
Induced Pluripotent Stem Cells Derived from a CLN5 Patient Manifest Phenotypic Characteristics of Neuronal Ceroid Lipofuscinoses

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
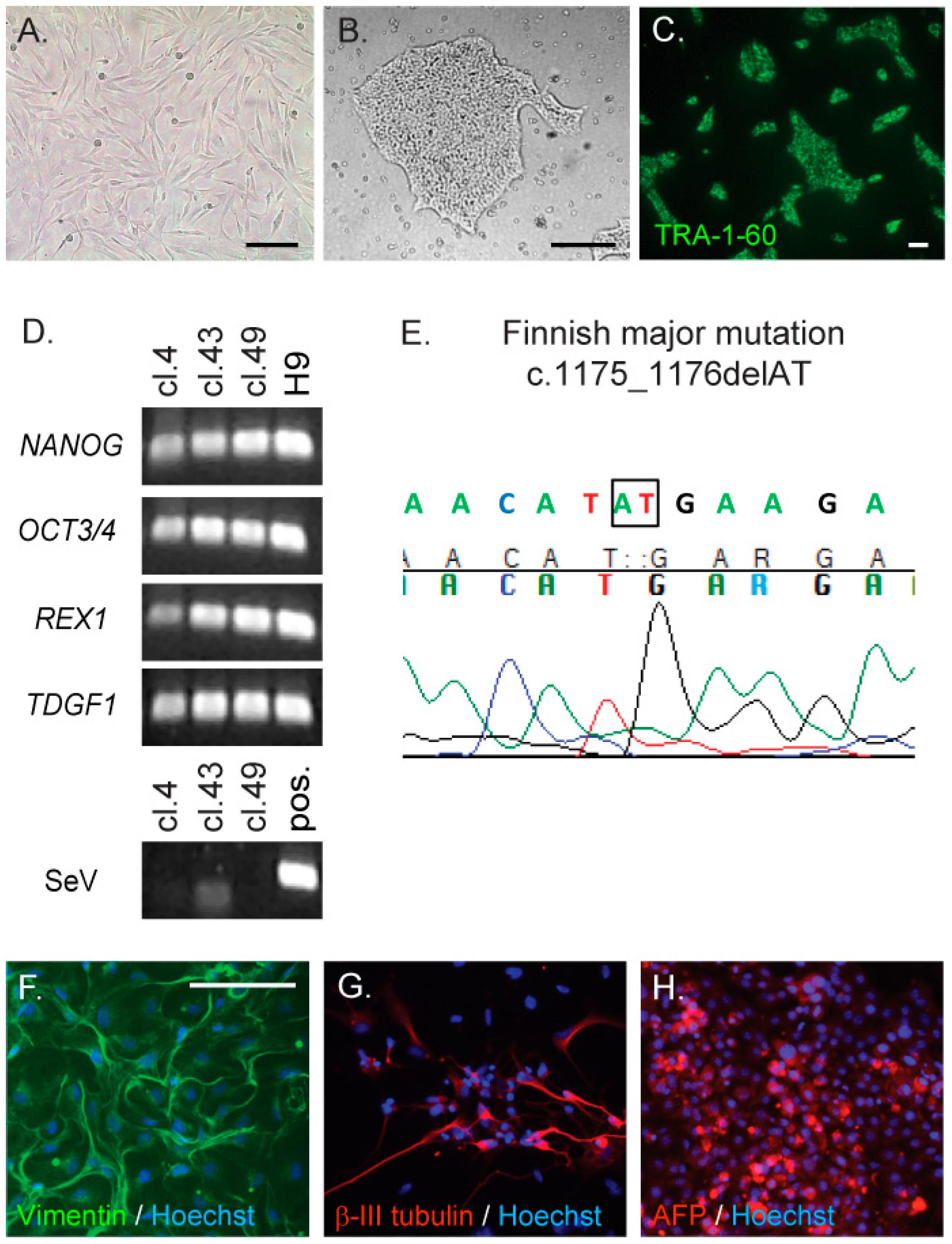
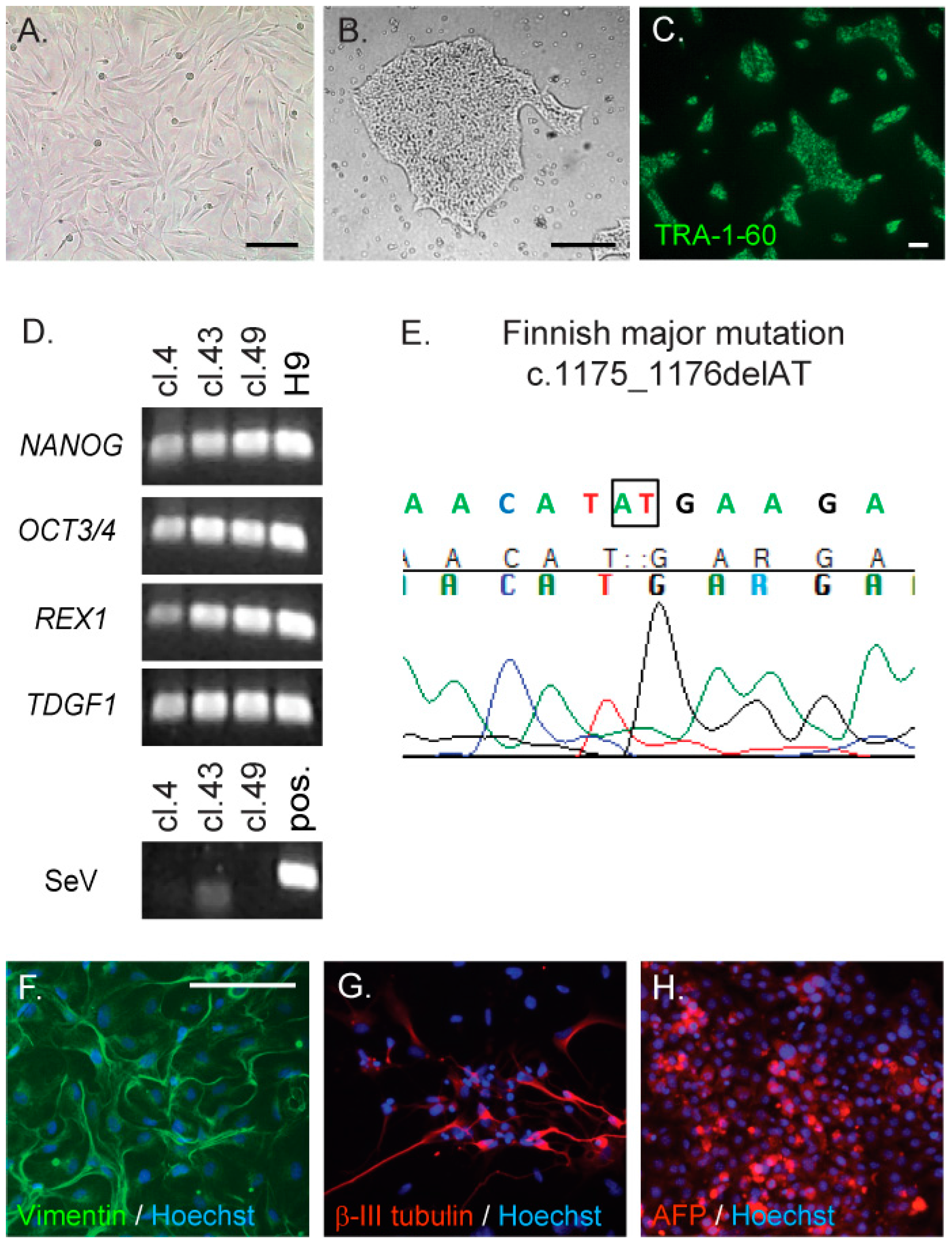
2.1. Generation and Characterisation of CLN5Y392X iPS Cells
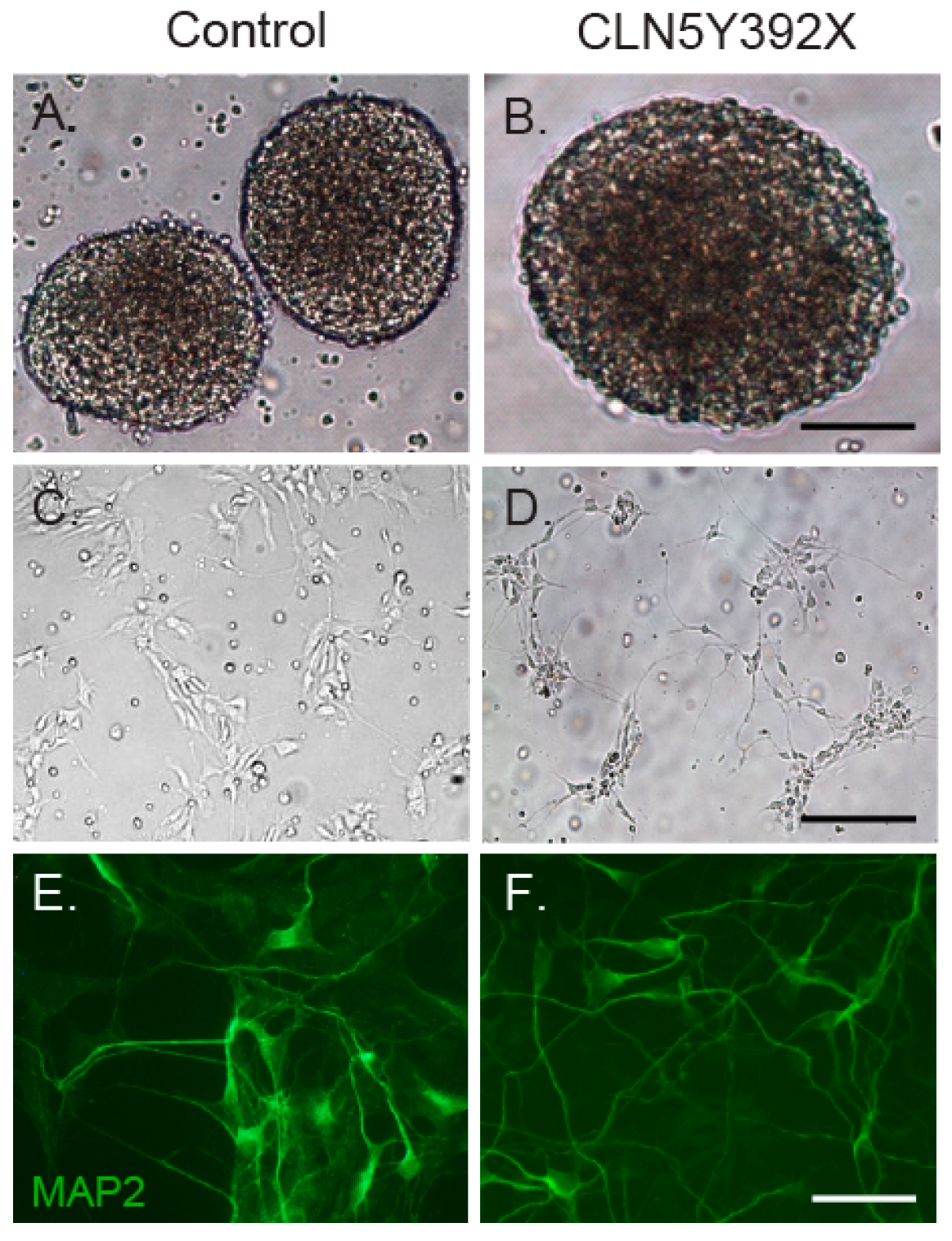
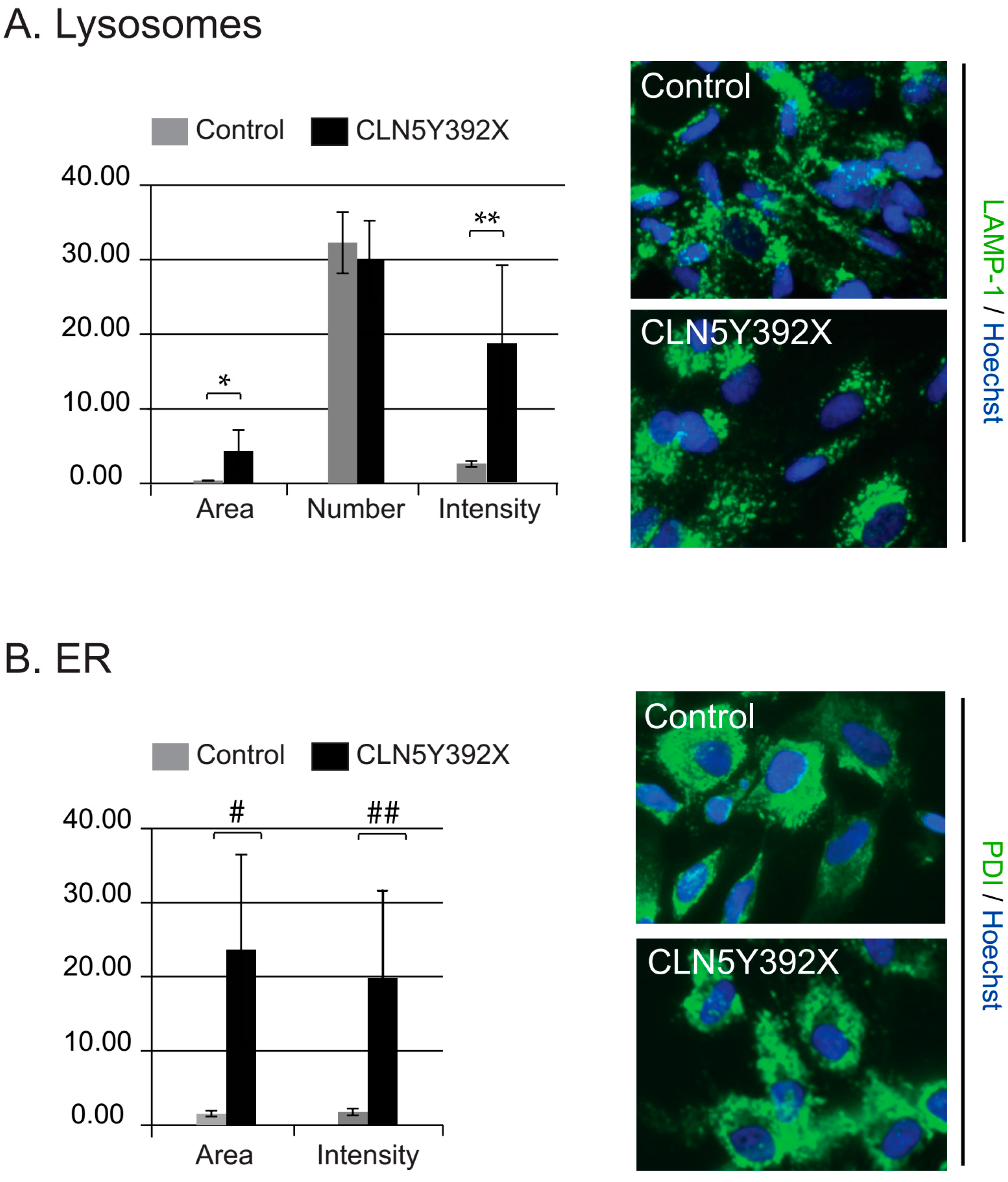
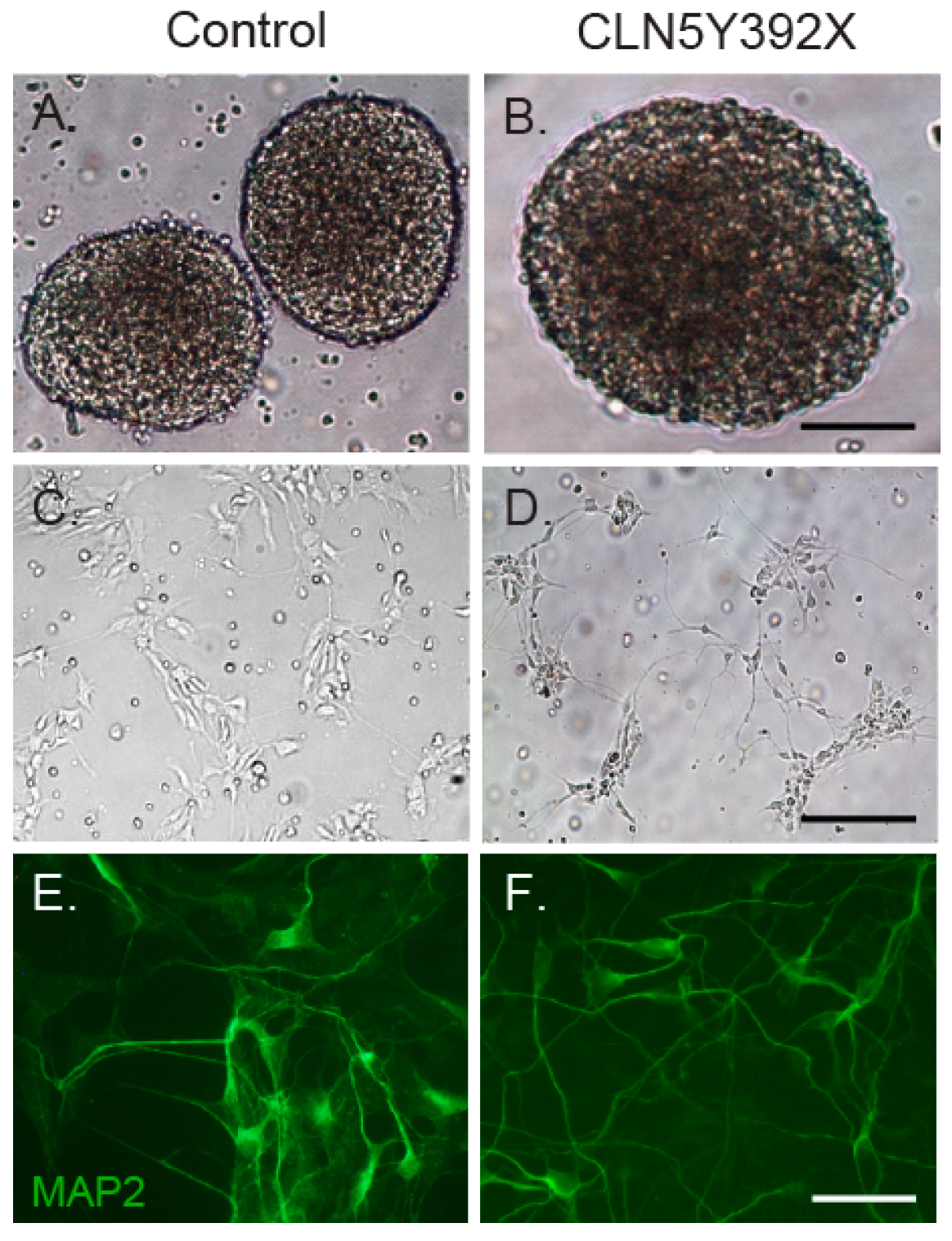
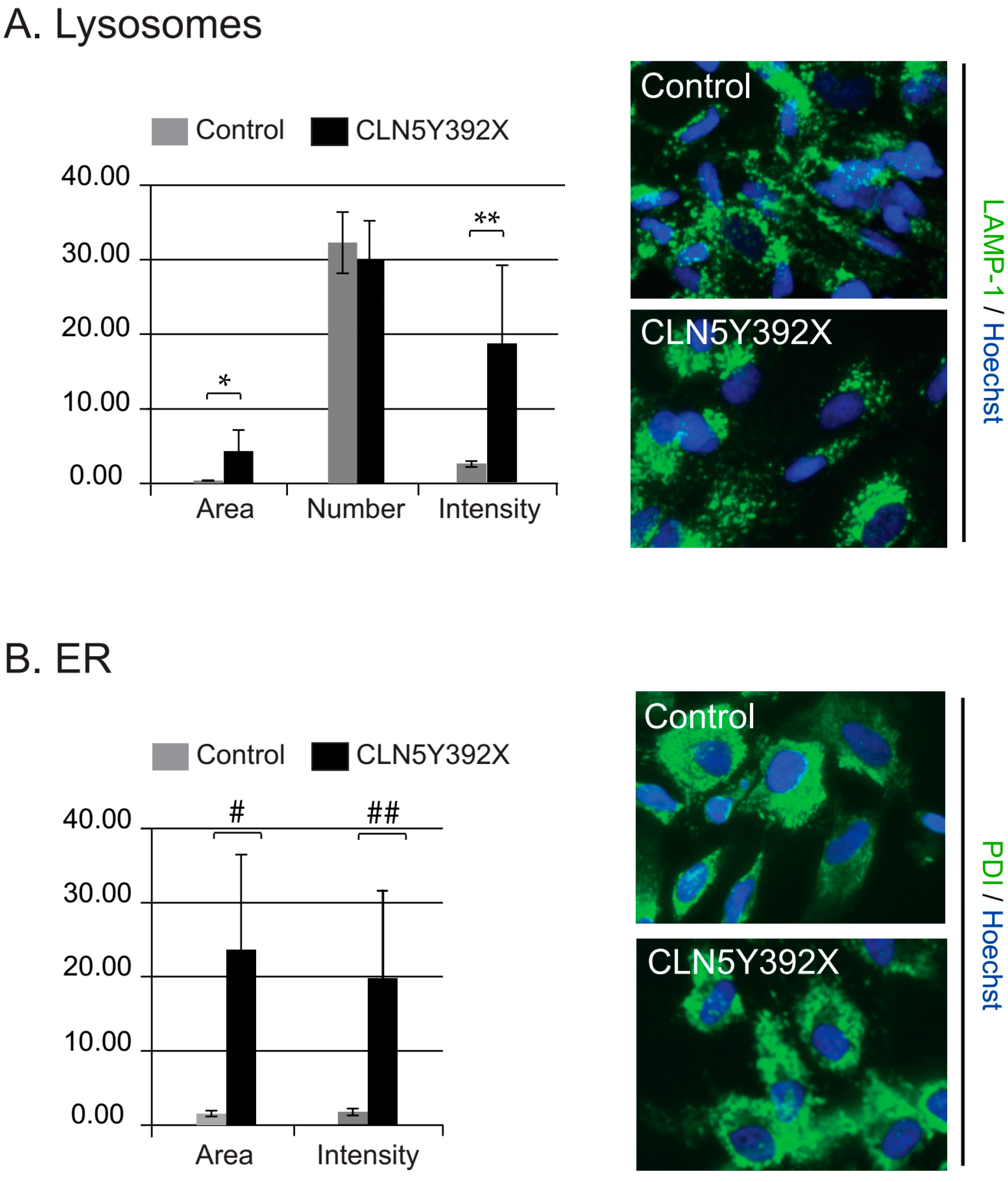
2.2. CLN5Y392X iPSC Differentiate into Neural Lineage Cells and Show Lysosomal and Endoplasmic Reticulum Abnormalities
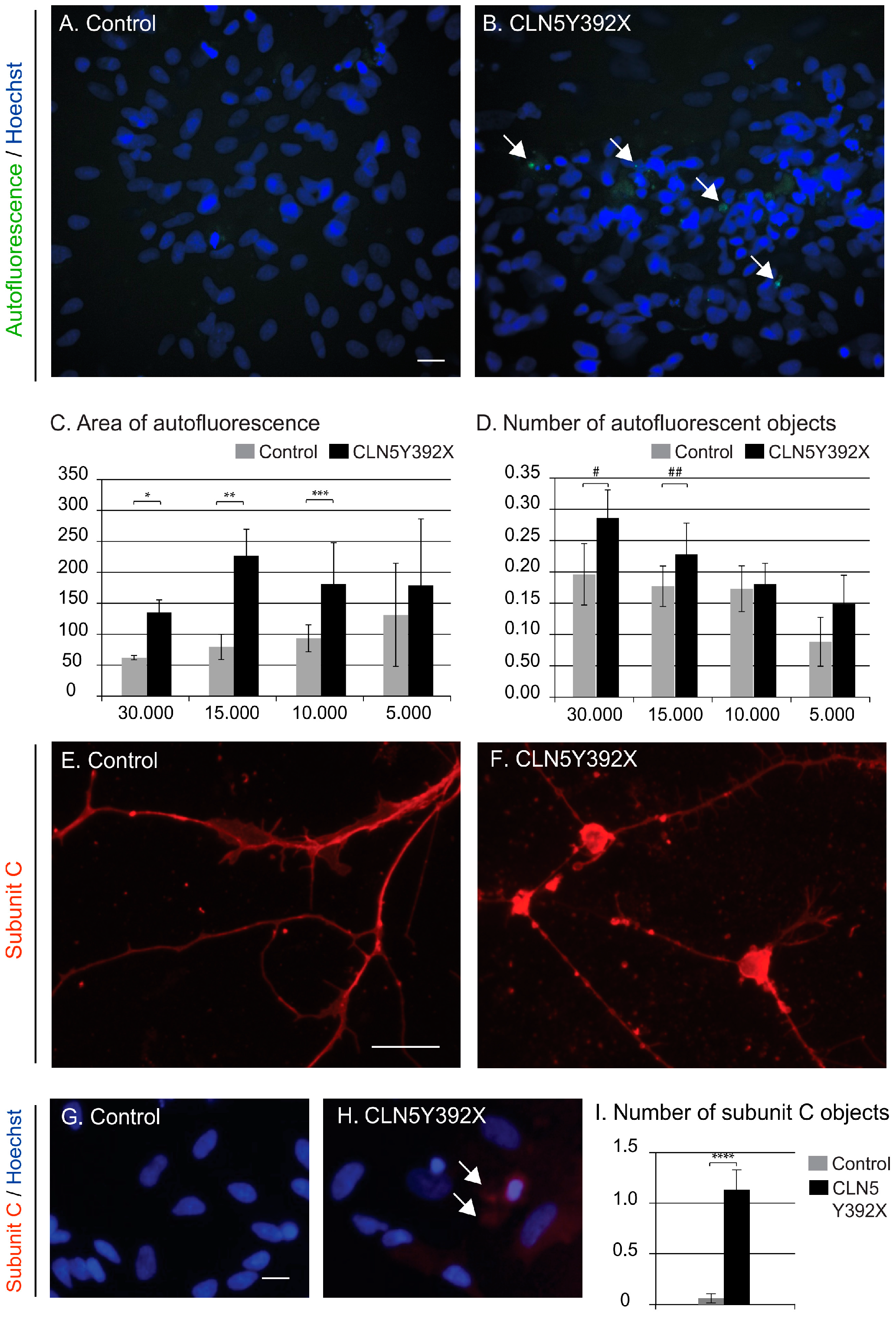
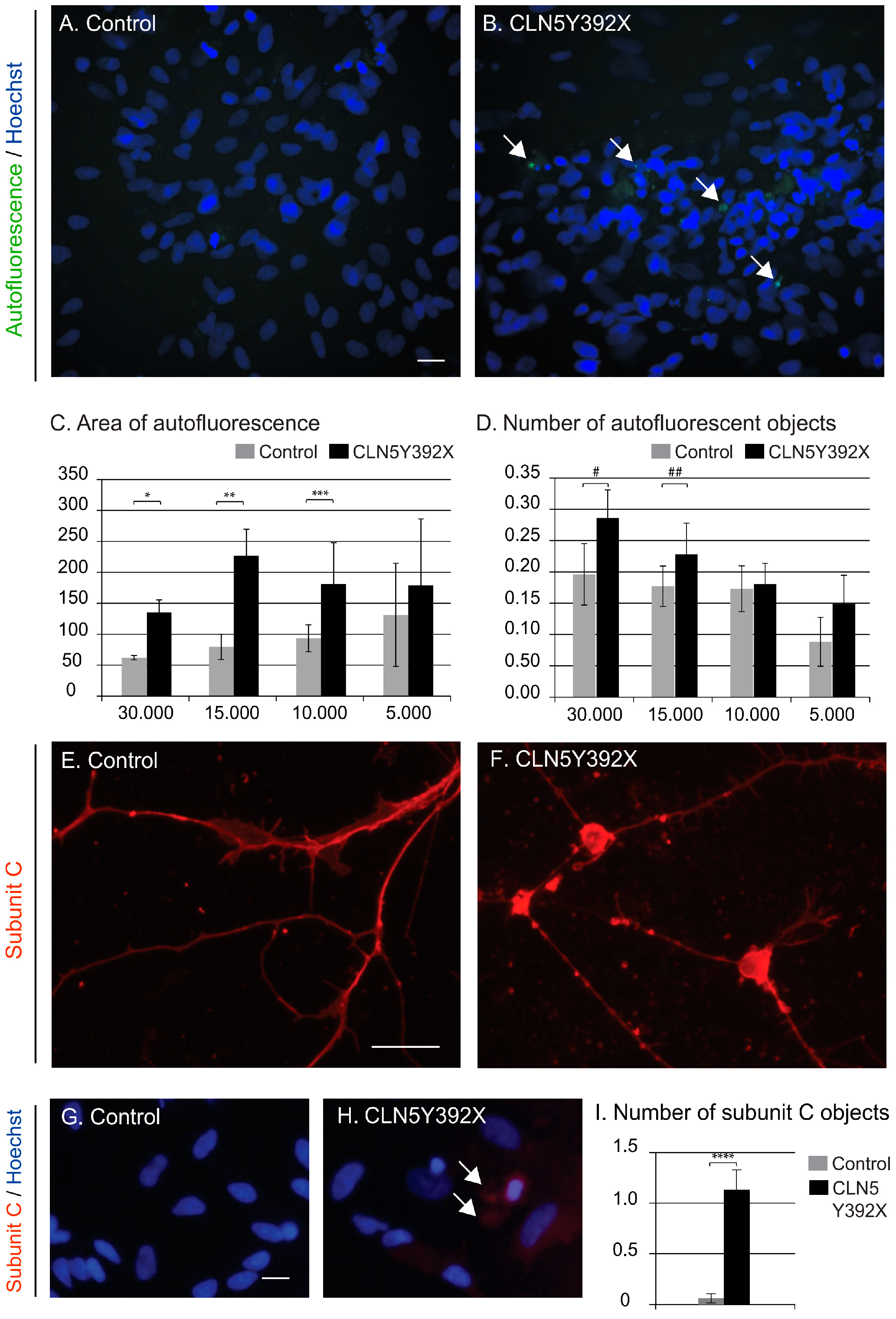
CLN5Y392X iPSC-Derived Neural Lineage Cells Show Accumulation of Storage Material
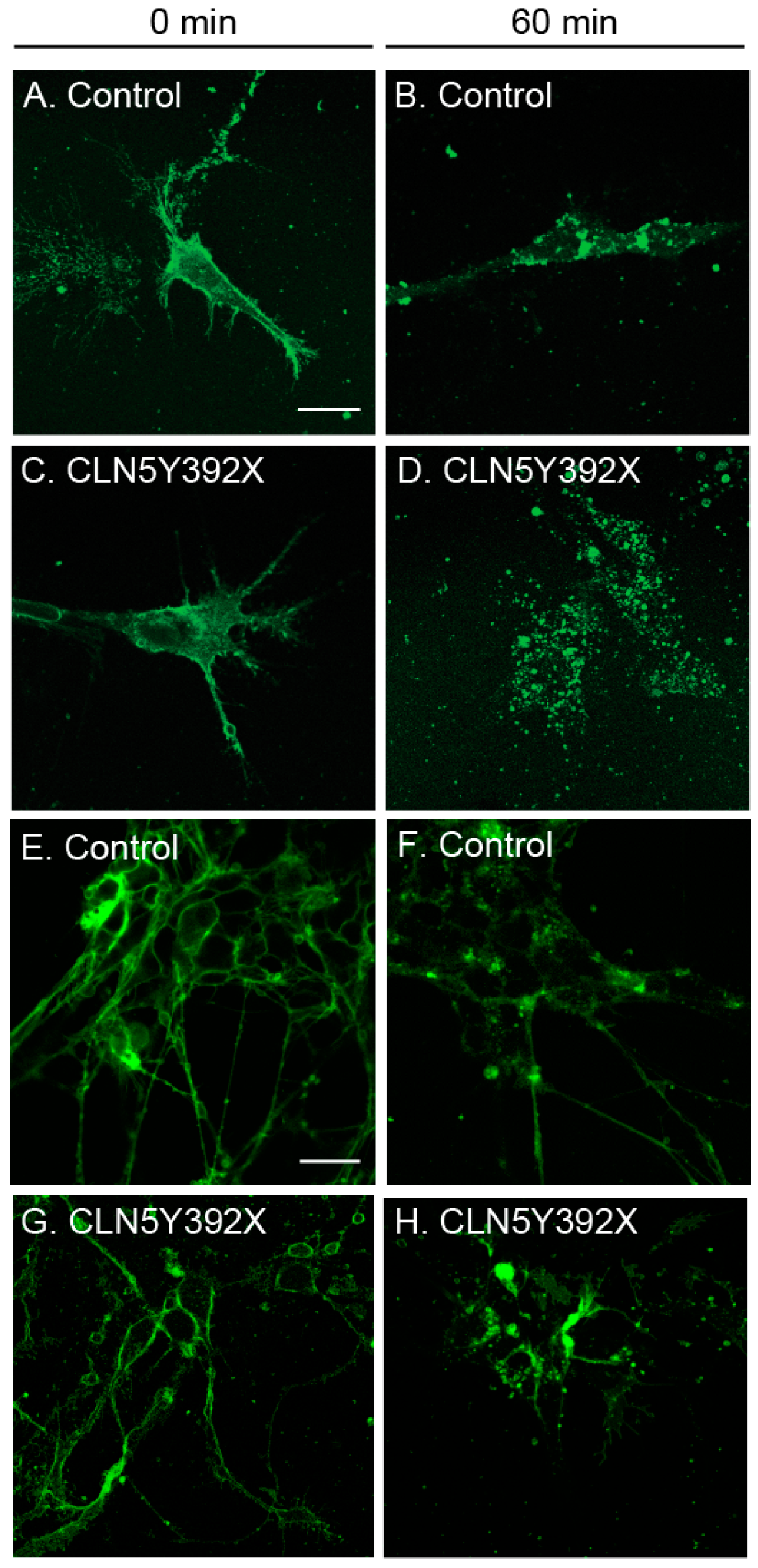
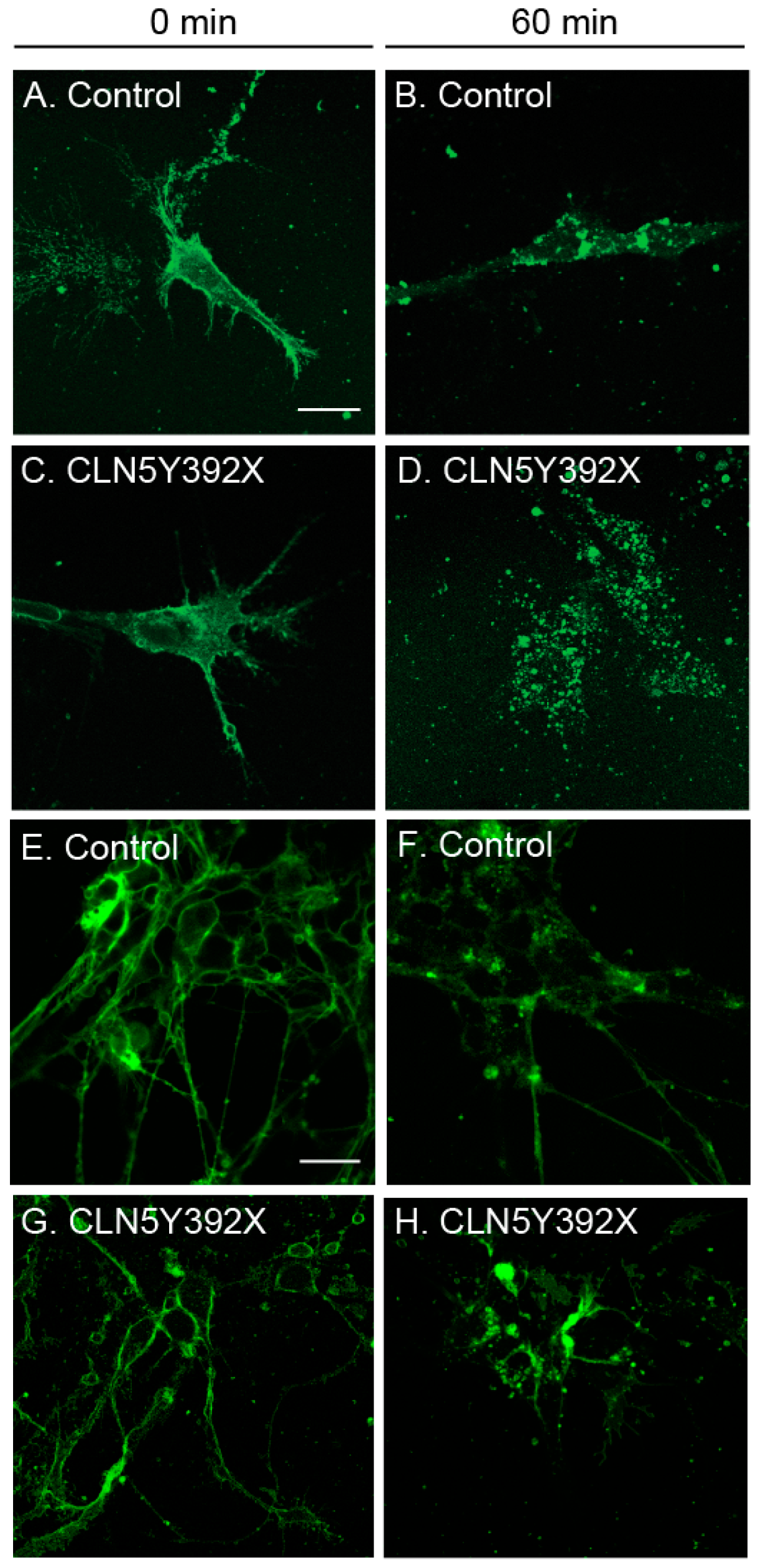
2.3. CLN5Y392X iPSC-Derived Neural Lineage Cells Show Changes in Sphingolipid Transport
3. Discussion
4. Materials and Methods
4.1. Ethical Issues
4.2. Cell Lines
4.3. Generation and Characterisation of CLN5Y392X iPSC Lines
4.4. Pluripotency of CLN5Y392X iPS Cells
4.5. Differentiation of iPS Cells towards Neural Lineage Cells
4.6. Immunofluorescence Analyses of iPSC-Derived Neural Lineage Cells
4.7. Quantitative Image Analyses
4.8. Analyses of Lipid Transport in iPSC-Derived Neural Lineage Cells
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| NCL | Neuronal ceroid lipofuscinoses |
| CLN | Ceroid lipofuscinosis neuronal |
| iPSC | Induced pluripotent stem cell |
| hESC | Human embryonic stem cell |
| NPC | Neural progenitor cell |
| EB | Embryoid body |
References
- Kousi, M.; Lehesjoki, A.E.; Mole, S.E. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum. Mutat. 2012, 33, 42–63. [Google Scholar] [CrossRef] [PubMed]
- Haltia, M. The neuronal ceroid-lipofuscinoses: From past to present. Biochim. Biophys. Acta 2006, 1762, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Santavuori, P. Neuronal ceroid-lipofuscinoses in childhood. Brain Dev. 1988, 10, 80–83. [Google Scholar] [CrossRef]
- Mole, S.E.; Cotman, S.L. Genetics of the neuronal ceroid lipofuscinoses (batten disease). Biochim. Biophys. Acta 2015, 1852, 2237–2241. [Google Scholar] [CrossRef] [PubMed]
- Savukoski, M.; Klockars, T.; Holmberg, V.; Santavuori, P.; Lander, E.S.; Peltonen, L. CLN5, a novel gene encoding a putative transmembrane protein mutated in finnish variant late infantile neuronal ceroid lipofuscinosis. Nat. Genet. 1998, 19, 286–288. [Google Scholar] [PubMed]
- Santavuori, P.; Rapola, J.; Raininko, R.; Autti, T.; Lappi, M.; Nuutila, A.; Launes, J.; Sainio, K. Early juvenile neuronal ceroid-lipofuscinosis or variant Jansky–Bielschowsky disease: Diagnostic criteria and nomenclature. J. Inherit. Metab. Dis. 1993, 16, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Santavuori, P.; Rapola, J.; Sainio, K.; Raitta, C. A variant of Jansky–Bielschowsky disease. Neuropediatrics 1982, 13, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Mancini, C.; Nassani, S.; Guo, Y.; Chen, Y.; Giorgio, E.; Brussino, A.; di Gregorio, E.; Cavalieri, S.; Lo Buono, N.; Funaro, A.; et al. Adult-onset autosomal recessive ataxia associated with neuronal ceroid lipofuscinosis type 5 gene (CLN5) mutations. J. Neurol. 2015, 262, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Setty, G.; Saleem, R.; Khan, A.; Hussain, N. Atypical juvenile neuronal ceroid lipofuscinosis: A report of three cases. J. Pediatr. Neurosci. 2013, 8, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Santavuori, P.; Rapola, J.; Nuutila, A.; Raininko, R.; Lappi, M.; Launes, J.; Herva, R.; Sainio, K. The spectrum of Jansky–Bielschowsky disease. Neuropediatrics 1991, 22, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Tyynela, J.; Cooper, J.D.; Khan, M.N.; Shemilts, S.J.; Haltia, M. Hippocampal pathology in the human neuronal ceroid-lipofuscinoses: Distinct patterns of storage deposition, neurodegeneration and glial activation. Brain Pathol. 2004, 14, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Autti, T.; Raininko, R.; Launes, J.; Nuutila, A.; Santavuori, P. Jansky–Bielschowsky variant disease: CT, MRI, and SPECT findings. Pediatr. Neurol. 1992, 8, 121–126. [Google Scholar] [CrossRef]
- Tyynela, J.; Suopanki, J.; Santavuori, P.; Baumann, M.; Haltia, M. Variant late infantile neuronal ceroid-lipofuscinosis: Pathology and biochemistry. J. Neuropathol. Exp. Neurol. 1997, 56, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Kopra, O.; Vesa, J.; von Schantz, C.; Manninen, T.; Minye, H.; Fabritius, A.L.; Rapola, J.; van Diggelen, O.P.; Saarela, J.; Jalanko, A.; et al. A mouse model for Finnish variant late infantile neuronal ceroid lipofuscinosis, CLN5, reveals neuropathology associated with early aging. Hum. Mol. Genet. 2004, 13, 2893–2906. [Google Scholar] [CrossRef] [PubMed]
- Schmiedt, M.L.; Blom, T.; Blom, T.; Kopra, O.; Wong, A.; von Schantz-Fant, C.; Ikonen, E.; Kuronen, M.; Jauhiainen, M.; Cooper, J.D.; et al. CLN5-deficiency in mice leads to microglial activation, defective myelination and changes in lipid metabolism. Neurobiol. Dis. 2012, 46, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Von Schantz, C.; Kielar, C.; Hansen, S.N.; Pontikis, C.C.; Alexander, N.A.; Kopra, O.; Jalanko, A.; Cooper, J.D. Progressive thalamocortical neuron loss in CLN5 deficient mice: Distinct effects in finnish variant late infantile NCL. Neurobiol. Dis. 2009, 34, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Schmiedt, M.L.; Bessa, C.; Heine, C.; Ribeiro, M.G.; Jalanko, A.; Kyttala, A. The neuronal ceroid lipofuscinosis protein CLN5: New insights into cellular maturation, transport, and consequences of mutations. Hum. Mutat. 2010, 31, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Vesa, J.; Peltonen, L. Mutated genes in juvenile and variant late infantile neuronal ceroid lipofuscinoses encode lysosomal proteins. Curr. Mol. Med. 2002, 2, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, V.; Jalanko, A.; Isosomppi, J.; Fabritius, A.L.; Peltonen, L.; Kopra, O. The mouse ortholog of the neuronal ceroid lipofuscinosis CLN5 gene encodes a soluble lysosomal glycoprotein expressed in the developing brain. Neurobiol. Dis. 2004, 16, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Von Schantz, C.; Saharinen, J.; Kopra, O.; Cooper, J.D.; Gentile, M.; Hovatta, I.; Peltonen, L.; Jalanko, A. Brain gene expression profiles of CLN1 and CLN5 deficient mice unravels common molecular pathways underlying neuronal degeneration in NCL diseases. BMC Genom. 2008, 9, 146. [Google Scholar] [CrossRef] [PubMed]
- Scifo, E.; Szwajda, A.; Debski, J.; Uusi-Rauva, K.; Kesti, T.; Dadlez, M.; Gingras, A.C.; Tyynela, J.; Baumann, M.H.; Jalanko, A.; et al. Drafting the CLN3 protein interactome in SH-SY5Y human neuroblastoma cells: A label-free quantitative proteomics approach. J. Proteome Res. 2013, 12, 2101–2115. [Google Scholar] [CrossRef] [PubMed]
- Scifo, E.; Szwajda, A.; Soliymani, R.; Pezzini, F.; Bianchi, M.; Dapkunas, A.; Debski, J.; Uusi-Rauva, K.; Dadlez, M.; Gingras, A.C.; et al. Proteomic analysis of the palmitoyl protein thioesterase 1 interactome in SH-SY5Y human neuroblastoma cells. J. Proteom. 2015, 123, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Vesa, J.; Chin, M.H.; Oelgeschlager, K.; Isosomppi, J.; DellAngelica, E.C.; Jalanko, A.; Peltonen, L. Neuronal ceroid lipofuscinoses are connected at molecular level: Interaction of CLN5 protein with CLN2 and CLN3. Mol. Biol. Cell 2002, 13, 2410–2420. [Google Scholar] [CrossRef] [PubMed]
- Lyly, A.; von Schantz, C.; Heine, C.; Schmiedt, M.L.; Sipila, T.; Jalanko, A.; Kyttala, A. Novel interactions of CLN5 support molecular networking between neuronal ceroid lipofuscinosis proteins. BMC Cell Biol. 2009, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Lyly, A.; Marjavaara, S.K.; Kyttala, A.; Uusi-Rauva, K.; Luiro, K.; Kopra, O.; Martinez, L.O.; Tanhuanpaa, K.; Kalkkinen, N.; Suomalainen, A.; et al. Deficiency of the INCL protein ppt1 results in changes in ectopic F1-ATP synthase and altered cholesterol metabolism. Hum. Mol. Genet. 2008, 17, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- Martinez, L.O.; Jacquet, S.; Esteve, J.P.; Rolland, C.; Cabezon, E.; Champagne, E.; Pineau, T.; Georgeaud, V.; Walker, J.E.; Terce, F.; et al. Ectopic β-chain of ATP synthase is an apolipoprotein A-I receptor in hepatic HDL endocytosis. Nature 2003, 421, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Ahtiainen, L.; Kolikova, J.; Mutka, A.L.; Luiro, K.; Gentile, M.; Ikonen, E.; Khiroug, L.; Jalanko, A.; Kopra, O. Palmitoyl protein thioesterase 1 (PPT1)-deficient mouse neurons show alterations in cholesterol metabolism and calcium homeostasis prior to synaptic dysfunction. Neurobiol. Dis. 2007, 28, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Haddad, S.E.; Khoury, M.; Daoud, M.; Kantar, R.; Harati, H.; Mousallem, T.; Alzate, O.; Meyer, B.; Boustany, R.M. CLN5 and CLN8 protein association with ceramide synthase: Biochemical and proteomic approaches. Electrophoresis 2012, 33, 3798–3809. [Google Scholar] [CrossRef] [PubMed]
- Mamo, A.; Jules, F.; Dumaresq-Doiron, K.; Costantino, S.; Lefrancois, S. The role of ceroid lipofuscinosis neuronal protein 5 (CLN5) in endosomal sorting. Mol. Cell. Biol. 2012, 32, 1855–1866. [Google Scholar] [CrossRef] [PubMed]
- Uusi-Rauva, K.; Kyttala, A.; van der Kant, R.; Vesa, J.; Tanhuanpaa, K.; Neefjes, J.; Olkkonen, V.M.; Jalanko, A. Neuronal ceroid lipofuscinosis protein CLN3 interacts with motor proteins and modifies location of late endosomal compartments. Cell. Mol. Life Sci. 2012, 69, 2075–2089. [Google Scholar] [CrossRef] [PubMed]
- Faller, K.M.; Gutierrez-Quintana, R.; Mohammed, A.; Rahim, A.A.; Tuxworth, R.I.; Wager, K.; Bond, M. The neuronal ceroid lipofuscinoses: Opportunities from model systems. Biochim. Biophys. Acta 2015, 1852, 2267–2278. [Google Scholar] [CrossRef] [PubMed]
- Bond, M.; Holthaus, S.M.; Tammen, I.; Tear, G.; Russell, C. Use of model organisms for the study of neuronal ceroid lipofuscinosis. Biochim. Biophys. Acta 2013, 1832, 1842–1865. [Google Scholar] [CrossRef] [PubMed]
- Trokovic, R.; Weltner, J.; Otonkoski, T. Generation of iPSC line HEL24.3 from human neonatal foreskin fibroblasts. Stem Cell Res. 2015, 15, 266–268. [Google Scholar] [CrossRef] [PubMed]
- Trokovic, R.; Weltner, J.; Nishimura, K.; Ohtaka, M.; Nakanishi, M.; Salomaa, V.; Jalanko, A.; Otonkoski, T.; Kyttala, A. Advanced feeder-free generation of induced pluripotent stem cells directly from blood cells. Stem Cells Transl. Med. 2014, 3, 1402–1409. [Google Scholar] [CrossRef] [PubMed]
- McCormick, P.J.; Dumaresq-Doiron, K.; Pluviose, A.S.; Pichette, V.; Tosato, G.; Lefrancois, S. Palmitoylation controls recycling in lysosomal sorting and trafficking. Traffic 2008, 9, 1984–1997. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.S.; Patterson, M.C.; Wheatley, C.L.; O’Brien, J.F.; Pagano, R.E. Broad screening test for sphingolipid-storage diseases. Lancet 1999, 354, 901–905. [Google Scholar] [CrossRef]
- Holmberg, V.; Lauronen, L.; Autti, T.; Santavuori, P.; Savukoski, M.; Uvebrant, P.; Hofman, I.; Peltonen, L.; Jarvela, I. Phenotype-genotype correlation in eight patients with Finnish variant late infantile NCL (CLN5). Neurology 2000, 55, 579–581. [Google Scholar] [CrossRef] [PubMed]
- Staropoli, J.F.; Xin, W.; Barone, R.; Cotman, S.L.; Sims, K.B. An atypical case of neuronal ceroid lipofuscinosis with co-inheritance of a variably penetrant polg1 mutation. BMC Med. Genet. 2012, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Poet, M.; Kornak, U.; Schweizer, M.; Zdebik, A.A.; Scheel, O.; Hoelter, S.; Wurst, W.; Schmitt, A.; Fuhrmann, J.C.; Planells-Cases, R.; et al. Lysosomal storage disease upon disruption of the neuronal chloride transport protein CLC-6. Proc. Natl. Acad. Sci. USA 2006, 103, 13854–13859. [Google Scholar] [CrossRef] [PubMed]
- Best, H.L.; Neverman, N.J.; Wicky, H.E.; Mitchell, N.L.; Leitch, B.; Hughes, S.M. Characterisation of early changes in ovine CLN5 and CLN6 batten disease neural cultures for the rapid screening of therapeutics. Neurobiol. Dis. 2017, 100, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Lojewski, X.; Staropoli, J.F.; Biswas-Legrand, S.; Simas, A.M.; Haliw, L.; Selig, M.K.; Coppel, S.H.; Goss, K.A.; Petcherski, A.; Chandrachud, U.; et al. Human iPSC models of neuronal ceroid lipofuscinosis capture distinct effects of tpp1 and CLN3 mutations on the endocytic pathway. Hum. Mol. Genet. 2014, 23, 2005–2022. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Zhang, Z.; Hitomi, E.; Lee, Y.C.; Mukherjee, A.B. Endoplasmic reticulum stress-induced caspase-4 activation mediates apoptosis and neurodegeneration in INCL. Hum. Mol. Genet. 2006, 15, 1826–1834. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lee, Y.C.; Kim, S.J.; Choi, M.S.; Tsai, P.C.; Xu, Y.; Xiao, Y.J.; Zhang, P.; Heffer, A.; Mukherjee, A.B. Palmitoyl-protein thioesterase-1 deficiency mediates the activation of the unfolded protein response and neuronal apoptosis in iNCL. Hum. Mol. Genet. 2006, 15, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Galizzi, G.; Russo, D.; Deidda, I.; Cascio, C.; Passantino, R.; Guarneri, R.; Bigini, P.; Mennini, T.; Drago, G.; Guarneri, P. Different early ER-stress responses in the CLN8(mnd) mouse model of neuronal ceroid lipofuscinosis. Neurosci. Lett. 2011, 488, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Kim, S.J.; Zhang, Z.; Tsai, P.C.; Wisniewski, K.E.; Mukherjee, A.B. ER and oxidative stresses are common mediators of apoptosis in both neurodegenerative and non-neurodegenerative lysosomal storage disorders and are alleviated by chemical chaperones. Hum. Mol. Genet. 2008, 17, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Kollmann, K.; Uusi-Rauva, K.; Scifo, E.; Tyynela, J.; Jalanko, A.; Braulke, T. Cell biology and function of neuronal ceroid lipofuscinosis-related proteins. Biochim. Biophys. Acta 2013, 1832, 1866–1881. [Google Scholar] [CrossRef] [PubMed]
- McMahon, A.P.; Bradley, A. The Wnt-1 (int-1) proto-oncogene is required for development of a large region of the mouse brain. Cell 1990, 62, 1073–1085. [Google Scholar] [CrossRef]
- Vuoristo, S.; Toivonen, S.; Weltner, J.; Mikkola, M.; Ustinov, J.; Trokovic, R.; Palgi, J.; Lund, R.; Tuuri, T.; Otonkoski, T. A novel feeder-free culture system for human pluripotent stem cell culture and induced pluripotent stem cell derivation. PLoS ONE 2013, 8, e76205. [Google Scholar] [CrossRef] [PubMed]
- Trokovic, R.; Weltner, J.; Manninen, T.; Mikkola, M.; Lundin, K.; Hamalainen, R.; Suomalainen, A.; Otonkoski, T. Small molecule inhibitors promote efficient generation of induced pluripotent stem cells from human skeletal myoblasts. Stem Cells Dev. 2013, 22, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Nat, R.; Nilbratt, M.; Narkilahti, S.; Winblad, B.; Hovatta, O.; Nordberg, A. Neurogenic neuroepithelial and radial glial cells generated from six human embryonic stem cell lines in serum-free suspension and adherent cultures. Glia 2007, 55, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Puttonen, K.A.; Ruponen, M.; Kauppinen, R.; Wojciechowski, S.; Hovatta, O.; Koistinaho, J. Improved method of producing human neural progenitor cells of high purity and in large quantities from pluripotent stem cells for transplantation studies. Cell Transplant. 2013, 22, 1753–1766. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.N.; Bayliss, S.L.; Westlake, V.J. Batten disease and the ATP synthase subunit c turnover pathway: Raising antibodies to subunit c. Am. J. Med. Genet. 1995, 57, 260–265. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uusi-Rauva, K.; Blom, T.; Von Schantz-Fant, C.; Blom, T.; Jalanko, A.; Kyttälä, A. Induced Pluripotent Stem Cells Derived from a CLN5 Patient Manifest Phenotypic Characteristics of Neuronal Ceroid Lipofuscinoses. Int. J. Mol. Sci. 2017, 18, 955. https://doi.org/10.3390/ijms18050955
Uusi-Rauva K, Blom T, Von Schantz-Fant C, Blom T, Jalanko A, Kyttälä A. Induced Pluripotent Stem Cells Derived from a CLN5 Patient Manifest Phenotypic Characteristics of Neuronal Ceroid Lipofuscinoses. International Journal of Molecular Sciences. 2017; 18(5):955. https://doi.org/10.3390/ijms18050955
Chicago/Turabian StyleUusi-Rauva, Kristiina, Tea Blom, Carina Von Schantz-Fant, Tomas Blom, Anu Jalanko, and Aija Kyttälä. 2017. "Induced Pluripotent Stem Cells Derived from a CLN5 Patient Manifest Phenotypic Characteristics of Neuronal Ceroid Lipofuscinoses" International Journal of Molecular Sciences 18, no. 5: 955. https://doi.org/10.3390/ijms18050955
APA StyleUusi-Rauva, K., Blom, T., Von Schantz-Fant, C., Blom, T., Jalanko, A., & Kyttälä, A. (2017). Induced Pluripotent Stem Cells Derived from a CLN5 Patient Manifest Phenotypic Characteristics of Neuronal Ceroid Lipofuscinoses. International Journal of Molecular Sciences, 18(5), 955. https://doi.org/10.3390/ijms18050955