba-Brzózka, A.; Franczyk, B.; Olszewski, R.; Banach, M.; Rysz, J. Personalized Medicine: New Perspectives for the Diagnosis and the Treatment of Renal Diseases. Int. J. Mol. Sci. 2017, 18, 1248. https://doi.org/10.3390/ijms18061248
Gluba-Brzózka A, Franczyk B, Olszewski R, Banach M, Rysz J. Personalized Medicine: New Perspectives for the Diagnosis and the Treatment of Renal Diseases. International Journal of Molecular Sciences. 2017; 18(6):1248. https://doi.org/10.3390/ijms18061248
Chicago/Turabian StyleGluba-Brzózka, Anna, Beata Franczyk, Robert Olszewski, Maciej Banach, and Jacek Rysz. 2017. "Personalized Medicine: New Perspectives for the Diagnosis and the Treatment of Renal Diseases" International Journal of Molecular Sciences 18, no. 6: 1248. https://doi.org/10.3390/ijms18061248
APA StyleGluba-Brzózka, A., Franczyk, B., Olszewski, R., Banach, M., & Rysz, J. (2017). Personalized Medicine: New Perspectives for the Diagnosis and the Treatment of Renal Diseases. International Journal of Molecular Sciences, 18(6), 1248. https://doi.org/10.3390/ijms18061248