
Characterization of Copy Number Variation’s Potential Role in Marek’s Disease

 , , , and
, , , and 
Abstract
:
1. Introduction
2. Results
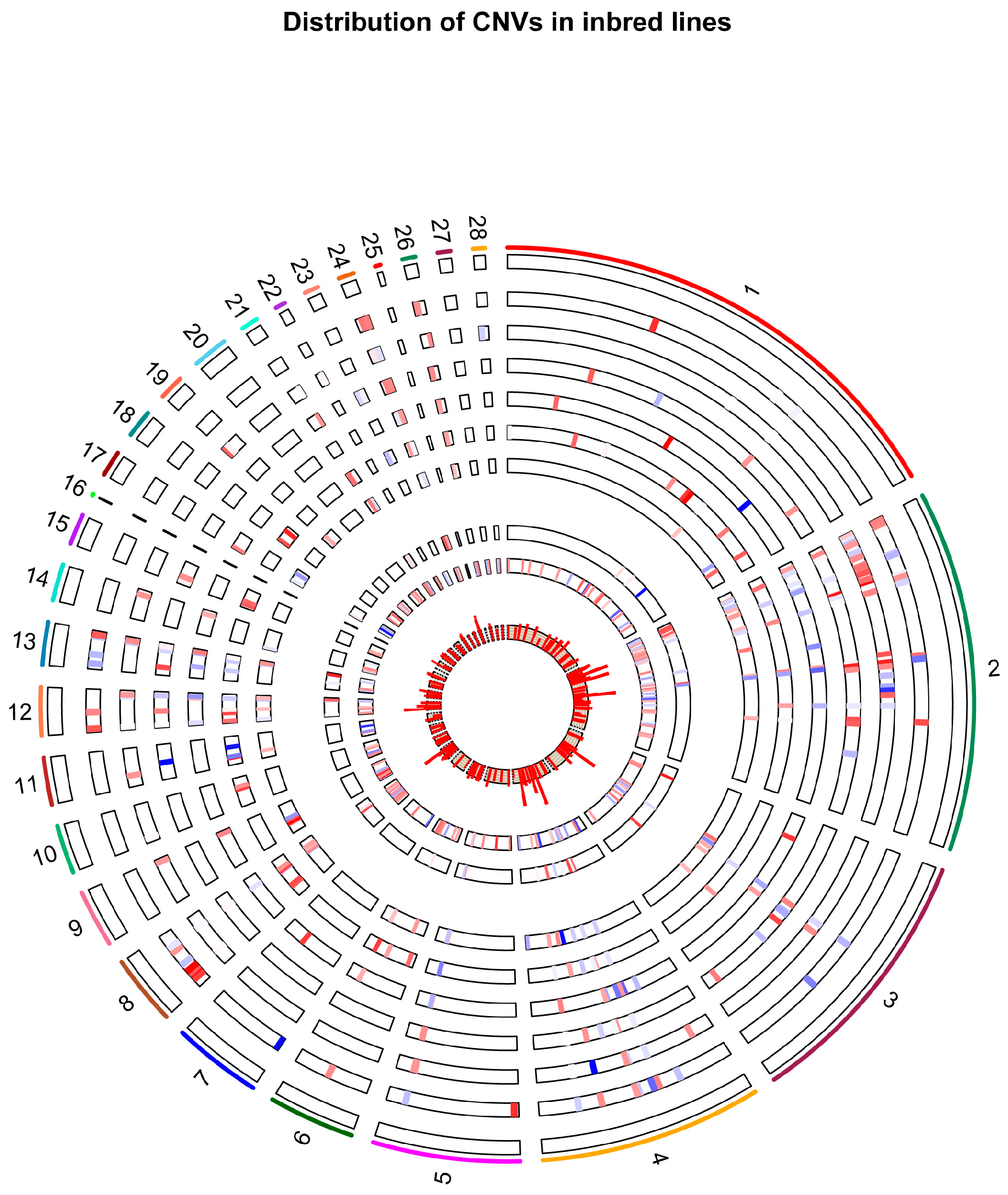
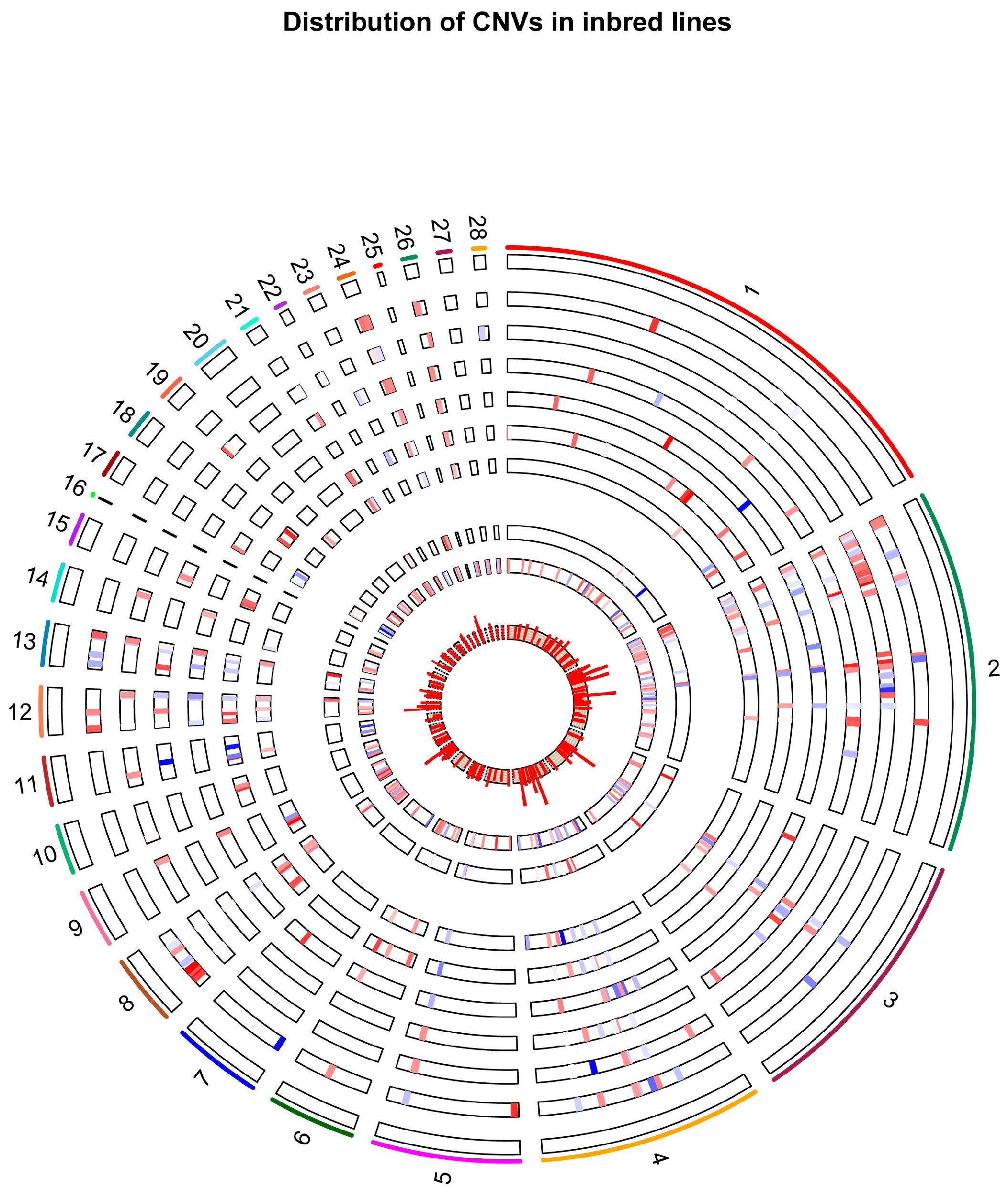
2.1. Identification of CNV Segments
2.2. Gene Annotation of Chicken CNV Segments
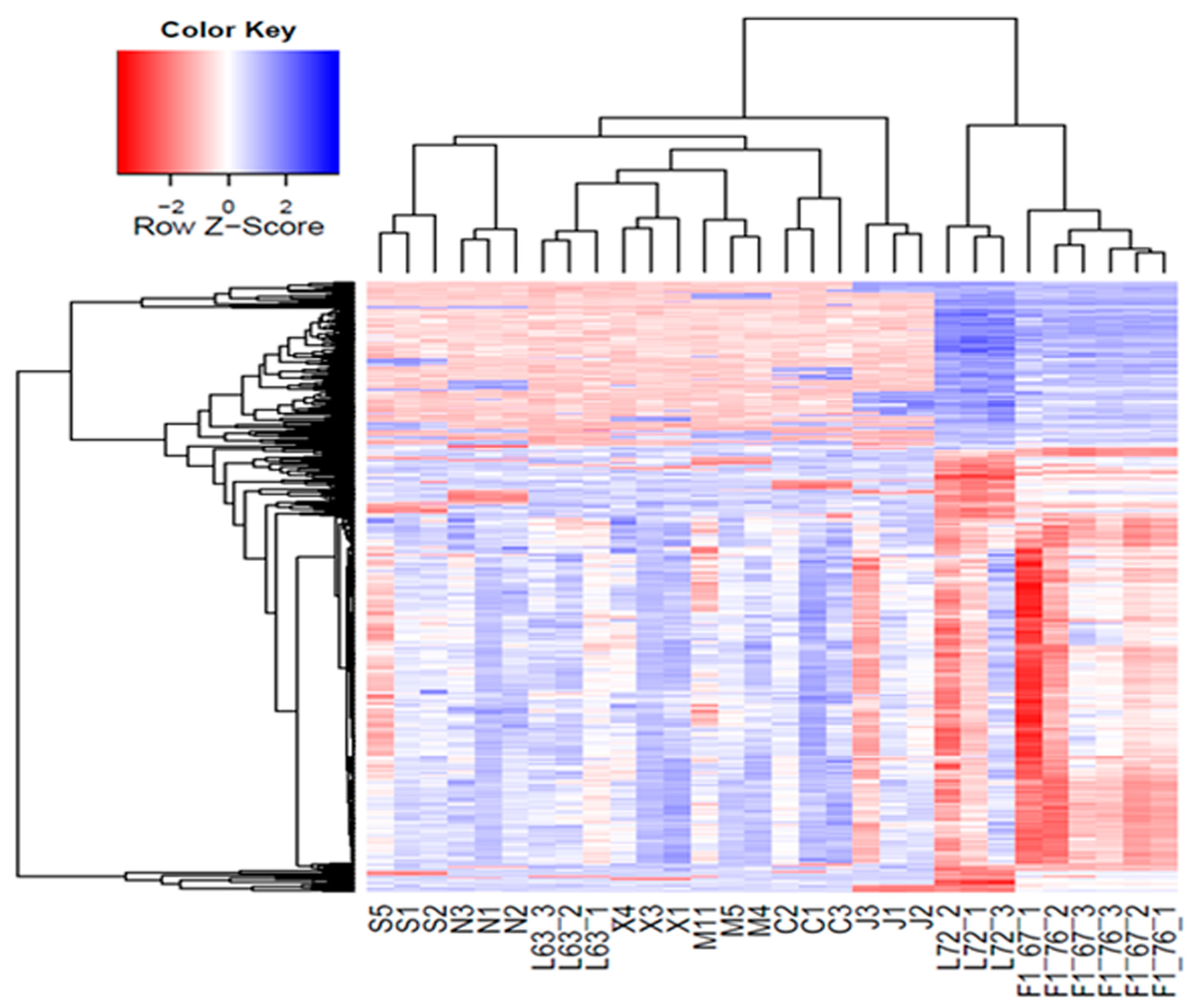
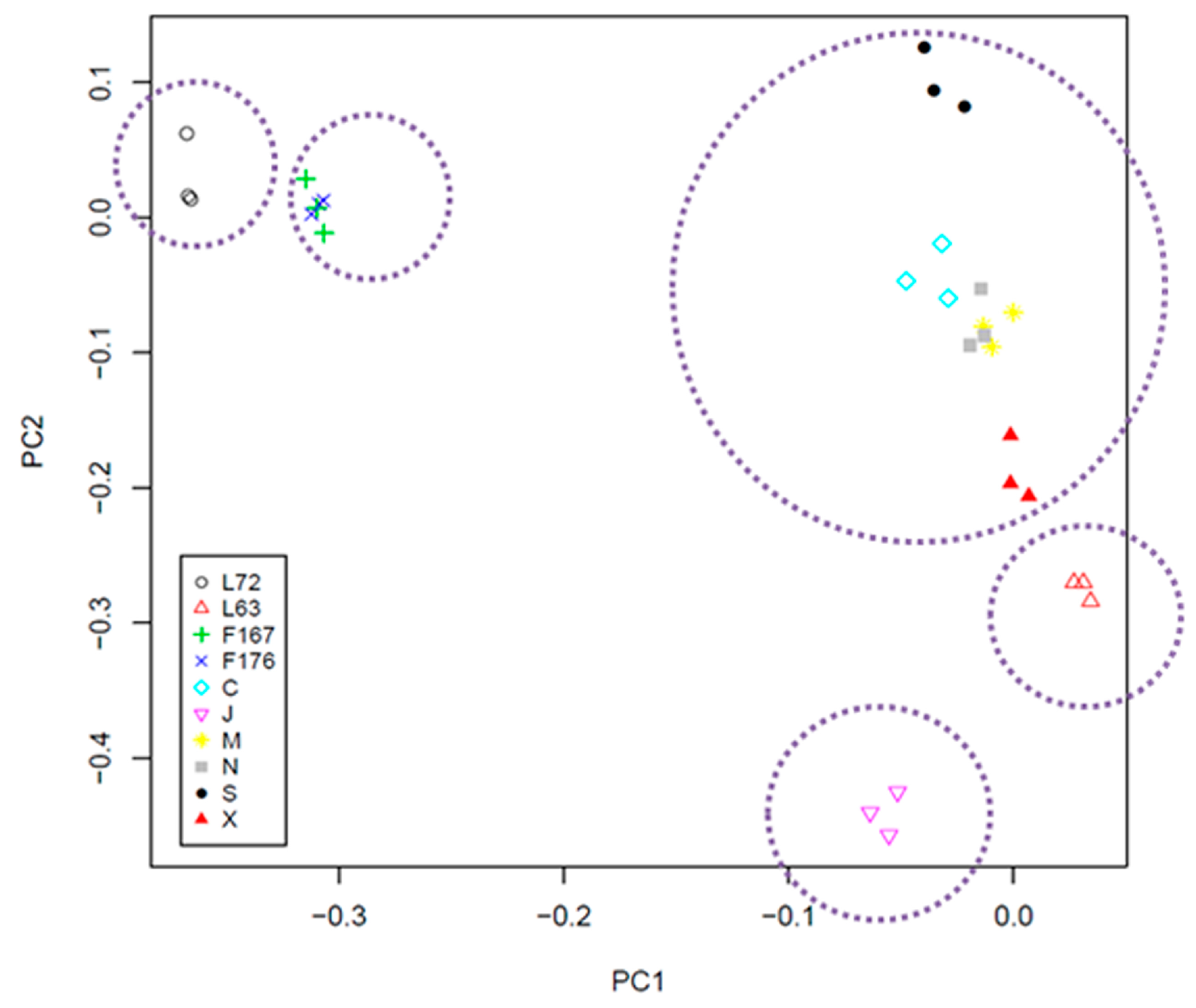
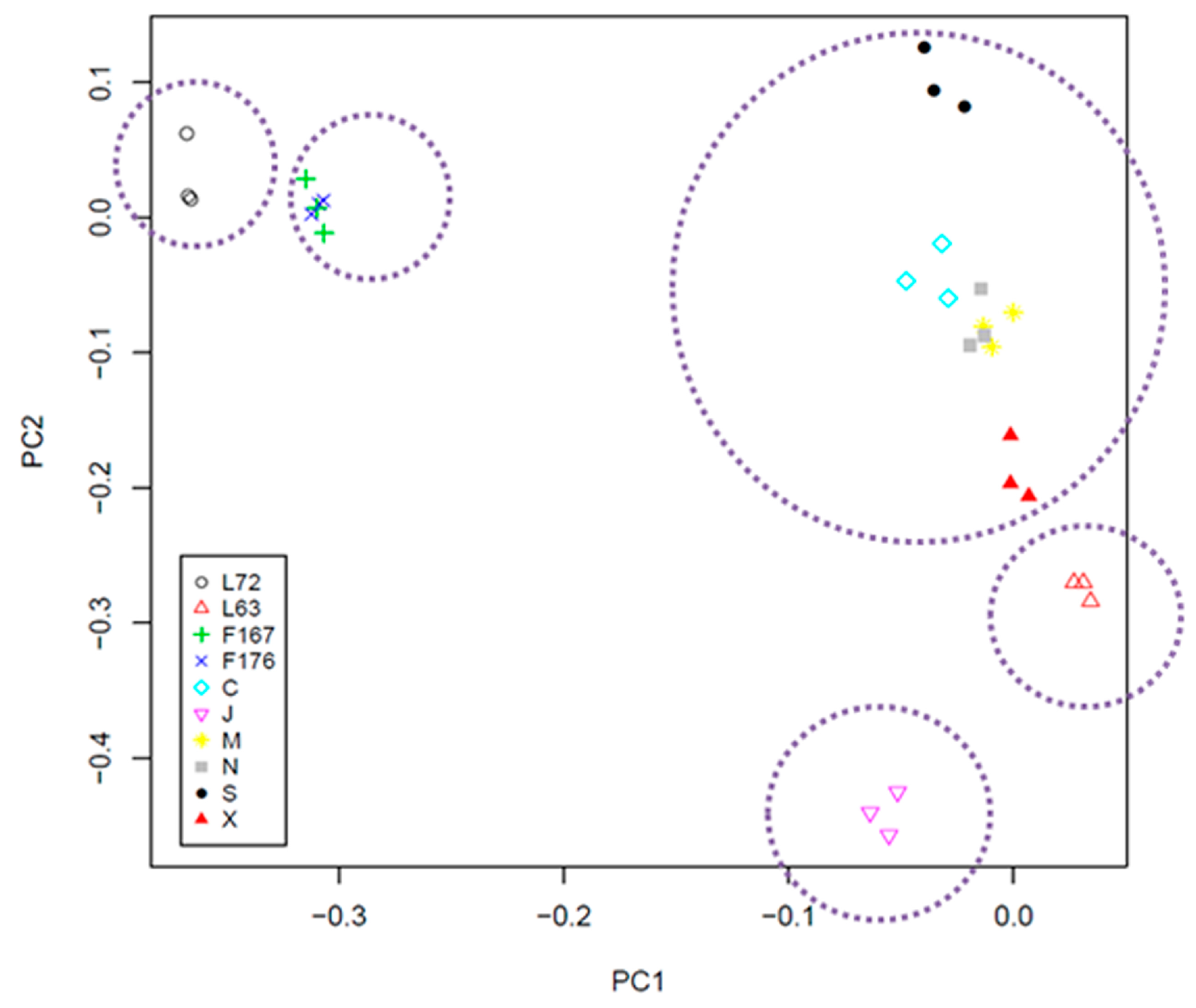
2.3. Cluster and PCA
2.4. Shared Versus Lineage-Specific Events
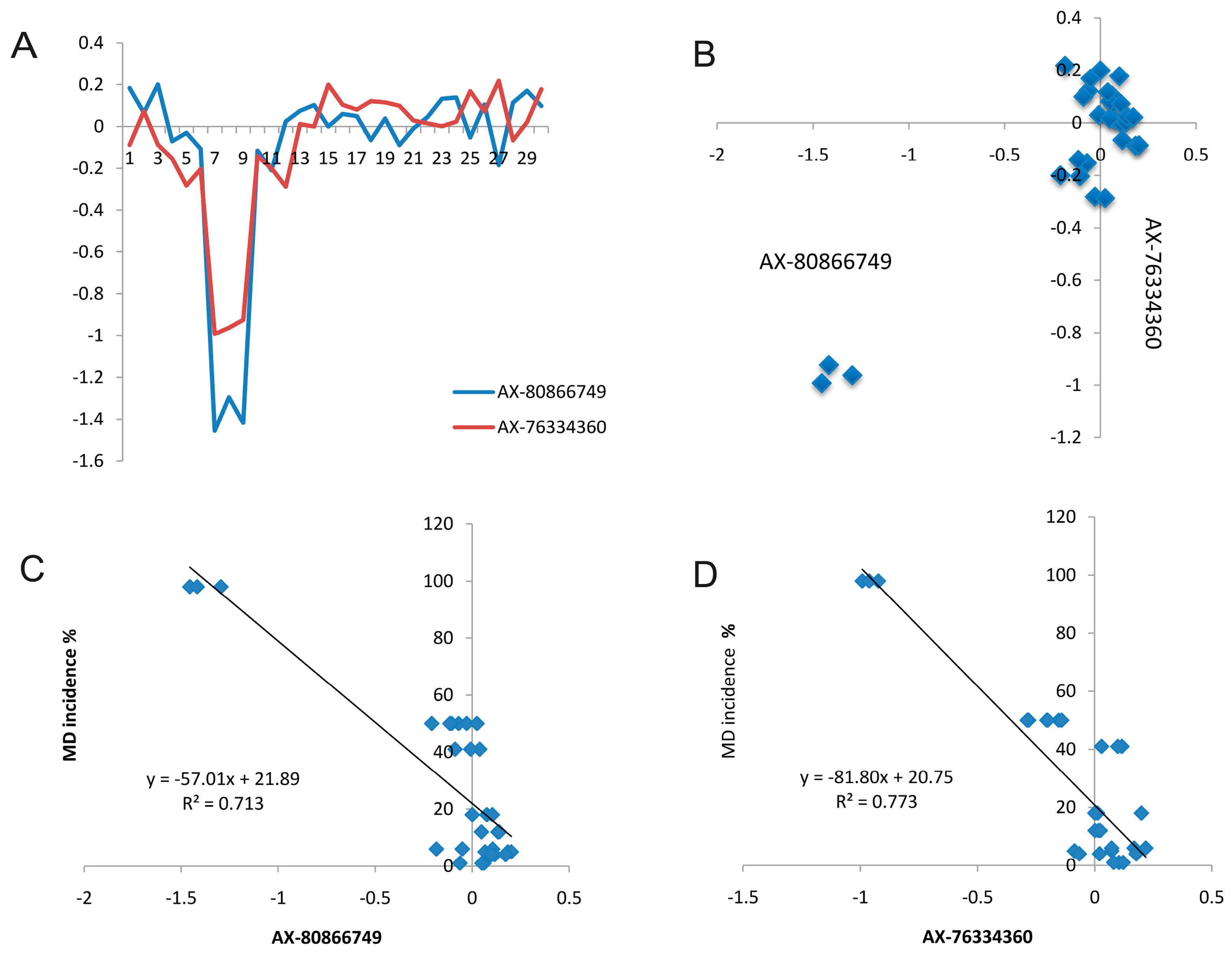
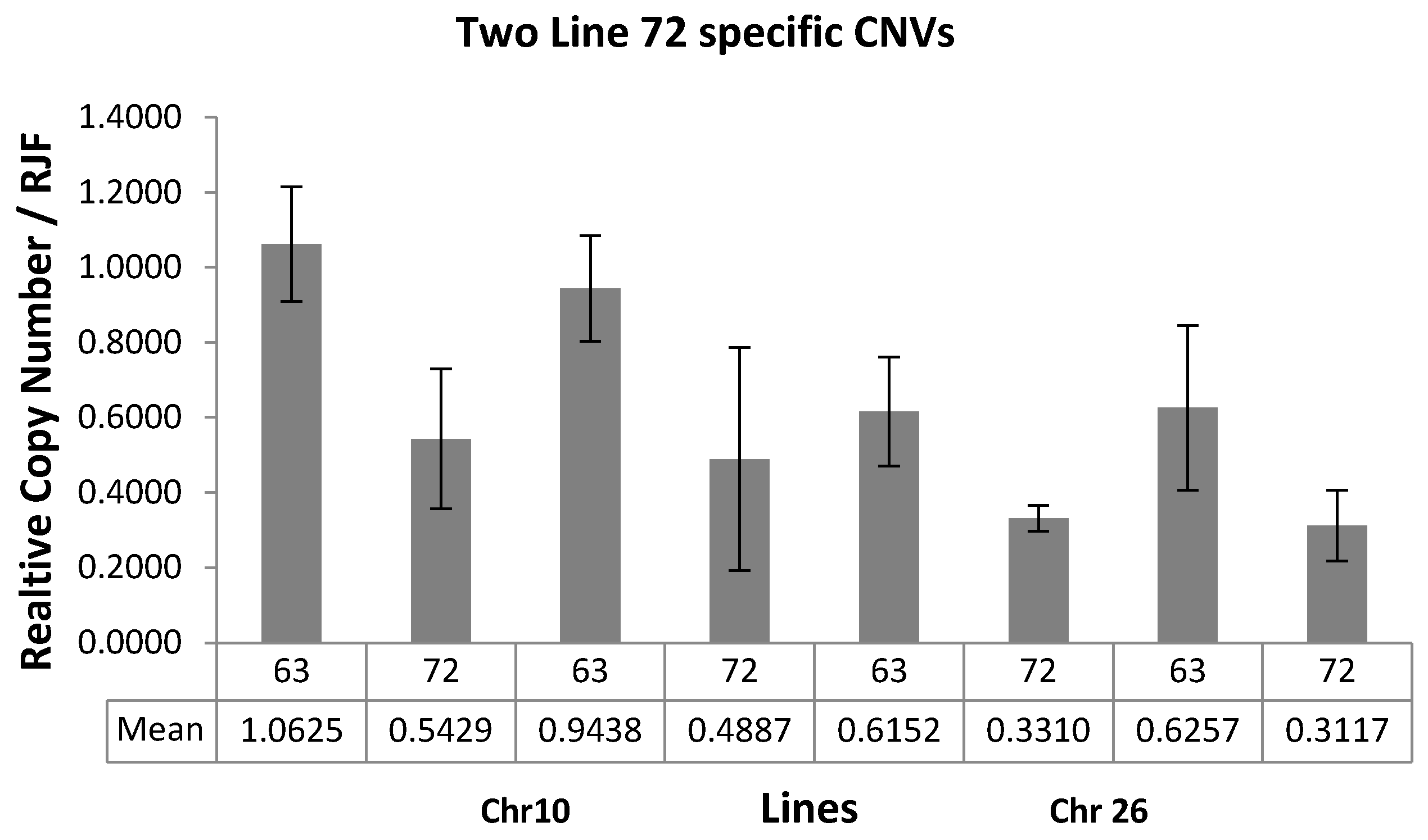
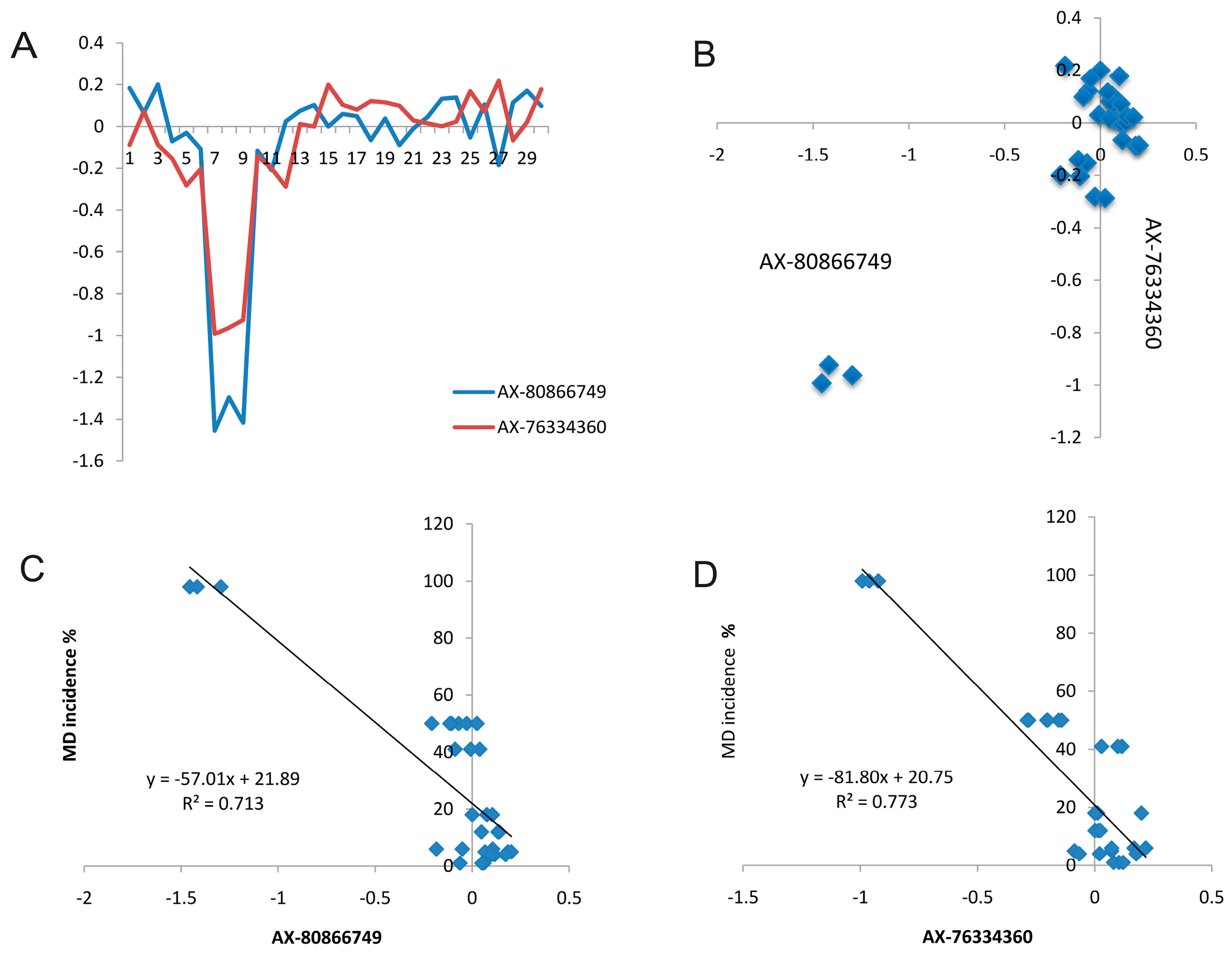
2.5. Candidate CNVs Conferring Marek’s Disease
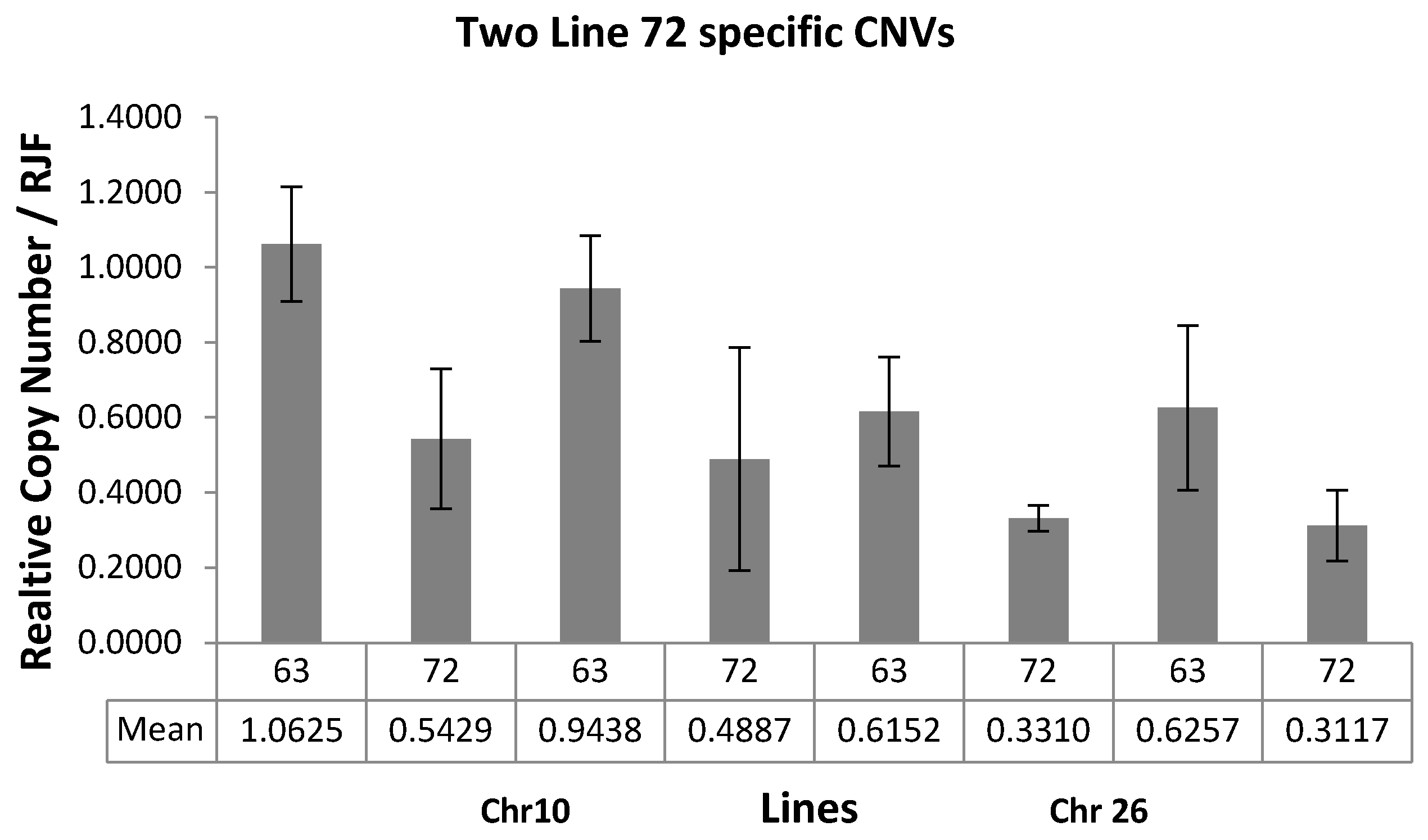
2.6. Validation of CNVs
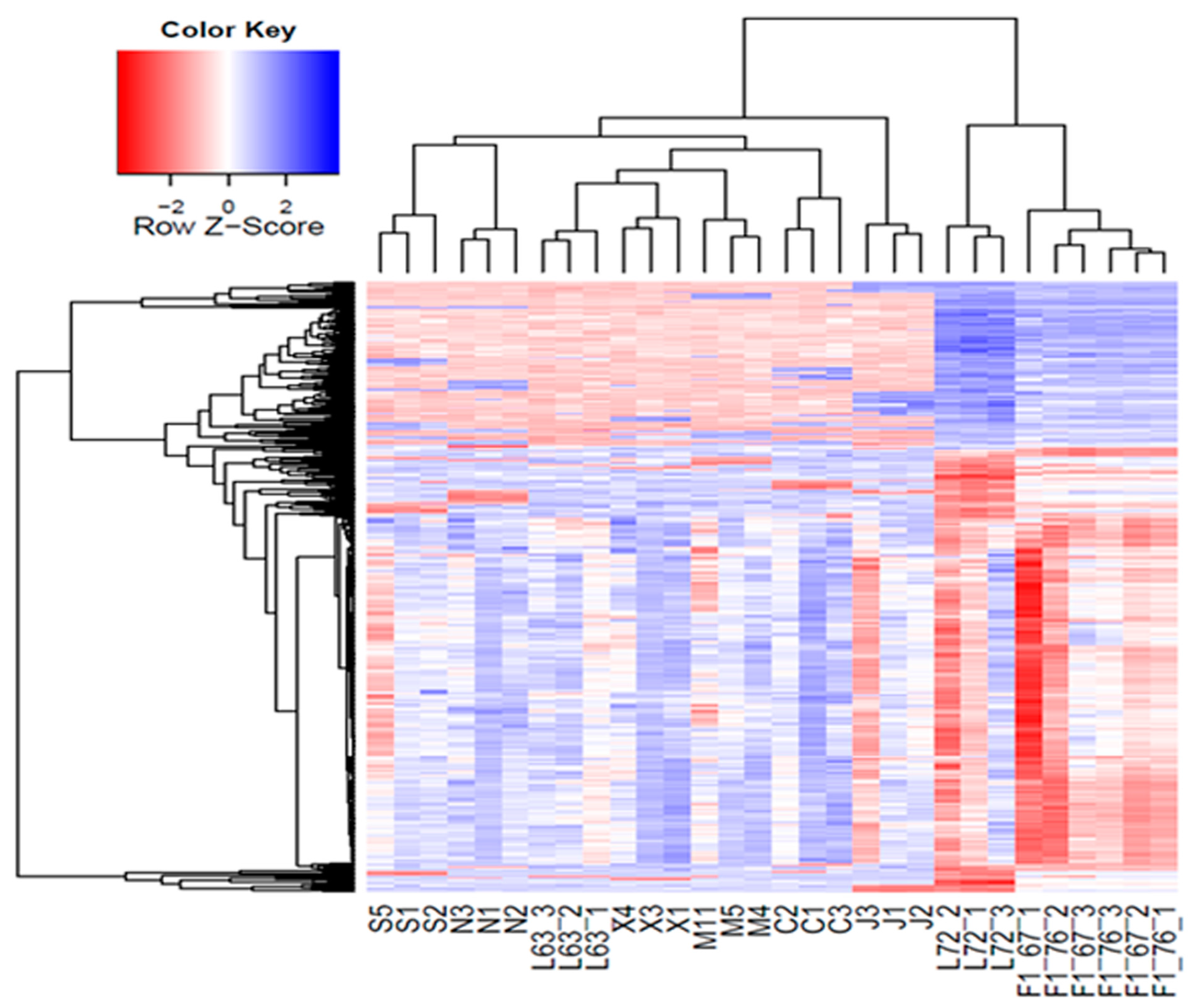
2.7. Influence of Deletions on Local Gene Expression
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. Phenotypic and MD Incidence
4.3. Copy Number Variation Analysis
4.4. Validation of CNVs by Quantitative Real-Time Polymerase Chain Reaction (qPCR)
4.5. PATHER Classification and IPA Network Analysis
4.6. Clustering and Population Genetics Analyses
4.7. RNA Extraction, RNA-Sequencing, and Differential Expression Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Davison, F.; Nair, V. Marek’s Disease: An Evolving Problem; Academic Press: Waltham, MA, USA, 2004. [Google Scholar]
- Osterrieder, N.; Kamil, J.P.; Schumacher, D.; Tischer, B.K.; Trapp, S. Marek’s disease virus: From miasma to model. Nat. Rev. Microbiol. 2006, 4, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Weischenfeldt, J.; Symmons, O.; Spitz, F.; Korbel, J.O. Phenotypic impact of genomic structural variation: Insights from and for human disease. Nat. Rev. Genet. 2013, 14, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Henrichsen, C.N.; Chaignat, E.; Reymond, A. Copy number variants, diseases and gene expression. Hum. Mol. Genet. 2009, 18, R1–R8. [Google Scholar] [CrossRef] [PubMed]
- Asmundson, V.S.; Biely, J. Inheritance and resistance to fowl paralysis (neuro-lymphomatosis gallinarum). I. Difference in susceptibility. Can. J. Res. 1932, 6, 171–176. [Google Scholar] [CrossRef]
- Gavora, J.S.; Kuhnlein, U.; Spencer, J.L. Absence of endogenous viral genes in an inbred line of leghorn chickens selected for high egg productions and Marek’s disease resistance. J. Anim. Breed. Genet. 1989, 106, 217–224. [Google Scholar] [CrossRef]
- Kuhnlein, U.; Sabour, M.; Gavora, J.S.; Fairfull, R.W.; Bernon, D.E. Influence of selection for egg production and Marek’s disease resistance on the incidence of endogenous viral genes in White Leghorns. Poult. Sci. 1989, 68, 1161–1167. [Google Scholar] [CrossRef] [PubMed]
- Kuhnlein, U.; Gavora, J.S.; Spencer, J.L.; Bernon, D.E.; Sabour, M. Incidence of endogenous viral genes in two strains of White Leghorn chickens selected for egg production and susceptibility or resistance to Marek’s disease. Theor. Appl. Genet. 1989, 77, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Aggrey, S.E.; Kuhnlein, U.; Gavora, J.S.; Zadworny, D. Association of endogenous viral genes with quantitative traits in chickens selected for high egg production and susceptibility or resistance to Marek’s disease. Br. Poult. Sci. 1998, 39, 39–41. [Google Scholar] [CrossRef] [PubMed]
- Bacon, L.D.; Hunt, H.D.; Cheng, H.H. Genetic resistance to Marek’s disease. Curr. Top. Microbiol. Immunol. 2001, 255, 121–141. [Google Scholar] [PubMed]
- Bacon, L.D.; Hunt, H.D.; Cheng, H.H. A review of the development of chicken lines to resolve genes determining resistance to diseases. Poult. Sci. 2000, 79, 1082–1093. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Dunn, J.R.; Heidari, M.; Lee, L.F.; Song, J.; Ernst, C.W.; Ding, Z.; Bacon, L.D.; Zhang, H. Genetics and vaccine efficacy: Host genetic variation affecting Marek’s disease vaccine efficacy in White Leghorn chickens. Poult. Sci. 2010, 89, 2083–2091. [Google Scholar] [CrossRef] [PubMed]
- Heifetz, E.M.; Fulton, J.E.; O’Sullivan, N.P.; Arthur, J.A.; Wang, J.; Dekkers, J.C.; Soller, M. Mapping quantitative trait loci affecting susceptibility to Marek’s disease virus in a backcross population of layer chickens. Genetics 2007, 177, 2417–2431. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.C.; Kung, H.J.; Fulton, J.E.; Morgan, R.W.; Cheng, H.H. Growth hormone interacts with the Marek’s disease virus SORF2 protein and is associated with disease resistance in chicken. Proc. Natl. Acad. Sci. USA 2001, 98, 9203–9208. [Google Scholar] [CrossRef] [PubMed]
- Heifetz, E.M.; Fulton, J.E.; O’Sullivan, N.P.; Arthur, J.A.; Cheng, H.; Wang, J.; Soller, M.; Dekkers, J. Mapping QTL affecting resistance to Marek’s disease in an F6 advanced intercross population of commercial layer chickens. BMC Genom. 2009, 10, 20. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.C.; Basaran, B.H.; Davison, T.F. Resistance to Marek’s disease herpesvirus-induced lymphoma is multiphasic and dependent on host genotype. Vet. Pathol. 2001, 38, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Darvasi, A.; Soller, M. A simple method to calculate resolving power and confidence interval of QTL map location. Behav. Genet. 1997, 27, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Rocha, J.L.; Eisen, E.J.; Van Vleck, L.D.; Pomp, D. A large-sample QTL study in mice: I. Growth. Mamm. Genome 2004, 15, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.C.; Cheng, H.H.; Tirunagaru, V.; Sofer, L.; Burnside, J. A strategy to identify positional candidate genes conferring Marek’s disease resistance by integrating DNA microarrays and genetic mapping. Anim. Genet. 2001, 32, 351–359. [Google Scholar] [CrossRef] [PubMed]
- McCarroll, S.A.; Altshuler, D.M. Copy-number variation and association studies of human disease. Nat. Genet. 2007, 39, S37–S42. [Google Scholar] [CrossRef] [PubMed]
- McCarroll, S.A. Extending genome-wide association studies to copy-number variation. Hum. Mole Genet. 2008, 17, R135–R142. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Conrad, D.F.; Pinto, D.; Redon, R.; Feuk, L.; Gokcumen, O.; Zhang, Y.; Aerts, J.; Andrews, T.D.; Barnes, C.; Campbell, P.; et al. Origins and functional impact of copy number variation in the human genome. Nature 2010, 464, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Karyadi, D.M.; Karlins, E.; Decker, B.; von Holdt, B.M.; Carpintero-Ramirez, G.; Parker, H.G.; Wayne, R.K.; Ostrander, E.A. A copy number variant at the KITLG locus likely confers risk for canine squamous cell carcinoma of the digit. PLoS Genet. 2013, 9, e1003409. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.; Meadows, J.R. S.; Truve, K.; Pielberg, G.R.; Puppo, F.; Mauceli, E.; Quilez, J.; Tonomura, N.; Zanna, G.; Docampo, M.J.; et al. A Novel Unstable Duplication Upstream of HAS2 Predisposes to a Breed-Defining Skin Phenotype and a Periodic Fever Syndrome in Chinese Shar-Pei Dogs. PLoS Genet. 2011, 7, e1001332. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Shi, N.; Lu, H.; Zhang, J.; Ma, Y.; Qiao, Y.; Mao, Y.; Jia, K.; Han, L.; Liu, F.; et al. ABCC4 copy number variation is associated with susceptibility to esophageal squamous cell carcinoma. Carcinogenesis 2014, 35, 1941–1950. [Google Scholar] [CrossRef] [PubMed]
- Clop, A.; Vidal, O.; Amills, M. Copy number variation in the genomes of domestic animals. Anim. Genet. 2012, 43, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.E.; Bickhart, D.M. Copy number variation in the cattle genome. Funct. Integr. Genom. 2012, 12, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Bickhart, D.M.; Liu, G.E. The challenges and importance of structural variation detection in livestock. Front. Genet. 2014, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Dorshorst, B.; Molin, A.M.; Rubin, C.J.; Johansson, A.M.; Stromstedt, L.; Pham, M.H.; Chen, C.F.; Hallbook, F.; Ashwell, C.; Andersson, L. A Complex Genomic Rearrangement Involving the Endothelin 3 Locus Causes Dermal Hyperpigmentation in the Chicken. PLoS Genet. 2011, 7, e1002412. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.; Boije, H.; Meadows, J.R.; Bed’hom, B.; Gourichon, D.; Vieaud, A.; Tixier-Boichard, M.; Rubin, C.J.; Imsland, F.; Hallbook, F.; et al. Copy number variation in intron 1 of SOX5 causes the Pea-comb phenotype in chickens. PLoS Genet. 2009, 5, e1000512. [Google Scholar] [CrossRef] [PubMed]
- Gunnarsson, U.; Kerje, S.; Bed’hom, B.; Sahlqvist, A.S.; Ekwall, O.; Tixier-Boichard, M.; Kampe, O.; Andersson, L. The Dark brown plumage color in chickens is caused by an 8.3-kb deletion upstream of SOX10. Pigment. Cell. Melanoma Res. 2011, 24, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Elferink, M.G.; Vallee, A.A.A.; Jungerius, A.P.; Crooijmans, R.P.M.A.; Groenen, M.A.M. Partial duplication of the PRLR and SPEF2 genes at the late feathering locus in chicken. BMC Genom. 2008, 9, 391. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Luo, J.; Mitra, A.; Chang, S.; Tian, F.; Zhang, H.; Yuan, P.; Zhou, H.; Song, J. Temporal transcriptome changes induced by MDV in Marek’s disease-resistant and -susceptible inbred chickens. BMC Genom. 2011, 12, 501. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.H.; Kaiser, P.; Lamont, S.J. Integrated Genomic Approaches to Enhance Genetic Resistance in Chickens. Annu. Rev. Anim. Biosci. 2013, 1, 239–260. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Yu, Y.; Mitra, A.; Chang, S.; Zhang, H.; Liu, G.; Yang, N.; Song, J. Genome-wide copy number variant analysis in inbred chickens lines with different susceptibility to Marek’s disease. G3 (Bethesda) 2013, 3, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Yang, N.; Cheng, H.H.; Song, J.; Qu, L. Genome-wide identification of copy number variations between two chicken lines that differ in genetic resistance to Marek’s disease. BMC Genom. 2015, 16, 843. [Google Scholar] [CrossRef] [PubMed]
- Perng, M.D.; Quinlan, R.A. Seeing is believing! The optical properties of the eye lens are dependent upon a functional intermediate filament cytoskeleton. Exp. Cell. Res. 2005, 305, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.D.; Perumalsamy, V.; Hejtmancik, J.F. Autosomal recessive juvenile onset cataract associated with mutation in BFSP1. Hum. Genet. 2007, 121, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, R.; Gloriam, D.E. I.; Hoglund, P.J.; Lagerstrom, M.C.; Schioth, H.B. There exist at least 30 human G-protein-coupled receptors with long Ser/Thr-rich N-termini. Biochem. Biophys. Res. Commun. 2003, 301, 725–734. [Google Scholar] [CrossRef]
- Monk, K.R.; Oshima, K.; Jors, S.; Heller, S.; Talbot, W.S. Gpr126 is essential for peripheral nerve development and myelination in mammals. Development 2011, 138, 2673–2680. [Google Scholar] [CrossRef] [PubMed]
- Korosec, B.; Glavac, D.; Volavsek, M.; Ravnik-Glavac, M. ATP2A3 gene is involved in cancer susceptibility. Cancer Genet. Cytogenet. 2009, 188, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Uzawa, K.; Mochida, Y.; Shiiba, M.; Bukawa, H.; Yokoe, H.; Tanzawa, H. Sarcoendoplasmic reticulum Ca2+ ATPase type 2 downregulated in human oral squamous cell carcinoma. Int. J. Cancer 2004, 110, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Korosec, B.; Glavac, D.; Rott, T.; Ravnik-Glavac, M. Alterations in the ATP2A2 gene in correlation with colon and lung cancer. Cancer Genet. Cytogenet. 2006, 171, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Li, D.Q.; Pakala, S.B.; Nair, S.S.; Eswaran, J.; Kumar, R. Metastasis-Associated Protein 1/Nucleosome Remodeling and Histone Deacetylase Complex in Cancer. Cancer Res. 2012, 72, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Pakala, S.B.; Rayala, S.K.; Wang, R.A.; Ohshiro, K.; Mudvari, P.; Reddy, S.D. N.; Zheng, Y.; Pires, R.; Casimiro, S.; Pillai, M.R.; et al. MTA1 Promotes STAT3 Transcription and Pulmonary Metastasis in Breast Cancer. Cancer Res. 2013, 73, 3761–3770. [Google Scholar] [CrossRef] [PubMed]
- Poliak, S.; Peles, E. The local differentiation of myelinated axons at nodes of Ranvier. Nat. Rev. Neurosci. 2003, 4, 968–980. [Google Scholar] [CrossRef] [PubMed]
- Stogmann, E.; Reinthaler, E.; ElTawil, S.; El Etribi, M.A.; Hemeda, M.; El Nahhas, N.; Gaber, A.M.; Fouad, A.; Edris, S.; Benet-Pages, A.; et al. Autosomal recessive cortical myoclonic tremor and epilepsy: Association with a mutation in the potassium channel associated gene CNTN2. Brain 2013, 136, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Buitenhuis, A.J.; Rodenburg, T.B.; van Hierden, Y.M.; Siwek, M.; Cornelissen, S.J.; Nieuwland, M.G.; Crooijmans, R.P.; Groenen, M.A.; Koene, P.; Korte, S.M.; et al. Mapping quantitative trait loci affecting feather pecking behavior and stress response in laying hens. Poult. Sci. 2003, 82, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Park, H.B.; Jacobsson, L.; Wahlberg, P.; Siegel, P.B.; Andersson, L. QTL analysis of body composition and metabolic traits in an intercross between chicken lines divergently selected for growth. Physiol. Genom. 2006, 25, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.F.; Nahashon, S.; Feaster, T.K.; Bohannon-Stewart, A.; Adefope, N. An initial map of chromosomal segmental copy number variations in the chicken. BMC Genom. 2010, 11, 351. [Google Scholar] [CrossRef] [PubMed]
- Volker, M.; Backstrom, N.; Skinner, B.M.; Langley, E.J.; Bunzey, S.K.; Ellegren, H.; Griffin, D.K. Copy number variation, chromosome rearrangement, and their association with recombination during avian evolution. Genome Res. 2010, 20, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gu, X.; Feng, C.; Song, C.; Hu, X.; Li, N. A genome-wide survey of copy number variation regions in various chicken breeds by array comparative genomic hybridization method. Anim. Genet. 2012, 43, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Chen, S.; Zhou, H.; Li, D.; Liu, W.; Yang, N. Copy number variations identified in the chicken using a 60K SNP BeadChip. Anim. Genet. 2013, 44, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Wang, Y.Q.; Gu, X.R.; Feng, C.G.; Fang, S.Y.; Hu, X.X.; Li, N. Copy number variants in locally raised Chinese chicken genomes determined using array comparative genomic hybridization. BMC Genom. 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Byers, S. Copy Number Variation in Chickens: A Review and Future Prospects. Microarrays 2014, 3, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Crooijmans, R.P.; Fife, M.S.; Fitzgerald, T.W.; Strickland, S.; Cheng, H.H.; Kaiser, P.; Redon, R.; Groenen, M.A. Large scale variation in DNA copy number in chicken breeds. BMC Genom. 2013, 14, 398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Du, Z.Q.; Dong, J.Q.; Wang, H.X.; Shi, H.Y.; Wang, N.; Wang, S.Z.; Li, H. Detection of genome-wide copy number variations in two chicken lines divergently selected for abdominal fat content. BMC Genom. 2014, 15, 517. [Google Scholar] [CrossRef] [PubMed]
- Strillacci, M.G.; Cozzi, M.C.; Gorla, E.; Mosca, F.; Schiavini, F.; Roman-Ponce, S.I.; Ruiz Lopez, F.J.; Schiavone, A.; Marzoni, M.; Cerolini, S.; et al. Genomic and genetic variability of six chicken populations using single nucleotide polymorphism and copy number variants as markers. Animal 2017, 11, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Mills, R.E.; Walter, K.; Stewart, C.; Handsaker, R.E.; Chen, K.; Alkan, C.; Abyzov, A.; Yoon, S.C.; Ye, K.; Cheetham, R.K.; et al. Mapping copy number variation by population-scale genome sequencing. Nature 2011, 470, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.H.; Dobrinski, K.P.; Lee, A.S.; Gokcumen, O.; Mills, R.E.; Shi, X.; Chong, W.W.; Chen, J.Y.; Yoo, P.; David, S.; et al. Extensive genetic diversity and substructuring among zebrafish strains revealed through copy number variant analysis. Proc. Natl. Acad. Sci. USA 2012, 109, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Henrichsen, C.N.; Vinckenbosch, N.; Zollner, S.; Chaignat, E.; Pradervand, S.; Schutz, F.; Ruedi, M.; Kaessmann, H.; Reymond, A. Segmental copy number variation shapes tissue transcriptomes. Nat. Genet. 2009, 41, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Massouras, A.; Waszak, S.M.; Albarca-Aguilera, M.; Hens, K.; Holcombe, W.; Ayroles, J.F.; Dermitzakis, E.T.; Stone, E.A.; Jensen, J.D.; Mackay, T.F.; et al. Genomic variation and its impact on gene expression in Drosophila melanogaster. PLoS Genet. 2012, 8, e1003055. [Google Scholar] [CrossRef] [PubMed]
- Stranger, B.E.; Forrest, M.S.; Dunning, M.; Ingle, C.E.; Beazley, C.; Thorne, N.; Redon, R.; Bird, C.P.; de Grassi, A.; Lee, C.; et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 2007, 315, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Lemos, B.; Dopman, E.B.; Hartl, D.L. Copy-number variation: The balance between gene dosage and expression in Drosophila melanogaster. Genome Biol. Evol. 2011, 3, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Luo, J.; Zhang, H.; Cui, K.; Zhao, K.; Song, J. Marek’s disease virus infection induces widespread differential chromatin marks in inbred chicken lines. BMC Genom. 2012, 13, 557. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Mitra, A.; Tian, F.; Chang, S.; Zhang, H.; Cui, K.; Yu, Y.; Zhao, K.; Song, J. Histone methylation analysis and pathway predictions in chickens after MDV infection. PLoS ONE 2012, 7, e41849. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.F.; Powell, P.C.; Rennie, M.; Ross, L.J.; Payne, L.N. Nature of genetic resistance to Marek’s disease in chickens. J. Natl. Cancer Inst. 1981, 66, 789–796. [Google Scholar] [PubMed]
- Vallejo, R.L.; Pharr, G.T.; Liu, H.C.; Cheng, H.H.; Witter, R.L.; Bacon, L.D. Non-association between Rfp-Y major histocompatibility complex-like genes and susceptibility to Marek’s disease virus-induced tumours in 6(3) x 7(2) F2 intercross chickens. Anim. Genet. 1997, 28, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Vallejo, R.L.; Bacon, L.D.; Liu, H.C.; Witter, R.L.; Groenen, M.A.; Hillel, J.; Cheng, H.H. Genetic mapping of quantitative trait loci affecting susceptibility to Marek’s disease virus induced tumors in F2 intercross chickens. Genetics 1998, 148, 349–360. [Google Scholar] [PubMed]
- Yonash, N.; Bacon, L.D.; Witter, R.L.; Cheng, H.H. High resolution mapping and identification of new quantitative trait loci (QTL) affecting susceptibility to Marek’s disease. Anim. Genet. 1999, 30, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Kranis, A.; Gheyas, A.A.; Boschiero, C.; Turner, F.; Yu, L.; Smith, S.; Talbot, R.; Pirani, A.; Brew, F.; Kaiser, P.; et al. Development of a high density 600K SNP genotyping array for chicken. BMC Genom. 2013, 14, 59. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Liu, G.E.; Bickhart, D.M.; Cardone, M.F.; Wang, K.; Kim, E.S.; Matukumalli, L.K.; Ventura, M.; Song, J.; VanRaden, P.M.; et al. Genomic characteristics of cattle copy number variations. BMC Genom. 2011, 12, 127. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Bickhart, D.M.; Hvinden, M.L.; Li, C.; Song, J.; Boichard, D.A.; Fritz, S.; Eggen, A.; Denise, S.; Wiggans, G.R.; et al. Fine mapping of copy number variations on two cattle genome assemblies using high density SNP array. BMC Genom. 2012, 13, 376. [Google Scholar] [CrossRef] [PubMed]
- Bickhart, D.M.; Hou, Y.; Schroeder, S.G.; Alkan, C.; Cardone, M.F.; Matukumalli, L.K.; Song, J.; Schnabel, R.D.; Ventura, M.; Taylor, J.F.; et al. Copy number variation of individual cattle genomes using next-generation sequencing. Genome Res. 2012, 22, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Zehner, Z.E.; Paterson, B.M. Characterization of the chicken vimentin gene: Single copy gene producing multiple mRNAs. Proc. Natl. Acad. Sci. USA 1983, 80, 911–915. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013, 8, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chr | Start | End | Length | Genes |
|---|---|---|---|---|
| 1 | 84,250,750 | 84,254,670 | 3920 | TBC1D23 |
| 2 | 69,574,893 | 69,579,418 | 4525 | CDH6 |
| 2 | 74,099,026 | 74,100,933 | 1907 | |
| 2 | 81,592,992 | 81,594,465 | 1473 | GRB10 |
| 2 | 115,475,473 | 115,484,664 | 9191 | CPA6 |
| 2 | 116,094,599 | 116,097,298 | 2699 | COPS5 |
| 3 | 43,999,153 | 44,003,340 | 4187 | |
| 3 | 53,309,153 | 53,311,383 | 2230 | TXLNB |
| 4 | 88,905,580 | 88,906,840 | 1260 | GFRA4 |
| 10 | 5,716,844 | 5,717,897 | 1053 | TARSL2 |
| 17 | 7,100,919 | 7,102,218 | 1299 | VAV2 |
| 26 | 1,927,477 | 1,931,663 | 4186 | CNTN2 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, L.; He, Y.; Ding, Y.; Sun, G.; Carrillo, J.A.; Li, Y.; Ghaly, M.M.; Ma, L.; Zhang, H.; Liu, G.E.; et al. Characterization of Copy Number Variation’s Potential Role in Marek’s Disease. Int. J. Mol. Sci. 2017, 18, 1020. https://doi.org/10.3390/ijms18051020
Xu L, He Y, Ding Y, Sun G, Carrillo JA, Li Y, Ghaly MM, Ma L, Zhang H, Liu GE, et al. Characterization of Copy Number Variation’s Potential Role in Marek’s Disease. International Journal of Molecular Sciences. 2017; 18(5):1020. https://doi.org/10.3390/ijms18051020
Chicago/Turabian StyleXu, Lingyang, Yanghua He, Yi Ding, Guirong Sun, Jose Adrian Carrillo, Yaokun Li, Mona M. Ghaly, Li Ma, Huanmin Zhang, George E. Liu, and et al. 2017. "Characterization of Copy Number Variation’s Potential Role in Marek’s Disease" International Journal of Molecular Sciences 18, no. 5: 1020. https://doi.org/10.3390/ijms18051020
APA StyleXu, L., He, Y., Ding, Y., Sun, G., Carrillo, J. A., Li, Y., Ghaly, M. M., Ma, L., Zhang, H., Liu, G. E., & Song, J. (2017). Characterization of Copy Number Variation’s Potential Role in Marek’s Disease. International Journal of Molecular Sciences, 18(5), 1020. https://doi.org/10.3390/ijms18051020