
Unraveling Prion Protein Interactions with Aptamers and Other PrP-Binding Nucleic Acids


Abstract
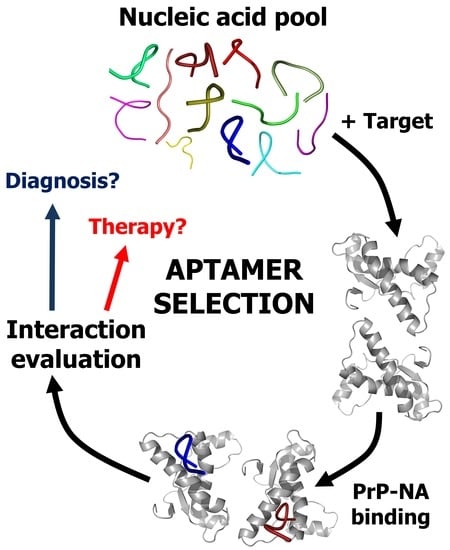
:
1. Introduction
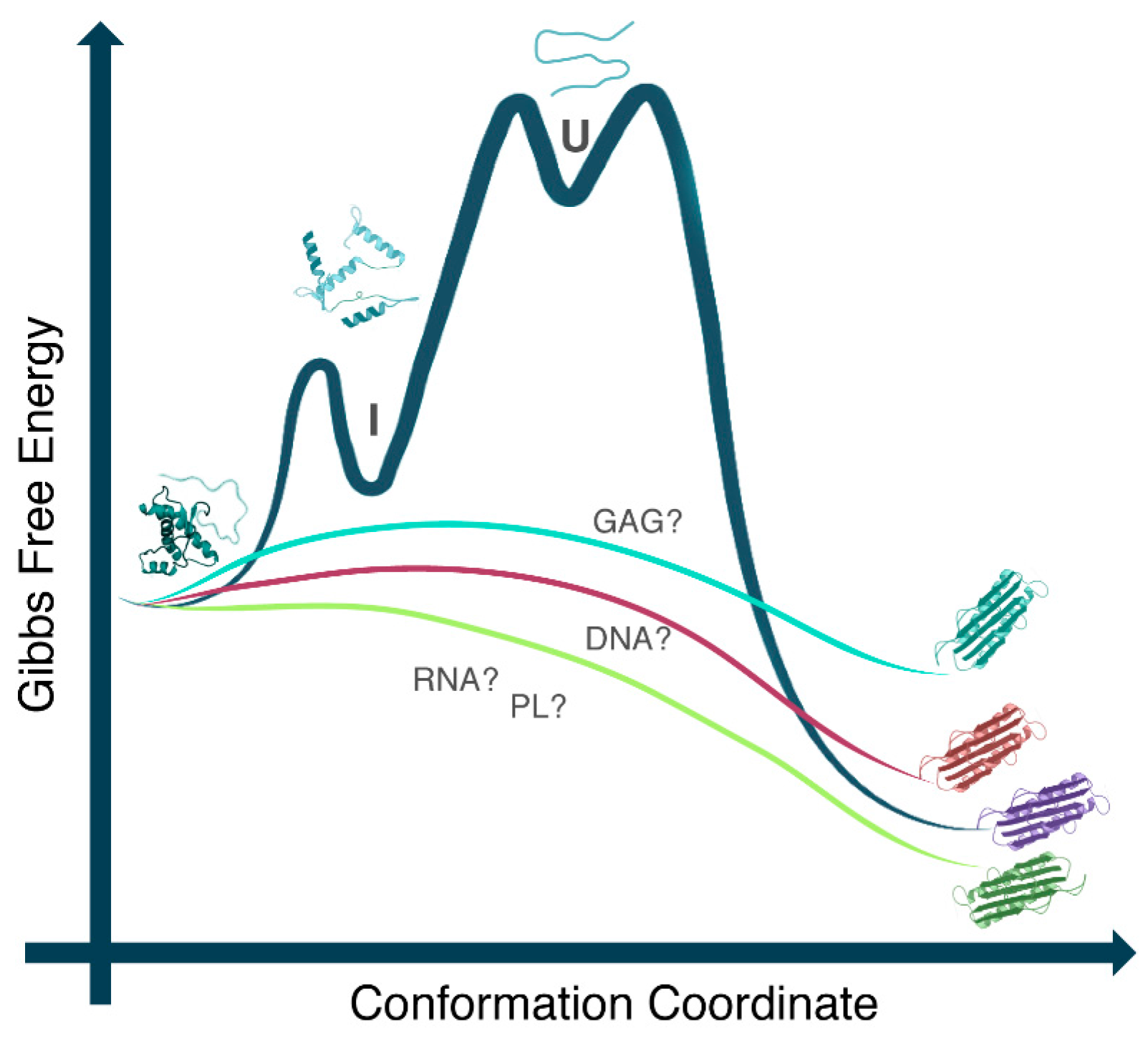
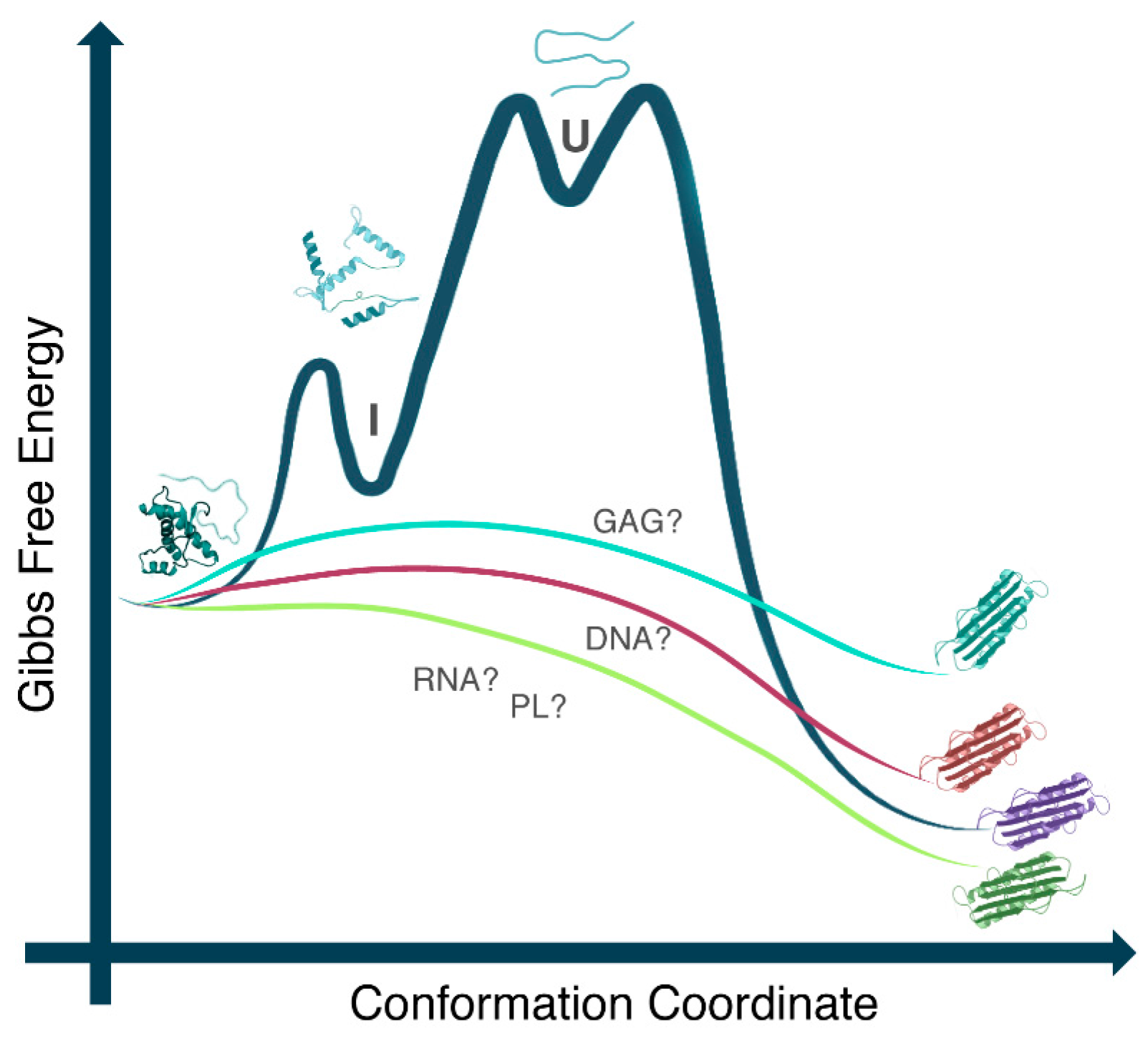
2. PrP and Nucleic Acids Interactions
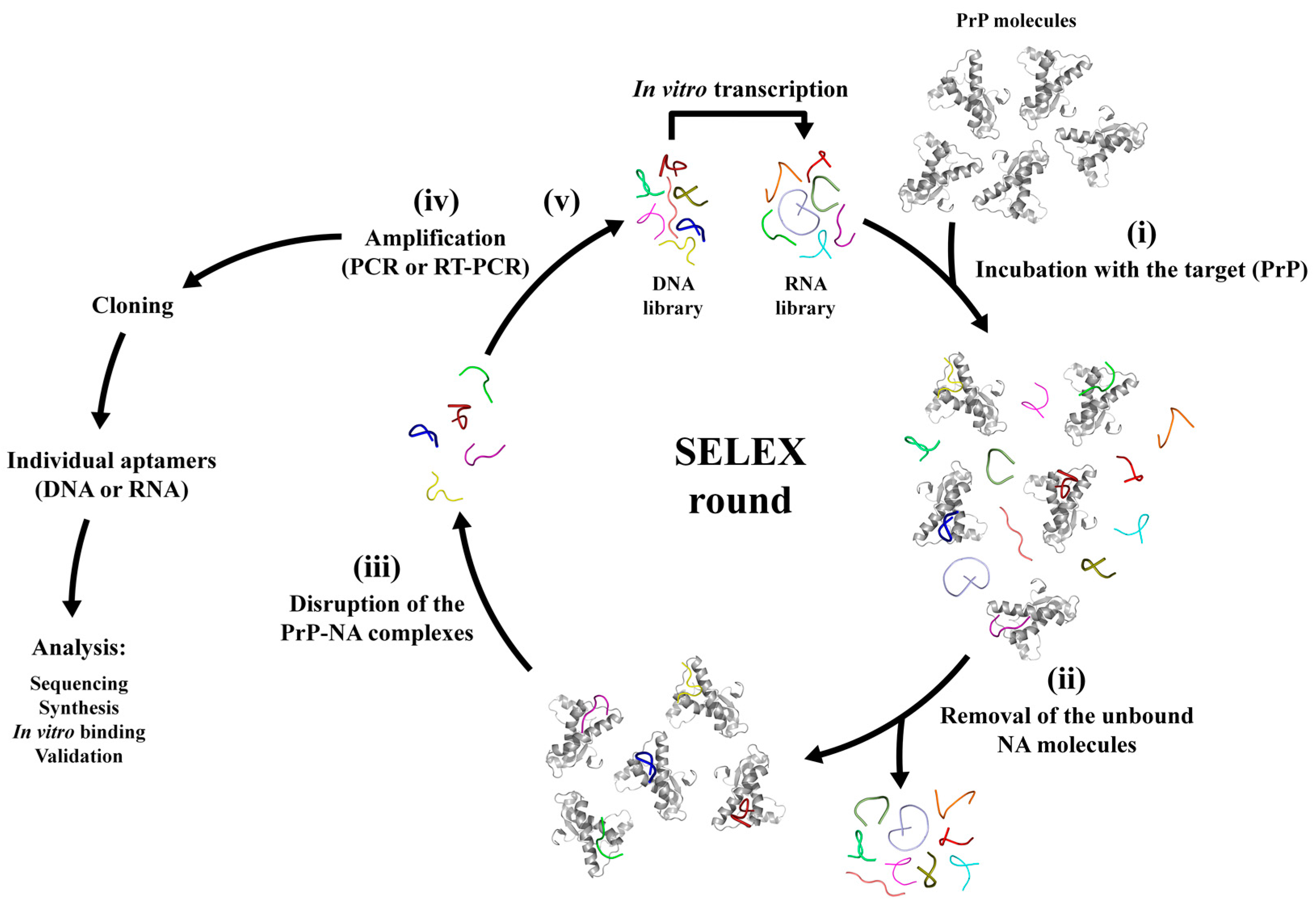
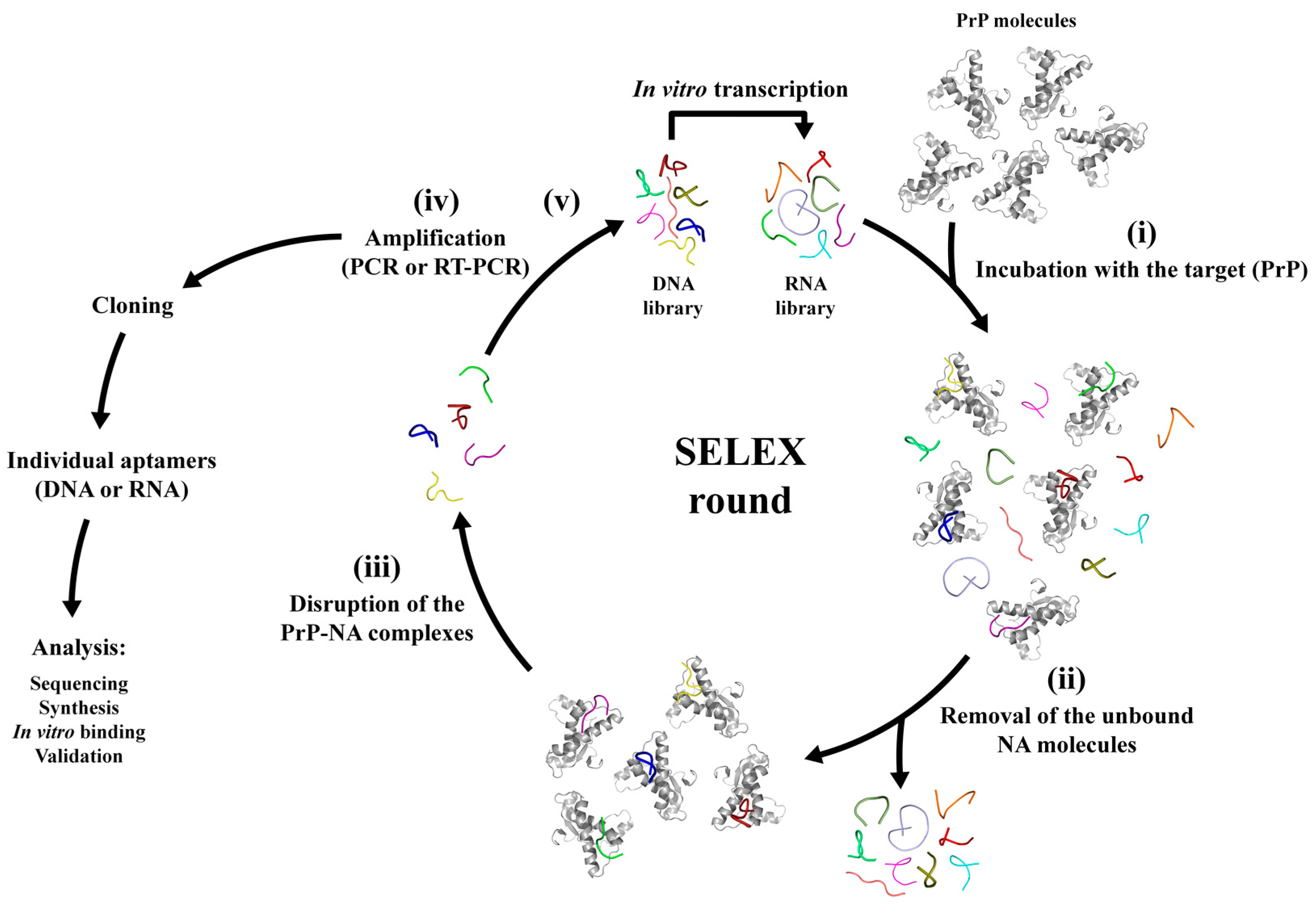
3. SELEX Technique and the Aptamer Discovery
4. Nucleic Acids Aptamers against PrP
5. Aptamers against Other Amyloidogenic Proteins
6. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Prusiner, S.B. Nobel Prize Lecture: Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Lakkaraju, A.K.K. Cell Biology of Prions and Prionoids: A Status Report. Trends Cell Biol. 2016, 26, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J. Prion diseases of humans and animals: Their causes and molecular basis. Annu. Rev. Neurosci. 2001, 24, 519–550. [Google Scholar] [CrossRef] [PubMed]
- Riek, R.; Hornemann, S.; Wider, G.; Billeter, M.; Glockshuber, R.; Wüthrich, K. NMR structure of the mouse prion protein domain PrP121–231. Nature 1996, 382, 180–182. [Google Scholar] [CrossRef] [PubMed]
- Knaus, K.J.; Morillas, M.; Swietnicki, W.; Malone, M.; Surewicz, W.K.; Yee, V.C. Crystal structure of the human prion protein reveals a mechanism for oligomerization. Nat. Struct. Biol. 2001, 8, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Baron, G.S.; Chesebro, B.; Jeffrey, M. Getting a grip on prions: Oligomers, amyloids, and pathological membrane interactions. Annu. Rev. Biochem. 2009, 78, 177–204. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.J.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Lee, V.M.Y. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 2014, 20, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.L.; Cordeiro, Y. The “Jekyll and Hyde” Actions of Nucleic Acids on the Prion-like Aggregation of Proteins. J. Biol. Chem. 2016, 291, 15482–15490. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Functional Amyloid, and Human Disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta Proteins Proteom. 2010, 1804, 1231–1264. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.-F.; Harischandra, D.S.; Kanthasamy, A.; Sivasankar, S. Copper-induced structural conversion templates prion protein oligomerization and neurotoxicity. Sci. Adv. 2016, 2, e1600014. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.L.; Gomes, M.P.; Vieira, T.C.; Cordeiro, Y. PrP interactions with nucleic acids and glycosaminoglycans in function and disease. Front. Biosci. 2010, 15, 132–150. [Google Scholar] [CrossRef]
- Vieira, T.C.R.G.; Reynaldo, D.P.; Gomes, M.P.B.; Almeida, M.S.; Cordeiro, Y.; Silva, J.L. Heparin binding by murine recombinant prion protein leads to transient aggregation and formation of rna-resistant species. J. Am. Chem. Soc. 2011, 133, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, Y. Nucleic acid-mediated protein aggregation and assembly. Adv. Protein Chem. Struct. Biol. 2011, 84, 1–40. [Google Scholar] [PubMed]
- Supattapone, S. Elucidating the role of cofactors in mammalian prion propagation. Prion 2014, 8, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Critchley, P.; Kazlauskaite, J.; Eason, R.; Pinheiro, T.J.T. Binding of prion proteins to lipid membranes. Biochem. Biophys. Res. Commun. 2004, 313, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Deleault, N.R.; Harris, B.T.; Rees, J.R.; Supattapone, S. Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. USA 2007, 104, 9741–9746. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.B.; Wang, D.W.; Wang, F.; Noble, G.P.; Ma, J.; Woods, V.L.; Li, S.; Supattapone, S. Cofactor molecules induce structural transformation during infectious prion formation. Structure 2013, 21, 2061–2068. [Google Scholar] [CrossRef] [PubMed]
- Soto, C. Prion hypothesis: The end of the controversy? Trends Biochem. Sci. 2011, 36, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, Y.; Machado, F.; Juliano, L.; Juliano, M.A.; Brentani, R.R.; Foguel, D.; Silva, J.L. DNA Converts Cellular Prion Protein into the β-Sheet Conformation and Inhibits Prion Peptide Aggregation. J. Biol. Chem. 2001, 276, 49400–49409. [Google Scholar] [CrossRef] [PubMed]
- Adler, V.; Zeiler, B.; Kryukov, V.; Kascsak, R.; Rubenstein, R.; Grossman, A. Small, highly structured RNAs participate in the conversion of human recombinant PrPSen to PrPRes in vitro. J. Mol. Biol. 2003, 332, 47–57. [Google Scholar] [CrossRef]
- Deleault, N.R.; Lucassen, R.W.; Supattapone, S. RNA molecules stimulate prion protein conversion. Nature 2003, 425, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Macedo, B.; Millen, T.A.; Braga, C.A.C.A.; Gomes, M.P.B.; Ferreira, P.S.; Kraineva, J.; Winter, R.; Silva, J.L.; Cordeiro, Y. Nonspecific prion protein-nucleic acid interactions lead to different aggregates and cytotoxic species. Biochemistry 2012, 51, 5402–5413. [Google Scholar] [CrossRef] [PubMed]
- Chaves, J.A.P.; Sanchez-López, C.; Gomes, M.P.B.; Sisnande, T.; Macedo, B.; de Oliveira, V.E.; Braga, C.A.C.; Rangel, L.P.; Silva, J.L.; Quintanar, L.; et al. Biophysical and morphological studies on the dual interaction of non-octarepeat prion protein peptides with copper and nucleic acids. J. Biol. Inorg. Chem. 2014, 19, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.-Q.; Zheng, J.; Gray, D.M.; Gambetti, P.; Chen, S.G. Antibody to DNA detects scrapie but not normal prion protein. Proc. Natl. Acad. Sci. USA 2004, 101, 1380–1385. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Nandi, P.K. Interaction of prion peptide HuPrP106–126 with nucleic acid: Brief report. Arch. Virol. 1997, 142, 2537–2545. [Google Scholar] [CrossRef] [PubMed]
- Nandi, P.K. Polymerization of human prion peptide HuPrP 106–126 to amyloid in nucleic acid solution. Arch. Virol. 1998, 143, 1251–1263. [Google Scholar] [CrossRef] [PubMed]
- Nandi, P.K.; Leclerc, E. Polymerization of murine recombinant prion protein in nucleic acid solution. Arch. Virol. 1999, 144, 1751–1763. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, Y.; Silva, J.L. The hypothesis of the catalytic action of nucleic acid on the conversion of prion protein. Protein Pept. Lett. 2005, 12, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.; Proske, D.; Neumann, M.; Groschup, M.H.; Kretzschmar, H.A.; Famulok, M.; Winnacker, E.L. RNA aptamers specifically interact with the prion protein PrP. J. Virol. 1997, 71, 8790–8797. [Google Scholar] [PubMed]
- Lima, L.M.T.R.; Cordeiro, Y.; Tinoco, L.W.; Marques, A.F.; Oliveira, C.L.P.; Sampath, S.; Kodali, R.; Choi, G.; Foguel, D.; Torriani, I.; et al. Structural insights into the interaction between prion protein and nucleic acid. Biochemistry 2006, 45, 9180–9187. [Google Scholar] [CrossRef] [PubMed]
- Mercey, R.; Lantier, I.; Maurel, M.C.; Grosclaude, J.; Lantier, F.; Marc, D. Fast, reversible interaction of prion protein with RNA aptamers containing specific sequence patterns. Arch. Virol. 2006, 151, 2197–2214. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, P.; Pagano, B.; Granata, V.; Prigent, S.; Rezaei, H.; Giancola, C.; Zagari, A. Cross-talk between prion protein and quadruplex-forming nucleic acids: A dynamic complex formation. Nucleic Acids Res. 2013, 41, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Gabus, C.; Derrington, E.; Leblanc, P.; Chnaiderman, J.; Dormont, D.; Swietnicki, W.; Morillas, M.; Surewicz, W.K.; Marc, D.; Nandi, P.; et al. The Prion Protein Has RNA Binding and Chaperoning Properties Characteristic of Nucleocapsid Protein NCp7 of HIV-1. J. Biol. Chem. 2001, 276, 19301–19309. [Google Scholar] [CrossRef] [PubMed]
- Guichard, C.; Ivanyi-Nagy, R.; Sharma, K.K.; Gabus, C.; Marc, D.; Mély, Y.; Darlix, J.L. Analysis of nucleic acid chaperoning by the prion protein and its inhibition by oligonucleotides. Nucleic Acids Res. 2011, 39, 8544–8558. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yu, S.; Yang, J.; Yin, X.; Zhao, D. RNA and CuCl2 induced conformational changes of the recombinant ovine prion protein. Mol. Cell. Biochem. 2007, 294, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.P.B.; Millen, T.A.; Ferreira, P.S.; Cunha E Silva, N.L.; Vieira, T.C.R.G.; Almeida, M.S.; Silva, J.L.; Cordeiro, Y. Prion protein complexed to N2a cellular RNAs through its N-terminal domain forms aggregates and is toxic to murine neuroblastoma cells. J. Biol. Chem. 2008, 283, 19616–19625. [Google Scholar] [CrossRef] [PubMed]
- Saborio, G.P.; Permanne, B.; Soto, C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001, 411, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Lucassen, R.; Nishina, K.; Supattapone, S. In vitro amplification of protease-resistant prion protein requires free sulfhydryl groups. Biochemistry 2003, 42, 4127–4135. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, Y.; Kraineva, J.; Gomes, M.P.B.; Lopes, M.H.; Martins, V.R.; Lima, L.M.T.R.; Foguel, D.; Winter, R.; Silva, J.L. The amino-terminal PrP domain is crucial to modulate prion misfolding and aggregation. Biophys. J. 2005, 89, 2667–2676. [Google Scholar] [CrossRef] [PubMed]
- Strom, A.; Wang, G.S.; Picketts, D.J.; Reimer, R.; Stuke, A.W.; Scott, F.W. Cellular prion protein localizes to the nucleus of endocrine and neuronal cells and interacts with structural chromatin components. Eur. J. Cell. Biol. 2011, 90, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Mangé, A.; Crozet, C.; Lehmann, S.; Béranger, F. Scrapie-like prion protein is translocated to the nuclei of infected cells independently of proteasome inhibition and interacts with chromatin. J. Cell. Sci. 2004, 117, 2411–2416. [Google Scholar] [CrossRef] [PubMed]
- Marijanovic, Z.; Caputo, A.; Campana, V.; Zurzolo, C. Identification of an intracellular site of prion conversion. PLoS Pathog. 2009, 5, e1000426. [Google Scholar] [CrossRef] [PubMed]
- Yim, Y.-I.; Park, B.-C.; Yadavalli, R.; Zhao, X.; Eisenberg, E.; Greene, L.E. The multivesicular body is the major internal site of prion conversion. J. Cell. Sci. 2015, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, S.; Vanderperre, B.; Grenier, C.; Tremblay, I.; Leduc, F.; Roucou, X. A large ribonucleoprotein particle induced by cytoplasmic PrP shares striking similarities with the chromatoid body, an RNA granule predicted to function in posttranscriptional gene regulation. Biochim. Biophys. Acta Mol. Cell. Res. 2009, 1793, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Baron, G.S.; Magalhães, A.C.; Prado, M.A.M.; Caughey, B. Mouse-adapted scrapie infection of SN56 cells: Greater efficiency with microsome-associated versus purified PrP-res. J. Virol. 2006, 80, 2106–2117. [Google Scholar] [CrossRef] [PubMed]
- Rouvinski, A.; Karniely, S.; Kounin, M.; Moussa, S.; Goldberg, M.D.; Warburg, G.; Lyakhovetsky, R.; Papy-Garcia, D.; Kutzsche, J.; Korth, C.; et al. Live imaging of prions reveals nascent PrPSc in cellsurface, raft-associated amyloid strings and webs. J. Cell. Biol. 2014, 204, 423–441. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Yu, S.; Zheng, Y.; Zheng, Y.; Yang, H.; Zhang, J. Aptamer and its applications in neurodegenerative diseases. Cell. Mol. Life Sci. 2017, 74, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [PubMed]
- Shangguan, D.; Li, Y.; Tang, Z.; Cao, Z.C.; Chen, H.W.; Mallikaratchy, P.; Sefah, K.; Yang, C.J.; Tan, W. Aptamers evolved from live cells as effective molecular probes for cancer study. Proc. Natl. Acad. Sci. USA 2006, 103, 11838–11843. [Google Scholar] [CrossRef] [PubMed]
- Gogtay, N.J.; Sridharan, K. Therapeutic Nucleic Acids: Current clinical status. Br. J. Clin. Pharmacol. 2016, 82, 659–672. [Google Scholar]
- Famulok, M. Molecular Recognition of Amino Acids by RNA-Aptamers: An l-Citrulline Binding RNA Motif and Its Evolution into an l-Arginine Binder. J. Am. Chem. Soc. 1994, 116, 1698–1706. [Google Scholar] [CrossRef]
- Lupold, S.E.; Hicke, B.J.; Lin, Y.; Coffey, D.S. Identification and characterization of nuclease-stabilized RNA molecules that bind human prostate cancer cells via the prostate-specific membrane antigen. Cancer Res. 2002, 62, 4029–4033. [Google Scholar] [PubMed]
- Tabarzad, M.; Jafari, M. Trends in the Design and Development of Specific Aptamers Against Peptides and Proteins. Protein J. 2016, 35, 81–99. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.P.; Liu, X.H.; Schumacher, T.N.; Lin, H.Y.; Ausiello, D.A.; Kim, P.S.; Bartel, D.P. Bioactive and nuclease-resistant l-DNA ligand of vasopressin. Proc. Natl. Acad. Sci. USA 1997, 94, 11285–11290. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Shangguan, D.; Wang, K.; Shi, H.; Sefah, K.; Mallikratchy, P.; Chen, H.W.; Li, Y.; Tan, W. Selection of aptamers for molecular recognition and characterization of cancer cells. Anal. Chem. 2007, 79, 4900–4907. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liang, C.; Lv, Q.; Li, D.; Xu, X.; Lui, B.; Lu, A.; Zhang, G. Molecular Selection, Modification and Development of Therapeutic Oligonucleotide Aptamers. Int. J. Mol. Sci. 2016, 17, 358. [Google Scholar] [CrossRef] [PubMed]
- Marc, D. Aptamers to explore prion protein interactions with nucleic acids. Front. Biosci. 2010, 15, 550–563. [Google Scholar] [CrossRef]
- Proske, D.; Gilch, S.; Wopfner, F.; Schatzl, H.M.; Winnacker, E.L.; Famulok, M. Prion-protein-specific aptamer reduces PrPSc formation. ChemBioChem 2002, 3, 717–725. [Google Scholar] [CrossRef]
- Rhie, A.; Kirby, L.; Sayer, N.; Wellesley, R.; Disterer, P.; Sylvester, I.; Gill, A.; Hope, J.; James, W.; Tahiri-Alaoui, A. Characterization of 2'-fluoro-RNA aptamers that bind preferentially to disease-associated conformations of prion protein and inhibit conversion. J. Biol. Chem. 2003, 278, 39697–39705. [Google Scholar] [CrossRef] [PubMed]
- Kouassi, G.K.; Wang, P.; Sreevatan, S.; Irudayaraj, J. Aptamer-mediated magnetic and gold-coated magnetic nanoparticles as detection assay for prion protein assessment. Biotechnol. Prog. 2007, 23, 1239–1244. [Google Scholar] [CrossRef] [PubMed]
- Takemura, K.; Wang, P.; Vorberg, I.; Surewicz, W.; Priola, S.A.; Kanthasamy, A.; Pottathil, R.; Chen, S.G.; Sreevatsan, S. DNA aptamers that bind to PrPC and not PrPSc show sequence and structure specificity. Exp. Biol. Med. 2006, 231, 204–214. [Google Scholar]
- King, D.J.; Safar, J.G.; Legname, G.; Prusiner, S.B. Thioaptamer Interactions with Prion Proteins: Sequence-specific and Non-specific Binding Sites. J. Mol. Biol. 2007, 369, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Kocisko, D.A.; Vaillant, A.; Lee, K.S.; Arnold, K.M.; Bertholet, N.; Race, R.E.; Olsen, E.A.; Juteau, J.M.; Caughey, B. Potent antiscrapie activities of degenerate phosphorothioate oligonucleotides. Antimicrob. Agents Chemother. 2006, 50, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Karpuj, M.V.; Giles, K.; Gelibter-Niv, S.; Scott, M.R.; Lingappa, V.R.; Szoka, F.C.; Peretz, D.; Denetclaw, W.; Prusiner, S.B. Phosphorothioate oligonucleotides reduce PrP levels and prion infectivity in cultured cells. Mol. Med. 2007, 13, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Mashima, T.; Matsugami, A.; Nishikawa, F.; Nishikawa, S.; Katahira, M. Unique quadruplex structure and interaction of an RNA aptamer against bovine prion protein. Nucleic Acids Res. 2009, 37, 6249–6258. [Google Scholar] [CrossRef] [PubMed]
- Burge, S.; Parkinson, G.N.; Hazel, P.; Todd, A.K.; Neidle, S. Quadruplex DNA: Sequence, topology and structure. Nucleic Acids Res. 2006, 34, 5402–5415. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Nishikawa, F.; Noda, K.; Yokoyama, T.; Nishikawa, S. Anti-bovine prion protein RNA aptamer containing tandem GGA repeat interacts both with recombinant bovine prion protein and its beta isoform with high affinity. Prion 2008, 2, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Mashima, T.; Nishikawa, F.; Kamatari, Y.O.; Fujiwara, H.; Saimura, M.; Nagata, T.; Kodaki, T.; Nishikawa, S.; Kuwata, K.; Katahira, M. Anti-prion activity of an RNA aptamer and its structural basis. Nucleic Acids Res. 2013, 41, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Oshima, H.; Mashima, T.; Nagata, T.; Katahira, M.; Kinoshita, M. Binding of an RNA aptamer and a partial peptide of a prion protein: Crucial importance of water entropy in molecular recognition. Nucleic Acids Res. 2014, 42, 6861–6875. [Google Scholar] [CrossRef] [PubMed]
- Yakovchuk, P.; Protozanova, E.; Frank-Kamenetskii, M.D. Base-stacking and base-pairing contributions into thermal stability of the DNA double helix. Nucleic Acids Res. 2006, 34, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Olsthoorn, R.C.L. G-quadruplexes within prion mRNA: The missing link in prion disease? Nucleic Acids Res. 2014, 42, 9327–9333. [Google Scholar] [CrossRef] [PubMed]
- Gabus, C.; Auxilien, S.; Péchoux, C.; Dormont, D.; Swietnicki, W.; Morillas, M.; Surewicz, W.; Nandi, P.; Darlix, J.L. The prion protein has DNA strand transfer properties similar to retroviral nucleocapsid protein. J. Mol. Biol. 2001, 307, 1011–1021. [Google Scholar] [CrossRef] [PubMed]
- Sayer, N.M.; Cubin, M.; Rhie, A.; Bullock, M.; Tahiri-Alaoui, A.; James, W. Structural Determinants of Conformationally Selective, Prion-binding Aptamers. J. Biol. Chem. 2004, 279, 13102–13109. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, S.; Nishikawa, F.; Noda, K.; Kumar, P.K.R.; Yokoyama, T.; Nishikawa, S. In vitro selection of RNA aptamers against cellular and abnormal isoform of mouse prion protein. Nucleic Acids Symp. Ser. 2005, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, S.; Noda, K.; Nishikawa, F.; Yokoyama, T.; Kumar, P.K.R.; Nishikawa, S. Characterization and application of a novel RNA aptamer against the mouse prion protein. J. Biochem. 2006, 139, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, D.; Hasegawa, H.; Kaneko, K.; Sode, K.; Ikebukuro, K. Screening of DNA aptamer against mouse prion protein by competitive selection. Prion 2007, 1, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Bibby, D.F.; Gill, A.C.; Kirby, L.; Farquhar, C.F.; Bruce, M.E.; Garson, J.A. Application of a novel in vitro selection technique to isolate and characterise high affinity DNA aptamers binding mammalian prion proteins. J. Virol. Methods 2008, 151, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Hatcher, K.L.; Bartz, J.C.; Chen, S.G.; Skinner, P.; Richt, J.; Liu, H.; Sreevatsan, S. Selection and characterization of DNA aptamers against PrPSc. Exp. Biol. Med. 2011, 236, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Jucker, M. The amyloid state of proteins in human diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef] [PubMed]
- Ylera, F.; Lurz, R.; Erdmann, V.A.; Fürste, J.P. Selection of RNA aptamers to the Alzheimer’s disease amyloid peptide. Biochem. Biophys. Res. Commun. 2002, 290, 1583–1588. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, F.; Murakami, K.; Summers, J.L.; Chen, C.H.B.; Bitan, G. RNA aptamers generated against oligomeric Aβ40 recognize common amyloid aptatopes with low specificity but high sensitivity. PLoS ONE 2009, 4, e7694. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, F.; Bitan, G. Selection of aptamers for amyloid β-protein, the causative agent of Alzheimer’s disease. J. Vis. Exp. 2010, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bitan, G.; Teplow, B. Preparation of Aggregate-Free, Low Molecular Weight Amyloid beta for Assembly and Toxicity Assays. Methods Mol. Biol. Protein 2005, 299, 3–9. [Google Scholar]
- Pesarrodona, M.; Unzueta, U.; Vázquez, E. Dialysis: A characterization method of aggregation tendency. In Insoluble Proteins: Methods and Protocols; Springer: Berlin, Germany, 2014; pp. 321–330. [Google Scholar]
- Hamada, H.; Arakawa, T.; Shiraki, K. Effect of additives on protein aggregation. Curr. Pharm. Biotechnol. 2009, 10, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Bunka, D.H.J.; Mantle, B.J.; Morten, I.J.; Tennent, G.A.; Radford, S.E.; Stockley, P.G. Production and characterization of RNA aptamers specific for amyloid fibril epitopes. J. Biol. Chem. 2007, 282, 34500–34509. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Sarsoza, F.; Saing, T.; Cotman, C.W.; Necula, M.; Margol, L.; Wu, J.; Breydo, L.; Thompson, J.L.; et al. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener. 2007, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Glabe, C.G. Conformation-Dependent Anti-Amyloid Oligomer Antibodies. Methods Enzymol. 2006, 413, 326–344. [Google Scholar] [PubMed]
- O’Nuallain, B.; Wetzel, R. Conformational Abs recognizing a generic amyloid fibril epitope. Proc. Natl. Acad. Sci. USA 2002, 99, 1485–1490. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.P.; Velasco, P.T.; Chang, L.; Viola, K.L.; Fernandez, S.; Lacor, P.N.; Khuon, D.; Gong, Y.; Bigio, E.H.; Shaw, P.; et al. Monoclonal antibodies that target pathological assemblies of Aβ. J. Neurochem. 2007, 100, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Lacor, P.N.; Buniel, M.C.; Chang, L.; Fernandez, S.J.; Gong, Y.; Viola, K.L.; Lambert, M.P.; Velasco, P.T.; Bigio, E.H.; Finch, C.E.; et al. Synaptic targeting by Alzheimer’s-related amyloid β oligomers. J. Neurosci. 2004, 24, 10191–10200. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.B.; Leng, L.Z.; Zhang, B.; Kwong, L.; Trojanowski, J.Q.; Abel, T.; Lee, V.M.Y. Targeting amyloid-β peptide (Aβ) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Aβ precursor protein (APP) transgenic mice. J. Biol. Chem. 2006, 281, 4292–4299. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, Y.; Macedo, B.; Silva, J.L.; Gomes, M.P.B. Pathological implications of nucleic acid interactions with proteins associated with neurodegenerative diseases. Biophys. Rev. 2014, 6, 97–110. [Google Scholar] [CrossRef]
- Calamai, M.; Taddei, N.; Stefani, M.; Ramponi, G.; Chiti, F. Relative Influence of Hydrophobicity and Net Charge in the Aggregation of Two Homologous Proteins. Biochemistry 2003, 42, 15078–15083. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F. Relative Importance of Hydrophobicity, Net Charge, and Secondary Structure Propensities in Protein Aggregation. Analysis 2004, 4, 43–59. [Google Scholar]
- Groveman, B.R.; Kraus, A.; Raymond, L.D.; Dolan, M.A.; Anson, K.J.; Dorward, D.W.; Caughey, B. Charge neutralization of the central lysine cluster in prion protein (PrP) promotes PrPSc-Like folding of recombinant PrP amyloids. J. Biol. Chem. 2015, 290, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, S.D.; Galvin, J.E.; Chiu, T.S.; Lee, V.M.Y.; Masliah, E.; Trojanowski, J.Q. RNA sequestration to pathological lesions of neurodegenerative diseases. Acta Neuropathol. 1998, 96, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, S.D.; Crino, P.B.; Hemby, S.E.; Weingarten, J.A.; Lee, V.M.Y.; Eberwine, J.H.; Trojanowski, J.Q. Predominance of neuronal mRNAs in individual Alzheimer’s disease senile plaques. Ann. Neurol. 1999, 45, 174–181. [Google Scholar] [CrossRef]
- Marcinkiewicz, M. BetaAPP and furin mRNA concentrates in immature senile plaques in the brain of Alzheimer patients. J. Neuropathol. Exp. Neurol. 2002, 61, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Manuelidis, L. Nuclease resistant circular DNAs copurify with infectivity in scrapie and CJD. J. Neurovirol. 2011, 17, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Botsios, S.; Manuelidis, L. CJD and Scrapie Require Agent-Associated Nucleic Acids for Infection. J. Cell. Biochem. 2016, 117, 1947–1958. [Google Scholar] [CrossRef] [PubMed]
- Simoneau, S.; Thomzig, A.; Ruchoux, M.-M.; Vignier, N.; Daus, M.L.; Poleggi, A.; Lebon, P.; Freire, S.; Durand, V.; Graziano, S.; et al. Synthetic Scrapie Infectivity: Interaction between Recombinant PrP and Scrapie Brain-Derived RNA. Virulence 2015, 6, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Maury, C.P.J. Self-Propagating beta-sheet polypeptide structures as prebiotic informational molecular entities: The amyloid world. Orig. Life Evol. Biosph. 2009, 39, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Si, K. Prions: What Are They Good For? Annu. Rev. Cell. Dev. Biol 2015, 31, 149–169. [Google Scholar] [CrossRef] [PubMed]
- Maury, C.P.J. The emerging concept of functional amyloid. J. Intern. Med. 2009, 265, 329–334. [Google Scholar] [PubMed]
- Takahashi, T.; Tada, K.; Mihara, H. RNA aptamers selected against amyloid β-peptide (Aβ) inhibit the aggregation of Abeta. Mol. Biosyst. 2009, 5, 986–991. [Google Scholar] [PubMed]
- Burchell, J.T.; Panegyres, P.K. Prion diseases: Immunotargets and therapy. ImmunoTargets Ther. 2016, 5, 57–68. [Google Scholar] [PubMed]
- Cordeiro, Y.; Ferreira, N.C. New approaches for the selection and evaluation of anti-prion organic compounds. Mini Rev. Med. Chem. 2015, 15, 84–92. [Google Scholar] [PubMed]
- Hanss, B.; Leal-Pinto, E.; Bruggeman, L.; Copeland, T.; Klotman, P. Identification and characterization of a cell membrane nucleic acid channel. Proc. Natl. Acad. Sci. USA 1998, 95, 1921–1926. [Google Scholar] [PubMed]
- Reyes-Reyes, E.M.; Teng, Y.; Bates, P.J. A new paradigm for aptamer therapeutic AS1411 action: Uptake by macropinocytosis and its stimulation by a nucleolin-dependent mechanism. Cancer Res. 2010, 70, 8617–8629. [Google Scholar] [PubMed]
- Cheng, C.; Chen, Y.H.; Lennox, K.A.; Behlke, M.A.; Davidson, B.L. In vivo SELEX for Identification of Brain-penetrating Aptamers. Mol. Ther. Nucleic Acids 2013, 2, E67. [Google Scholar] [PubMed]
- Kim, Y.; Wu, Q.; Hamerlik, P.; Hitomi, M.; Sloan, A.E.; Barnett, G.H.; Weil, R.J.; Leahy, P.; Hjelmeland, A.B.; Rich, J.N. Aptamer identification of brain tumor-initiating cells. Cancer Res. 2013, 73, 4923–4936. [Google Scholar] [PubMed]
- Xiao, S.J.; Hu, P.P.; Wu, X.D.; Zou, Y.L.; Chen, L.Q.; Peng, L.; Ling, J.; Zhen, S.J.; Zhan, L.; Li, Y.F.; et al. Sensitive discrimination and detection of prion disease-associated isoform with a dual-aptamer strategy by developing a sandwich structure of magnetic microparticles and quantum dots. Anal. Chem. 2010, 82, 9736–9742. [Google Scholar] [PubMed]
- Zeiler, B.; Adler, V.; Kryukov, V.; Grossman, A. Concentration and removal of prion proteins from biological solutions. Biotechnol. Appl. Biochem. 2003, 37, 173–182. [Google Scholar] [PubMed]
- Yu, P.; Zhang, X.; Xiong, E.; Zhou, J.; Li, X.; Chen, J. A label-free and cascaded dual-signaling amplified electrochemical aptasensing platform for sensitive prion assay. Biosens. Bioelectron. 2016, 85, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Battig, M.R.; Wang, Y. Aptamer-based molecular recognition for biosensor development. Anal. Bioanal. Chem. 2010, 398, 2471–2480. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Author, Year (Ref.) | Nucleic Acid Type | KD 1 (nM) | Binding Assay | PrP SELEX Target | PrPs Recognized | PrP Binding Region(s) |
|---|---|---|---|---|---|---|
| Weiss, 1997 [33] | RNA-aptamer | ND | Gel-shift of labeled aptamer | Hamster rPrP23−231 | Mouse, hamster, cow (PrP in brain homogenates) | (23–36) |
| Nandi, 1997 [29,30,31] | Plasmid DNA | 250 | Fluorescent dye displacement | NS | Human rPrP106–126 and rPrP23–231 | ND |
| Cordeiro, 2001 [21] | Short dsDNAs | 25 | Fluorescence polarization | NS | Murine rPrP23–231 | N-terminal and C-terminal domains |
| Gabus, 2001 [76] | HIV-1 LTR DNA (1000 bp) | ND | Gel-shift assay | NS | Human rPrP23–231 or 23–144 | N-terminal |
| Gabus, 2001 [37] | HIV-1 5’-leader RNA (415 nt) | ND | Gel shift assay | NS | Human rPrP23–231; Ovine rPrP25–234 | N-terminal |
| Proske, 2002 [62] | RNA-aptamer | 100 | Filter-binding assay | Human PrP90−129 | Hamster, mouse or human rPrP | (90–129) |
| Adler, 2003 [22] | Small, highly structured RNAs | 3.8 | Gel shift, filter-binding assay | NS | Human rPrP, PrP from brain homogenates of mouse, rat and hamster | N-terminal domain |
| Rhie, 2003 [63] | RNA-aptamer | 16 | Homologous competition binding assay | SAF material from infected brain homogenates | Bovine rPrP in b-oligomeric or a-helical form, PK-untreated SAF, PK-treated SAF | N-terminal and SAF conformation-specific site in (110–230) |
| Sayer, 2004 [77] | RNA-aptamer | 6.8 | Equilibrium binding | Bovine rPrP23−230 | Bovine rPrP | ND |
| Sekiya, 2005 [78] | RNA-aptamer | ND | ND | Murine rPrP23−231 and murine SAF infected material | Murine rPrP23–231 and mouse SAF | (23–108) of Murine rPrP and mouse SAF |
| Sekiya, 2006 [79] | RNA-aptamer | 5.6 | Filter-binding assay | Murine rPrP23–231 with competitive selection | Murine rPrP23–231, Bovine rPrP, Mouse PrP in brain homogenate | (23–108) and (23–88) |
| Mercey, 2006 [35] | RNA-aptamer | 15 | Surface plasmon resonance, filter-binding assay | Ovine PrP23–231 with mutations associated with disease | Ovine rPrP(ARR, VRQ, AHQ, ARQ), Murine rPrP, Bovine rPrP | (25–34) and (101–110) |
| Lima, 2006 [34] | Short dsDNAs | 90 | Fluorescence polarization and SAXS | NS | Murine rPrP23–231 | N-terminal and C-terminal |
| Takemura, 2006 [65] | DNA-aptamer | 16 | End-point titration method in microplate, gel-shift, and dot-blot assays | Human rPrP23−231 | Murine rPrP23–231, PrP from brain homogenates of sheep, calves, pigs, deer, PK-untreated PrP from ScN2a cells | (23–89) |
| Ogasawara, 2007 [80] | DNA-aptamer | 100 | Surface plasmon resonance, dot-blot, competitive selection and fluorescence measurements | Murine rPrP23−231 | Murine rPrP23–231 | ND |
| Murakami, 2008 [71] | RNA-aptamer | 31 | Surface plasmon resonance | Bovine PrP23−231 | Bovine rPrP23–231 | (125–231) |
| Bibby, 2008 [81] | DNA-aptamer | 18 | Saturation binding using PrP-coated Ni-NTA beads | Ni-NTA beads coated Murine PrP90−231 | Murine rPrP90–231, Ovine rPrP and Human rPrP90–231 | (90–230) |
| Mashima, 2009 [69] | G4 RNA-aptamer | 8.5 | Northwestern blotting assay | Bovine PrP23−231 | Bovine PrPC | (25–34) and (110–118) |
| G4 RNA-aptamer | 280 | Amyloidogenic bovine PrP-β | ND | |||
| G4 DNA-aptamer | 85 | Bovine PrPC | (25–34) and (110–118) | |||
| G4 DNA-aptamer | >280 | Amyloidogenic Bovine PrP-β | ND | |||
| Wang, 2011 [82] | DNA-aptamer biosensor immobilized | 22 | Surface plasmon resonance | PrPSc from brain tissues of scrapie-infected animals with counter-selection with PrPC | Pathological isoforms of PrP from distinct species | ND |
| Macedo, 2012 [24] | Small dsDNAs | ND | Fluorescence measurements | NS | murine rPrP23–231 and rPrP109–149 | N-terminal and C-terminal domains |
| Cavaliere, 2013 [36] | G4 DNA-aptamer | 62 | Surface plasmon resonance and Isothermal Titration Calorimetry (ITC) | Ovine rPrP-23–231 | Ovine rPrP23–24 | 23-134 |
| G4 RNA-aptamer | 75 | Ovine rPrP23–24 | ||||
| G4 DNA-aptamer | 300 | Amyloidogenic Ovine PrP-β | ND | |||
| G4 RNA-aptamer | 400 | Amyloidogenic Ovine PrP-β |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macedo, B.; Cordeiro, Y. Unraveling Prion Protein Interactions with Aptamers and Other PrP-Binding Nucleic Acids. Int. J. Mol. Sci. 2017, 18, 1023. https://doi.org/10.3390/ijms18051023
Macedo B, Cordeiro Y. Unraveling Prion Protein Interactions with Aptamers and Other PrP-Binding Nucleic Acids. International Journal of Molecular Sciences. 2017; 18(5):1023. https://doi.org/10.3390/ijms18051023
Chicago/Turabian StyleMacedo, Bruno, and Yraima Cordeiro. 2017. "Unraveling Prion Protein Interactions with Aptamers and Other PrP-Binding Nucleic Acids" International Journal of Molecular Sciences 18, no. 5: 1023. https://doi.org/10.3390/ijms18051023
APA StyleMacedo, B., & Cordeiro, Y. (2017). Unraveling Prion Protein Interactions with Aptamers and Other PrP-Binding Nucleic Acids. International Journal of Molecular Sciences, 18(5), 1023. https://doi.org/10.3390/ijms18051023