
Diverging Concepts and Novel Perspectives in Regenerative Medicine


 , ,
, ,  and
and 
Abstract
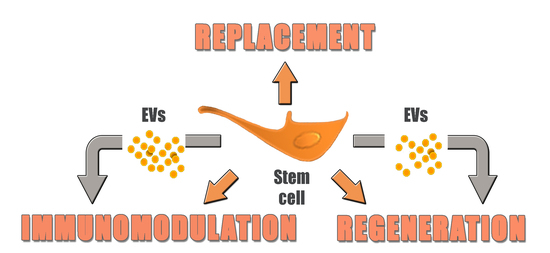
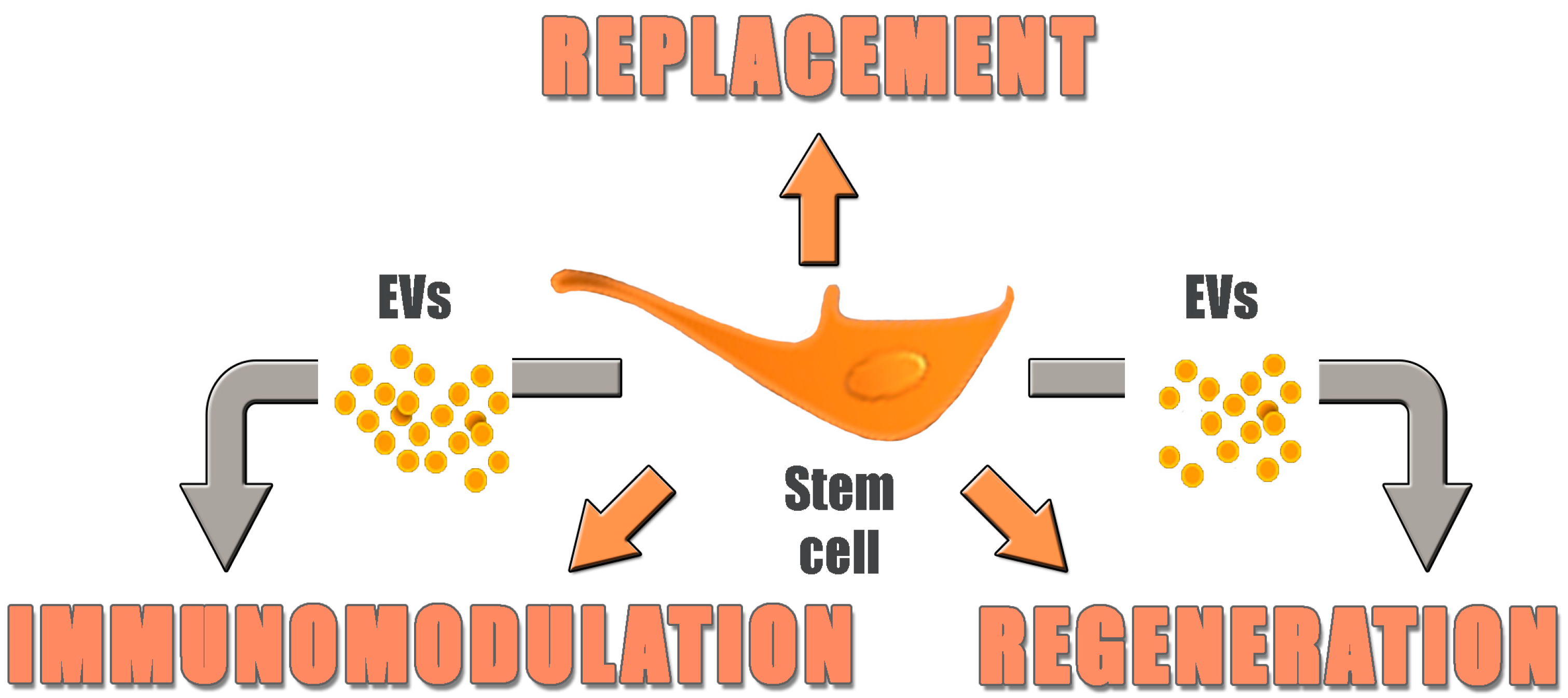
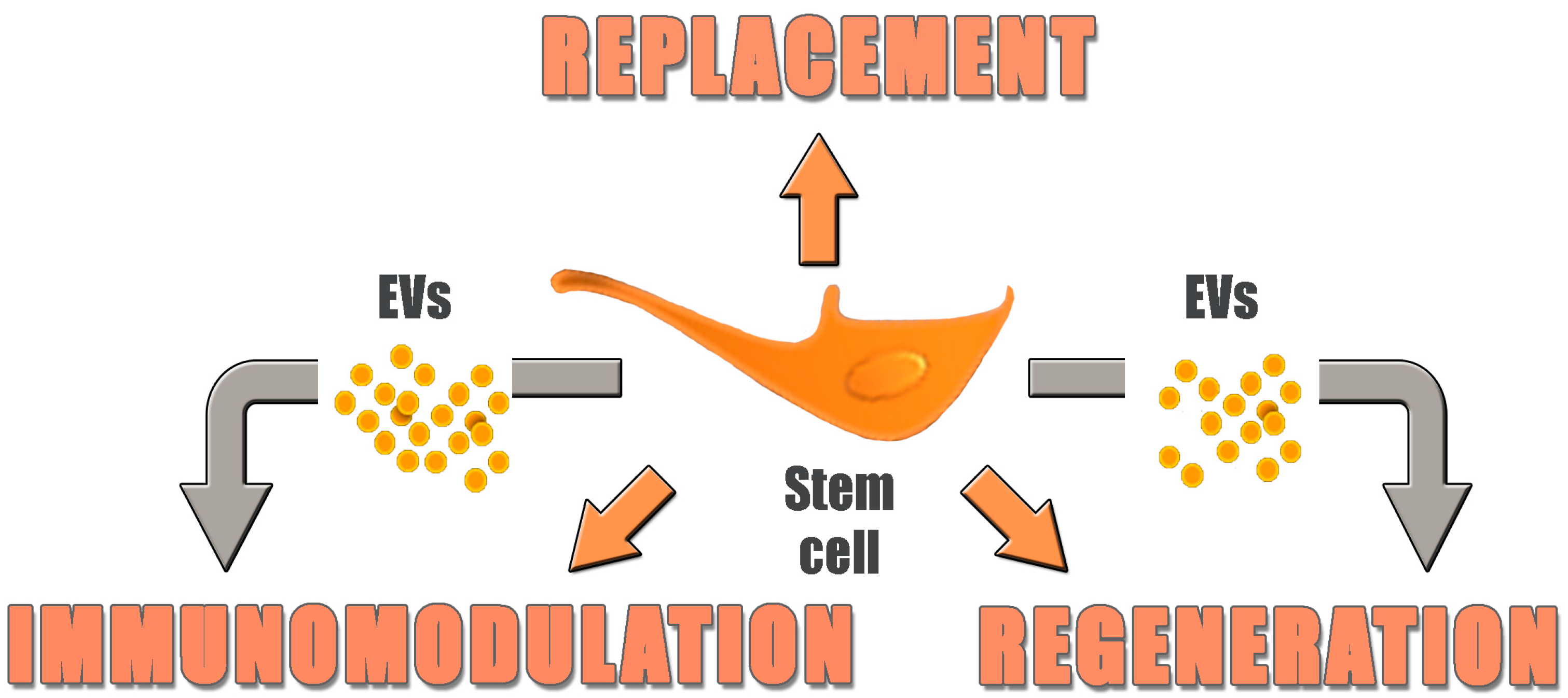
:
1. Introduction
2. Pluripotent vs. Somatic Stem Cells
3. Allogenic vs. Autologous Cells
4. Replacement vs. Regeneration
5. Novel Technologies for Bioprocessing of Advanced Cellular Therapies
5.1. Automated vs. Non-Automated Procedures for Cell Culture
5.2. Defined vs. Non-Defined Culture Media
6. From Cells to Cell Products: Introducing Extracellular Vesicles
Acknowledgments
Conflicts of Interest
References
- Maguire, G. Therapeutics from adult stem cells and the hype curve. ACS Med. Chem. Lett. 2016, 7, 441–443. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Kimbrel, E.A.; Lanza, R. Current status of pluripotent stem cells: Moving the first therapies to the clinic. Nat. Rev. Drug Discov. 2015, 14, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.D.; Regillo, C.D.; Lam, B.L.; Eliott, D.; Rosenfeld, P.J.; Gregori, N.Z.; Hubschman, J.P.; Davis, J.L.; Heilwell, G.; Spirn, M.; et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and stargardt’s macular dystrophy: Follow-up of two open-label phase 1/2 studies. Lancet 2015, 385, 509–516. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Termination of Subject Enrollment for the “Clinical Study of Autologous Induced Pluripotent Stem Cell-Derived Retinal Pigment Epithelium (RPE) Cell Sheets for Exudative Age-Related Macular Degeneration (AMD)”. Pilot Saf Study Ipsc-Based Interv Wet-Type Amd. Available online: http://www.riken-ibri.jp/AMD/img/20151125en.pdf (accessed on 6 March 2017).
- Kumar, D.; Anand, T.; Kues, W.A. Clinical potential of human-induced pluripotent stem cells: Perspectives of induced pluripotent stem cells. Cell Biol. Toxicol. 2017, 33, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Neofytou, E.; O’Brien, C.G.; Couture, L.A.; Wu, J.C. Hurdles to clinical translation of human induced pluripotent stem cells. J. Clin. Investig. 2015, 125, 2551–2557. [Google Scholar] [CrossRef] [PubMed]
- Herberts, C.A.; Kwa, M.S.; Hermsen, H.P. Risk factors in the development of stem cell therapy. J. Transl. Med. 2011, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Garitaonandia, I.; Amir, H.; Boscolo, F.S.; Wambua, G.K.; Schultheisz, H.L.; Sabatini, K.; Morey, R.; Waltz, S.; Wang, Y.C.; Tran, H.; et al. Increased risk of genetic and epigenetic instability in human embryonic stem cells associated with specific culture conditions. PLoS ONE 2015, 10, e0118307. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Kalsan, M.; Kumar, N.; Saini, A.; Chandra, R. Induced pluripotent stem cells: Applications in regenerative medicine, disease modeling, and drug discovery. Front. Cell Dev. Biol. 2015, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Shang, B.; Yang, P.; Cao, Z.; Pan, Y.; Zhou, Q. Induced pluripotent stem cell consensus genes: Implication for the risk of tumorigenesis and cancers in induced pluripotent stem cell therapy. Stem Cells Dev. 2012, 21, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Schlaeger, T.M.; Daheron, L.; Brickler, T.R.; Entwisle, S.; Chan, K.; Cianci, A.; DeVine, A.; Ettenger, A.; Fitzgerald, K.; Godfrey, M.; et al. A comparison of non-integrating reprogramming methods. Nat. Biotechnol. 2015, 33, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Luni, C.; Giulitti, S.; Serena, E.; Ferrari, L.; Zambon, A.; Gagliano, O.; Giobbe, G.G.; Michielin, F.; Knobel, S.; Bosio, A.; et al. High-efficiency cellular reprogramming with microfluidics. Nat. Methods 2016, 13, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Lee, A.S.; Volkmer, J.P.; Sahoo, D.; Nag, D.; Mosley, A.R.; Inlay, M.A.; Ardehali, R.; Chavez, S.L.; Pera, R.R.; et al. An antibody against SSEA-5 glycan on human pluripotent stem cells enables removal of teratoma-forming cells. Nat. Biotechnol. 2011, 29, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Yea, C.H.; Jeong, H.C.; Moon, S.H.; Lee, M.O.; Kim, K.J.; Choi, J.W.; Cha, H.J. In situ label-free quantification of human pluripotent stem cells with electrochemical potential. Biomaterials 2016, 75, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Gardien, K.L.; Middelkoop, E.; Ulrich, M.M. Progress towards cell-based burn wound treatments. Regen. Med. 2014, 9, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Rama, P.; Matuska, S.; Paganoni, G.; Spinelli, A.; De Luca, M.; Pellegrini, G. Limbal stem-cell therapy and long-term corneal regeneration. N. Engl. J. Med. 2010, 363, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Le Blanc, K.; Ringden, O. Immunomodulation by mesenchymal stem cells and clinical experience. J. Intern. Med. 2007, 262, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.B.; Moncivais, K.; Caplan, A.I. Mesenchymal stem cells: Environmentally responsive therapeutics for regenerative medicine. Exp. Mol. Med. 2013, 45, e54. [Google Scholar] [CrossRef] [PubMed]
- Lalu, M.M.; McIntyre, L.; Pugliese, C.; Fergusson, D.; Winston, B.W.; Marshall, J.C.; Granton, J.; Stewart, D.J. Canadian Critical Care Trials Group. Safety of cell therapy with mesenchymal stromal cells (safecell): A systematic review and meta-analysis of clinical trials. PLoS ONE 2012, 7, e47559. [Google Scholar] [CrossRef] [PubMed]
- Taddio, A.; Tommasini, A.; Valencic, E.; Biagi, E.; Decorti, G.; de Iudicibus, S.; Cuzzoni, E.; Gaipa, G.; Badolato, R.; Prandini, A.; et al. Failure of interferon-γ pre-treated mesenchymal stem cell treatment in a patient with Crohn’s disease. World J. Gastroenterol. 2015, 21, 4379–4384. [Google Scholar] [CrossRef] [PubMed]
- Trounson, A.; McDonald, C. Stem cell therapies in clinical trials: Progress and challenges. Cell Stem Cell 2015, 17, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.J.; Uberti, J.P.; Soiffer, R.J.; Klingemann, H.; Waller, E.K.; Daly, A.S.; Herrmann, R.P.; Kebriaei, P. Prochymal improves response rates in patients with steroid-refractory acute graft versus host disease (SR-GVHD) involving the liver and gut: Results of a randomized, placebo-controlled, multicenter phase III trial in GVHD. Biol. Blood Marrow Transplant. 2010, 16, S169–S170. [Google Scholar] [CrossRef]
- Kurtzberg, J.; Prockop, S.; Teira, P.; Bittencourt, H.; Lewis, V.; Chan, K.W.; Horn, B.; Yu, L.; Talano, J.A.; Nemecek, E.; et al. Allogeneic human mesenchymal stem cell therapy (remestemcel-l, prochymal) as a rescue agent for severe refractory acute graft-versus-host disease in pediatric patients. Biol. Blood Marrow Transplant. 2014, 20, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Panes, J.; Garcia-Olmo, D.; van Assche, G.; Colombel, J.F.; Reinisch, W.; Baumgart, D.C.; Dignass, A.; Nachury, M.; Ferrante, M.; Kazemi-Shirazi, L.; et al. Expanded allogeneic adipose-derived mesenchymal stem cells (Cx601) for complex perianal fistulas in Crohn’s disease: A phase 3 randomised, double-blind controlled trial. Lancet 2016, 388, 1281–1290. [Google Scholar] [CrossRef]
- Duijvestein, M.; Molendijk, I.; Roelofs, H.; Vos, A.C.; Verhaar, A.P.; Reinders, M.E.; Fibbe, W.E.; Verspaget, H.W.; van den Brink, G.R.; Wildenberg, M.E.; et al. Mesenchymal stromal cell function is not affected by drugs used in the treatment of inflammatory bowel disease. Cytotherapy 2011, 13, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Butler, J. Safety and Efficacy of Intravenous Infusion of Ischemia-Tolerant Allogeneic Mesenchymal Stem Cells in Patients with Non-Ischemic Cardiomyopathy. In Proceedings of the ESC Congress 2016, Rome, Italy, 27–31 August 2016. [Google Scholar]
- Steinberg, G.K.; Kondziolka, D.; Wechsler, L.R.; Lunsford, L.D.; Coburn, M.L.; Billigen, J.B.; Kim, A.S.; Johnson, J.N.; Bates, D.; King, B.; et al. Clinical outcomes of transplanted modified bone marrow-derived mesenchymal stem cells in stroke: A phase 1/2a study. Stroke 2016, 47, 1817–1824. [Google Scholar] [CrossRef] [PubMed]
- Bartholomew, A.; Sturgeon, C.; Siatskas, M.; Ferrer, K.; McIntosh, K.; Patil, S.; Hardy, W.; Devine, S.; Ucker, D.; Deans, R.; et al. Mesenchymal stem cells suppress lymphocyte proliferation in vitro and prolong skin graft survival in vivo. Exp. Hematol. 2002, 30, 42–48. [Google Scholar] [CrossRef]
- Comoli, P.; Ginevri, F.; Maccario, R.; Avanzini, M.A.; Marconi, M.; Groff, A.; Cometa, A.; Cioni, M.; Porretti, L.; Barberi, W.; et al. Human mesenchymal stem cells inhibit antibody production induced in vitro by allostimulation. Nephrol. Dial. Transplant. 2008, 23, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Di Nicola, M.; Carlo-Stella, C.; Magni, M.; Milanesi, M.; Longoni, P.D.; Matteucci, P.; Grisanti, S.; Gianni, A.M. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood 2002, 99, 3838–3843. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Reinhard, A.; Seckinger, A.; Katus, H.A.; Kuecherer, H.; Hansen, A. Dose-dependent effects of intravenous allogeneic mesenchymal stem cells in the infarcted porcine heart. Stem Cells Dev. 2009, 18, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Le Blanc, K.; Tammik, C.; Rosendahl, K.; Zetterberg, E.; Ringden, O. Hla expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp. Hematol. 2003, 31, 890–896. [Google Scholar] [CrossRef]
- Schu, S.; Nosov, M.; O’Flynn, L.; Shaw, G.; Treacy, O.; Barry, F.; Murphy, M.; O’Brien, T.; Ritter, T. Immunogenicity of allogeneic mesenchymal stem cells. J. Cell. Mol. Med. 2012, 16, 2094–2103. [Google Scholar] [CrossRef] [PubMed]
- Spaggiari, G.M.; Capobianco, A.; Becchetti, S.; Mingari, M.C.; Moretta, L. Mesenchymal stem cell-natural killer cell interactions: Evidence that activated NK cells are capable of killing MSCs, whereas MSCs can inhibit IL-2-induced NK-cell proliferation. Blood 2006, 107, 1484–1490. [Google Scholar] [CrossRef] [PubMed]
- Amsalem, Y.; Mardor, Y.; Feinberg, M.S.; Landa, N.; Miller, L.; Daniels, D.; Ocherashvilli, A.; Holbova, R.; Yosef, O.; Barbash, I.M.; et al. Iron-oxide labeling and outcome of transplanted mesenchymal stem cells in the infarcted myocardium. Circulation 2007, 116, I38–I45. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Lu, M.; Ma, N.; Yin, G.; Cui, C.; Zhao, S. Dynamic tracking of injected mesenchymal stem cells after myocardial infarction in rats: A serial 7T MRI study. Stem Cells Int. 2016, 2016, 4656539. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, R.P.; Sturm, M.J. Adult human mesenchymal stromal cells and the treatment of graft versus host disease. Stem Cells Cloning Adv. Appl. 2014, 7, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, F.; Tokunaga, K.; Nakatsuji, N. Human leukocyte antigen matching estimations in a hypothetical bank of human embryonic stem cell lines in the Japanese population for use in cell transplantation therapy. Stem Cells 2007, 25, 983–985. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Xie, Y.; Ouyang, Q.; Qian, X.; Xie, P.; Zhou, X.; Xiong, B.; Tan, Y.; Li, W.; Deng, L.; et al. HLA-matching potential of an established human embryonic stem cell bank in china. Cell Stem Cell 2009, 5, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Hildreth, C. ORIG3N’s Crowdsourced Cell Repository, Lifecapsule, Contains Donor Matches for 90 Percent of U.S. Residents. Available online: http://www.bioinformant.com/orig3n-lifecapsule/ (accessed on 6 May 2017).
- Sipe, J.D. Tissue engineering and reparative medicine. Ann. N. Y. Acad. Sci. 2002, 961, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Murry, C.E.; Soonpaa, M.H.; Reinecke, H.; Nakajima, H.; Nakajima, H.O.; Rubart, M.; Pasumarthi, K.B.; Virag, J.I.; Bartelmez, S.H.; Poppa, V.; et al. Haematopoietic stem cells do not transdifferentiate into cardiac myocytes in myocardial infarcts. Nature 2004, 428, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Gnecchi, M.; He, H.; Noiseux, N.; Liang, O.D.; Zhang, L.; Morello, F.; Mu, H.; Melo, L.G.; Pratt, R.E.; Ingwall, J.S.; et al. Evidence supporting paracrine hypothesis for Akt-modified mesenchymal stem cell-mediated cardiac protection and functional improvement. FASEB J. 2006, 20, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Doorn, J.; Moll, G.; le Blanc, K.; van Blitterswijk, C.; de Boer, J. Therapeutic applications of mesenchymal stromal cells: Paracrine effects and potential improvements. Tissue Eng. Part B Rev. 2012, 18, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Moerkamp, A.T.; Goumans, M.J. Cardiac regeneration: Stem cells and beyond. Curr. Med. Chem. 2012, 19, 5993–6002. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Shen, R.; Song, L.; Lu, M.; Wang, J.; Zhao, S.; Tang, Y.; Meng, X.; Li, Z.; He, Z.X. Bone marrow mesenchymal stem cells (BM-MSCs) improve heart function in swine myocardial infarction model through paracrine effects. Sci. Rep. 2016, 6, 28250. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, L.; Angioni, R.; Cali, B.; Soldani, C.; Ploia, C.; Moalli, F.; Gargesha, M.; D’Amico, G.; Elliman, S.; Tedeschi, G.; et al. Mouse mesenchymal stem cells inhibit high endothelial cell activation and lymphocyte homing to lymph nodes by releasing TIMP-1. Leukemia 2016, 30, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Chiu, S.M.; Motan, D.A.; Zhang, Z.; Chen, L.; Ji, H.L.; Tse, H.F.; Fu, Q.L.; Lian, Q. Mesenchymal stem cells and immunomodulation: Current status and future prospects. Cell Death Dis. 2016, 7, e2062. [Google Scholar] [CrossRef] [PubMed]
- Martin, I.; Simmons, P.J.; Williams, D.F. Manufacturing challenges in regenerative medicine. Sci. Transl. Med. 2014, 6, 232fs216. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Ding, S.; Rando, T.A.; Trounson, A. Translational strategies and challenges in regenerative medicine. Nat. Med. 2014, 20, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Grunseth, M.; Chen, C.; Kelly, R.; Cook, S.B.; Megaw, G.; Couture, L. Surveying the best in translation. Nat. Biotechnol. 2014, 32, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, F.; Campbell, A.; Fernandes-Platzgummer, A.; Andrade, P.Z.; Gimble, J.M.; Wen, Y.; Boucher, S.; Vemuri, M.C.; da Silva, C.L.; Cabral, J.M. A xenogeneic-free bioreactor system for the clinical-scale expansion of human mesenchymal stem/stromal cells. Biotechnol. Bioeng. 2014, 111, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Lotvall, J.; Hill, A.F.; Hochberg, F.; Buzas, E.I.; di Vizio, D.; Gardiner, C.; Gho, Y.S.; Kurochkin, I.V.; Mathivanan, S.; Quesenberry, P.; et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: A position statement from the international society for extracellular vesicles. J. Extracell. Vesicles 2014, 3, 26913. [Google Scholar] [CrossRef] [PubMed]
- Thery, C. Exosomes: Secreted vesicles and intercellular communications. F1000 Biol. Rep. 2011, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Yanez-Mo, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borras, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed]
- Kordelas, L.; Rebmann, V.; Ludwig, A.K.; Radtke, S.; Ruesing, J.; Doeppner, T.R.; Epple, M.; Horn, P.A.; Beelen, D.W.; Giebel, B. MSC-derived exosomes: A novel tool to treat therapy-refractory graft-versus-host disease. Leukemia 2014, 28, 970–973. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V. Microvesicles from mesenchymal stromal cells protect against acute kidney injury. J. Am. Soc. Nephrol. 2009, 20, 927–928. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.C.; Arslan, F.; Lee, M.M.; Sze, N.S.; Choo, A.; Chen, T.S.; Salto-Tellez, M.; Timmers, L.; Lee, C.N.; el Oakley, R.M.; et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010, 4, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Gatti, S.; Bruno, S.; Deregibus, M.C.; Sordi, A.; Cantaluppi, V.; Tetta, C.; Camussi, G. Microvesicles derived from human adult mesenchymal stem cells protect against ischaemia-reperfusion-induced acute and chronic kidney injury. Nephrol. Dial. Transplant. 2011, 26, 1474–1483. [Google Scholar] [CrossRef] [PubMed]
- Bruno, S.; Grange, C.; Collino, F.; Deregibus, M.C.; Cantaluppi, V.; Biancone, L.; Tetta, C.; Camussi, G. Microvesicles derived from mesenchymal stem cells enhance survival in a lethal model of acute kidney injury. PLoS ONE 2012, 7, e33115. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.C.; Liu, X.B.; Huang, S.; Bi, X.Y.; Wang, H.X.; Xie, L.X.; Wang, Y.Q.; Cao, X.F.; Lv, J.; Xiao, F.J.; et al. Microvesicles derived from human umbilical cord mesenchymal stem cells stimulated by hypoxia promote angiogenesis both in vitro and in vivo. Stem Cells Dev. 2012, 21, 3289–3297. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Mitsialis, S.A.; Aslam, M.; Vitali, S.H.; Vergadi, E.; Konstantinou, G.; Sdrimas, K.; Fernandez-Gonzalez, A.; Kourembanas, S. Exosomes mediate the cytoprotective action of mesenchymal stromal cells on hypoxia-induced pulmonary hypertension. Circulation 2012, 126, 2601–2611. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xu, H.; Xu, W.; Wang, B.; Wu, H.; Tao, Y.; Zhang, B.; Wang, M.; Mao, F.; Yan, Y.; et al. Exosomes released by human umbilical cord mesenchymal stem cells protect against cisplatin-induced renal oxidative stress and apoptosis in vivo and in vitro. Stem Cell Res. Ther. 2013, 4, 34. [Google Scholar] [CrossRef] [PubMed]
- Bian, S.; Zhang, L.; Duan, L.; Wang, X.; Min, Y.; Yu, H. Extracellular vesicles derived from human bone marrow mesenchymal stem cells promote angiogenesis in a rat myocardial infarction model. J. Mol. Med. 2014, 92, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Yin, Y.; Lai, R.C.; Tan, S.S.; Choo, A.B.; Lim, S.K. Mesenchymal stem cells secrete immunologically active exosomes. Stem Cells Dev. 2014, 23, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Del Fattore, A.L.R.; Fierabracci, A.; Muraca, M. Mesenchymal Stem/Stromal Cell-Derived Microparticles Show Anti-Inflammatory Activity in an Animal Model Of Ulcerative Colitis. In Proceedings of the 20th Annual ISCT meeting, Paris, France, 23–26 April 2014. [Google Scholar]
- Yang, J.; Liu, X.X.; Fan, H.; Tang, Q.; Shou, Z.X.; Zuo, D.M.; Zou, Z.; Xu, M.; Chen, Q.Y.; Peng, Y.; et al. Extracellular vesicles derived from bone marrow mesenchymal stem cells protect against experimental colitis via attenuating colon inflammation, oxidative stress and apoptosis. PLoS ONE 2015, 10, e0140551. [Google Scholar] [CrossRef] [PubMed]
- Ophelders, D.R.; Wolfs, T.G.; Jellema, R.K.; Zwanenburg, A.; Andriessen, P.; Delhaas, T.; Ludwig, A.K.; Radtke, S.; Peters, V.; Janssen, L.; et al. Mesenchymal stromal cell-derived extracellular vesicles protect the fetal brain after hypoxia-ischemia. Stem Cells Transl. Med. 2016, 5, 754–763. [Google Scholar] [CrossRef] [PubMed]
- Kilpinen, L.; Impola, U.; Sankkila, L.; Ritamo, I.; Aatonen, M.; Kilpinen, S.; Tuimala, J.; Valmu, L.; Levijoki, J.; Finckenberg, P.; et al. Extracellular membrane vesicles from umbilical cord blood-derived MSC protect against ischemic acute kidney injury, a feature that is lost after inflammatory conditioning. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Wiklander, O.P.; Nordin, J.Z.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mager, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles 2015, 4, 26316. [Google Scholar] [CrossRef] [PubMed]
- Le Pecq, J.B. Dexosomes as a therapeutic cancer vaccine: From bench to bedside. Blood Cells Mol. Dis. 2005, 35, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Morse, M.A.; Garst, J.; Osada, T.; Khan, S.; Hobeika, A.; Clay, T.M.; Valente, N.; Shreeniwas, R.; Sutton, M.A.; Delcayre, A.; et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J. Transl. Med. 2005, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Drummen, G.P.; Kuroda, M. Focus on extracellular vesicles: Development of extracellular vesicle-based therapeutic systems. Int. J. Mol. Sci. 2016, 17, 172. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Dorval, T.; Chaput, N.; Andre, F.; Caby, M.P.; Novault, S.; Flament, C.; Leboulaire, C.; Borg, C.; Amigorena, S.; et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: Results of thefirst phase i clinical trial. J. Transl. Med. 2005, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Wei, D.; Wu, Z.; Zhou, X.; Wei, X.; Huang, H.; Li, G. Phase i clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol. Ther. 2008, 16, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Viaud, S.; Ploix, S.; Lapierre, V.; Thery, C.; Commere, P.H.; Tramalloni, D.; Gorrichon, K.; Virault-Rocroy, P.; Tursz, T.; Lantz, O.; et al. Updated technology to produce highly immunogenic dendritic cell-derived exosomes of clinical grade: A critical role of interferon-γ. J. Immunother. 2011, 34, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Nickoloff, E.; Abramova, T.; Johnson, J.; Verma, S.K.; Krishnamurthy, P.; Mackie, A.R.; Vaughan, E.; Garikipati, V.N.; Benedict, C.; et al. Embryonic stem cell-derived exosomes promote endogenous repair mechanisms and enhance cardiac function following myocardial infarction. Circ. Res. 2015, 117, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.W.; Li, Q.; Niu, X.; Hu, B.; Liu, J.; Zhou, S.M.; Guo, S.C.; Lang, H.L.; Zhang, C.Q.; Wang, Y.; et al. Exosomes secreted by human-induced pluripotent stem cell-derived mesenchymal stem cells attenuate limb ischemia by promoting angiogenesis in mice. Stem Cell Res. Ther. 2015, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, L.; Li, Y.; Chen, L.; Wang, X.; Guo, W.; Zhang, X.; Qin, G.; He, S.H.; Zimmerman, A.; et al. Exosomes/microvesicles from induced pluripotent stem cells deliver cardioprotective mirnas and prevent cardiomyocyte apoptosis in the ischemic myocardium. Int. J. Cardiol. 2015, 192, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guan, J.; Niu, X.; Hu, G.; Guo, S.; Li, Q.; Xie, Z.; Zhang, C.; Wang, Y. Exosomes released from human induced pluripotent stem cells-derived MSCs facilitate cutaneous wound healing by promoting collagen synthesis and angiogenesis. J. Transl. Med. 2015, 13, 49. [Google Scholar] [CrossRef] [PubMed]
- Van der Pol, E.; Hoekstra, A.G.; Sturk, A.; Otto, C.; van Leeuwen, T.G.; Nieuwland, R. Optical and non-optical methods for detection and characterization of microparticles and exosomes. J. Thromb. Haemost. 2010, 8, 2596–2607. [Google Scholar] [CrossRef] [PubMed]
- Krampera, M.; Galipeau, J.; Shi, Y.; Tarte, K.; Sensebe, L. Immunological characterization of multipotent mesenchymal stromal cells—The international society for cellular therapy (ISCT) working proposal. Cytotherapy 2013, 15, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Wuchter, P.; Bieback, K.; Schrezenmeier, H.; Bornhauser, M.; Muller, L.P.; Bonig, H.; Wagner, W.; Meisel, R.; Pavel, P.; Tonn, T.; et al. Standardization of good manufacturing practice-compliant production of bone marrow-derived human mesenchymal stromal cells for immunotherapeutic applications. Cytotherapy 2015, 17, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Kanada, M.; Bachmann, M.H.; Hardy, J.W.; Frimannson, D.O.; Bronsart, L.; Wang, A.; Sylvester, M.D.; Schmidt, T.L.; Kaspar, R.L.; Butte, M.J.; et al. Differential fates of biomolecules delivered to target cells via extracellular vesicles. Proc. Natl. Acad. Sci. USA 2015, 112, E1433–E1442. [Google Scholar] [CrossRef] [PubMed]
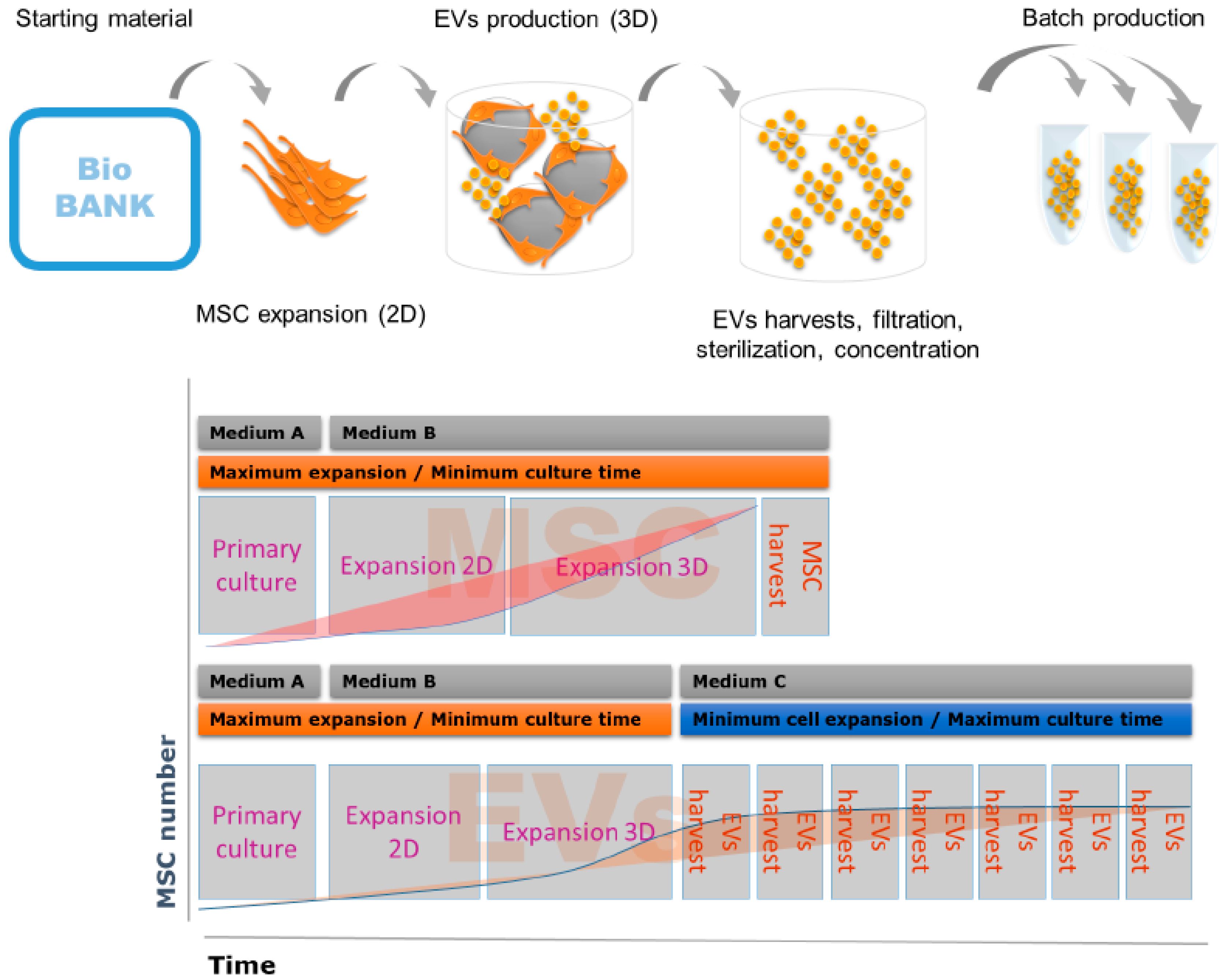

{kind=link}
{kind=link}
{kind=link}
| ATMP | Batch Volume | Primary Culture | Upstream | Downstream | Cost per Cell Batch |
|---|---|---|---|---|---|
| Tissue engineering | ++ | Manual | NA | NA | ++++ |
| Autologous MSCs | ++ | Manual | NA | NA | +++ |
| Allogeneic MSCs | +++ | Manual | Automated | Semi-automated | ++ |
| MSC-EVs | ++++ | Manual | Automated | Automated | + |
| Source of EVs | Methodology | Results | Reference |
|---|---|---|---|
| Rat BM-derived MSCs | Rat Acute Kidney Injury (AKI) induced by gentamicin (G). | Improved renal histology and function. | [ 60] |
| Human embryonic stem cell-derived MSCs | Myocardial infarction in mice. | Reduced infarct area. | [ 61] |
| Human BM-derived MSCs | AKI induced by ischemia-reperfusion injury in rats. | Improved renal histology and function. | [ 62] |
| Human BM-derived MSCs | In vitro: EVs on cisplatin-induced apoptosis of human renal tubular epithelial cells. | In vitro: EVs up-regulated in cisplatin-treated human tubular epithelial cells anti-apoptotic genes, and down-regulated genes leading to cell apoptosis. | [63] |
| In vivo: Cisplatin-induced AKI. | In vivo: Improved renal function and histology, improved survival. | ||
| Human UC-derived MSCs cultured under hypoxia | Rat hindlimb ischemia model. | Improved blood flow recovery. | [ 64] |
| Murine BM-derived MSCs | Hypoxia-induced pulmonary hypertension in rats. | Inhibition of vascular remodeling and of hypoxic pulmonary hypertension. | [ 65] |
| Human umbilical cord mesenchymal stem cells | In vitro: treatment with cisplatin alone in NRK-52E cells. | In vitro: Reversal of cisplatin induced apoptosis and oxidative. | [66] |
| In vivo: cisplatin-induced Acute Kidnay Injury (AKI) rat models. At 24 h after treatment with cisplatin, EVs injected into the kidneys. | In vivo: Improved kidney histology and biochemical parameters of kidney function. | ||
| Human BM-derived MSCs cultured under hypoxia | Acute myocardial infarction rat model. | Improved blood flow recovery, reduced infarct size and preserved cardiac systolic and diastolic performance. | [ 67] |
| Human UC-derived MSCs | Rat model of skin deep second-degree burn wound. | EV-treated wounds exhibited accelerated re-epithelialization, with increased expression of CK19, PCNA, collagen I (compared to collagen III). Activation of Wnt/β-catenin by hucMSC-; Wnt4 was found in MSC-EVs, and promoted β-catenin nuclear translocation and activity to enhance proliferation and migration of skin cells. | [ 68] |
| Human BM-derived MSCs | DSS-induced colitis in mice. | Improved body weight and clinical score, reduced colon shortening, reduced TNFα, IL-1β and COX-2 expression in colon mucosa. | [ 69] |
| Rat BM-derived MSCs | TNBS-induced colitis in rats. | Improved body weight and clinical score, reduced colon shortening, improved histology, reduced TNFα, IL-1β, COX-2 and increased IL-10 in colon mucosa. | [ 70] |
| Human BM-derived MSCs | Hypoxic-ischemic injury of the preterm brain in lamb fetuses. | Improved brain function by reducing the total number and duration of seizures, and by preserving baroreceptor reflex sensitivity, tendency to prevent hypomyelination. | [ 71] |
| Disease (No. of Patients) | EV Source | Outcome | Side Effects | Reference |
|---|---|---|---|---|
| Metastatic melanoma (15) | DCs autologous | MART1–HLA-A2 T-cell response and tumor shrinkage (1); minor response (1); mixed response (1); stabilization (2) | No major toxicity; minor inflammation, Grade 1 fever (5) | [78] |
| Non-small cell lung cancer (9) | DCs autologous | MAGE-specific T cell responses (3); NK cell lysis (2) | No major toxicity; moderate pain (1), swelling at injection site (8); mild fever (1) | [76] |
| Colorectal cancer (37) | Ascitic Fluid autologous | EVs + GM-CSF: cytotoxic T cell resp. to CAP-1 (76.9%); stabilization (1); minor response (1) | No major toxicity; moderate pain, swelling, pruritus at injection site (37); fever (1), fatigue (3) and nausea (1) | [79] |
| GVHD (1) | MSCs allogenic | GVHD symptoms improved; stabilization for several months. Patient died of pneumonia 7 months post exosome application | No major side-effects | [59] |
| Non-small cell lung cancer (41) | DCs autologous | N.D. | N.D. | [80] |
| QC Number | Tests |
|---|---|
| Starting material | |
| QC1 |
|
| QC2 |
|
| Master bank | |
| QC3 |
|
| QC4 |
|
| Working bank | |
| QC5 |
|
| QC6 |
|
| QC7 |
|
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muraca, M.; Piccoli, M.; Franzin, C.; Tolomeo, A.M.; Jurga, M.; Pozzobon, M.; Perilongo, G. Diverging Concepts and Novel Perspectives in Regenerative Medicine. Int. J. Mol. Sci. 2017, 18, 1021. https://doi.org/10.3390/ijms18051021
Muraca M, Piccoli M, Franzin C, Tolomeo AM, Jurga M, Pozzobon M, Perilongo G. Diverging Concepts and Novel Perspectives in Regenerative Medicine. International Journal of Molecular Sciences. 2017; 18(5):1021. https://doi.org/10.3390/ijms18051021
Chicago/Turabian StyleMuraca, Maurizio, Martina Piccoli, Chiara Franzin, Anna Maria Tolomeo, Marcin Jurga, Michela Pozzobon, and Giorgio Perilongo. 2017. "Diverging Concepts and Novel Perspectives in Regenerative Medicine" International Journal of Molecular Sciences 18, no. 5: 1021. https://doi.org/10.3390/ijms18051021
APA StyleMuraca, M., Piccoli, M., Franzin, C., Tolomeo, A. M., Jurga, M., Pozzobon, M., & Perilongo, G. (2017). Diverging Concepts and Novel Perspectives in Regenerative Medicine. International Journal of Molecular Sciences, 18(5), 1021. https://doi.org/10.3390/ijms18051021