
Prostaglandin E2-Induced COX-2 Expressions via EP2 and EP4 Signaling Pathways in Human LoVo Colon Cancer Cells


Abstract
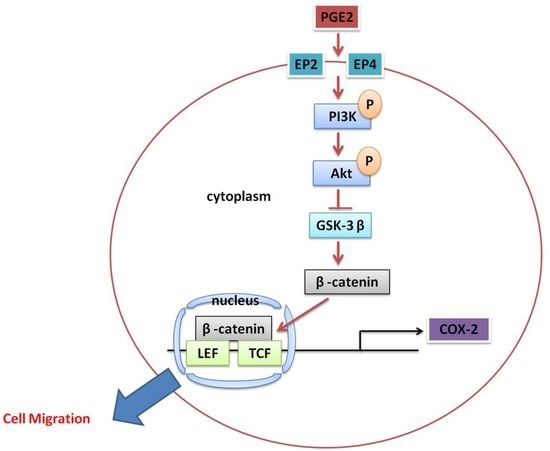
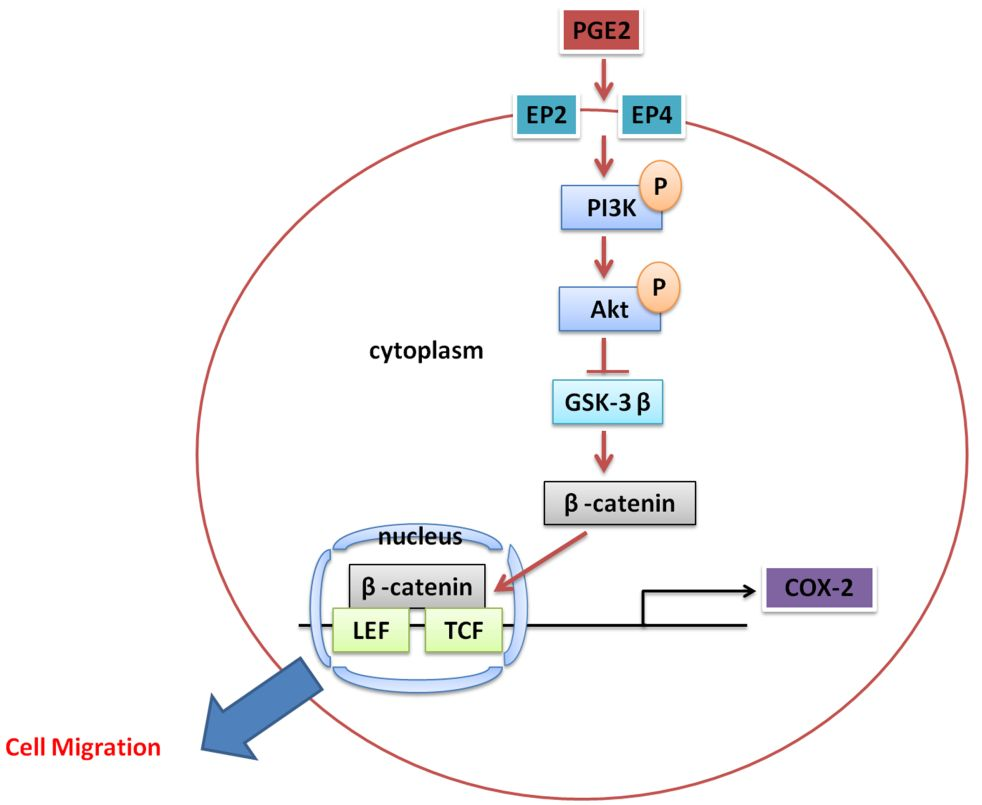
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
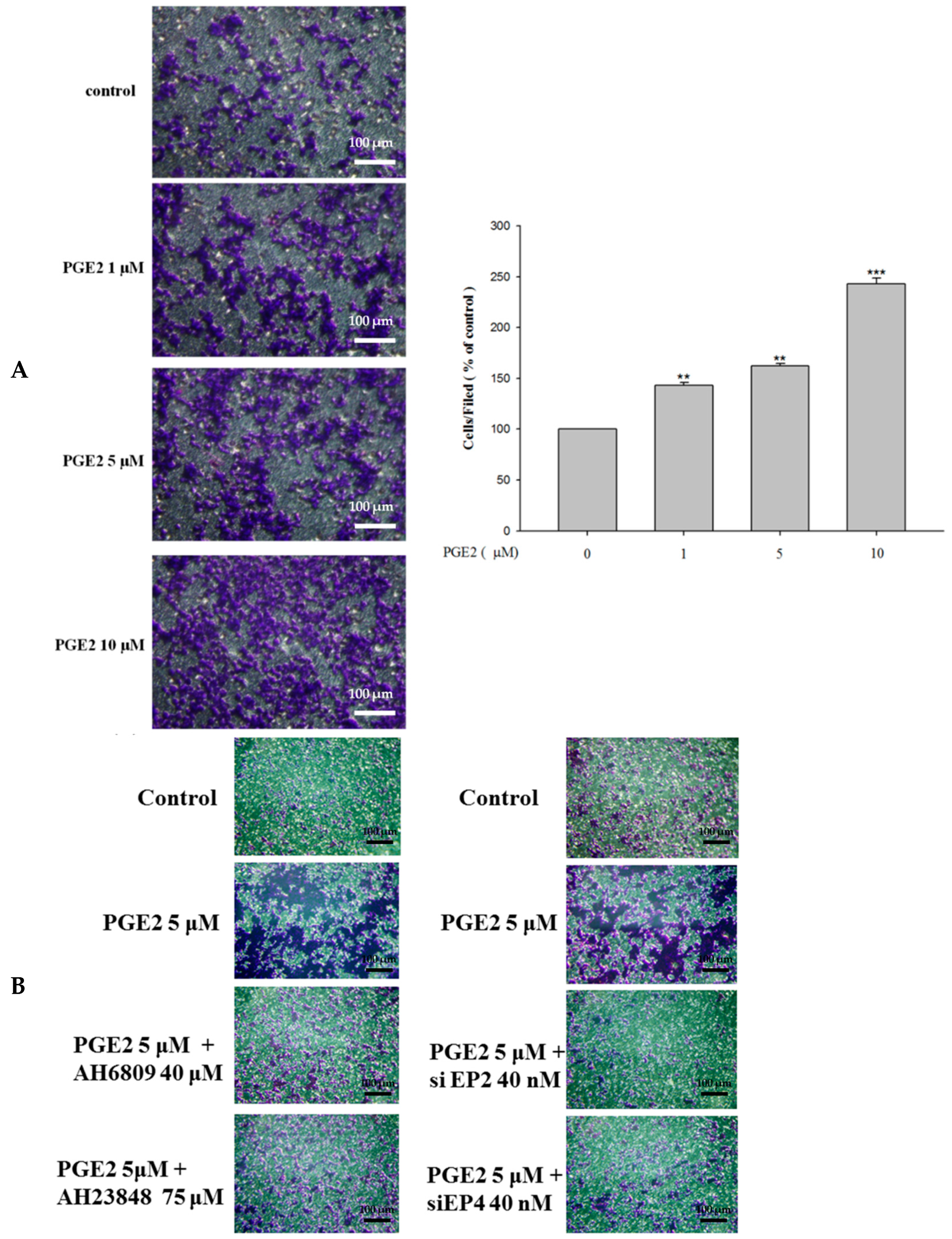
2.1. Effect of PGE2 on Viability of LoVo Cells
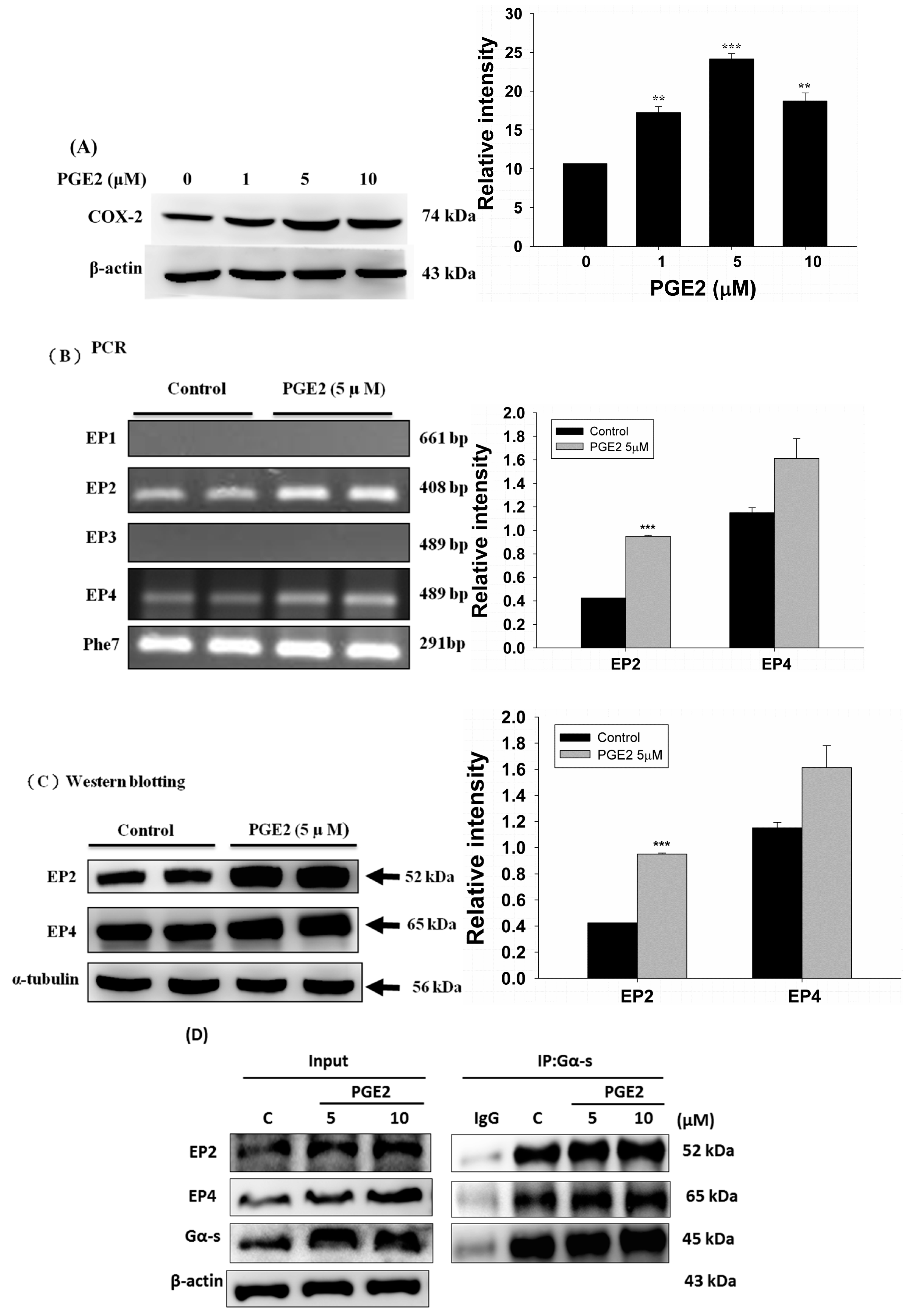
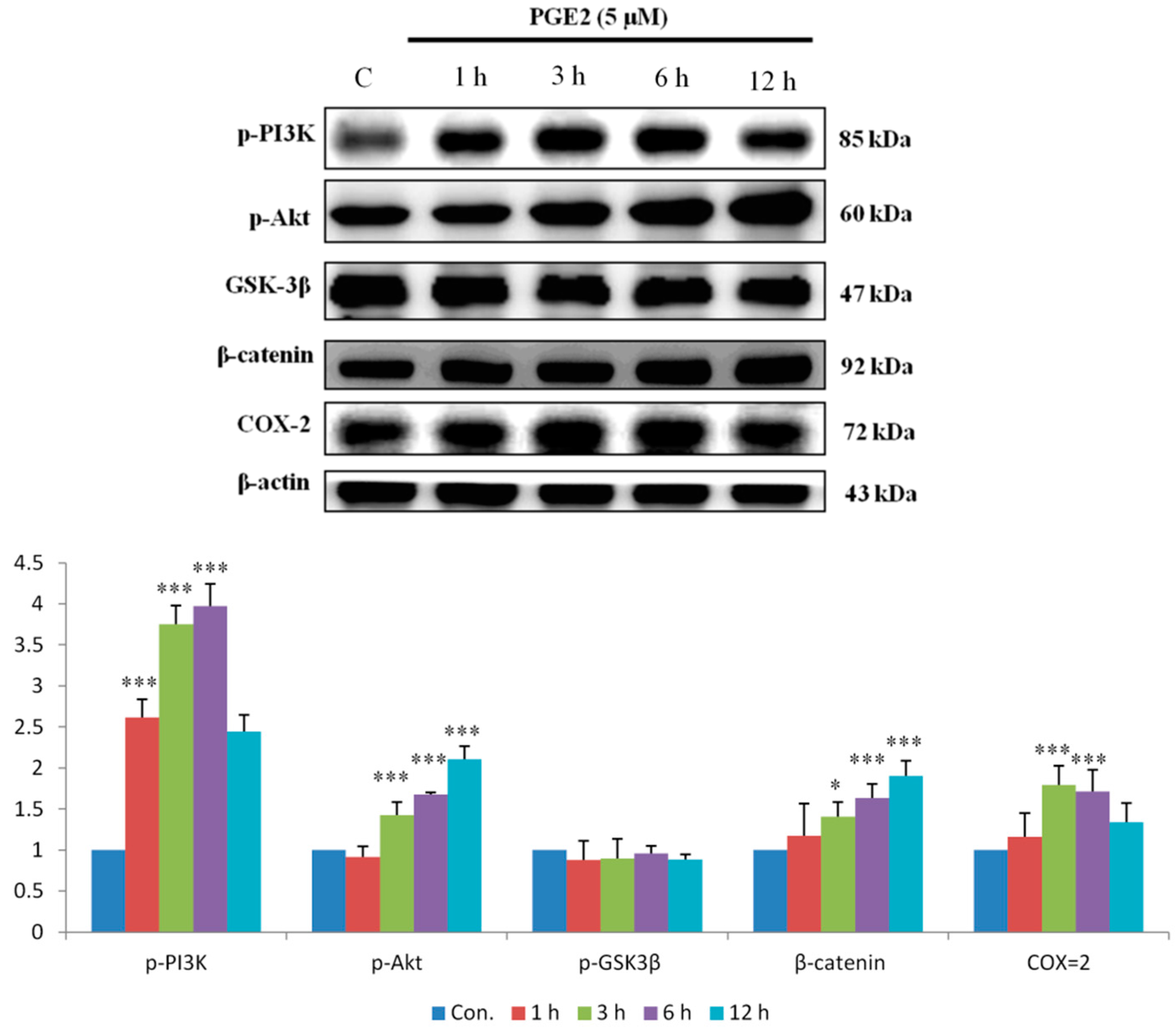
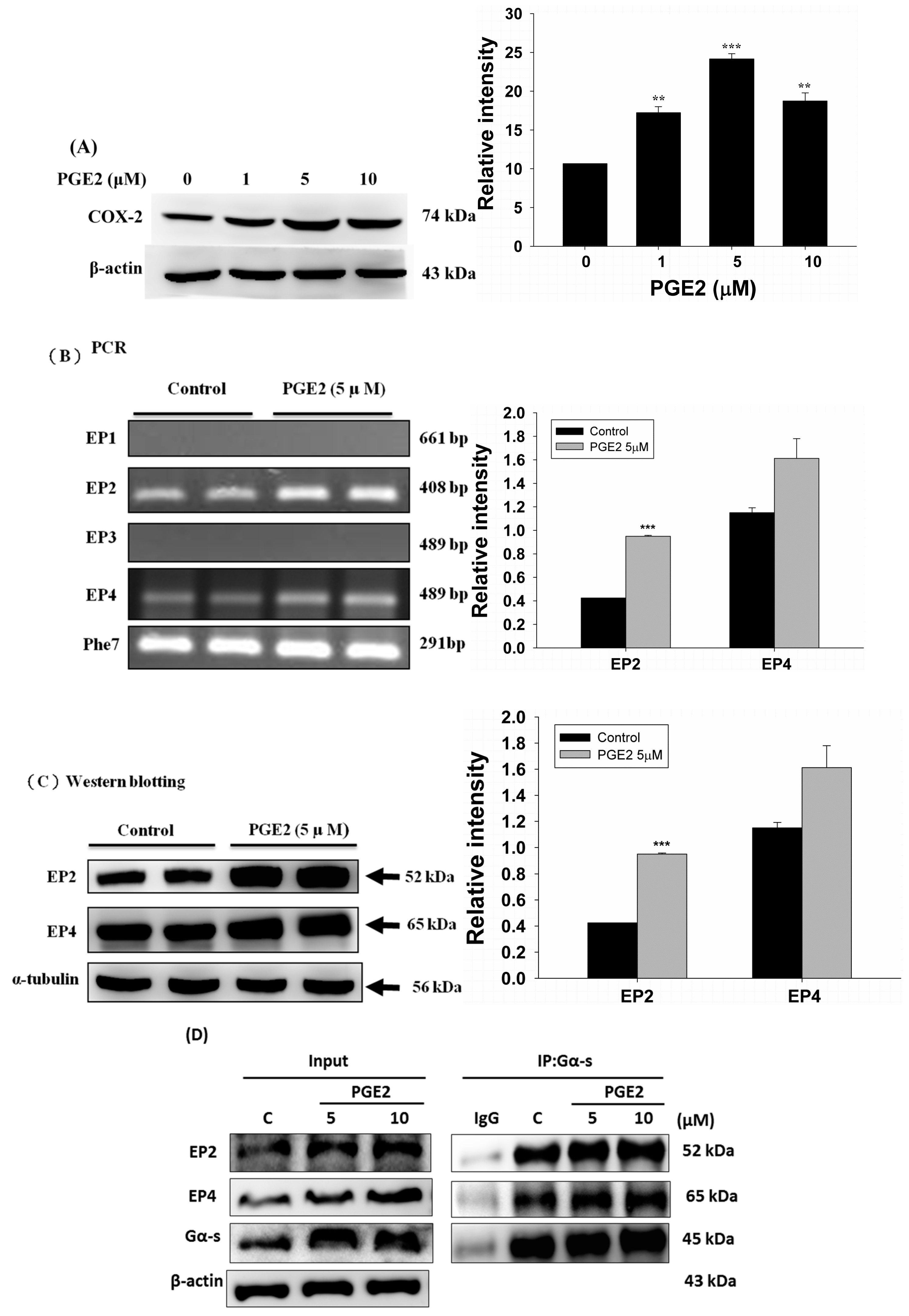
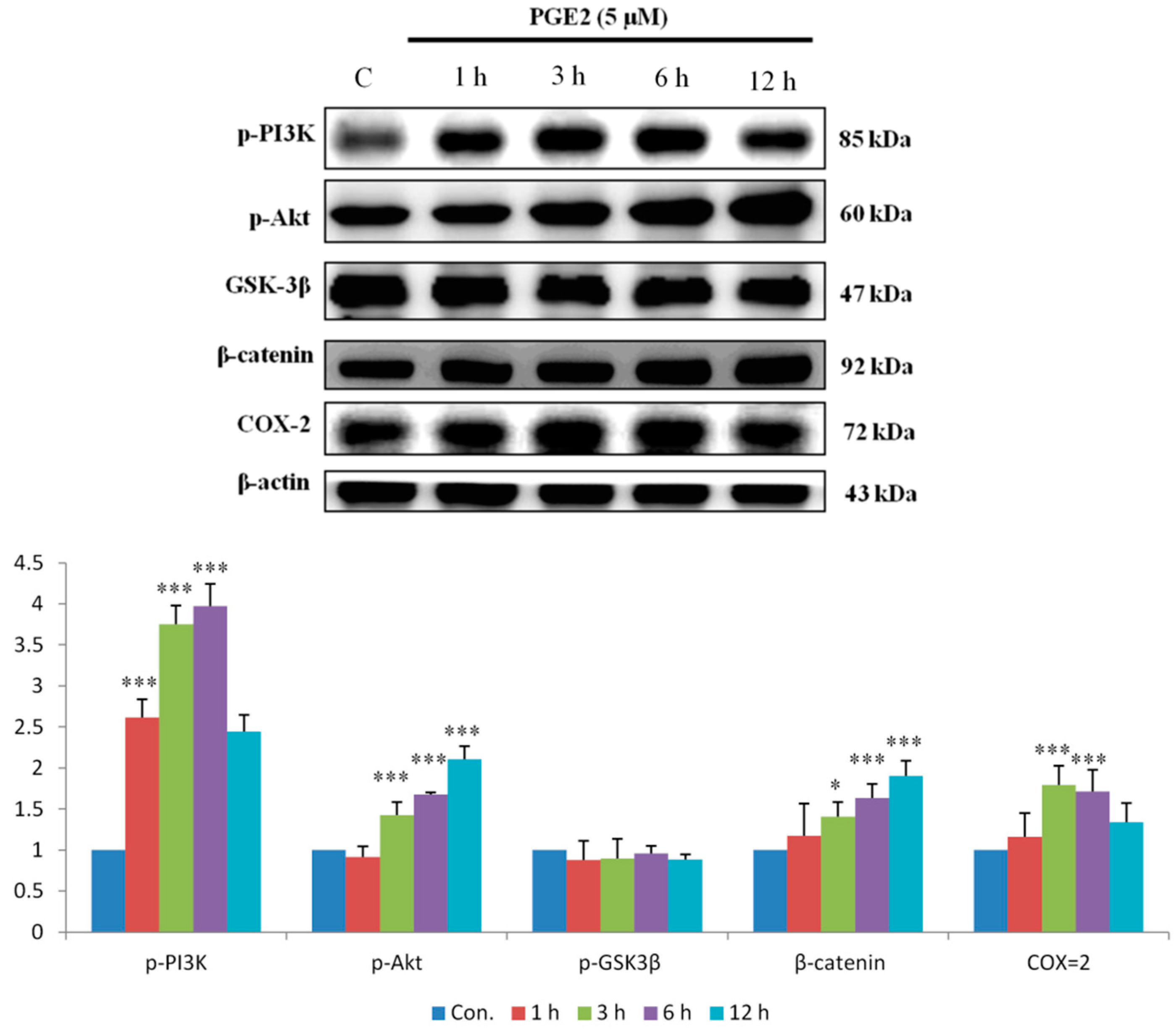
2.2. PGE2 Treatment Upregulated the Expression of COX2 and Proteins in the PI3K/Akt and p-GSK3β/β-Catenin Pathways
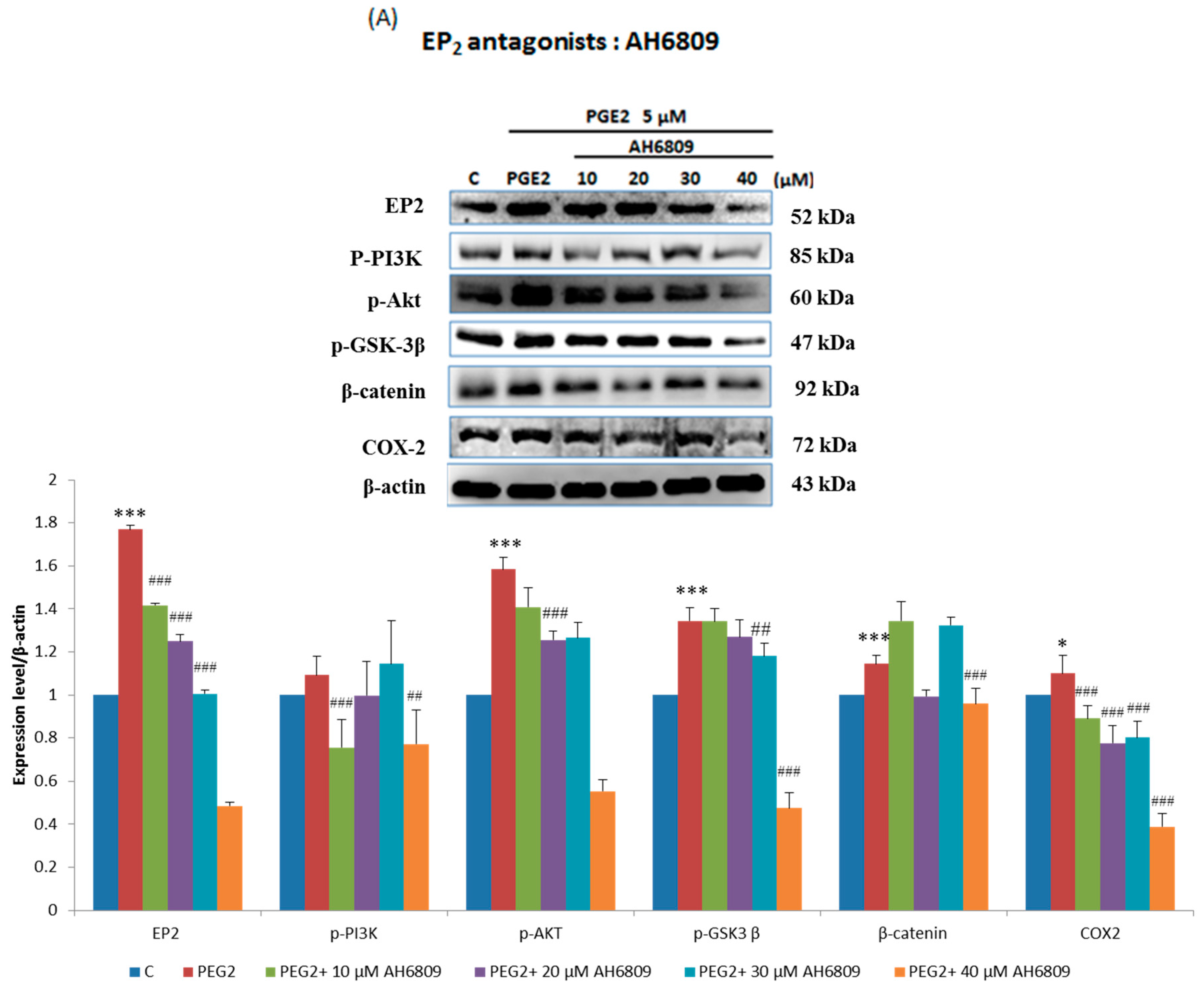
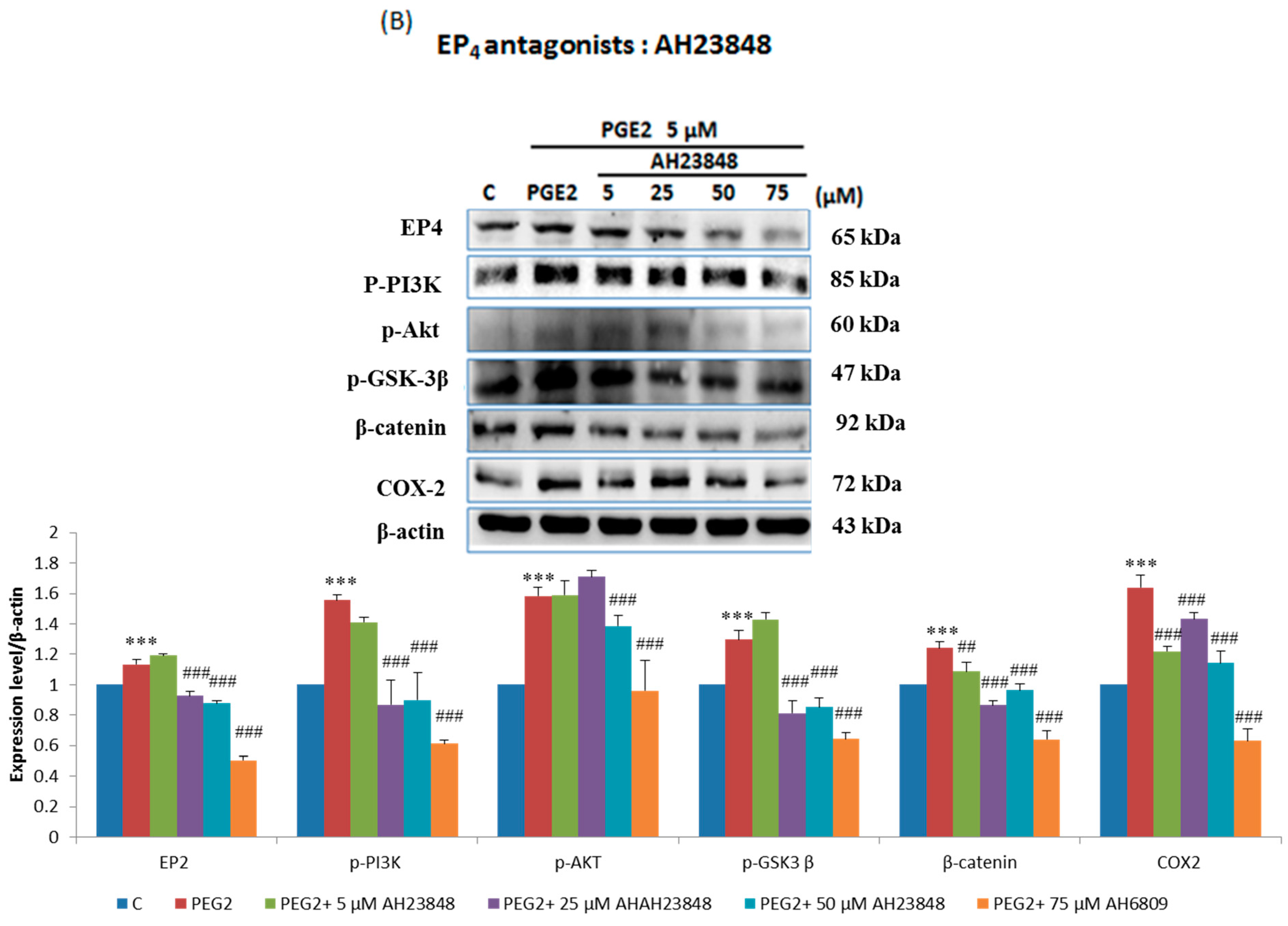
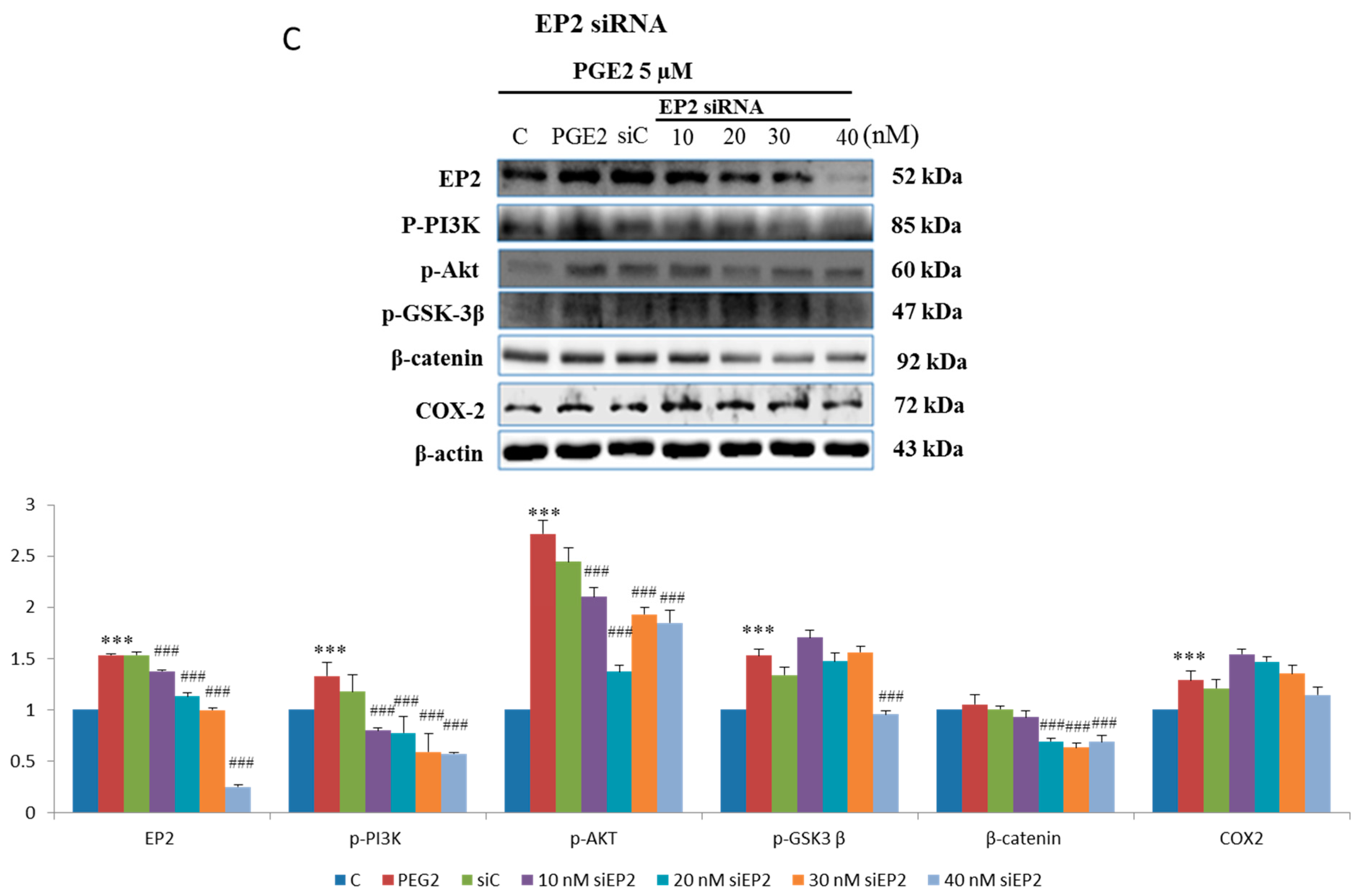
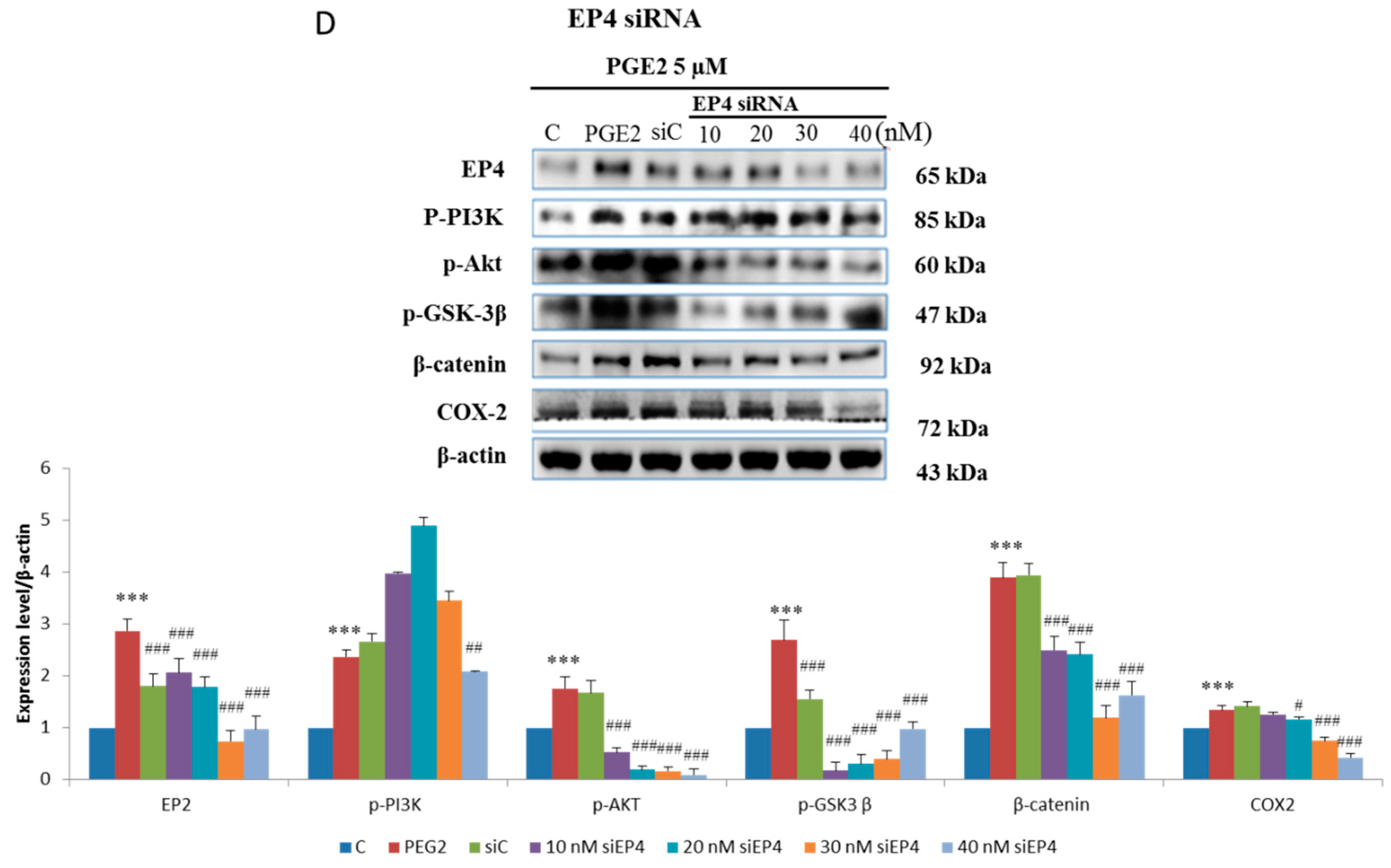
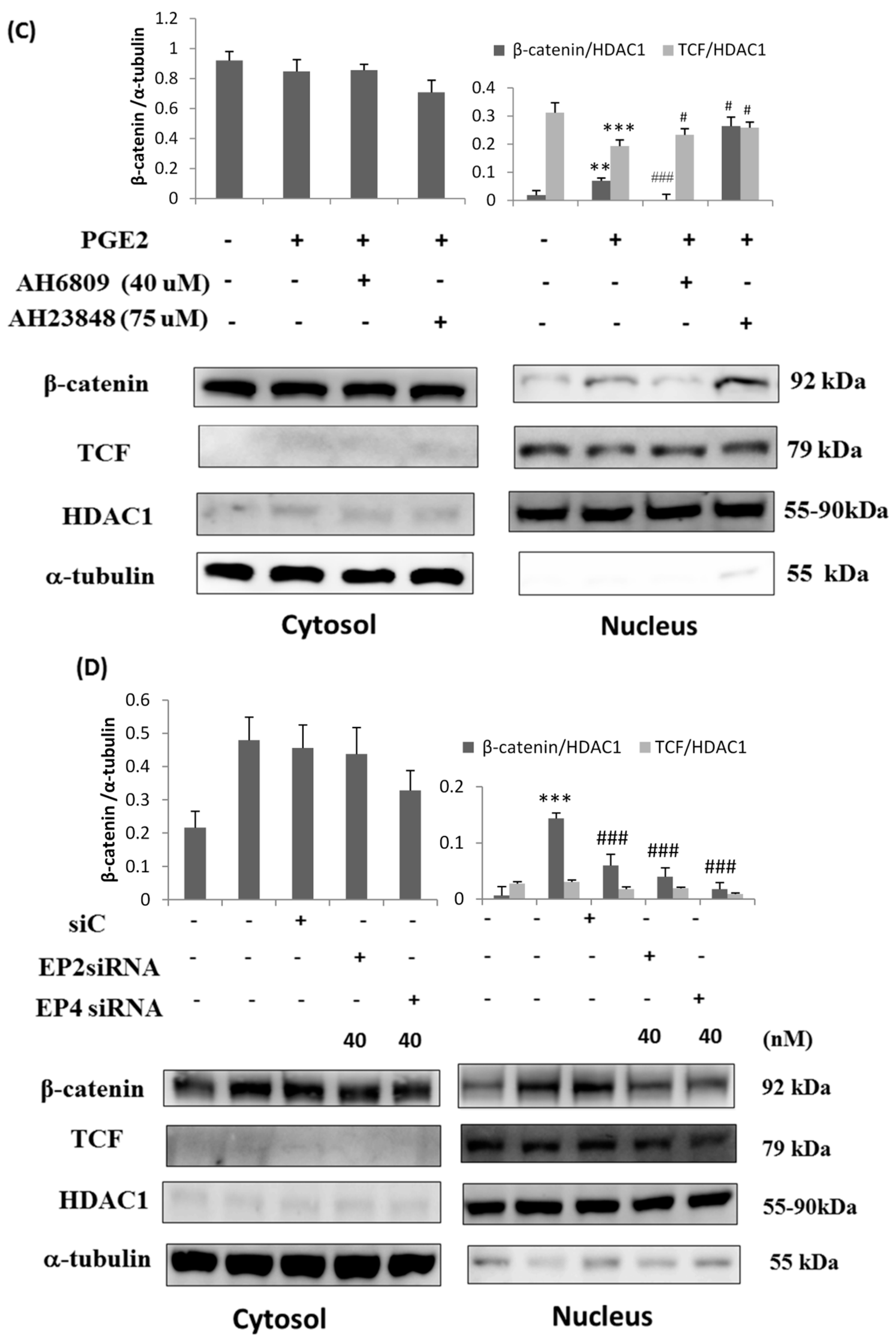
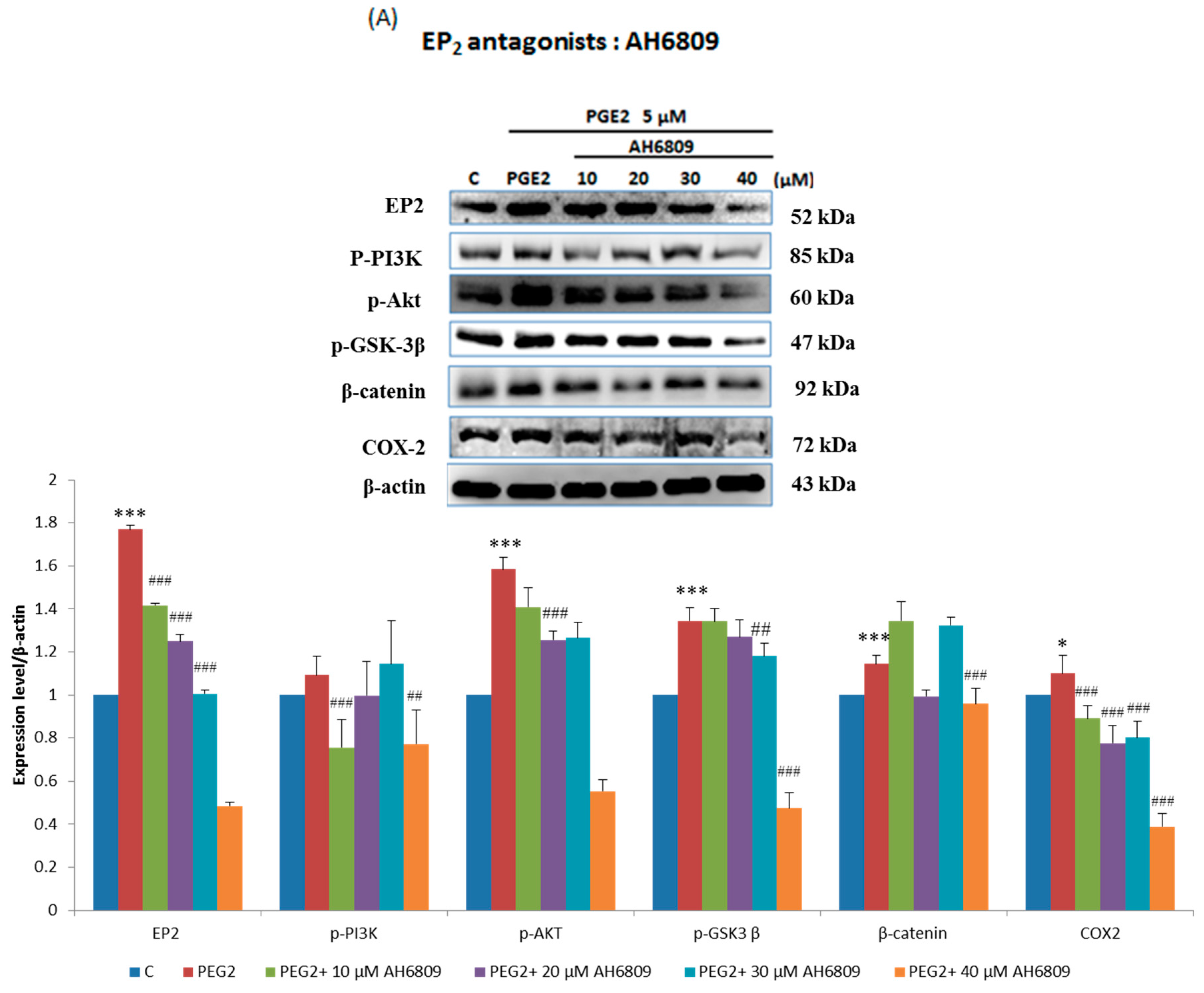
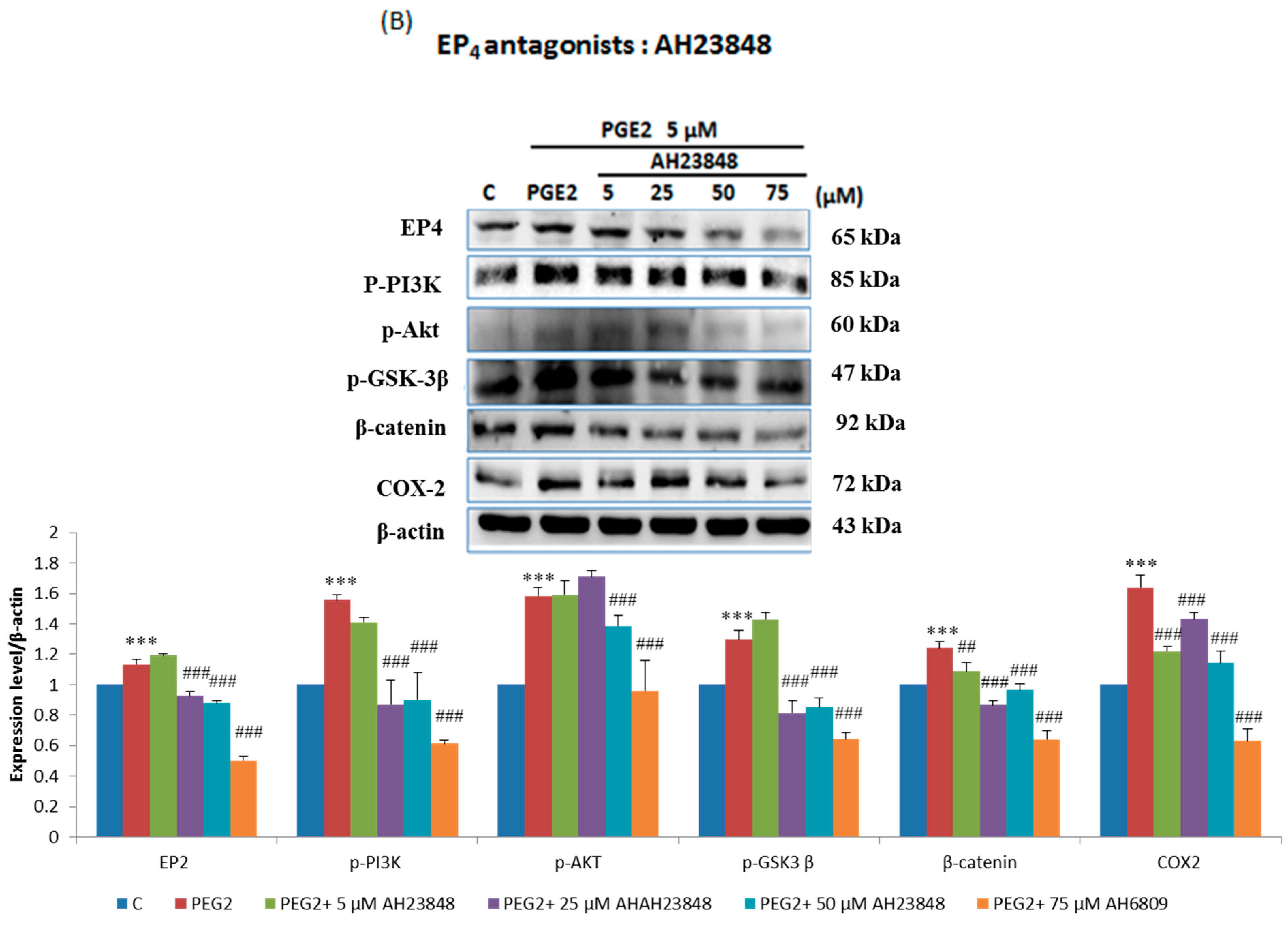
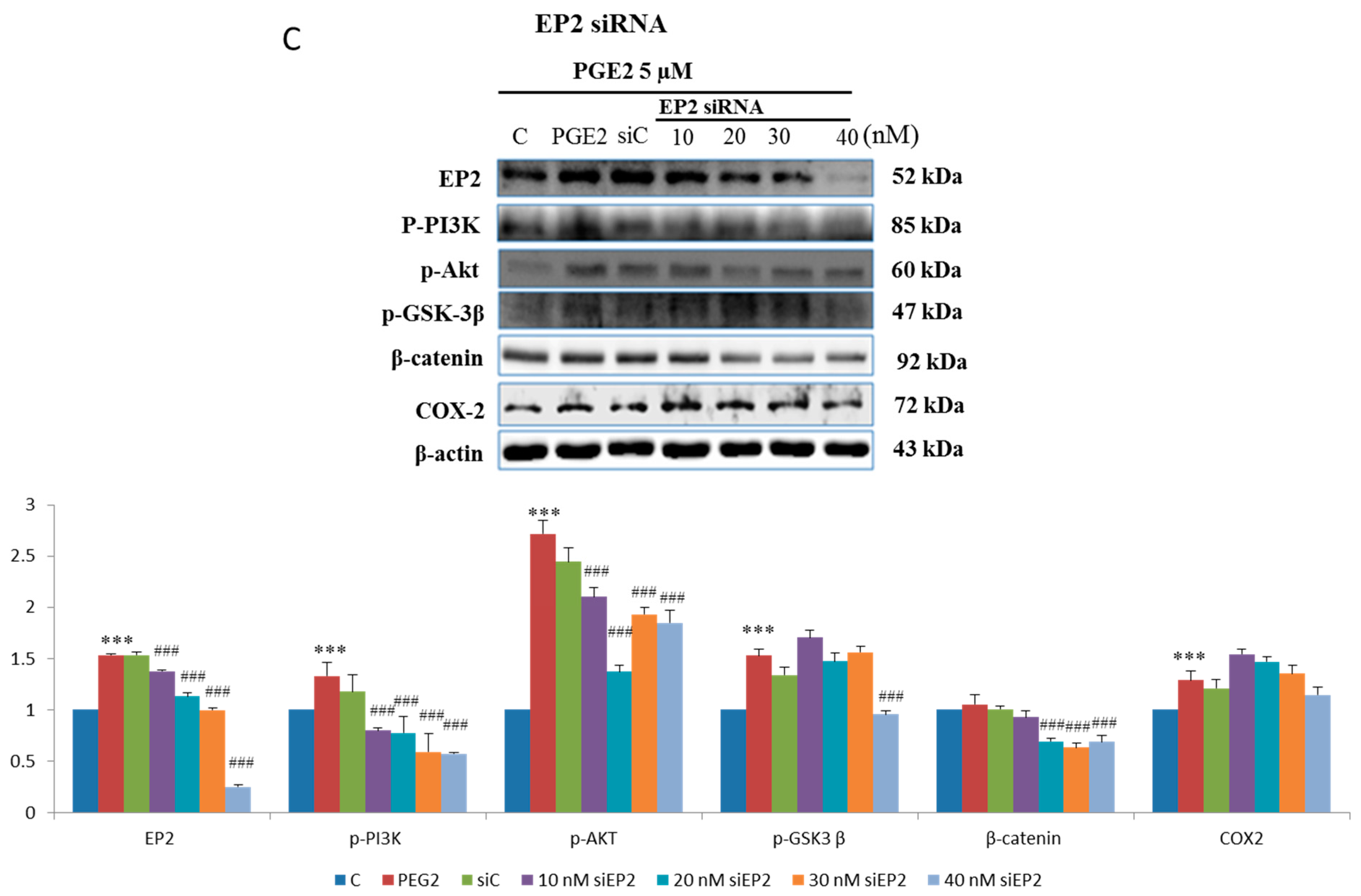
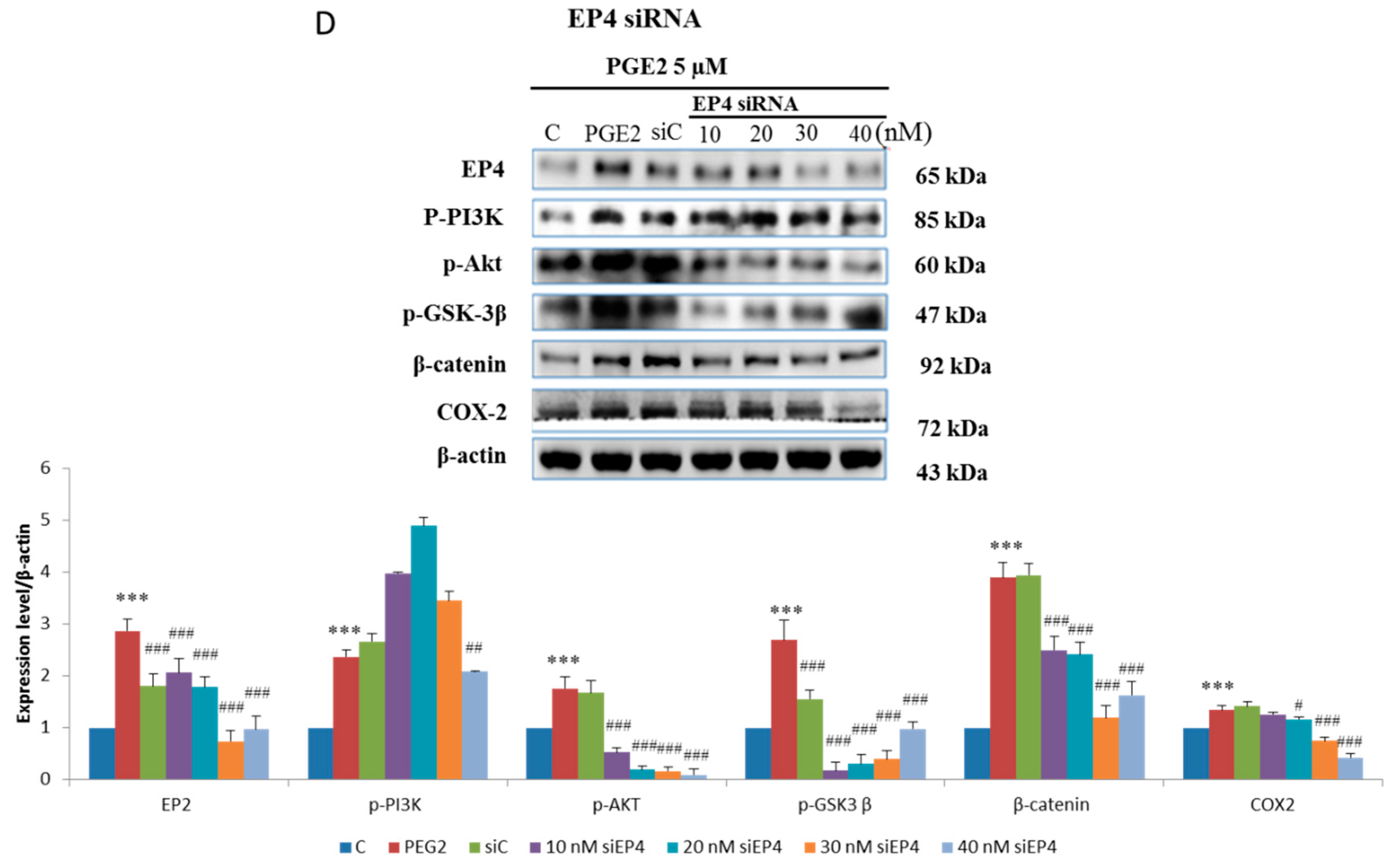
2.3. EP2- or EP4-Specific siRNA and Receptor Antagonists Inhibit the Induction of PI3K, Akt, β-Catenin and COX2 in PGE2-Treated LoVo Cells
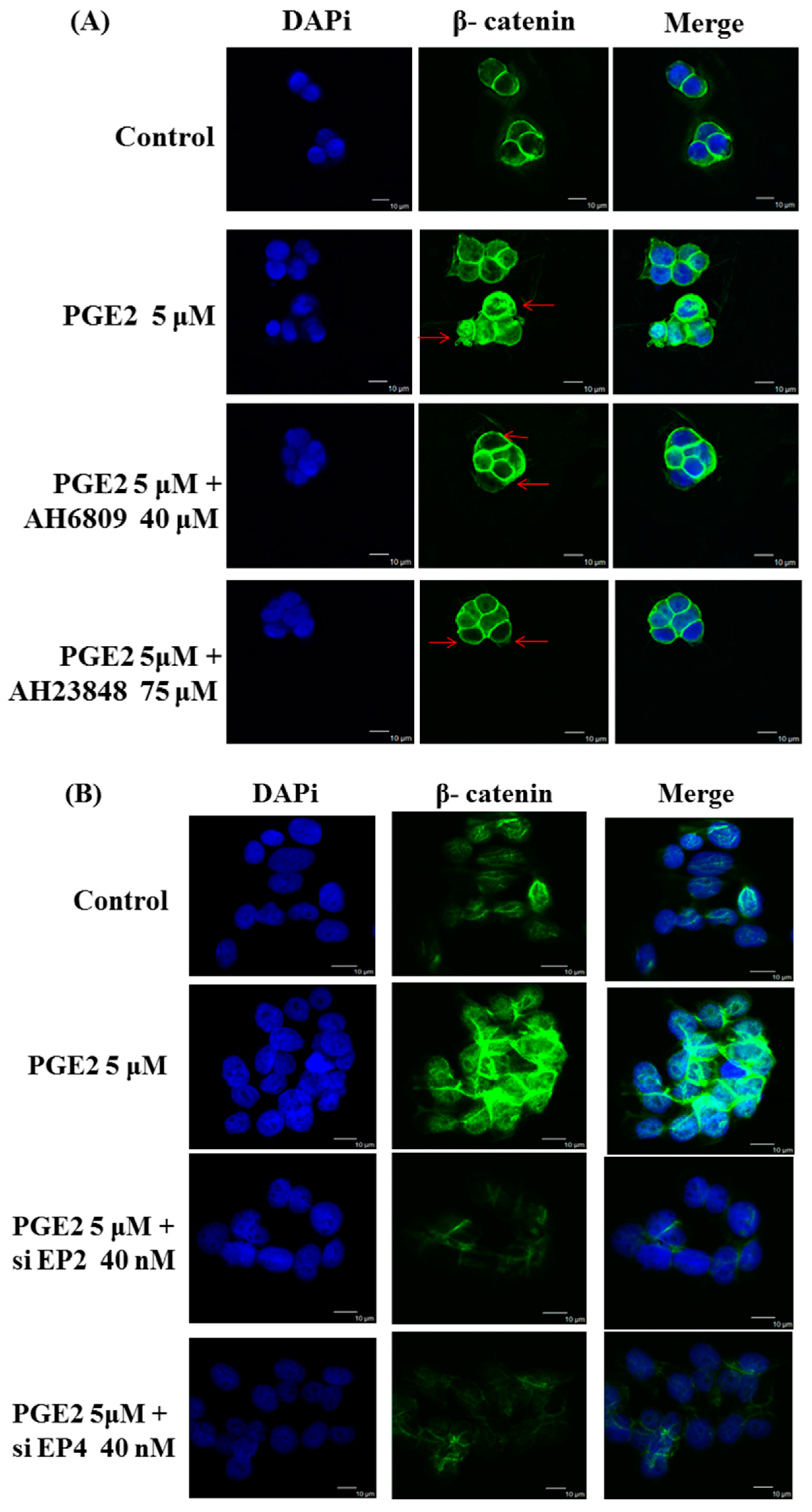
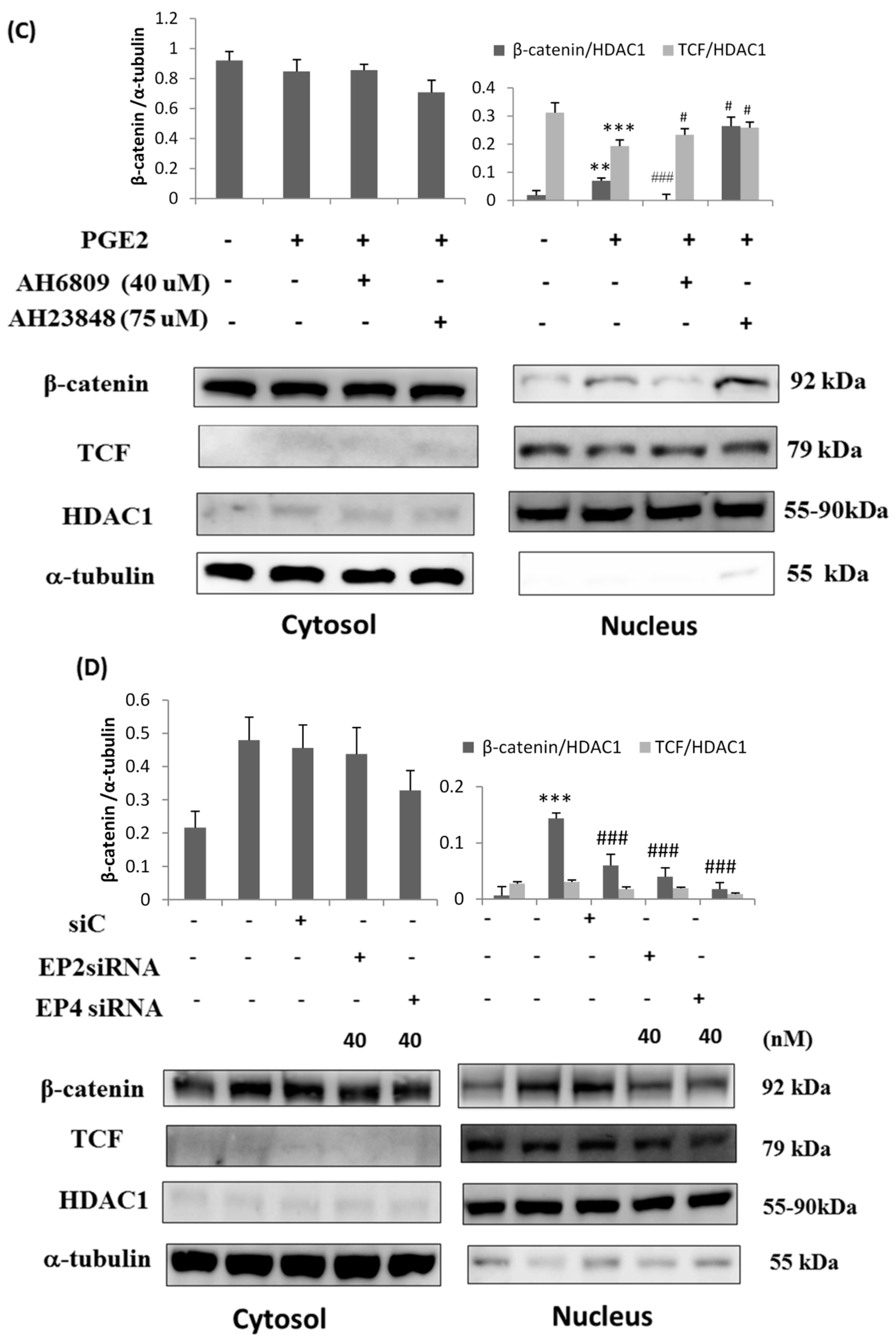
2.4. Immunofluorescence Assays and Analysis of Cytosolic and Nuclear Protein Fractions
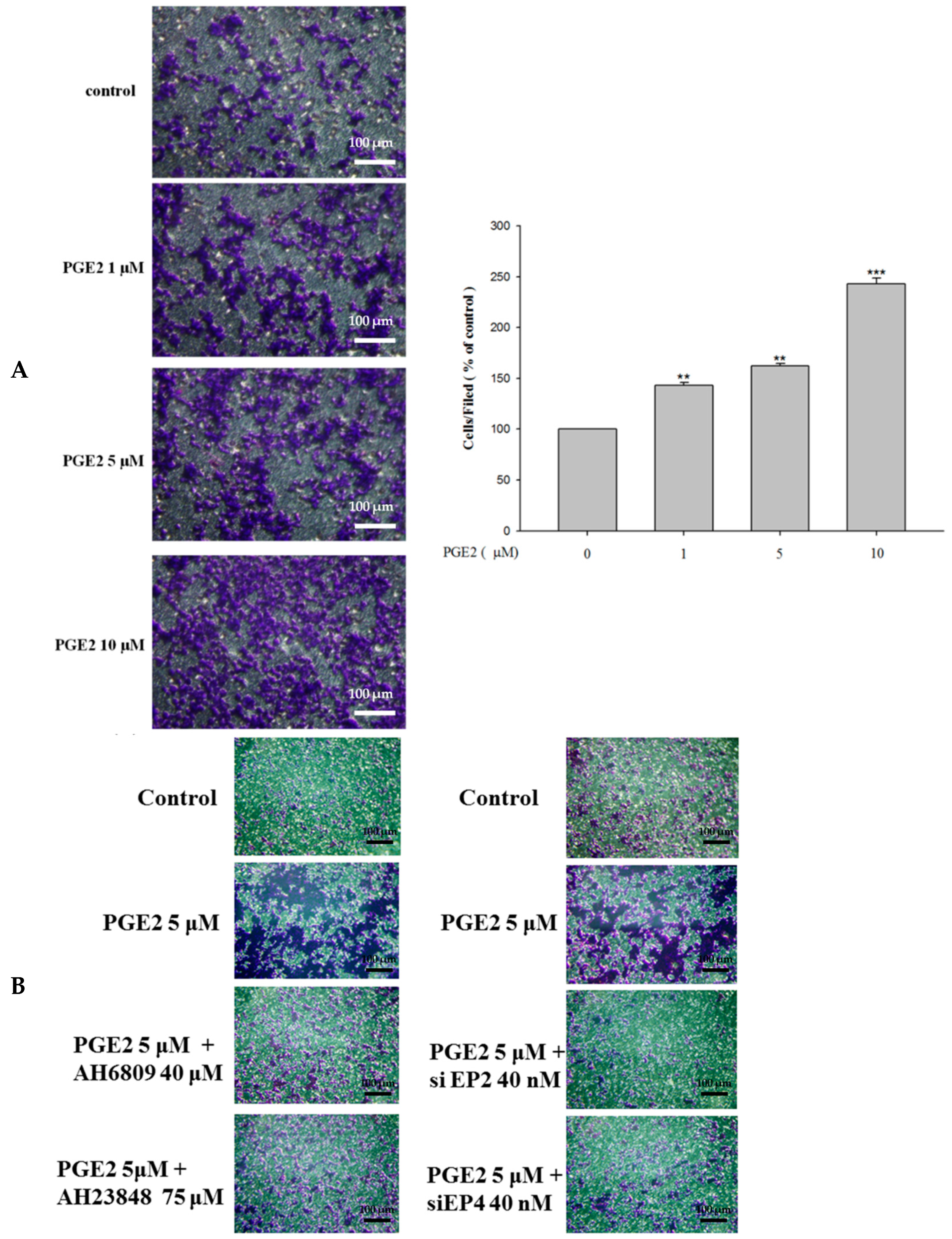
2.5. Inhibition of PGE2-Induced Cell Migration by Antagonist and siRNA Treatment
3. Discussion
4. Materials and Methods
4.1. Cells, Antibodies, Reagents and Enzymes
4.2. Cell Culture
4.3. Total RNA Extraction and Reverse Transcription
The Expression of EP1–EP4 in LoVo Colon Cancer Cells Was Detected by Reverse Transcription PCR (RT-PCR)
4.4. Immunoblotting Assay
4.5. Co-Immunoprecipitation Assay (Co-IP)
4.6. Gene Knockdown Using siRNA
4.7. Migration Assay
4.8. Nuclear Extraction
4.9. Immunofluorescence Assay
4.10. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jemal, A.; Murray, T.; Ward, E.; Samuels, A.; Tiwari, R.C.; Ghafoor, A.; Feuer, E.J.; Thun, M.J. Cancer statistics, 2005. CA Cancer J. Clin. 2005, 55, 10–30. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.Y.; Fan, C.W.; Maa, M.C.; Leu, T.H. Lipopolysaccharide-promoted proliferation of Caco-2 cells is mediated by c-Src induction and ERK activation. Biomedicine 2015, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, P. Colon cancer overview. Cancer 1992, 70, 1206–1215. [Google Scholar] [CrossRef]
- Jemal, A.; Thomas, A.; Murray, T.; Thun, M. Cancer statistics, 2002. CA Cancer J. Clin. 2002, 52, 23–47. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Thomas, A.; Murray, T.; Ghafoor, A.; Samuels, A.; Ward, E.; Feuer, E.J.; Thun, M.J.; American Cancer Society. Cancer statistics, 2004. CA Cancer J. Clin. 2004, 54, 8–29. [Google Scholar] [CrossRef] [PubMed]
- Liotta, L.A.; Stetler-Stevenson, W.G. Tumor invasion and metastasis: An imbalance of positive and negative regulation. Cancer Res. 1991, 51, 5054s–5059s. [Google Scholar] [PubMed]
- Yoon, W.H.; Jung, Y.J.; Kim, T.D.; Li, G.; Park, B.J.; Kim, J.Y.; Lee, Y.C.; Kim, J.M.; Park, J.I.; Park, H.D.; et al. Gabexate mesilate inhibits colon cancer growth, invasion, and metastasis by reducing matrix metalloproteinases and angiogenesis. Clin. Cancer Res. 2004, 10, 4517–4526. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Fingleton, B.; Matrisian, L.M. Matrix metalloproteinase inhibitors and cancer: Trials and tribulations. Science 2002, 295, 2387–2392. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.H.; Tsai, Y.S.; Lai, C.L.; Tang, C.H.; Lai, C.H.; Wu, H.C.; Hsieh, J.T.; Yang, C.R. Evolving Personalized Therapy for Castration-Resistant Prostate Cancer. Biomedicine 2014, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Mook, O.R.; Frederiks, W.M.; Van Noorden, C.J. The role of gelatinases in colorectal cancer progression and metastasis. Biochim. Biophys. Acta 2004, 1705, 69–89. [Google Scholar] [CrossRef] [PubMed]
- Eberhart, C.E.; Coffey, R.J.; Radhika, A.; Giardiello, F.M.; Ferrenbach, S.; DuBois, R.N. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology 1994, 107, 1183–1188. [Google Scholar] [CrossRef]
- Chandrasekharan, S.; Foley, N.A.; Jania, L.; Clark, P.; Audoly, L.P.; Koller, B.H. Coupling of COX-1 to mPGES1 for prostaglandin E2 biosynthesis in the murine mammary gland. J. Lipid Res. 2005, 46, 2636–2648. [Google Scholar] [CrossRef] [PubMed]
- Eruslanov, E.; Kaliberov, S.; Daurkin, I.; Kaliberova, L.; Buchsbaum, D.; Vieweg, J.; Kusmartsev, S. Altered expression of 15-hydroxyprostaglandin dehydrogenase in tumor-infiltrated CD11b myeloid cells: A mechanism for immune evasion in cancer. J. Immunol. 2009, 182, 7548–7557. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.H.; Hu, W.S.; Lin, Y.M.; Kuo, W.W.; Chen, L.M.; Chen, W.K.; Hwang, J.M.; Tsai, F.J.; Liu, C.J.; Huang, C.Y. JNK suppression is essential for 17beta-Estradiol inhibits prostaglandin E2-Induced uPA and MMP-9 expressions and cell migration in human LoVo colon cancer cells. J. Biomed. Sci. 2011, 18, 61. [Google Scholar] [CrossRef] [PubMed]
- Lord, A.M.; North, T.E.; Zon, L.I. Prostaglandin E2: Making more of your marrow. Cell Cycle 2007, 6, 3054–3057. [Google Scholar] [CrossRef] [PubMed]
- Cimino, P.J.; Keene, C.D.; Breyer, R.M.; Montine, K.S.; Montine, T.J. Therapeutic targets in prostaglandin E2 signaling for neurologic disease. Curr. Med. Chem. 2008, 15, 1863–1869. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Shih, Y.W.; Chang, C.H.; Ou, T.T.; Huang, C.C.; Hsu, J.D.; Wang, C.J. EP4 upregulation of Ras signaling and feedback regulation of Ras in human colon tissues and cancer cells. Arch. Toxicol. 2010, 84, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Dubois, R.N. Prostaglandins and cancer. Gut 2006, 55, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Kundu, N.; Rifat, S.; Walser, T.; Fulton, A.M. Prostaglandin E receptor EP4 antagonism inhibits breast cancer metastasis. Cancer Res. 2006, 66, 2923–2927. [Google Scholar] [CrossRef] [PubMed]
- Clasadonte, J.; Sharif, A.; Baroncini, M.; Prevot, V. Gliotransmission by prostaglandin E2: A prerequisite for GnRH neuronal function? Front. Endocrinol. 2011, 2, 91. [Google Scholar] [CrossRef] [PubMed]
- Banu, S.K.; Lee, J.; Speights, V.O., Jr.; Starzinski-Powitz, A.; Arosh, J.A. Selective inhibition of prostaglandin E2 receptors EP2 and EP4 induces apoptosis of human endometriotic cells through suppression of ERK1/2, AKT, NFkappaB, and beta-catenin pathways and activation of intrinsic apoptotic mechanisms. Mol. Endocrinol. 2009, 23, 1291–1305. [Google Scholar] [CrossRef] [PubMed]
- Yokoya, F.; Imamoto, N.; Tachibana, T.; Yoneda, Y. β-catenin can be transported into the nucleus in a Ran-unassisted manner. Mol. Biol. Cell 1999, 10, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Che, X.H.; Chen, C.L.; Ye, X.L.; Weng, G.B.; Guo, X.Z.; Yu, W.Y.; Tao, J.; Chen, Y.C.; Chen, X. Dual inhibition of COX-2/5-LOX blocks colon cancer proliferation, migration and invasion in vitro. Oncol. Rep. 2016, 35, 1680–1688. [Google Scholar] [CrossRef] [PubMed]
- Filipenko, I.; Schwalm, S.; Reali, L.; Pfeilschifter, J.; Fabbro, D.; Huwiler, A.; Zangemeister-Wittke, U. Upregulation of the S1P3 receptor in metastatic breast cancer cells increases migration and invasion by induction of PGE2 and EP2/EP4 activation. Biochim. Biophys. Acta 2016, 1861, 1840–1851. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Dubois, R.N. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene 2010, 29, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Voutsadakis, I.A. Pathogenesis of colorectal carcinoma and therapeutic implications: The roles of the ubiquitin-proteasome system and Cox-2. J. Cell. Mol. Med. 2007, 11, 252–285. [Google Scholar] [CrossRef] [PubMed]
- Balansky, R.; Ganchev, G.; Iltcheva, M.; Nikolov, M.; Maestra, S.L.; Micale, R.T.; D'Agostini, F.; Steele, V.E.; De Flora, S. Modulation by licofelone and celecoxib of experimentally induced cancer and preneoplastic lesions in mice exposed to cigarette smoke. Curr. Cancer Drug Targets 2015, 15, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.; Janakiram, N.B.; Li, Q.; Choi, C.I.; Zhang, Y.T.; Steele, V.E.; Rao, C.V. Chemoprevention of Colon and Small Intestinal Tumorigenesis in APC(Min/+) Mice by Licofelone, a Novel Dual 5-LOX/COX Inhibitor: Potential Implications for Human Colon Cancer Prevention. Cancer Prev. Res. 2011, 4, 2015–2026. [Google Scholar] [CrossRef] [PubMed]
- Perumal, V.; Banerjee, S.; Das, S.; Sen, R.K.; Mandal, M. Effect of liposomal celecoxib on proliferation of colon cancer cell and inhibition of DMBA-induced tumor in rat model. Cancer Nanotechnol. 2011, 2, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Tavolari, S.; Munarini, A.; Storci, G.; Laufer, S.; Chieco, P.; Guarnieri, T. The decrease of cell membrane fluidity by the non-steroidal anti-inflammatory drug Licofelone inhibits epidermal growth factor receptor signalling and triggers apoptosis in HCA-7 colon cancer cells. Cancer Lett. 2012, 321, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Vasen, H.F.A.; Mecklin, J.P.; Khan, P.M.; Lynch, H.T. Hereditary non-polyposis colorectal cancer. Lancet 1991, 338, 877. [Google Scholar] [CrossRef]
- Strate, L.L.; Syngal, S. Hereditary colon cancer syndromes. Semin. Oncol. 2011, 16, 201–213. [Google Scholar]
- Kohlmann, W.; Gruber, S.B. Lynch Syndrome. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1211/?report=reader (acessed on 22 May 2017).
- Kalady, M.F. Surgical management of hereditary nonpolyposis colorectal cancer. Adv. Surg. 2011, 45, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhang, S.; Aung, L.H.; Ouyang, J.; Wei, L. Lymph node harvested in laparoscopic versus open colorectal cancer approaches: A meta-analysis. Surg. Laparosc. Endosc. Percutan. Tech. 2012, 22, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Rakhila, H.; Bourcier, N.; Akoum, A.; Pouliot, M. Abnormal Expression of Prostaglandins E2 and F2alpha Receptors and Transporters in Patients with Endometriosis. BioMed Res. Int. 2015, 2015, 808146. [Google Scholar] [CrossRef] [PubMed]
- Tak, F.F. The PGE2-EP Receptor Axis in Colorectal Cancer and Angiogenesis. J. Tumor 2014, 2, 208–218. [Google Scholar]
- Fiebich, B.L.; Schleicher, S.; Spleiss, O.; Czygan, M.; Hull, M. Mechanisms of prostaglandin E2-induced interleukin-6 release in astrocytes: Possible involvement of EP4-like receptors, p38 mitogen-activated protein kinase and protein kinase C. J. Neurochem. 2001, 79, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, Y.; Narumiya, S. Prostaglandin E receptors. J. Biol. Chem. 2007, 282, 11613–11617. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, A.; Yan, X.; Macias-Perez, I.; Wei, S.; Hata, A.N.; Breyer, R.M.; Morrow, J.D.; Capdevila, J.H. Colon carcinoma cell growth is associated with prostaglandin E2/EP4 receptor-evoked ERK activation. J. Biol. Chem. 2004, 279, 29797–29804. [Google Scholar] [CrossRef] [PubMed]
- George, R.J.; Sturmoski, M.A.; Anant, S.; Houchen, C.W. EP4 mediates PGE2 dependent cell survival through the PI3 kinase/AKT pathway. Prostaglandins Other Lipid Mediat. 2007, 83, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.I.; Lakshmikanthan, V.; Frilot, N.; Daaka, Y. Prostaglandin E2 promotes lung cancer cell migration via EP4-betaArrestin1-c-Src signalsome. Mol. Cancer Res. 2010, 8, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Majumder, M.; Xin, X.; Liu, L.; Girish, G.V.; Lala, P.K. Prostaglandin E2 receptor EP4 as the common target on cancer cells and macrophages to abolish angiogenesis, lymphangiogenesis, metastasis, and stem-like cell functions. Cancer Sci. 2014, 105, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.N.; Wu, W.K.; Shin, V.Y.; Bruce, I.C.; Wong, B.C.; Cho, C.H. Dual inhibition of 5-LOX and COX-2 suppresses colon cancer formation promoted by cigarette smoke. Carcinogenesis 2005, 26, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Sumitani, K.; Kamijo, R.; Toyoshima, T.; Nakanishi, Y.; Takizawa, K.; Hatori, M.; Nagumo, M. Specific inhibition of cyclooxygenase-2 results in inhibition of proliferation of oral cancer cell lines via suppression of prostaglandin E2 production. J. Oral Pathol. Med. 2001, 30, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Thayele Purayil, H.; Black, J.B.; Fetto, F.; Lynch, L.D.; Masannat, J.N.; Daaka, Y. Prostaglandin E2 receptor 4 mediates renal cell carcinoma intravasation and metastasis. Cancer Lett. 2017, 391, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Boutaud, O.; Sosa, I.R.; Amin, T.; Oram, D.; Adler, D.; Hwang, H.S.; Crews, B.C.; Milne, G.; Harris, B.K.; Hoeksema, M.; et al. Inhibition of the Biosynthesis of Prostaglandin E2 By Low-Dose Aspirin: Implications for Adenocarcinoma Metastasis. Cancer Prev. Res. 2016, 9, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ji, Q.; Ye, N.; Sui, H.; Zhou, L.; Zhu, H.; Fan, Z.; Cai, J.; Li, Q. Berberine Inhibits Invasion and Metastasis of Colorectal Cancer Cells via COX-2/PGE2 Mediated JAK2/STAT3 Signaling Pathway. PLoS ONE 2015, 10, e0123478. [Google Scholar] [CrossRef] [PubMed]
- Barth, A.I.; Nathke, I.S.; Nelson, W.J. Cadherins, catenins and APC protein: Interplay between cytoskeletal complexes and signaling pathways. Curr. Opin. Cell Biol. 1997, 9, 683–690. [Google Scholar] [CrossRef]
- Nunez, F.; Bravo, S.; Cruzat, F.; Montecino, M.; De Ferrari, G.V. Wnt/beta-catenin signaling enhances cyclooxygenase-2 (COX2) transcriptional activity in gastric cancer cells. PLoS ONE 2011, 6, e18562. [Google Scholar] [CrossRef] [PubMed]
- Leone, V.; di Palma, A.; Ricchi, P.; Acquaviva, F.; Giannouli, M.; Di Prisco, A.M.; Iuliano, F.; Acquaviva, A.M. PGE2 inhibits apoptosis in human adenocarcinoma Caco-2 cell line through Ras-PI3K association and cAMP-dependent kinase A activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G673–G681. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.M.; Li, Y.M.; Semenov, M.; Han, C.; Baeg, G.H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 2002, 108, 837–847. [Google Scholar] [CrossRef]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, H.-H.; Lin, Y.-M.; Shen, C.-Y.; Shibu, M.A.; Li, S.-Y.; Chang, S.-H.; Lin, C.-C.; Chen, R.-J.; Viswanadha, V.P.; Shih, H.-N.; et al. Prostaglandin E2-Induced COX-2 Expressions via EP2 and EP4 Signaling Pathways in Human LoVo Colon Cancer Cells. Int. J. Mol. Sci. 2017, 18, 1132. https://doi.org/10.3390/ijms18061132
Hsu H-H, Lin Y-M, Shen C-Y, Shibu MA, Li S-Y, Chang S-H, Lin C-C, Chen R-J, Viswanadha VP, Shih H-N, et al. Prostaglandin E2-Induced COX-2 Expressions via EP2 and EP4 Signaling Pathways in Human LoVo Colon Cancer Cells. International Journal of Molecular Sciences. 2017; 18(6):1132. https://doi.org/10.3390/ijms18061132
Chicago/Turabian StyleHsu, Hsi-Hsien, Yueh-Min Lin, Chia-Yao Shen, Marthandam Asokan Shibu, Shin-Yi Li, Sheng-Huang Chang, Chien-Chung Lin, Ray-Jade Chen, Vijaya Padma Viswanadha, Hui-Nung Shih, and et al. 2017. "Prostaglandin E2-Induced COX-2 Expressions via EP2 and EP4 Signaling Pathways in Human LoVo Colon Cancer Cells" International Journal of Molecular Sciences 18, no. 6: 1132. https://doi.org/10.3390/ijms18061132
APA StyleHsu, H.-H., Lin, Y.-M., Shen, C.-Y., Shibu, M. A., Li, S.-Y., Chang, S.-H., Lin, C.-C., Chen, R.-J., Viswanadha, V. P., Shih, H.-N., & Huang, C.-Y. (2017). Prostaglandin E2-Induced COX-2 Expressions via EP2 and EP4 Signaling Pathways in Human LoVo Colon Cancer Cells. International Journal of Molecular Sciences, 18(6), 1132. https://doi.org/10.3390/ijms18061132