
The Cytokine Flt3-Ligand in Normal and Malignant Hematopoiesis


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. FL and Its Receptor, Flt3
3. The Role of FL in Normal Hematopoiesis
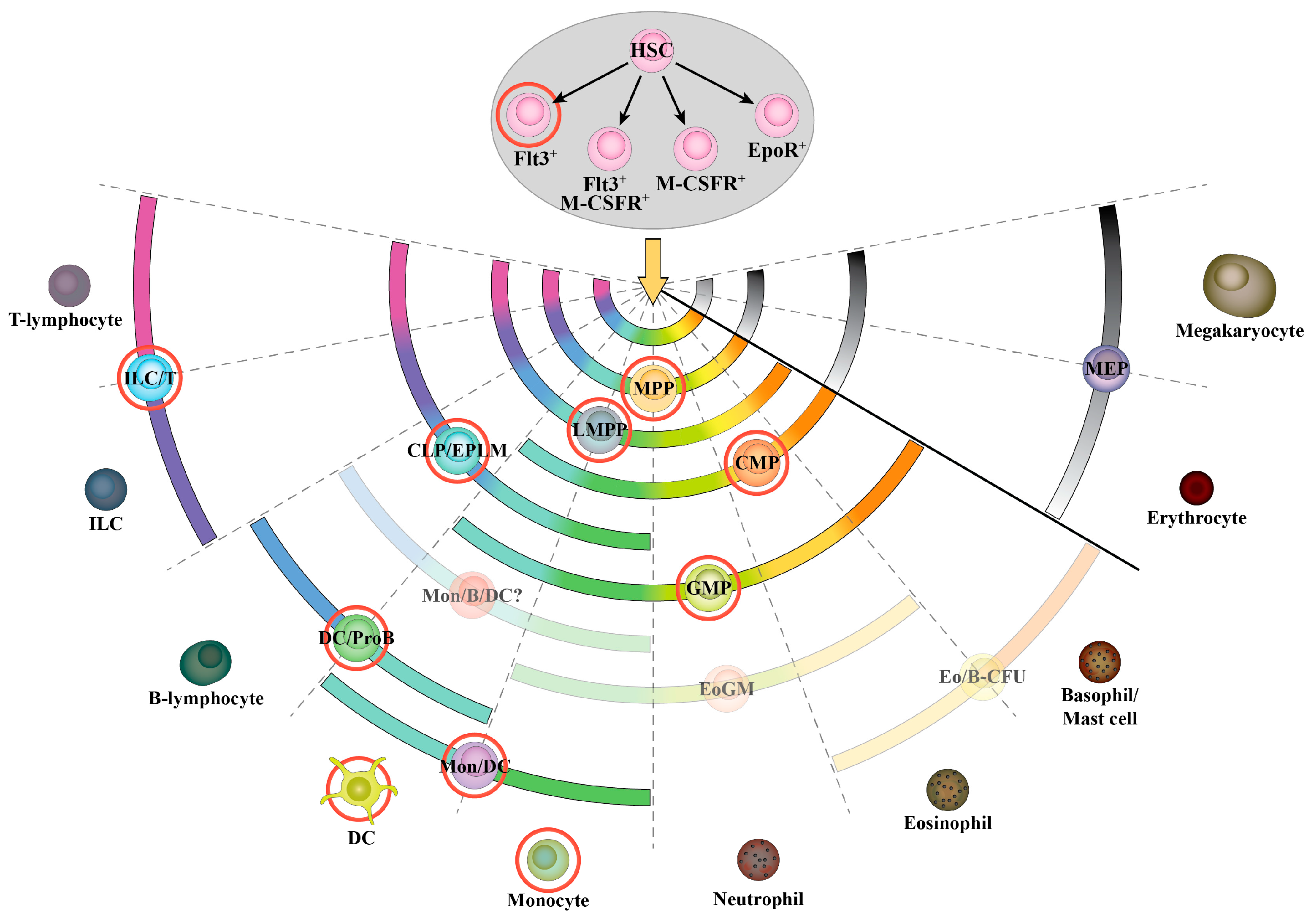
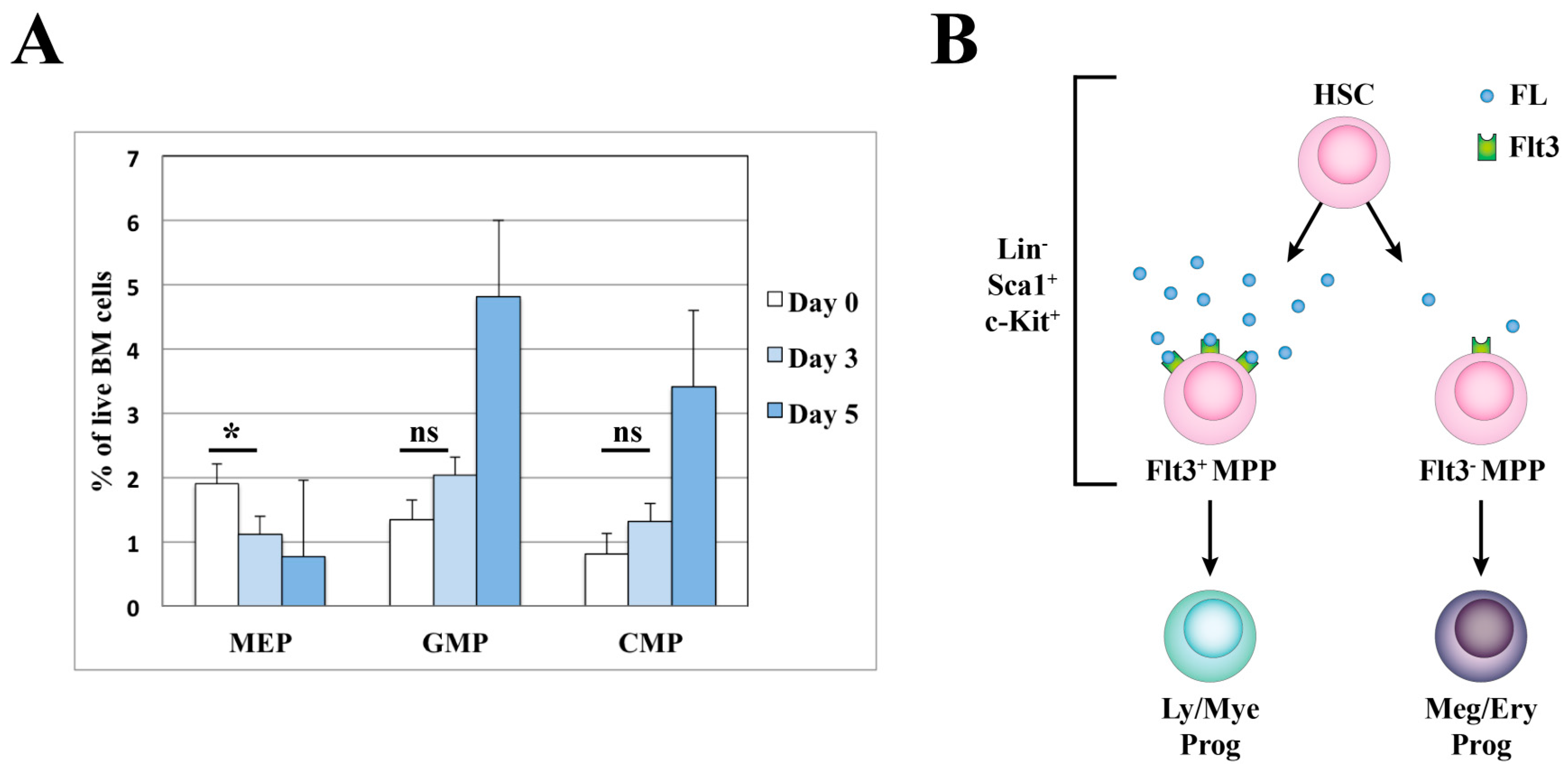
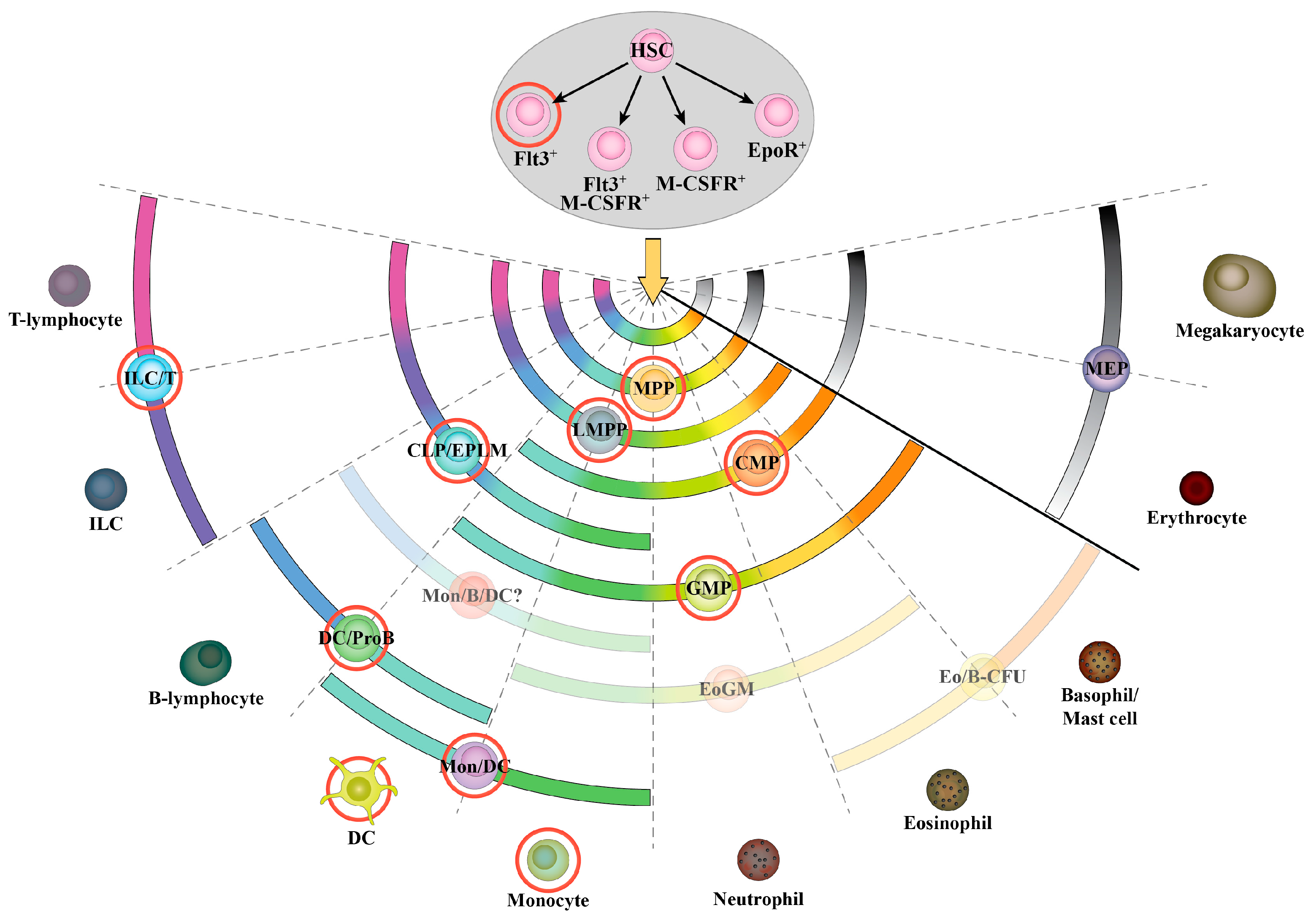
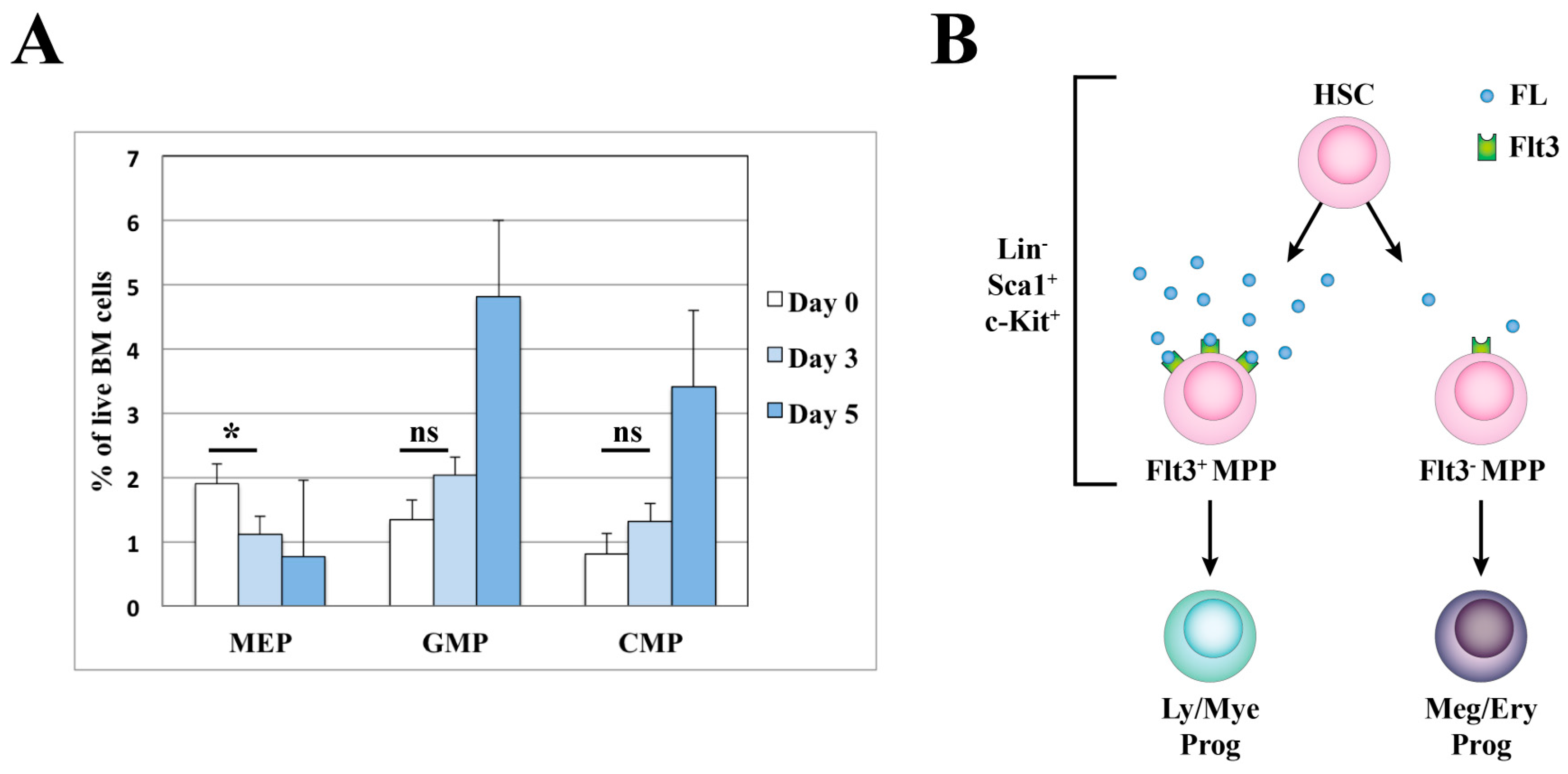
3.1. Hematopoietic Stem Cells and Early Progenitors
3.2. Dendritic Cells
3.3. B Cells
3.4. T Cells
3.5. Overview
4. FL and Flt3 in Hematopoietic Malignancies
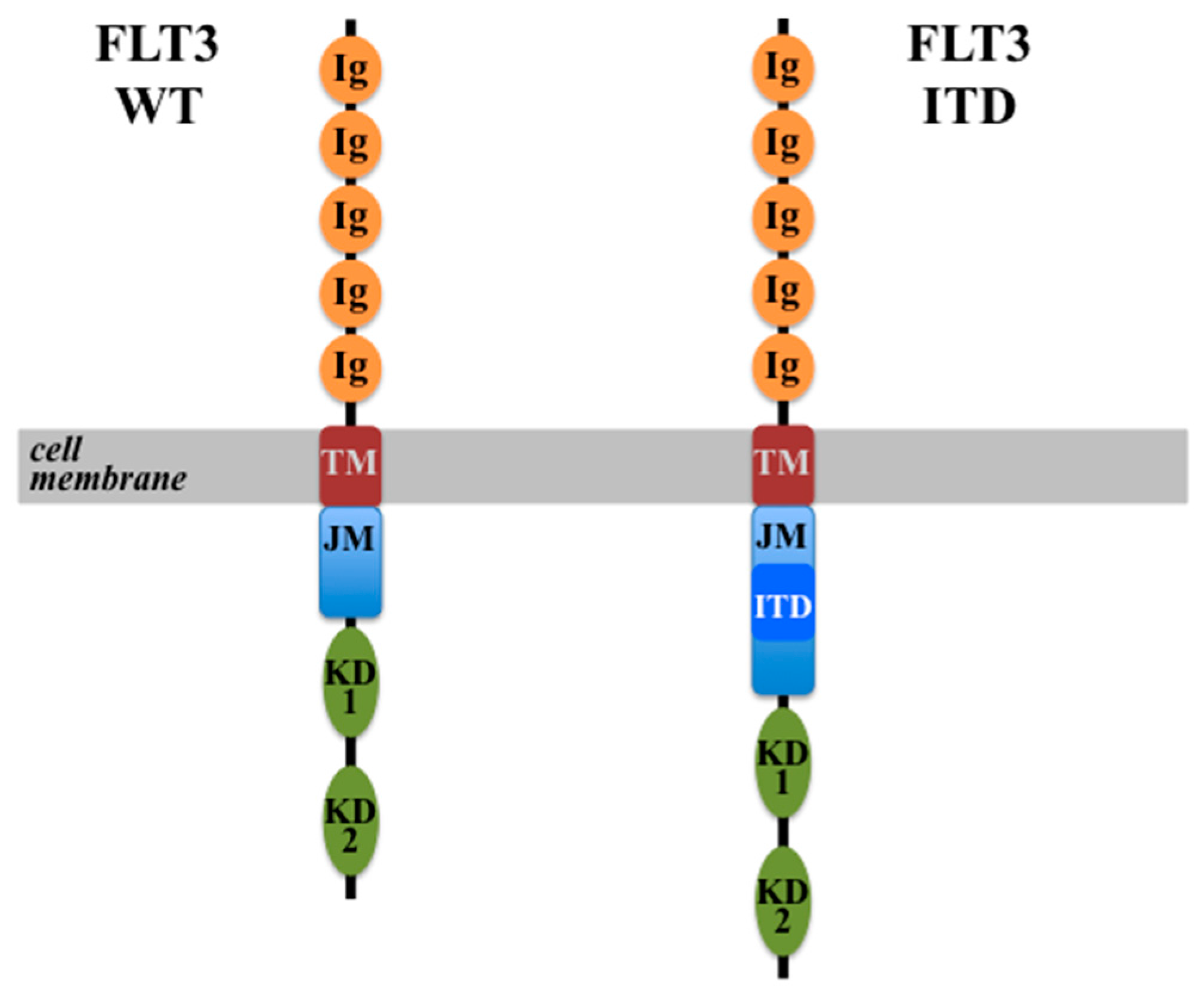
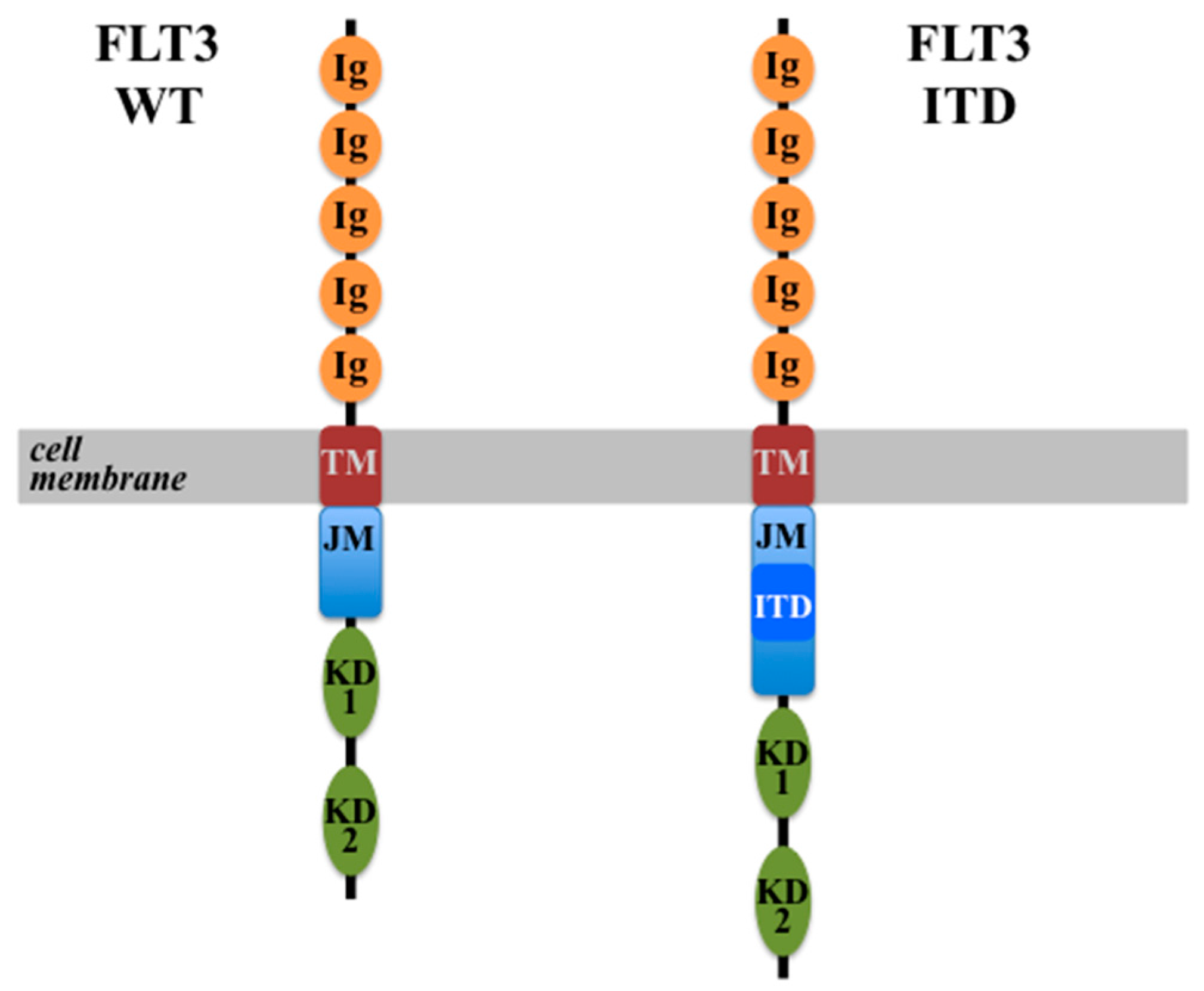
4.1. FLT3-ITD
4.2. FLT3-TKD
4.3. FLT3 Inhibitors
5. Open Questions and Future Challenges
Acknowledgments
Conflicts of Interest
References
- Metcalf, D. Hematopoietic cytokines. Blood 2008, 111, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Matthews, W.; Jordan, C.T.; Wiegand, G.W.; Pardoll, D.; Lemischka, I.R. A receptor tyrosine kinase specific to hematopoietic stem and progenitor cell-enriched populations. Cell 1991, 65, 1143–1152. [Google Scholar] [CrossRef]
- Rosnet, O.; Marchetto, S.; deLapeyriere, O.; Birnbaum, D. Murine Flt3, a gene encoding a novel tyrosine kinase receptor of the PDGFR/CSF1R family. Oncogene 1991, 6, 1641–1650. [Google Scholar] [PubMed]
- Rosnet, O.; Mattei, M.G.; Marchetto, S.; Birnbaum, D. Isolation and chromosomal localization of a novel FMS-like tyrosine kinase gene. Genomics 1991, 9, 380–385. [Google Scholar] [CrossRef]
- Small, D.; Levenstein, M.; Kim, E.; Carow, C.; Amin, S.; Rockwell, P.; Witte, L.; Burrow, C.; Ratajczak, M.Z.; Gewirtz, A.M.; et al. STK-1, the human homolog of Flk-2/Flt-3, is selectively expressed in CD34+ human bone marrow cells and is involved in the proliferation of early progenitor/stem cells. Proc. Natl. Acad. Sci. USA 1994, 91, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Rosnet, O.; Birnbaum, D. Hematopoietic receptors of class III receptor-type tyrosine kinases. Crit. Rev. Oncog. 1993, 4, 595–613. [Google Scholar] [PubMed]
- Lyman, S.D.; James, L.; Vanden Bos, T.; de Vries, P.; Brasel, K.; Gliniak, B.; Hollingsworth, L.T.; Picha, K.S.; McKenna, H.J.; Splett, R.R.; et al. Molecular cloning of a ligand for the flt3flk-2 tyrosine kinase receptor: A proliferative factor for primitive hematopoietic cells. Cell 1993, 75, 1157–1167. [Google Scholar] [CrossRef]
- Lyman, S.D.; James, L.; Escobar, S.; Downey, H.; de Vries, P.; Brasel, K.; Stocking, K.; Beckmann, M.P.; Copeland, N.G.; Cleveland, L.S.; et al. Identification of soluble and membrane-bound isoforms of the murine flt3 ligand generated by alternative splicing of mRNAs. Oncogene 1995, 10, 149–157. [Google Scholar] [PubMed]
- Lyman, S.D.; James, L.; Johnson, L.; Brasel, K.; de Vries, P.; Escobar, S.S.; Downey, H.; Splett, R.R.; Beckmann, M.P.; McKenna, H.J. Cloning of the human homologue of the murine flt3 ligand: A growth factor for early hematopoietic progenitor cells. Blood 1994, 83, 2795–2801. [Google Scholar] [PubMed]
- Turner, A.M.; Lin, N.L.; Issarachai, S.; Lyman, S.D.; Broudy, V.C. Flt3 receptor expression on the surface of normal and malignant human hematopoietic cells. Blood 1996, 88, 3383–3390. [Google Scholar] [PubMed]
- Dosil, M.; Wang, S.; Lemischka, I.R. Mitogenic signalling and substrate specificity of the Flk2/Flt3 receptor tyrosine kinase in fibroblasts and interleukin 3-dependent hematopoietic cells. Mol. Cell. Biol. 1993, 13, 6572–6585. [Google Scholar] [CrossRef] [PubMed]
- Rottapel, R.; Turck, C.W.; Casteran, N.; Liu, X.; Birnbaum, D.; Pawson, T.; Dubreuil, P. Substrate specificities and identification of a putative binding site for PI3K in the carboxy tail of the murine Flt3 receptor tyrosine kinase. Oncogene 1994, 9, 1755–1765. [Google Scholar] [PubMed]
- Lavagna-Sevenier, C.; Marchetto, S.; Birnbaum, D.; Rosnet, O. FLT3 signaling in hematopoietic cells involves CBL, SHC and an unknown P115 as prominent tyrosine-phosphorylated substrates. Leukemia 1998, 12, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Marchetto, S.; Fournier, E.; Beslu, N.; Aurran-Schleinitz, T.; Dubreuil, P.; Borg, J.P.; Birnbaum, D.; Rosnet, O. SHC and SHIP phosphorylation and interaction in response to activation of the FLT3 receptor. Leukemia 1999, 13, 1374–1382. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Broxmeyer, H.E. p85 subunit of pi3 kinase does not bind to human Flt3 receptor, but associates with SHP2, SHIP, and a tyrosine-phosphorylated 100-kDa protein in Flt3 ligand-stimulated hematopoietic cells. Biochem. Biophys. Res. Commun. 1999, 254, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Ahsberg, J.; Tsapogas, P.; Qian, H.; Zetterblad, J.; Zandi, S.; Mansson, R.; Jonsson, J.I.; Sigvardsson, M. Interleukin-7-induced Stat-5 acts in synergy with Flt-3 signaling to stimulate expansion of hematopoietic progenitor cells. J. Biol. Chem. 2010, 285, 36275–36284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Broxmeyer, H.E. Flt3 ligand induces tyrosine phosphorylation of gab1 and gab2 and their association with shp-2, grb2, and PI3 kinase. Biochem. Biophys. Res. Commun. 2000, 277, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Mantel, C.; Broxmeyer, H.E. Flt3 signaling involves tyrosyl-phosphorylation of SHP-2 and SHIP and their association with Grb2 and Shc in Baf3/Flt3 cells. J. Leukoc. Biol. 1999, 65, 372–380. [Google Scholar] [PubMed]
- Laouar, Y.; Welte, T.; Fu, X.Y.; Flavell, R.A. STAT3 is required for Flt3L-dependent dendritic cell differentiation. Immunity 2003, 19, 903–912. [Google Scholar] [CrossRef]
- Onai, N.; Obata-Onai, A.; Tussiwand, R.; Lanzavecchia, A.; Manz, M.G. Activation of the Flt3 signal transduction cascade rescues and enhances type I interferon-producing and dendritic cell development. J. Exp. Med. 2006, 203, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Fukuda, S.; Lee, Y.; Hangoc, G.; Cooper, S.; Spolski, R.; Leonard, W.J.; Broxmeyer, H.E. Essential role of signal transducer and activator of transcription (Stat)5a but not Stat5b for Flt3-dependent signaling. J. Exp. Med. 2000, 192, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Banu, N.; Deng, B.; Lyman, S.D.; Avraham, H. Modulation of haematopoietic progenitor development by FLT-3 ligand. Cytokine 1999, 11, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Brashem-Stein, C.; Flowers, D.A.; Bernstein, I.D. Regulation of colony forming cell generation by flt-3 ligand. Br. J. Haematol. 1996, 94, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Broxmeyer, H.E.; Lu, L.; Cooper, S.; Ruggieri, L.; Li, Z.H.; Lyman, S.D. Flt3 ligand stimulates/costimulates the growth of myeloid stem/progenitor cells. Exp. Hematol. 1995, 23, 1121–1129. [Google Scholar] [PubMed]
- Gabbianelli, M.; Pelosi, E.; Montesoro, E.; Valtieri, M.; Luchetti, L.; Samoggia, P.; Vitelli, L.; Barberi, T.; Testa, U.; Lyman, S.; et al. Multi-level effects of flt3 ligand on human hematopoiesis: Expansion of putative stem cells and proliferation of granulomonocytic progenitors/monocytic precursors. Blood 1995, 86, 1661–1670. [Google Scholar] [PubMed]
- Hirayama, F.; Lyman, S.D.; Clark, S.C.; Ogawa, M. The flt3 ligand supports proliferation of lymphohematopoietic progenitors and early B-lymphoid progenitors. Blood 1995, 85, 1762–1768. [Google Scholar] [PubMed]
- Hudak, S.; Hunte, B.; Culpepper, J.; Menon, S.; Hannum, C.; Thompson-Snipes, L.; Rennick, D. FLT3/FLK2 ligand promotes the growth of murine stem cells and the expansion of colony-forming cells and spleen colony-forming units. Blood 1995, 85, 2747–2755. [Google Scholar] [PubMed]
- Jacobsen, S.E.; Okkenhaug, C.; Myklebust, J.; Veiby, O.P.; Lyman, S.D. The FLT3 ligand potently and directly stimulates the growth and expansion of primitive murine bone marrow progenitor cells in vitro: Synergistic interactions with interleukin (IL) 11, IL-12, and other hematopoietic growth factors. J. Exp. Med. 1995, 181, 1357–1363. [Google Scholar] [CrossRef] [PubMed]
- Namikawa, R.; Muench, M.O.; de Vries, J.E.; Roncarolo, M.G. The FLK2/FLT3 ligand synergizes with interleukin-7 in promoting stromal-cell-independent expansion and differentiation of human fetal pro-B cells in vitro. Blood 1996, 87, 1881–1890. [Google Scholar] [PubMed]
- Ray, R.J.; Paige, C.J.; Furlonger, C.; Lyman, S.D.; Rottapel, R. Flt3 ligand supports the differentiation of early B cell progenitors in the presence of interleukin-11 and interleukin-7. Eur. J. Immunol. 1996, 26, 1504–1510. [Google Scholar] [CrossRef] [PubMed]
- Veiby, O.P.; Lyman, S.D.; Jacobsen, S.E. Combined signaling through interleukin-7 receptors and flt3 but not c-kit potently and selectively promotes B-cell commitment and differentiation from uncommitted murine bone marrow progenitor cells. Blood 1996, 88, 1256–1265. [Google Scholar] [PubMed]
- McKenna, H.J.; de Vries, P.; Brasel, K.; Lyman, S.D.; Williams, D.E. Effect of flt3 ligand on the ex vivo expansion of human CD34+ hematopoietic progenitor cells. Blood 1995, 86, 3413–3420. [Google Scholar] [PubMed]
- Rusten, L.S.; Lyman, S.D.; Veiby, O.P.; Jacobsen, S.E. The FLT3 ligand is a direct and potent stimulator of the growth of primitive and committed human CD34+ bone marrow progenitor cells in vitro. Blood 1996, 87, 1317–1325. [Google Scholar] [PubMed]
- Mackarehtschian, K.; Hardin, J.D.; Moore, K.A.; Boast, S.; Goff, S.P.; Lemischka, I.R. Targeted disruption of the flk2/flt3 gene leads to deficiencies in primitive hematopoietic progenitors. Immunity 1995, 3, 147–161. [Google Scholar] [CrossRef]
- McKenna, H.J.; Stocking, K.L.; Miller, R.E.; Brasel, K.; De Smedt, T.; Maraskovsky, E.; Maliszewski, C.R.; Lynch, D.H.; Smith, J.; Pulendran, B.; et al. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood 2000, 95, 3489–3497. [Google Scholar] [PubMed]
- Sitnicka, E.; Bryder, D.; Theilgaard-Monch, K.; Buza-Vidas, N.; Adolfsson, J.; Jacobsen, S.E. Key role of flt3 ligand in regulation of the common lymphoid progenitor but not in maintenance of the hematopoietic stem cell pool. Immunity 2002, 17, 463–472. [Google Scholar] [CrossRef]
- Von Muenchow, L.; Alberti-Servera, L.; Klein, F.; Capoferri, G.; Finke, D.; Ceredig, R.; Rolink, A.; Tsapogas, P. Permissive roles of cytokines interleukin-7 and Flt3 ligand in mouse B-cell lineage commitment. Proc. Natl. Acad. Sci. USA 2016, 113, E8122–E8130. [Google Scholar] [CrossRef] [PubMed]
- Beaudin, A.E.; Boyer, S.W.; Forsberg, E.C. Flk2/Flt3 promotes both myeloid and lymphoid development by expanding non-self-renewing multipotent hematopoietic progenitor cells. Exp. Hematol. 2014, 42, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Dolence, J.J.; Gwin, K.A.; Shapiro, M.B.; Medina, K.L. Flt3 signaling regulates the proliferation, survival, and maintenance of multipotent hematopoietic progenitors that generate B cell precursors. Exp. Hematol. 2014, 42, 380–393. [Google Scholar] [CrossRef] [PubMed]
- Balciunaite, G.; Ceredig, R.; Massa, S.; Rolink, A.G. A B220+ CD117+ CD19− hematopoietic progenitor with potent lymphoid and myeloid developmental potential. Eur. J. Immunol. 2005, 35, 2019–2030. [Google Scholar] [CrossRef] [PubMed]
- Karsunky, H.; Inlay, M.A.; Serwold, T.; Bhattacharya, D.; Weissman, I.L. Flk2+ common lymphoid progenitors possess equivalent differentiation potential for the B and T lineages. Blood 2008, 111, 5562–5570. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.; Mooney, C.J.; Alberti-Servera, L.; Muenchow, L.; Toellner, K.M.; Ceredig, R.; Rolink, A. Versatility of stem and progenitor cells and the instructive actions of cytokines on hematopoiesis. Crit. Rev. Clin. Lab. Sci. 2015, 52, 168–179. [Google Scholar] [PubMed]
- Ceredig, R.; Rolink, A.G.; Brown, G. Models of haematopoiesis: Seeing the wood for the trees. Nat. Rev. Immunol. 2009, 9, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Adolfsson, J.; Borge, O.J.; Bryder, D.; Theilgaard-Monch, K.; Astrand-Grundstrom, I.; Sitnicka, E.; Sasaki, Y.; Jacobsen, S.E. Upregulation of Flt3 expression within the bone marrow Lin−Sca1+c-kit+ stem cell compartment is accompanied by loss of self-renewal capacity. Immunity 2001, 15, 659–669. [Google Scholar] [CrossRef]
- Christensen, J.L.; Weissman, I.L. Flk-2 is a marker in hematopoietic stem cell differentiation: A simple method to isolate long-term stem cells. Proc. Natl. Acad. Sci. USA 2001, 98, 14541–14546. [Google Scholar] [CrossRef] [PubMed]
- Akashi, K.; Traver, D.; Miyamoto, T.; Weissman, I.L. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 2000, 404, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Kondo, M.; Weissman, I.L.; Akashi, K. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell 1997, 91, 661–672. [Google Scholar] [CrossRef]
- Adolfsson, J.; Mansson, R.; Buza-Vidas, N.; Hultquist, A.; Liuba, K.; Jensen, C.T.; Bryder, D.; Yang, L.; Borge, O.J.; Thoren, L.A.; et al. Identification of Flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential a revised road map for adult blood lineage commitment. Cell 2005, 121, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Boyer, S.W.; Schroeder, A.V.; Smith-Berdan, S.; Forsberg, E.C. All hematopoietic cells develop from hematopoietic stem cells through Flk2/Flt3-positive progenitor cells. Cell Stem Cell 2011, 9, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, E.C.; Serwold, T.; Kogan, S.; Weissman, I.L.; Passegue, E. New evidence supporting megakaryocyte-erythrocyte potential of flk2/flt3+ multipotent hematopoietic progenitors. Cell 2006, 126, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Luc, S.; Buza-Vidas, N.; Jacobsen, S.E. Biological and molecular evidence for existence of lymphoid-primed multipotent progenitors. Ann. N. Y. Acad. Sci. 2007, 1106, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Luc, S.; Anderson, K.; Kharazi, S.; Buza-Vidas, N.; Boiers, C.; Jensen, C.T.; Ma, Z.; Wittmann, L.; Jacobsen, S.E. Down-regulation of Mpl marks the transition to lymphoid-primed multipotent progenitors with gradual loss of granulocyte-monocyte potential. Blood 2008, 111, 3424–3434. [Google Scholar] [CrossRef] [PubMed]
- Buza-Vidas, N.; Woll, P.; Hultquist, A.; Duarte, S.; Lutteropp, M.; Bouriez-Jones, T.; Ferry, H.; Luc, S.; Jacobsen, S.E. Flt3 expression initiates in fully multipotent mouse hematopoietic progenitor cells. Blood 2011, 118, 1544–1548. [Google Scholar] [CrossRef] [PubMed]
- Buza-Vidas, N.; Woll, P.; Hultquist, A.; Duarte, S.; Lutteropp, M.; Bouriez-Jones, T.; Ferry, H.; Luc, S.; Jacobsen, S.E. Selective expression of Flt3 within the mouse hematopoietic stem cell compartment. Int. J. Mol. Sci. 2017, 18, 1037. [Google Scholar]
- Kiel, M.J.; Yilmaz, O.H.; Iwashita, T.; Yilmaz, O.H.; Terhorst, C.; Morrison, S.J. Slam family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005, 121, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Oguro, H.; Ding, L.; Morrison, S.J. SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell 2013, 13, 102–116. [Google Scholar] [CrossRef] [PubMed]
- Crisan, M.; Dzierzak, E. The many faces of hematopoietic stem cell heterogeneity. Development 2016, 143, 4571–4581. [Google Scholar] [CrossRef] [PubMed]
- Eaves, C.J. Hematopoietic stem cells: Concepts, definitions, and the new reality. Blood 2015, 125, 2605–2613. [Google Scholar] [CrossRef] [PubMed]
- Notta, F.; Zandi, S.; Takayama, N.; Dobson, S.; Gan, O.I.; Wilson, G.; Kaufmann, K.B.; McLeod, J.; Laurenti, E.; Dunant, C.F.; et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 2016, 351, aab2116. [Google Scholar] [CrossRef] [PubMed]
- Endele, M.; Etzrodt, M.; Schroeder, T. Instruction of hematopoietic lineage choice by cytokine signaling. Exp. Cell Res. 2014, 329, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, S.; Sieweke, M. Integration of cytokine and transcription factor signals in hematopoietic stem cell commitment. Semin Immunol. 2011, 23, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Ceredig, R.; Rauch, M.; Balciunaite, G.; Rolink, A.G. Increasing Flt3L availability alters composition of a novel bone marrow lymphoid progenitor compartment. Blood 2006, 108, 1216–1222. [Google Scholar] [CrossRef] [PubMed]
- Swee, L.K.; Bosco, N.; Malissen, B.; Ceredig, R.; Rolink, A. Expansion of peripheral naturally occurring T regulatory cells by Fms-like tyrosine kinase 3 ligand treatment. Blood 2009, 113, 6277–6287. [Google Scholar] [CrossRef] [PubMed]
- Tsapogas, P.; Swee, L.K.; Nusser, A.; Nuber, N.; Kreuzaler, M.; Capoferri, G.; Rolink, H.; Ceredig, R.; Rolink, A. In vivo evidence for an instructive role of fms-like tyrosine kinase-3 (FLT3) ligand in hematopoietic development. Haematologica 2014, 99, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Grover, A.; Mancini, E.; Moore, S.; Mead, A.J.; Atkinson, D.; Rasmussen, K.D.; O’Carroll, D.; Jacobsen, S.E.; Nerlov, C. Erythropoietin guides multipotent hematopoietic progenitor cells toward an erythroid fate. J. Exp. Med. 2014, 211, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Mossadegh-Keller, N.; Sarrazin, S.; Kandalla, P.K.; Espinosa, L.; Stanley, E.R.; Nutt, S.L.; Moore, J.; Sieweke, M.H. M-CSF instructs myeloid lineage fate in single haematopoietic stem cells. Nature 2013, 497, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Rieger, M.A.; Hoppe, P.S.; Smejkal, B.M.; Eitelhuber, A.C.; Schroeder, T. Hematopoietic cytokines can instruct lineage choice. Science 2009, 325, 217–218. [Google Scholar] [CrossRef] [PubMed]
- Waskow, C.; Liu, K.; Darrasse-Jeze, G.; Guermonprez, P.; Ginhoux, F.; Merad, M.; Shengelia, T.; Yao, K.; Nussenzweig, M. The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat. Immunol. 2008, 9, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Gilliet, M.; Boonstra, A.; Paturel, C.; Antonenko, S.; Xu, X.L.; Trinchieri, G.; O’Garra, A.; Liu, Y.J. The development of murine plasmacytoid dendritic cell precursors is differentially regulated by FLT3-ligand and granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 2002, 195, 953–958. [Google Scholar] [CrossRef] [PubMed]
- Karsunky, H.; Merad, M.; Cozzio, A.; Weissman, I.L.; Manz, M.G. Flt3 ligand regulates dendritic cell development from Flt3+ lymphoid and myeloid-committed progenitors to Flt3+ dendritic cells in vivo. J. Exp. Med. 2003, 198, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Brasel, K.; De Smedt, T.; Smith, J.L.; Maliszewski, C.R. Generation of murine dendritic cells from flt3-ligand-supplemented bone marrow cultures. Blood 2000, 96, 3029–3039. [Google Scholar] [PubMed]
- Brawand, P.; Fitzpatrick, D.R.; Greenfield, B.W.; Brasel, K.; Maliszewski, C.R.; De Smedt, T. Murine plasmacytoid pre-dendritic cells generated from Flt3 ligand-supplemented bone marrow cultures are immature APCs. J. Immunol. 2002, 169, 6711–6719. [Google Scholar] [CrossRef] [PubMed]
- Naik, S.H.; Proietto, A.I.; Wilson, N.S.; Dakic, A.; Schnorrer, P.; Fuchsberger, M.; Lahoud, M.H.; O’Keeffe, M.; Shao, Q.X.; Chen, W.F.; et al. Cutting edge: Generation of splenic CD8+ and CD8− dendritic cell equivalents in Fms-like tyrosine kinase 3 ligand bone marrow cultures. J. Immunol. 2005, 174, 6592–6597. [Google Scholar] [CrossRef] [PubMed]
- Daro, E.; Pulendran, B.; Brasel, K.; Teepe, M.; Pettit, D.; Lynch, D.H.; Vremec, D.; Robb, L.; Shortman, K.; McKenna, H.J.; et al. Polyethylene glycol-modified GM-CSF expands CD11bhighCD11chigh but notCD11blowCD11chigh murine dendritic cells in vivo: A comparative analysis with Flt3 ligand. J. Immunol. 2000, 165, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Maraskovsky, E.; Brasel, K.; Teepe, M.; Roux, E.R.; Lyman, S.D.; Shortman, K.; McKenna, H.J. Dramatic increase in the numbers of functionally mature dendritic cells in Flt3 ligand-treated mice: Multiple dendritic cell subpopulations identified. J. Exp. Med. 1996, 184, 1953–1962. [Google Scholar] [CrossRef] [PubMed]
- O’Keeffe, M.; Hochrein, H.; Vremec, D.; Pooley, J.; Evans, R.; Woulfe, S.; Shortman, K. Effects of administration of progenipoietin 1, Flt-3 ligand, granulocyte colony-stimulating factor, and pegylated granulocyte-macrophage colony-stimulating factor on dendritic cell subsets in mice. Blood 2002, 99, 2122–2130. [Google Scholar] [CrossRef] [PubMed]
- Manfra, D.J.; Chen, S.C.; Jensen, K.K.; Fine, J.S.; Wiekowski, M.T.; Lira, S.A. Conditional expression of murine Flt3 ligand leads to expansion of multiple dendritic cell subsets in peripheral blood and tissues of transgenic mice. J. Immunol. 2003, 170, 2843–2852. [Google Scholar] [CrossRef] [PubMed]
- Juan, T.S.; McNiece, I.K.; Van, G.; Lacey, D.; Hartley, C.; McElroy, P.; Sun, Y.; Argento, J.; Hill, D.; Yan, X.Q.; et al. Chronic expression of murine flt3 ligand in mice results in increased circulating white blood cell levels and abnormal cellular infiltrates associated with splenic fibrosis. Blood 1997, 90, 76–84. [Google Scholar] [PubMed]
- Liu, K.; Victora, G.D.; Schwickert, T.A.; Guermonprez, P.; Meredith, M.M.; Yao, K.; Chu, F.F.; Randolph, G.J.; Rudensky, A.Y.; Nussenzweig, M. In vivo analysis of dendritic cell development and homeostasis. Science 2009, 324, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Onai, N.; Kurabayashi, K.; Hosoi-Amaike, M.; Toyama-Sorimachi, N.; Matsushima, K.; Inaba, K.; Ohteki, T. A clonogenic progenitor with prominent plasmacytoid dendritic cell developmental potential. Immunity 2013, 38, 943–957. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, P.K.; Alfieri, A.; Thomas, E.K.; Beri, V.; Tanaka, K.E.; Vikram, B.; Guha, C. Flt3-ligand administration after radiation therapy prolongs survival in a murine model of metastatic lung cancer. Cancer Res. 1999, 59, 6028–6032. [Google Scholar] [PubMed]
- Chen, K.; Braun, S.; Lyman, S.; Fan, Y.; Traycoff, C.M.; Wiebke, E.A.; Gaddy, J.; Sledge, G.; Broxmeyer, H.E.; Cornetta, K. Antitumor activity and immunotherapeutic properties of Flt3-ligand in a murine breast cancer model. Cancer Res. 1997, 57, 3511–3516. [Google Scholar] [PubMed]
- Lynch, D.H.; Andreasen, A.; Maraskovsky, E.; Whitmore, J.; Miller, R.E.; Schuh, J.C. Flt3 ligand induces tumor regression and antitumor immune responses in vivo. Nat. Med. 1997, 3, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L.; Rinn, K.; Knutson, K.L.; Davis, D.; Caron, D.; dela Rosa, C.; Schiffman, K. Flt3 ligand as a vaccine adjuvant in association with HER-2/neu peptide-based vaccines in patients with HER-2/neu-overexpressing cancers. Blood 2002, 99, 2845–2850. [Google Scholar] [CrossRef] [PubMed]
- Fong, L.; Hou, Y.; Rivas, A.; Benike, C.; Yuen, A.; Fisher, G.A.; Davis, M.M.; Engleman, E.G. Altered peptide ligand vaccination with Flt3 ligand expanded dendritic cells for tumor immunotherapy. Proc. Natl. Acad. Sci. USA 2001, 98, 8809–8814. [Google Scholar] [CrossRef] [PubMed]
- Buza-Vidas, N.; Cheng, M.; Duarte, S.; Nozad, H.; Jacobsen, S.E.; Sitnicka, E. Crucial role of FLT3 ligand in immune reconstitution after bone marrow transplantation and high-dose chemotherapy. Blood 2007, 110, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Jensen, C.T.; Kharazi, S.; Boiers, C.; Cheng, M.; Lubking, A.; Sitnicka, E.; Jacobsen, S.E. FLT3 ligand and not TSLP is the key regulator of IL-7-independent B-1 and B-2 B lymphopoiesis. Blood 2008, 112, 2297–2304. [Google Scholar] [CrossRef] [PubMed]
- Nutt, S.L.; Heavey, B.; Rolink, A.G.; Busslinger, M. Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature 1999, 401, 556–562. [Google Scholar] [PubMed]
- Rolink, A.G.; Nutt, S.L.; Melchers, F.; Busslinger, M. Long-term in vivo reconstitution of T-cell development by Pax5-deficient B-cell progenitors. Nature 1999, 401, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Holmes, M.L.; Carotta, S.; Corcoran, L.M.; Nutt, S.L. Repression of Flt3 by Pax5 is crucial for B-cell lineage commitment. Genes Dev. 2006, 20, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Inlay, M.A.; Bhattacharya, D.; Sahoo, D.; Serwold, T.; Seita, J.; Karsunky, H.; Plevritis, S.K.; Dill, D.L.; Weissman, I.L. Ly6d marks the earliest stage of B-cell specification and identifies the branchpoint between B-cell and T-cell development. Genes Dev. 2009, 23, 2376–2381. [Google Scholar] [CrossRef] [PubMed]
- Mansson, R.; Zandi, S.; Welinder, E.; Tsapogas, P.; Sakaguchi, N.; Bryder, D.; Sigvardsson, M. Single-cell analysis of the common lymphoid progenitor compartment reveals functional and molecular heterogeneity. Blood 2010, 115, 2601–2609. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, L.A.; Huang, D.C.; Strasser, A. The cell death inhibitor Bcl-2 and its homologues influence control of cell cycle entry. EMBO J. 1996, 15, 6979–6990. [Google Scholar] [PubMed]
- Sitnicka, E.; Buza-Vidas, N.; Ahlenius, H.; Cilio, C.M.; Gekas, C.; Nygren, J.M.; Mansson, R.; Cheng, M.; Jensen, C.T.; Svensson, M.; et al. Critical role of FLT3 ligand in IL-7 receptor independent T lymphopoiesis and regulation of lymphoid-primed multipotent progenitors. Blood 2007, 110, 2955–2964. [Google Scholar] [CrossRef] [PubMed]
- Luc, S.; Luis, T.C.; Boukarabila, H.; Macaulay, I.C.; Buza-Vidas, N.; Bouriez-Jones, T.; Lutteropp, M.; Woll, P.S.; Loughran, S.J.; Mead, A.J.; et al. The earliest thymic T cell progenitors sustain B cell and myeloid lineage potential. Nat. Immunol. 2012, 13, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Wils, E.J.; Braakman, E.; Verjans, G.M.; Rombouts, E.J.; Broers, A.E.; Niesters, H.G.; Wagemaker, G.; Staal, F.J.; Lowenberg, B.; Spits, H.; et al. Flt3 ligand expands lymphoid progenitors prior to recovery of thymopoiesis and accelerates T cell reconstitution after bone marrow transplantation. J. Immunol. 2007, 178, 3551–3557. [Google Scholar] [CrossRef] [PubMed]
- Kenins, L.; Gill, J.W.; Boyd, R.L.; Hollander, G.A.; Wodnar-Filipowicz, A. Intrathymic expression of Flt3 ligand enhances thymic recovery after irradiation. J. Exp. Med. 2008, 205, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Kenins, L.; Gill, J.W.; Hollander, G.A.; Wodnar-Filipowicz, A. Flt3 ligand-receptor interaction is important for maintenance of early thymic progenitor numbers in steady-state thymopoiesis. Eur. J. Immunol. 2010, 40, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Baerenwaldt, A.; von Burg, N.; Kreuzaler, M.; Sitte, S.; Horvath, E.; Peter, A.; Voehringer, D.; Rolink, A.G.; Finke, D. Flt3 ligand regulates the development of innate lymphoid cells in fetal and adult mice. J. Immunol. 2016, 196, 2561–2571. [Google Scholar] [CrossRef] [PubMed]
- Shaw, S.G.; Maung, A.A.; Steptoe, R.J.; Thomson, A.W.; Vujanovic, N.L. Expansion of functional NK cells in multiple tissue compartments of mice treated with Flt3-ligand: Implications for anti-cancer and anti-viral therapy. J. Immunol. 1998, 161, 2817–2824. [Google Scholar] [PubMed]
- Kallies, A.; Hasbold, J.; Fairfax, K.; Pridans, C.; Emslie, D.; McKenzie, B.S.; Lew, A.M.; Corcoran, L.M.; Hodgkin, P.D.; Tarlinton, D.M.; et al. Initiation of plasma-cell differentiation is independent of the transcription factor Blimp-1. Immunity 2007, 26, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Svensson, M.N.; Andersson, K.M.; Wasen, C.; Erlandsson, M.C.; Nurkkala-Karlsson, M.; Jonsson, I.M.; Brisslert, M.; Bemark, M.; Bokarewa, M.I. Murine germinal center B cells require functional Fms-like tyrosine kinase 3 signaling for IgG1 class-switch recombination. Proc. Natl. Acad. Sci. USA 2015, 112, E6644–E6653. [Google Scholar] [CrossRef] [PubMed]
- Astier, A.L.; Beriou, G.; Eisenhaure, T.M.; Anderton, S.M.; Hafler, D.A.; Hacohen, N. RNA interference screen in primary human T cells reveals FLT3 as a modulator of IL-10 levels. J. Immunol. 2010, 184, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Gilliland, D.G.; Griffin, J.D. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002, 100, 1532–1542. [Google Scholar] [CrossRef] [PubMed]
- Annesley, C.E.; Brown, P. The biology and targeting of FLT3 in pediatric leukemia. Front. Oncol. 2014, 4, 263. [Google Scholar] [CrossRef] [PubMed]
- Nakao, M.; Yokota, S.; Iwai, T.; Kaneko, H.; Horiike, S.; Kashima, K.; Sonoda, Y.; Fujimoto, T.; Misawa, S. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia 1996, 10, 1911–1918. [Google Scholar] [PubMed]
- Meshinchi, S.; Woods, W.G.; Stirewalt, D.L.; Sweetser, D.A.; Buckley, J.D.; Tjoa, T.K.; Bernstein, I.D.; Radich, J.P. Prevalence and prognostic significance of Flt3 internal tandem duplication in pediatric acute myeloid leukemia. Blood 2001, 97, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Schnittger, S.; Schoch, C.; Dugas, M.; Kern, W.; Staib, P.; Wuchter, C.; Loffler, H.; Sauerland, C.M.; Serve, H.; Buchner, T.; et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: Correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood 2002, 100, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Thiede, C.; Steudel, C.; Mohr, B.; Schaich, M.; Schakel, U.; Platzbecker, U.; Wermke, M.; Bornhauser, M.; Ritter, M.; Neubauer, A.; et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: Association with FAB subtypes and identification of subgroups with poor prognosis. Blood 2002, 99, 4326–4335. [Google Scholar] [CrossRef] [PubMed]
- Kiyoi, H.; Ohno, R.; Ueda, R.; Saito, H.; Naoe, T. Mechanism of constitutive activation of FLT3 with internal tandem duplication in the juxtamembrane domain. Oncogene 2002, 21, 2555–2563. [Google Scholar] [CrossRef] [PubMed]
- Kiyoi, H.; Towatari, M.; Yokota, S.; Hamaguchi, M.; Ohno, R.; Saito, H.; Naoe, T. Internal tandem duplication of the FLT3 gene is a novel modality of elongation mutation which causes constitutive activation of the product. Leukemia 1998, 12, 1333–1337. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, F.; Towatari, M.; Kiyoi, H.; Tanimoto, M.; Kitamura, T.; Saito, H.; Naoe, T. Tandem-duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL-3-dependent cell lines. Oncogene 2000, 19, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Stirewalt, D.L.; Radich, J. The role of FLT3 in haematopoietic malignancies. Nat. Rev. Cancer 2003, 3, 650–665. [Google Scholar] [CrossRef] [PubMed]
- Mizuki, M.; Fenski, R.; Halfter, H.; Matsumura, I.; Schmidt, R.; Muller, C.; Gruning, W.; Kratz-Albers, K.; Serve, S.; Steur, C.; et al. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood 2000, 96, 3907–3914. [Google Scholar] [PubMed]
- Mizuki, M.; Schwable, J.; Steur, C.; Choudhary, C.; Agrawal, S.; Sargin, B.; Steffen, B.; Matsumura, I.; Kanakura, Y.; Bohmer, F.D.; et al. Suppression of myeloid transcription factors and induction of STAT response genes by AML-specific Flt3 mutations. Blood 2003, 101, 3164–3173. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Friedman, A.D.; Levis, M.; Li, L.; Weir, E.G.; Small, D. Internal tandem duplication mutation of FLT3 blocks myeloid differentiation through suppression of C/EBPα expression. Blood 2004, 103, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Li, L.; Small, D.; Rassool, F. Cells expressing FLT3/ITD mutations exhibit elevated repair errors generated through alternative NHEJ pathways: Implications for genomic instability and therapy. Blood 2010, 116, 5298–5305. [Google Scholar] [CrossRef] [PubMed]
- Sallmyr, A.; Fan, J.; Datta, K.; Kim, K.T.; Grosu, D.; Shapiro, P.; Small, D.; Rassool, F. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: Implications for poor prognosis in AML. Blood 2008, 111, 3173–3182. [Google Scholar] [CrossRef] [PubMed]
- Scheijen, B.; Ngo, H.T.; Kang, H.; Griffin, J.D. FLT3 receptors with internal tandem duplications promote cell viability and proliferation by signaling through Foxo proteins. Oncogene 2004, 23, 3338–3349. [Google Scholar] [CrossRef] [PubMed]
- Levis, M.; Allebach, J.; Tse, K.F.; Zheng, R.; Baldwin, B.R.; Smith, B.D.; Jones-Bolin, S.; Ruggeri, B.; Dionne, C.; Small, D. A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood 2002, 99, 3885–3891. [Google Scholar] [CrossRef] [PubMed]
- Grundler, R.; Miething, C.; Thiede, C.; Peschel, C.; Duyster, J. FLT3-ITD and tyrosine kinase domain mutants induce 2 distinct phenotypes in a murine bone marrow transplantation model. Blood 2005, 105, 4792–4799. [Google Scholar] [CrossRef] [PubMed]
- Kelly, L.M.; Liu, Q.; Kutok, J.L.; Williams, I.R.; Boulton, C.L.; Gilliland, D.G. FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood 2002, 99, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Williams, I.R.; Anastasiadou, E.; Boulton, C.L.; Joseph, S.W.; Amaral, S.M.; Curley, D.P.; Duclos, N.; Huntly, B.J.; Fabbro, D.; et al. FLT3 internal tandem duplication mutations induce myeloproliferative or lymphoid disease in a transgenic mouse model. Oncogene 2005, 24, 7882–7892. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Tothova, Z.; Levine, R.L.; Anderson, K.; Buza-Vidas, N.; Cullen, D.E.; McDowell, E.P.; Adelsperger, J.; Frohling, S.; Huntly, B.J.; et al. FLT3 mutations confer enhanced proliferation and survival properties to multipotent progenitors in a murine model of chronic myelomonocytic leukemia. Cancer Cell 2007, 12, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Piloto, O.; Nguyen, H.B.; Greenberg, K.; Takamiya, K.; Racke, F.; Huso, D.; Small, D. Knock-in of an internal tandem duplication mutation into murine FLT3 confers myeloproliferative disease in a mouse model. Blood 2008, 111, 3849–3858. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.H.; Heiser, D.; Li, L.; Kaplan, I.; Collector, M.; Huso, D.; Sharkis, S.J.; Civin, C.; Small, D. FLT3-ITD knockin impairs hematopoietic stem cell quiescence/homeostasis, leading to myeloproliferative neoplasm. Cell Stem Cell 2012, 11, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Abu-Duhier, F.M.; Goodeve, A.C.; Wilson, G.A.; Care, R.S.; Peake, I.R.; Reilly, J.T. Identification of novel FLT-3 Asp835 mutations in adult acute myeloid leukaemia. Br. J. Haematol. 2001, 113, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Kiyoi, H.; Nakano, Y.; Suzuki, R.; Kodera, Y.; Miyawaki, S.; Asou, N.; Kuriyama, K.; Yagasaki, F.; Shimazaki, C.; et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood 2001, 97, 2434–2439. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.; Black, J.; Faerman, C.; Swenson, L.; Wynn, M.; Lu, F.; Lippke, J.; Saxena, K. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol. Cell 2004, 13, 169–178. [Google Scholar] [CrossRef]
- Choudhary, C.; Schwable, J.; Brandts, C.; Tickenbrock, L.; Sargin, B.; Kindler, T.; Fischer, T.; Berdel, W.E.; Muller-Tidow, C.; Serve, H. AML-associated Flt3 kinase domain mutations show signal transduction differences compared with Flt3 ITD mutations. Blood 2005, 106, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Lacayo, N.J.; Meshinchi, S.; Kinnunen, P.; Yu, R.; Wang, Y.; Stuber, C.M.; Douglas, L.; Wahab, R.; Becton, D.L.; Weinstein, H.; et al. Gene expression profiles at diagnosis in de novo childhood AML patients identify FLT3 mutations with good clinical outcomes. Blood 2004, 104, 2646–2654. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Jacobi, A.; Ryser, M.; Ehninger, G.; Thiede, C. Abnormal localization and accumulation of FLT3-ITD, a mutant receptor tyrosine kinase involved in leukemogenesis. Cells Tissues Organs 2008, 188, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Olsen, J.V.; Brandts, C.; Cox, J.; Reddy, P.N.; Bohmer, F.D.; Gerke, V.; Schmidt-Arras, D.E.; Berdel, W.E.; Muller-Tidow, C.; et al. Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol. Cell 2009, 36, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.M. Differential signaling of Flt3 activating mutations in acute myeloid leukemia: A working model. Protein Cell 2011, 2, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.A.; Grundler, R.; Istvanffy, R.; Rudelius, M.; Hennighausen, L.; Illert, A.L.; Duyster, J. Lineage-specific STAT5 target gene activation in hematopoietic progenitor cells predicts the FLT3+-mediated leukemic phenotype. Leukemia 2016, 30, 1725–1733. [Google Scholar] [CrossRef] [PubMed]
- Bailey, E.; Li, L.; Duffield, A.S.; Ma, H.S.; Huso, D.L.; Small, D. FLT3/D835Y mutation knock-in mice display less aggressive disease compared with FLT3/internal tandem duplication (ITD) mice. Proc. Natl. Acad. Sci. USA 2013, 110, 21113–21118. [Google Scholar] [CrossRef] [PubMed]
- Birg, F.; Courcoul, M.; Rosnet, O.; Bardin, F.; Pebusque, M.J.; Marchetto, S.; Tabilio, A.; Mannoni, P.; Birnbaum, D. Expression of the FMS/KIT-like gene FLT3 in human acute leukemias of the myeloid and lymphoid lineages. Blood 1992, 80, 2584–2593. [Google Scholar] [PubMed]
- Carow, C.E.; Levenstein, M.; Kaufmann, S.H.; Chen, J.; Amin, S.; Rockwell, P.; Witte, L.; Borowitz, M.J.; Civin, C.I.; Small, D. Expression of the hematopoietic growth factor receptor FLT3 (STK-1/Flk2) in human leukemias. Blood 1996, 87, 1089–1096. [Google Scholar] [PubMed]
- Rosnet, O.; Buhring, H.J.; Marchetto, S.; Rappold, I.; Lavagna, C.; Sainty, D.; Arnoulet, C.; Chabannon, C.; Kanz, L.; Hannum, C.; et al. Human FLT3/FLK2 receptor tyrosine kinase is expressed at the surface of normal and malignant hematopoietic cells. Leukemia 1996, 10, 238–248. [Google Scholar] [PubMed]
- Stacchini, A.; Fubini, L.; Severino, A.; Sanavio, F.; Aglietta, M.; Piacibello, W. Expression of type III receptor tyrosine kinases FLT3 and KIT and responses to their ligands by acute myeloid leukemia blasts. Leukemia 1996, 10, 1584–1591. [Google Scholar] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Ozeki, K.; Kiyoi, H.; Hirose, Y.; Iwai, M.; Ninomiya, M.; Kodera, Y.; Miyawaki, S.; Kuriyama, K.; Shimazaki, C.; Akiyama, H.; et al. Biologic and clinical significance of the FLT3 transcript level in acute myeloid leukemia. Blood 2004, 103, 1901–1908. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Levis, M.; Piloto, O.; Brown, P.; Baldwin, B.R.; Gorin, N.C.; Beran, M.; Zhu, Z.; Ludwig, D.; Hicklin, D.; et al. FLT3 ligand causes autocrine signaling in acute myeloid leukemia cells. Blood 2004, 103, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Pratz, K.W.; Sato, T.; Murphy, K.M.; Stine, A.; Rajkhowa, T.; Levis, M. FLT3-mutant allelic burden and clinical status are predictive of response to FLT3 inhibitors in AML. Blood 2010, 115, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- Fabbro, D.; Buchdunger, E.; Wood, J.; Mestan, J.; Hofmann, F.; Ferrari, S.; Mett, H.; O’Reilly, T.; Meyer, T. Inhibitors of protein kinases: CGP 41251, a protein kinase inhibitor with potential as an anticancer agent. Pharmacol. Ther. 1999, 82, 293–301. [Google Scholar] [CrossRef]
- Hexner, E.O.; Serdikoff, C.; Jan, M.; Swider, C.R.; Robinson, C.; Yang, S.; Angeles, T.; Emerson, S.G.; Carroll, M.; Ruggeri, B.; et al. Lestaurtinib (CEP701) is a JAK2 inhibitor that suppresses JAK2/STAT5 signaling and the proliferation of primary erythroid cells from patients with myeloproliferative disorders. Blood 2008, 111, 5663–5671. [Google Scholar] [CrossRef] [PubMed]
- Mendel, D.B.; Laird, A.D.; Xin, X.; Louie, S.G.; Christensen, J.G.; Li, G.; Schreck, R.E.; Abrams, T.J.; Ngai, T.J.; Lee, L.B.; et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: Determination of a pharmacokinetic/pharmacodynamic relationship. Clin. Cancer Res. 2003, 9, 327–337. [Google Scholar] [PubMed]
- O’Farrell, A.M.; Abrams, T.J.; Yuen, H.A.; Ngai, T.J.; Louie, S.G.; Yee, K.W.; Wong, L.M.; Hong, W.; Lee, L.B.; Town, A.; et al. SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood 2003, 101, 3597–3605. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet 2006, 368, 1329–1338. [Google Scholar] [CrossRef]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Siebels, M.; Negrier, S.; Chevreau, C.; Solska, E.; Desai, A.A.; et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Rixe, O.; Oudard, S.; Negrier, S.; Szczylik, C.; Kim, S.T.; et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 115–124. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, A.M.; Foran, J.M.; Fiedler, W.; Serve, H.; Paquette, R.L.; Cooper, M.A.; Yuen, H.A.; Louie, S.G.; Kim, H.; Nicholas, S.; et al. An innovative phase I clinical study demonstrates inhibition of FLT3 phosphorylation by SU11248 in acute myeloid leukemia patients. Clin. Cancer Res. 2003, 9, 5465–5476. [Google Scholar] [PubMed]
- Fiedler, W.; Kayser, S.; Kebenko, M.; Janning, M.; Krauter, J.; Schittenhelm, M.; Gotze, K.; Weber, D.; Gohring, G.; Teleanu, V.; et al. A phase I/II study of sunitinib and intensive chemotherapy in patients over 60 years of age with acute myeloid leukaemia and activating FLT3 mutations. Br. J. Haematol. 2015, 169, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, W.; Serve, H.; Dohner, H.; Schwittay, M.; Ottmann, O.G.; O’Farrell, A.M.; Bello, C.L.; Allred, R.; Manning, W.C.; Cherrington, J.M.; et al. A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood 2005, 105, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Metzelder, S.; Wang, Y.; Wollmer, E.; Wanzel, M.; Teichler, S.; Chaturvedi, A.; Eilers, M.; Enghofer, E.; Neubauer, A.; Burchert, A. Compassionate use of sorafenib in FLT3-ITD-positive acute myeloid leukemia: Sustained regression before and after allogeneic stem cell transplantation. Blood 2009, 113, 6567–6571. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Ravandi, F.; Bayraktar, U.D.; Chiattone, A.; Bashir, Q.; Giralt, S.; Chen, J.; Qazilbash, M.; Kebriaei, P.; Konopleva, M.; et al. Treatment of FLT3-ITD-positive acute myeloid leukemia relapsing after allogeneic stem cell transplantation with sorafenib. Biol. Blood Marrow Transplant. 2011, 17, 1874–1877. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Konopleva, M.; Shi, Y.X.; McQueen, T.; Harris, D.; Ling, X.; Estrov, Z.; Quintas-Cardama, A.; Small, D.; Cortes, J.; et al. Mutant FLT3: A direct target of sorafenib in acute myelogenous leukemia. J. Natl. Cancer Inst. 2008, 100, 184–198. [Google Scholar] [CrossRef] [PubMed]
- Serve, H.; Krug, U.; Wagner, R.; Sauerland, M.C.; Heinecke, A.; Brunnberg, U.; Schaich, M.; Ottmann, O.; Duyster, J.; Wandt, H.; et al. Sorafenib in combination with intensive chemotherapy in elderly patients with acute myeloid leukemia: Results from a randomized, placebo-controlled trial. J. Clin. Oncol. 2013, 31, 3110–3118. [Google Scholar] [CrossRef] [PubMed]
- Ravandi, F.; Cortes, J.E.; Jones, D.; Faderl, S.; Garcia-Manero, G.; Konopleva, M.Y.; O’Brien, S.; Estrov, Z.; Borthakur, G.; Thomas, D.; et al. Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 1856–1862. [Google Scholar] [CrossRef] [PubMed]
- Rollig, C.; Serve, H.; Huttmann, A.; Noppeney, R.; Muller-Tidow, C.; Krug, U.; Baldus, C.D.; Brandts, C.H.; Kunzmann, V.; Einsele, H.; et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): A multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015, 16, 1691–1699. [Google Scholar] [CrossRef]
- Antar, A.; Kharfan-Dabaja, M.A.; Mahfouz, R.; Bazarbachi, A. Sorafenib Maintenance Appears Safe and Improves Clinical Outcomes in FLT3-ITD Acute Myeloid Leukemia After Allogeneic Hematopoietic Cell Transplantation. Clin. Lymphoma Myeloma Leuk. 2015, 15, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Metzelder, S.K.; Schroeder, T.; Finck, A.; Scholl, S.; Fey, M.; Gotze, K.; Linn, Y.C.; Kroger, M.; Reiter, A.; Salih, H.R.; et al. High activity of sorafenib in FLT3-ITD-positive acute myeloid leukemia synergizes with allo-immune effects to induce sustained responses. Leukemia 2012, 26, 2353–2359. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; DeAngelo, D.J.; Klimek, V.; Galinsky, I.; Estey, E.; Nimer, S.D.; Grandin, W.; Lebwohl, D.; Wang, Y.; Cohen, P.; et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood 2005, 105, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; Mandrekar, S.; Sanford, B.L.; Geyer, S.; Bloomfield, C.D.; Dohner, K.; Thiede, C.; Marcucci, G.; Lo-Coco, F.; Klisovic, R.B.; et al. The multi-kinase inhibitor midostaurin (M) prolongs survival compared with placebo (P) in combination with daunorubicin (D)/cytarabine (C) induction (ind), high-dose c consolidation (consol), and as maintenance (maint) therapy in newly diagnosed acute myeloid leukemia (AML) patients (pts) age 18–60 with FLT3 mutations (muts): An international prospective randomized (rand) P-controlled double-blind trial (CALGB 10603/RATIFY [Alliance]). Blood 2015, 126, 6. [Google Scholar]
- Knapper, S.; Russell, N.; Gilkes, A.; Hills, R.K.; Gale, R.E.; Cavenagh, J.D.; Jones, G.; Kjeldsen, L.; Grunwald, M.R.; Thomas, I.; et al. A randomized assessment of adding the kinase inhibitor lestaurtinib to first-line chemotherapy for FLT3-mutated AML. Blood 2017, 129, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Levis, M.; Ravandi, F.; Wang, E.S.; Baer, M.R.; Perl, A.; Coutre, S.; Erba, H.; Stuart, R.K.; Baccarani, M.; Cripe, L.D.; et al. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood 2011, 117, 3294–3301. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Perl, A.E.; Dombret, H.; Kayser, S.; Steffen, B.; Rousselot, P.; Martinelli, G.; Estey, E.H.; Burnett, A.K.; Gammon, G.; et al. Final results of a phase 2 open-label, monotherapy efficacy and safety study of quizartinib (AC220) in patients ≥ 60 years of age with FLT3 ITD positive or negative relapsed/refractory acute myeloid leukemia. Blood 2012, 120, 48. [Google Scholar]
- Levis, M.J.; Perl, A.E.; Dombret, H.; Döhner, H.; Steffen, B.; Rousselot, P.; Martinelli, G.; Estey, E.H.; Burnett, A.K.; Gammon, G.; et al. Final results of a phase 2 open-label, monotherapy efficacy and safety study of quizartinib (AC220) in patients with FLT3-ITD positive or negative relapsed/refractory acute myeloid leukemia after second-line chemotherapy or hematopoietic stem cell transplantation. Blood 2012, 120, 673. [Google Scholar]
- Burnett, A.K.; Bowen, D.; Russell, N.; Knapper, S.; Milligan, D.; Hunter, A.E.; Khwaja, A.; Clark, R.E.; Culligan, D.; Clark, H.; et al. AC220 (quizartinib) can be safely combined with conventional chemotherapy in older patients with newly diagnosed acute myeloid leukaemia: Experience from the AML 18 pilot trial. Blood 2013, 122, 622. [Google Scholar]
- Altman, J.K.; Perl, A.E.; Cortes, J.E.; Levis, M.J.; Smith, C.C.; Litzow, M.R.; Baer, M.R.; Claxton, D.F.; Erba, H.P.; Gill, S.C.; et al. Antileukemic activity and tolerability of ASP2215 80mg and greater in FLT3 mutation-positive subjects with relapsed or refractory acute myeloid leukemia: Results from a phase 1/2, open-label, dose-escalation/dose-response study. Blood 2015, 126, 321. [Google Scholar]
- Randhawa, J.K.; Kantarjian, H.M.; Borthakur, G.; Thompson, P.A.; Konopleva, M.; Daver, N.; Pemmaraju, N.; Jabbour, E.; Kadia, T.M.; Estrov, Z.; et al. Results of a phase II study of crenolanib in relapsed/refractory acute myeloid leukemia patients (Pts) with activating FLT3 mutations. Blood 2014, 124, 389. [Google Scholar]
- Galanis, A.; Ma, H.; Rajkhowa, T.; Ramachandran, A.; Small, D.; Cortes, J.; Levis, M. Crenolanib is a potent inhibitor of FLT3 with activity against resistance-conferring point mutants. Blood 2014, 123, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Lasater, E.A.; Lin, K.C.; Wang, Q.; McCreery, M.Q.; Stewart, W.K.; Damon, L.E.; Perl, A.E.; Jeschke, G.R.; Sugita, M.; et al. Crenolanib is a selective type I pan-FLT3 inhibitor. Proc. Natl. Acad. Sci. USA 2014, 111, 5319–5324. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, E.I.; Turner, D.C.; Buaboonnam, J.; Hu, S.; Orwick, S.; Roberts, M.S.; Janke, L.J.; Ramachandran, A.; Stewart, C.F.; Inaba, H.; et al. Crenolanib is active against models of drug-resistant FLT3-ITD-positive acute myeloid leukemia. Blood 2013, 122, 3607–3615. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.S.; Stone, R.M.; Tallman, M.S.; Walter, R.B.; Eckardt, J.R.; Collins, R. Crenolanib, a type I FLT3 TKI, can be safely combined with cytarabine and anthracycline induction chemotherapy and results in high response rates in patients with newly diagnosed FLT3 mutant acute myeloid leukemia (AML). Blood 2016, 128, 1071. [Google Scholar]
- Smith, C.C.; Lin, K.; Stecula, A.; Sali, A.; Shah, N.P. FLT3 D835 mutations confer differential resistance to type II FLT3 inhibitors. Leukemia 2015, 29, 2390–2392. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Zhang, C.; Lin, K.C.; Lasater, E.A.; Zhang, Y.; Massi, E.; Damon, L.E.; Pendleton, M.; Bashir, A.; Sebra, R.; et al. Characterizing and overriding the structural mechanism of the Quizartinib-Resistant FLT3 “Gatekeeper” F691L mutation with PLX3397. Cancer Discov. 2015, 5, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Yang, X.; Knapper, S.; White, P.; Smith, B.D.; Galkin, S.; Small, D.; Burnett, A.; Levis, M. FLT3 ligand impedes the efficacy of FLT3 inhibitors in vitro and in vivo. Blood 2011, 117, 3286–3293. [Google Scholar] [CrossRef] [PubMed]
- Borthakur, G.; Kantarjian, H.M.; O’Brien, S.; Garcia-Manero, G.; Jabbour, E.; Daver, N.; Kadia, T.M.; Gborogen, R.; Konopleva, M.; Andreeff, M.; et al. The combination of quizartinib with azacitidine or low dose cytarabine is highly active in patients (Pts) with FLT3-ITD mutated myeloid leukemias: Interim report of a phase I/II trial. Blood 2014, 124, 388. [Google Scholar]
- Hofmann, M.; Grosse-Hovest, L.; Nubling, T.; Pyz, E.; Bamberg, M.L.; Aulwurm, S.; Buhring, H.J.; Schwartz, K.; Haen, S.P.; Schilbach, K.; et al. Generation, selection and preclinical characterization of an Fc-optimized FLT3 antibody for the treatment of myeloid leukemia. Leukemia 2012, 26, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G., Jr.; Hethcote, H.W.; Brown, B.W. Mutation and childhood cancer: A probabilistic model for the incidence of retinoblastoma. Proc. Natl. Acad. Sci. USA 1975, 72, 5116–5120. [Google Scholar] [CrossRef] [PubMed]
- Kelly, L.M.; Kutok, J.L.; Williams, I.R.; Boulton, C.L.; Amaral, S.M.; Curley, D.P.; Ley, T.J.; Gilliland, D.G. PML/RARα and FLT3-ITD induce an APL-like disease in a mouse model. Proc. Natl. Acad. Sci. USA 2002, 99, 8283–8288. [Google Scholar] [CrossRef] [PubMed]
- Mead, A.J.; Kharazi, S.; Atkinson, D.; Macaulay, I.; Pecquet, C.; Loughran, S.; Lutteropp, M.; Woll, P.; Chowdhury, O.; Luc, S.; et al. FLT3-ITDs instruct a myeloid differentiation and transformation bias in lymphomyeloid multipotent progenitors. Cell Rep. 2013, 3, 1766–1776. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsapogas, P.; Mooney, C.J.; Brown, G.; Rolink, A. The Cytokine Flt3-Ligand in Normal and Malignant Hematopoiesis. Int. J. Mol. Sci. 2017, 18, 1115. https://doi.org/10.3390/ijms18061115
Tsapogas P, Mooney CJ, Brown G, Rolink A. The Cytokine Flt3-Ligand in Normal and Malignant Hematopoiesis. International Journal of Molecular Sciences. 2017; 18(6):1115. https://doi.org/10.3390/ijms18061115
Chicago/Turabian StyleTsapogas, Panagiotis, Ciaran James Mooney, Geoffrey Brown, and Antonius Rolink. 2017. "The Cytokine Flt3-Ligand in Normal and Malignant Hematopoiesis" International Journal of Molecular Sciences 18, no. 6: 1115. https://doi.org/10.3390/ijms18061115
APA StyleTsapogas, P., Mooney, C. J., Brown, G., & Rolink, A. (2017). The Cytokine Flt3-Ligand in Normal and Malignant Hematopoiesis. International Journal of Molecular Sciences, 18(6), 1115. https://doi.org/10.3390/ijms18061115