
Growth Hormone Resistance—Special Focus on Inflammatory Bowel Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
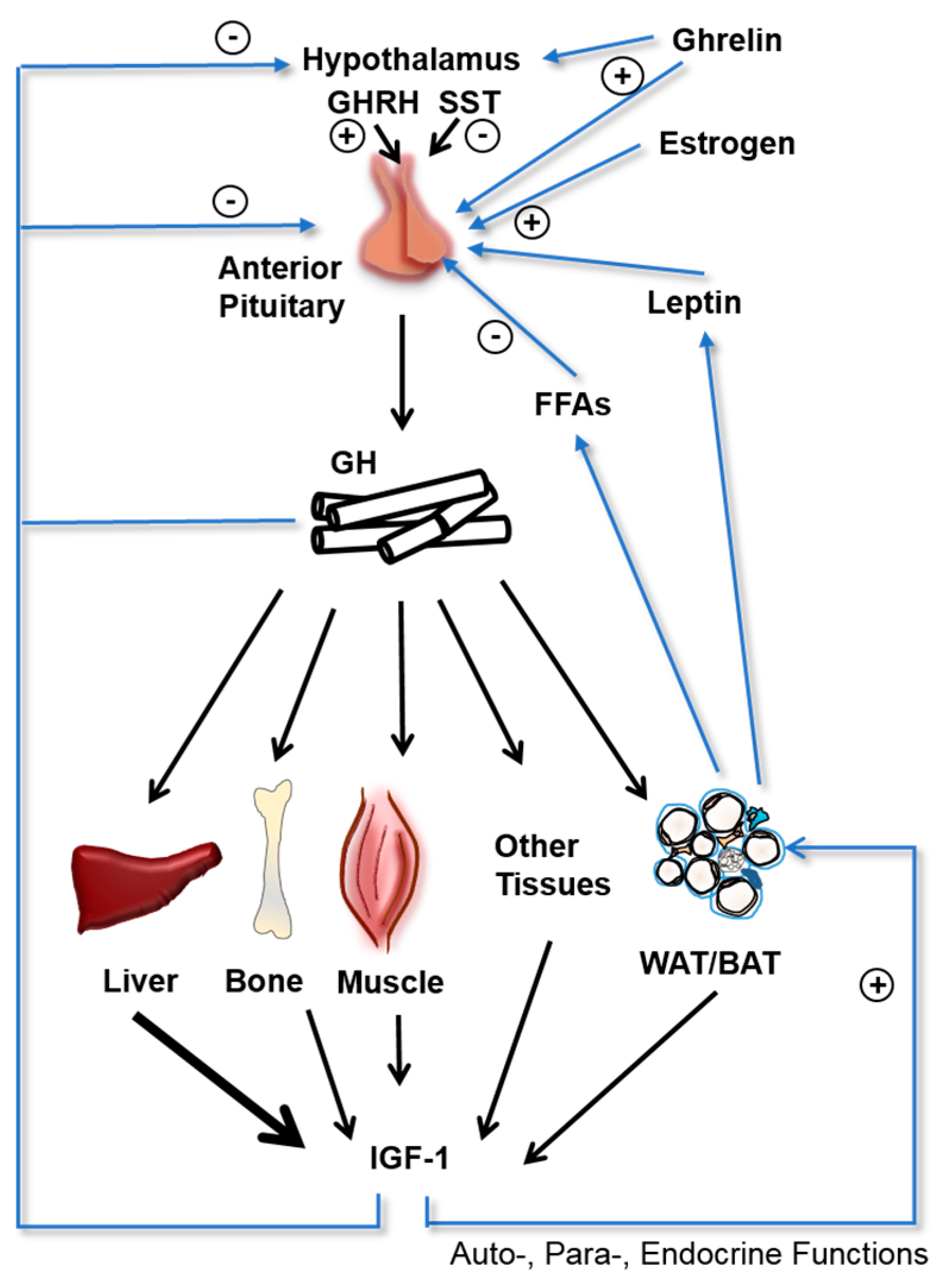
1. Introduction
2. Inflammatory Bowel Diseases
3. Mechanisms of Inflammation-Driven Growth Hormone (GH) Resistance
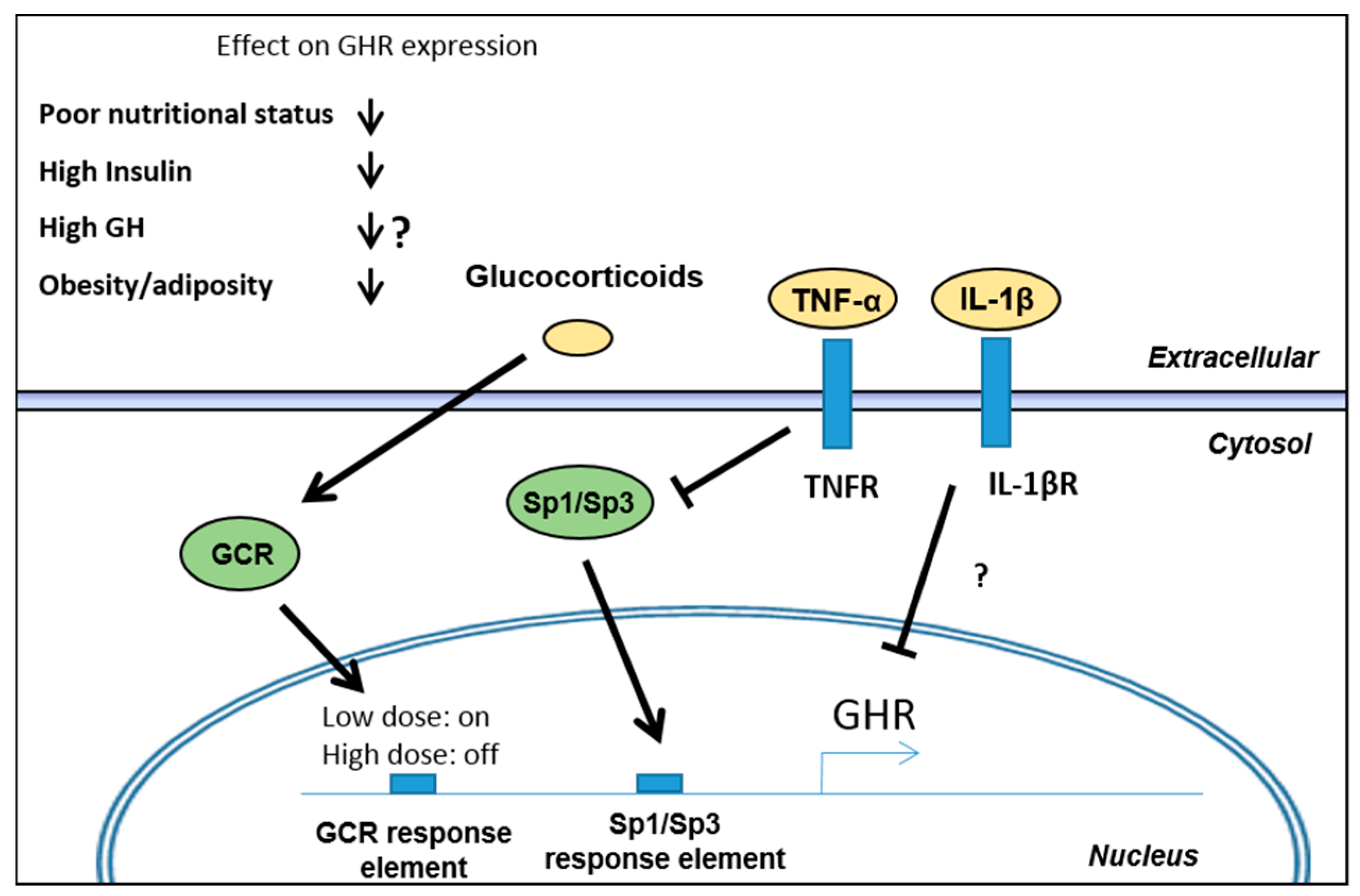
3.1. Regulation of GH Receptor (GHR) Expression
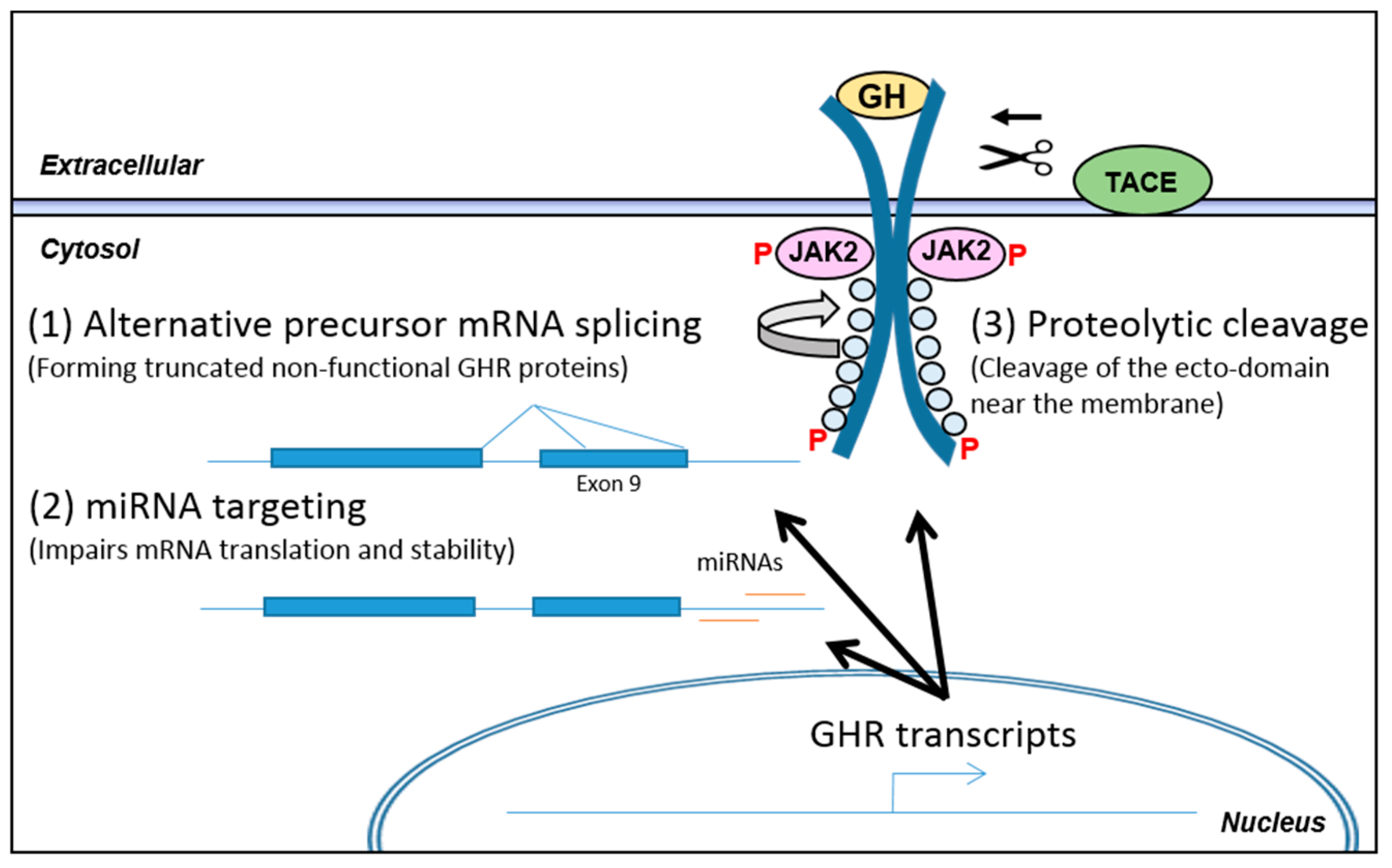
3.2. Post-Transcriptional Regulation of GHR Levels
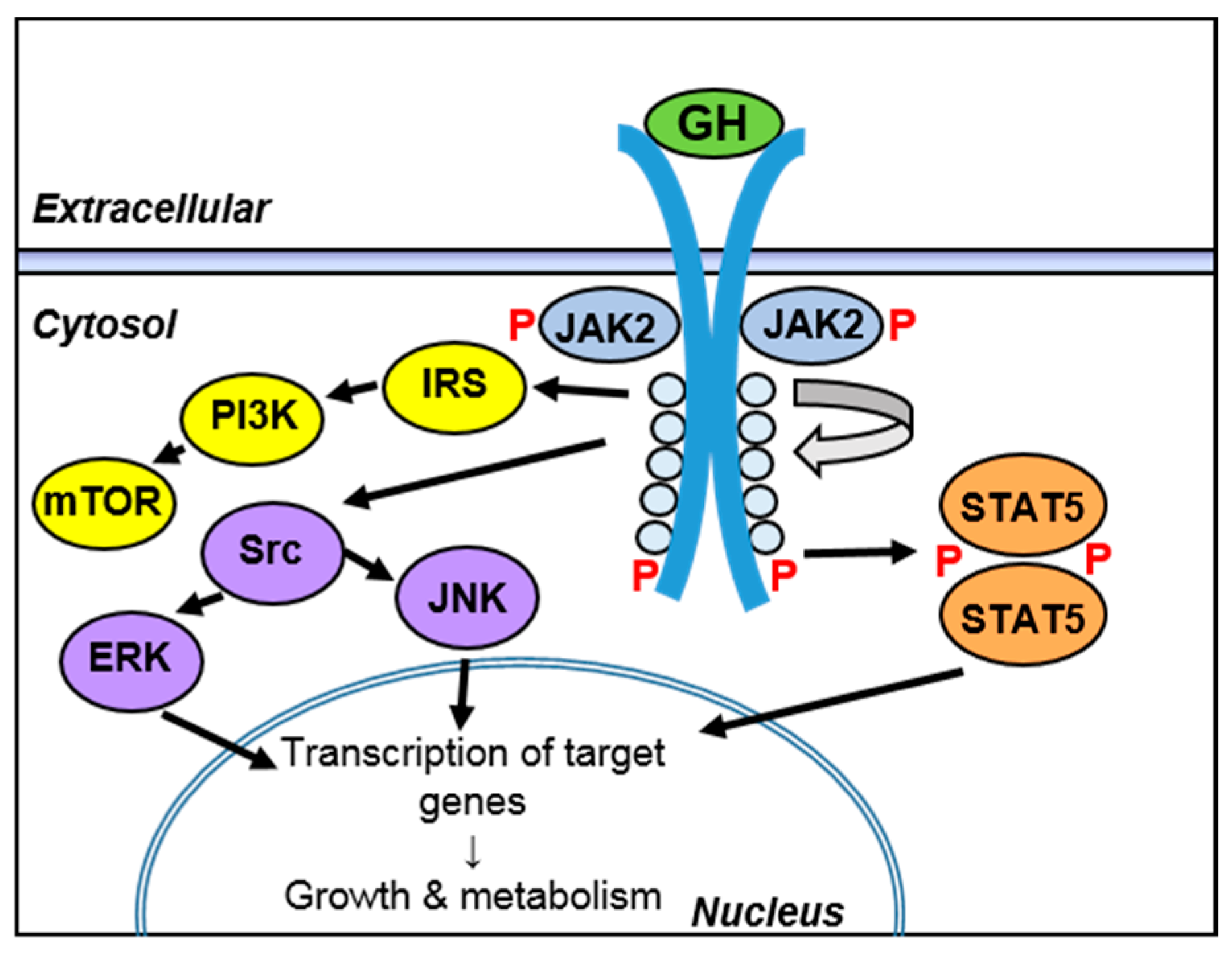
3.3. Regulation of Intracellular Signaling during Inflammation
4. Effects of Growth Hormone in the Intestine during Inflammation
4.1. Effects of GH on Mucosal Integrity
4.2. Effects of GH on Immune Cells
4.3. Effects of GH in Experimental Models of Colitis and Short Bowel Syndrome (SBS)
4.4. Clinical Evaluation of GH in Inflammatory Bowel Diseases (IBD) Patients
5. Discussion and Conclusions
Author Contributions
Conflicts of Interest
References
- Green, H.; Morikawa, M.; Nixon, T. A dual effector theory of growth-hormone action. Differentiation 1985, 29, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Lupu, F.; Terwilliger, J.D.; Lee, K.; Segre, G.V.; Efstratiadis, A. Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth. Dev. Biol. 2001, 229, 141–162. [Google Scholar] [CrossRef] [PubMed]
- Steyn, F.J.; Tolle, V.; Chen, C.; Epelbaum, J. Neuroendocrine regulation of growth hormone secretion. Compr. Physiol. 2016, 6, 687–735. [Google Scholar] [PubMed]
- Waters, M.J. The growth hormone receptor. Growth Horm. IGF Res. 2016, 28, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; He, K.; Gerhart, M.; Huang, Y.; Jiang, J.; Paxton, R.J.; Yang, S.; Lu, C.; Menon, R.K.; Black, R.A.; et al. Metalloprotease-mediated GH receptor proteolysis and GHBP shedding. Determination of extracellular domain stem region cleavage site. J. Biol. Chem. 2002, 277, 50510–50519. [Google Scholar] [CrossRef] [PubMed]
- Lanning, N.J.; Carter-Su, C. Recent advances in growth hormone signaling. Rev. Endocr. Metab. Disord. 2006, 7, 225–235. [Google Scholar] [CrossRef] [PubMed]
- D’Ercole, A.J.; Stiles, A.D.; Underwood, L.E. Tissue concentrations of somatomedin C: Further evidence for multiple sites of synthesis and paracrine or autocrine mechanisms of action. Proc. Natl. Acad. Sci. USA 1984, 81, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Houssay, B.A.; Smyth, F.S.; Foglia, V.G.; Houssay, A.B. Comparative diabetogenic action of the hypophysis from various animals. J. Exp. Med. 1942, 75, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, D.; Klassen, G.A.; Zierler, K.L. Effect of human growth hormone on muscle and adipose tissue metabolism in the forearm of man. J. Clin. Investig. 1965, 44, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Roberts-Thomson, I.C.; Fon, J.; Uylaki, W.; Cummins, A.G.; Barry, S. Cells, cytokines and inflammatory bowel disease: A clinical perspective. Expert Rev. Gastroenterol. Hepatol. 2011, 5, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Soendergaard, C.; Nielsen, O.H.; Seidelin, J.B.; Kvist, P.H.; Bjerrum, J.T. α-1 antitrypsin and granulocyte colony-stimulating factor as serum biomarkers of disease severity in ulcerative colitis. Inflamm. Bowel Dis. 2015, 21, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.J.; Mayer, L. The immune response in inflammatory bowel disease. Am. J. Gastroenterol. 2007, 102, 2058–2069. [Google Scholar] [CrossRef] [PubMed]
- Abraham, C.; Medzhitov, R. Interactions between the host innate immune system and microbes in inflammatory bowel disease. Gastroenterology 2011, 140, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, S.C.; Barbara, G.; Buurman, W.; Ockhuizen, T.; Schulzke, J.D.; Serino, M.; Tilg, H.; Watson, A.; Wells, J.M. Intestinal permeability—A new target for disease prevention and therapy. BMC Gastroenterol. 2014, 14, 189. [Google Scholar] [CrossRef] [PubMed]
- Hering, N.A.; Fromm, M.; Schulzke, J.D. Determinants of colonic barrier function in inflammatory bowel disease and potential therapeutics. J. Physiol. 2012, 590, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Boltin, D.; Perets, T.T.; Vilkin, A.; Niv, Y. Mucin function in inflammatory bowel disease: An update. J. Clin. Gastroenterol. 2013, 47, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Simms, L.A.; Doecke, J.D.; Walsh, M.D.; Huang, N.; Fowler, E.V.; Radford-Smith, G.L. Reduced α-defensin expression is associated with inflammation and not NOD2 mutation status in ileal Crohn’s disease. Gut 2008, 57, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Mijac, D.D.; Jankovic, G.L.; Jorga, J.; Krstic, M.N. Nutritional status in patients with active inflammatory bowel disease: Prevalence of malnutrition and methods for routine nutritional assessment. Eur. J. Intern. Med. 2010, 21, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Valentini, L.; Schulzke, J.D. Mundane, yet challenging: The assessment of malnutrition in inflammatory bowel disease. Eur. J. Intern. Med. 2011, 22, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Lochs, H.; Dejong, C.; Hammarqvist, F.; Hebuterne, X.; Leon-Sanz, M.; Schutz, T.; van Gemert, W.; van Gossum, A.; Valentini, L. German Society for Nutritional Medicine; et al. Espen guidelines on enteral nutrition: Gastroenterology. Clin. Nutr. 2006, 25, 260–274. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.G.; Miller, V.; Taylor, F.; Maycock, P.; Scrimgeour, C.M.; Rennie, M.J. Whole body protein turnover in childhood Crohn’s disease. Gut 1992, 33, 675–677. [Google Scholar] [CrossRef] [PubMed]
- Ockenga, J.; Borchert, K.; Stuber, E.; Lochs, H.; Manns, M.P.; Bischoff, S.C. Glutamine-enriched total parenteral nutrition in patients with inflammatory bowel disease. Eur. J. Clin. Nutr. 2005, 59, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Motil, K.J.; Grand, R.J.; Davis-Kraft, L.; Ferlic, L.L.; Smith, E.O. Growth failure in children with inflammatory bowel disease: A prospective study. Gastroenterology 1993, 105, 681–691. [Google Scholar] [CrossRef]
- Wong, S.C.; Dobie, R.; Altowati, M.A.; Werther, G.A.; Farquharson, C.; Ahmed, S.F. Growth and the growth hormone-insulin like growth factor 1 axis in children with chronic inflammation: Current evidence, gaps in knowledge, and future directions. Endocr. Rev. 2016, 37, 62–110. [Google Scholar] [CrossRef] [PubMed]
- Fumery, M.; Duricova, D.; Gower-Rousseau, C.; Annese, V.; Peyrin-Biroulet, L.; Lakatos, P.L. Review article: The natural history of paediatric-onset ulcerative colitis in population-based studies. Aliment. Pharmacol. Ther. 2016, 43, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, I.R. Growth problems in children with IBD. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Mason, A.; Malik, S.; McMillan, M.; McNeilly, J.D.; Bishop, J.; McGrogan, P.; Russell, R.K.; Ahmed, S.F. A prospective longitudinal study of growth and pubertal progress in adolescents with inflammatory bowel disease. Horm. Res. Paediatr. 2015, 83, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F.; Travis, S.P. Mucosal healing in inflammatory bowel diseases: A systematic review. Gut 2012, 61, 1619–1635. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, O.H.; Ainsworth, M.A. Tumor necrosis factor inhibitors for inflammatory bowel disease. N. Engl. J. Med. 2013, 369, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, J.; Coskun, M.; Soendergaard, C.; Salem, M.; Nielsen, O.H. Inflammatory pathways of importance for management of inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Frolkis, A.D.; Dykeman, J.; Negron, M.E.; Debruyn, J.; Jette, N.; Fiest, K.M.; Frolkis, T.; Barkema, H.W.; Rioux, K.P.; Panaccione, R.; et al. Risk of surgery for inflammatory bowel diseases has decreased over time: A systematic review and meta-analysis of population-based studies. Gastroenterology 2013, 145, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Carroll, R.E.; Benedetti, E.; Schowalter, J.P.; Buchman, A.L. Management and complications of short bowel syndrome: An updated review. Curr. Gastroenterol. Rep. 2016, 18, 40. [Google Scholar] [CrossRef] [PubMed]
- Misra, M.; Klibanski, A. Endocrine consequences of anorexia nervosa. Lancet Diabetes Endocrinol. 2014, 2, 581–592. [Google Scholar] [CrossRef]
- Erman, A.; Veilleux, A.; Tchernof, A.; Goodyer, C.G. Human growth hormone receptor (GHR) expression in obesity: I. GHR mRNA expression in omental and subcutaneous adipose tissues of obese women. Int. J. Obes. Lond. 2011, 35, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Schwartzbauer, G.; Menon, R.K. Regulation of growth hormone receptor gene expression. Mol. Genet. Metab. 1998, 63, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Birzniece, V.; Sata, A.; Ho, K.K. Growth hormone receptor modulators. Rev. Endocr. Metab. Disord. 2009, 10, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Straus, D.S.; Takemoto, C.D. Effect of fasting on insulin-like growth factor-I (IGF-I) and growth hormone receptor mRNA levels and IGF-I gene transcription in rat liver. Mol. Endocrinol. 1990, 4, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Beauloye, V.; Willems, B.; de Coninck, V.; Frank, S.J.; Edery, M.; Thissen, J.P. Impairment of liver GH receptor signaling by fasting. Endocrinology 2002, 143, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Fazeli, P.K.; Lawson, E.A.; Prabhakaran, R.; Miller, K.K.; Donoho, D.A.; Clemmons, D.R.; Herzog, D.B.; Misra, M.; Klibanski, A. Effects of recombinant human growth hormone in anorexia nervosa: A randomized, placebo-controlled study. J. Clin. Endocrinol. Metab. 2010, 95, 4889–4897. [Google Scholar] [CrossRef] [PubMed]
- Thissen, J.P.; Ketelslegers, J.M.; Underwood, L.E. Nutritional regulation of the insulin-like growth factors. Endocr. Rev. 1994, 15, 80–101. [Google Scholar] [PubMed]
- Gao, X.; Tian, F.; Wang, X.; Zhao, J.; Wan, X.; Zhang, L.; Wu, C.; Li, N.; Li, J. Leucine supplementation improves acquired growth hormone resistance in rats with protein-energy malnutrition. PLoS ONE 2015, 10, e0125023. [Google Scholar] [CrossRef]
- Bornfeldt, K.E.; Arnqvist, H.J.; Enberg, B.; Mathews, L.S.; Norstedt, G. Regulation of insulin-like growth factor-I and growth hormone receptor gene expression by diabetes and nutritional state in rat tissues. J. Endocrinol. 1989, 122, 651–656. [Google Scholar] [CrossRef] [PubMed]
- VandeHaar, M.J.; Moats-Staats, B.M.; Davenport, M.L.; Walker, J.L.; Ketelslegers, J.M.; Sharma, B.K.; Underwood, L.E. Reduced serum concentrations of insulin-like growth factor-I (IGF-I) in protein-restricted growing rats are accompanied by reduced IGF-I mRNA levels in liver and skeletal muscle. J. Endocrinol. 1991, 130, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Tannenbaum, G.S.; Rorstad, O.; Brazeau, P. Effects of prolonged food deprivation on the ultradian growth hormone rhythm and immunoreactive somatostatin tissue levels in the rat. Endocrinology 1979, 104, 1733–1738. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.Y.; Veldhuis, J.D.; Johnson, M.L.; Furlanetto, R.; Evans, W.S.; Alberti, K.G.; Thorner, M.O. Fasting enhances growth hormone secretion and amplifies the complex rhythms of growth hormone secretion in man. J. Clin. Investig. 1988, 81, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Merimee, T.J.; Fineberg, S.E. Growth hormone secretion in starvation: A reassessment. J. Clin. Endocrinol. Metab. 1974, 39, 385–386. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.R.; Edgar, P.J.; Pozefsky, T.; Chhetri, M.K.; Prout, T.E. Growth hormone in adults with protein-calorie malnutrition. J. Clin. Endocrinol. Metab. 1974, 39, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Iguchi, G.; Fukuoka, H.; Suda, K.; Bando, H.; Takahashi, M.; Nishizawa, H.; Seino, S.; Takahashi, Y. SIRT1 regulates adaptive response of the growth hormone-insulin-like growth factor-I axis under fasting conditions in liver. Proc. Natl. Acad. Sci. USA 2013, 110, 14948–14953. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Lin, V.Y.; Goetz, R.; Mohammadi, M.; Mangelsdorf, D.J.; Kliewer, S.A. Inhibition of growth hormone signaling by the fasting-induced hormone FGF21. Cell Metab. 2008, 8, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Fazeli, P.K.; Klibanski, A. Determinants of GH resistance in malnutrition. J. Endocrinol. 2014, 220, R57–R65. [Google Scholar] [CrossRef] [PubMed]
- Horner, J.M.; Kemp, S.F.; Hintz, R.L. Growth hormone and somatomedin in insulin-dependent diabetes mellitus. J. Clin. Endocrinol. Metab. 1981, 53, 1148–1153. [Google Scholar] [CrossRef] [PubMed]
- Lundbaek, K.; Christensen, N.J.; Jensen, V.A.; Johansen, K.; Olsen, T.S.; Hansen, A.P.; Orskov, H.; Osterby, R. Diabetes, diabetic angiopathy, and growth hormone. Lancet 1970, 2, 131–133. [Google Scholar] [CrossRef]
- Baxter, R.C.; Turtle, J.R. Regulation of hepatic growth hormone receptors by insulin. Biochem. Biophys. Res. Commun. 1978, 84, 350–357. [Google Scholar] [CrossRef]
- Leung, K.C.; Doyle, N.; Ballesteros, M.; Waters, M.J.; Ho, K.K. Insulin regulation of human hepatic growth hormone receptors: Divergent effects on biosynthesis and surface translocation. J. Clin. Endocrinol. Metab. 2000, 85, 4712–4720. [Google Scholar] [CrossRef] [PubMed]
- Bennett, W.L.; Keeton, A.B.; Ji, S.; Xu, J.; Messina, J.L. Insulin regulation of growth hormone receptor gene expression: Involvement of both the PI-3 kinase and MEK/ERK signaling pathways. Endocrine 2007, 32, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Bennett, W.L.; Ji, S.; Messina, J.L. Insulin regulation of growth hormone receptor gene expression. Evidence for a transcriptional mechanism of down-regulation in rat hepatoma cells. Mol. Cell. Endocrinol. 2007, 274, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Katsuki, A.; Urakawa, H.; Gabazza, E.C.; Murashima, S.; Nakatani, K.; Togashi, K.; Yano, Y.; Adachi, Y.; Sumida, Y. Circulating levels of active ghrelin is associated with abdominal adiposity, hyperinsulinemia and insulin resistance in patients with type 2 diabetes mellitus. Eur. J. Endocrinol. 2004, 151, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Thimmarayappa, J.; Sun, J.; Schultz, L.E.; Dejkhamron, P.; Lu, C.; Giallongo, A.; Merchant, J.L.; Menon, R.K. Inhibition of growth hormone receptor gene expression by saturated fatty acids: Role of kruppel-like zinc finger factor, ZBP-89. Mol. Endocrinol. 2006, 20, 2747–2760. [Google Scholar] [CrossRef] [PubMed]
- Meinhardt, U.; Eble, A.; Besson, A.; Strasburger, C.J.; Sraer, J.D.; Mullis, P.E. Regulation of growth-hormone-receptor gene expression by growth hormone and pegvisomant in human mesangial cells. Kidney Int. 2003, 64, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Sun, D.; Jiang, J.; Deng, L.; Zhang, Y.; Yu, H.; Bahl, D.; Langenheim, J.F.; Chen, W.Y.; Fuchs, S.Y.; et al. The role of prolactin receptor in GH signaling in breast cancer cells. Mol. Endocrinol. 2013, 27, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Yumet, G.; Shumate, M.L.; Bryant, P.; Lin, C.M.; Lang, C.H.; Cooney, R.N. Tumor necrosis factor mediates hepatic growth hormone resistance during sepsis. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E472–E481. [Google Scholar] [CrossRef] [PubMed]
- Denson, L.A.; Menon, R.K.; Shaufl, A.; Bajwa, H.S.; Williams, C.R.; Karpen, S.J. TNF-α downregulates murine hepatic growth hormone receptor expression by inhibiting Sp1 and Sp3 binding. J. Clin. Investig. 2001, 107, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, N.; Li, J.S.; Li, W.Q. The role of endotoxin, TNF-α, and IL-6 in inducing the state of growth hormone insensitivity. World J. Gastroenterol. 2002, 8, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xiao, X.; Frank, S.J.; Lin, H.Y.; Xia, Y. Distinct mechanisms of induction of hepatic growth hormone resistance by endogenous IL-6, TNF-α, and IL-1β. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E186–E198. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.; Yumet, G.; Shumate, M.; Lang, C.H.; Rotwein, P.; Cooney, R.N. Tumor necrosis factor inhibits growth hormone-mediated gene expression in hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G35–G44. [Google Scholar] [CrossRef] [PubMed]
- Difedele, L.M.; He, J.; Bonkowski, E.L.; Han, X.; Held, M.A.; Bohan, A.; Menon, R.K.; Denson, L.A. Tumor necrosis factor α blockade restores growth hormone signaling in murine colitis. Gastroenterology 2005, 128, 1278–1291. [Google Scholar] [CrossRef] [PubMed]
- Erman, A.; Wabitsch, M.; Goodyer, C.G. Human growth hormone receptor (GHR) expression in obesity: II. Regulation of the human GHR gene by obesity-related factors. Int. J. Obes. Lond. 2011, 35, 1520–1529. [Google Scholar] [CrossRef] [PubMed]
- Shumate, M.L.; Yumet, G.; Ahmed, T.A.; Cooney, R.N. Interleukin-1 inhibits the induction of insulin-like growth factor-I by growth hormone in CWSV-1 hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G227–G239. [Google Scholar] [CrossRef] [PubMed]
- Martensson, K.; Chrysis, D.; Savendahl, L. Interleukin-1β and TNF-α act in synergy to inhibit longitudinal growth in fetal rat metatarsal bones. J. Bone Miner. Res. 2004, 19, 1805–1812. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Benight, N.; Osuntokun, B.; Loesch, K.; Frank, S.J.; Denson, L.A. Tumour necrosis factor α blockade induces an anti-inflammatory growth hormone signalling pathway in experimental colitis. Gut 2007, 56, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Eivindson, M.; Gronbaek, H.; Skogstrand, K.; Thorsen, P.; Frystyk, J.; Flyvbjerg, A.; Dahlerup, J.F. The insulin-like growth factor (IGF) system and its relation to infliximab treatment in adult patients with crohn’s disease. Scand. J. Gastroenterol. 2007, 42, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Vespasiani Gentilucci, U.; Caviglia, R.; Picardi, A.; Carotti, S.; Ribolsi, M.; Galati, G.; Petitti, T.; Afeltra, A.; Cicala, M. Infliximab reverses growth hormone resistance associated with inflammatory bowel disease. Aliment. Pharmacol. Ther. 2005, 21, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Mazziotti, G.; Giustina, A. Glucocorticoids and the regulation of growth hormone secretion. Nat. Rev. Endocrinol. 2013, 9, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, M.; Leung, K.C.; Ross, R.J.; Iismaa, T.P.; Ho, K.K. Distribution and abundance of messenger ribonucleic acid for growth hormone receptor isoforms in human tissues. J. Clin. Endocrinol. Metab. 2000, 85, 2865–2871. [Google Scholar] [CrossRef] [PubMed]
- Dastot, F.; Sobrier, M.L.; Duquesnoy, P.; Duriez, B.; Goossens, M.; Amselem, S. Alternatively spliced forms in the cytoplasmic domain of the human growth hormone (GH) receptor regulate its ability to generate a soluble GH-binding protein. Proc. Natl. Acad. Sci. USA 1996, 93, 10723–10728. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.J.; Esposito, N.; Shen, X.Y.; von Laue, S.; Chew, S.L.; Dobson, P.R.; Postel-Vinay, M.C.; Finidori, J. A short isoform of the human growth hormone receptor functions as a dominant negative inhibitor of the full-length receptor and generates large amounts of binding protein. Mol. Endocrinol. 1997, 11, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Ayling, R.M.; Ross, R.; Towner, P.; Von Laue, S.; Finidori, J.; Moutoussamy, S.; Buchanan, C.R.; Clayton, P.E.; Norman, M.R. A dominant-negative mutation of the growth hormone receptor causes familial short stature. Nat. Genet. 1997, 16, 13–14. [Google Scholar] [CrossRef] [PubMed]
- Walenkamp, M.J.; Wit, J.M. Genetic disorders in the growth hormone—Insulin-like growth factor-I axis. Horm. Res. 2006, 66, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Elzein, S.; Goodyer, C.G. Regulation of human growth hormone receptor expression by microRNAs. Mol. Endocrinol. 2014, 28, 1448–1459. [Google Scholar] [CrossRef] [PubMed]
- Edens, A.; Southard, J.N.; Talamantes, F. Mouse growth hormone-binding protein and growth hormone receptor transcripts are produced from a single gene by alternative splicing. Endocrinology 1994, 135, 2802–2805. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jiang, J.; Black, R.A.; Baumann, G.; Frank, S.J. Tumor necrosis factor-α converting enzyme (TACE) is a growth hormone binding protein (GHBP) sheddase: The metalloprotease TACE/ADAM-17 is critical for (PMA-induced) GH receptor proteolysis and GHBP generation. Endocrinology 2000, 141, 4342–4348. [Google Scholar] [CrossRef] [PubMed]
- Loesch, K.; Deng, L.; Cowan, J.W.; Wang, X.; He, K.; Jiang, J.; Black, R.A.; Frank, S.J. Janus kinase 2 influences growth hormone receptor metalloproteolysis. Endocrinology 2006, 147, 2839–2849. [Google Scholar] [CrossRef] [PubMed]
- Lisi, S.; D’Amore, M.; Sisto, M. Adam17 at the interface between inflammation and autoimmunity. Immunol. Lett. 2014, 162, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Peiretti, F.; Canault, M.; Deprez-Beauclair, P.; Berthet, V.; Bonardo, B.; Juhan-Vague, I.; Nalbone, G. Intracellular maturation and transport of tumor necrosis factor α converting enzyme. Exp. Cell Res. 2003, 285, 278–285. [Google Scholar] [CrossRef]
- Cesaro, A.; Abakar-Mahamat, A.; Brest, P.; Lassalle, S.; Selva, E.; Filippi, J.; Hebuterne, X.; Hugot, J.P.; Doglio, A.; Galland, F.; et al. Differential expression and regulation of ADAM17 and TIMP3 in acute inflamed intestinal epithelia. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G1332–G1343. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, M.; Horiuchi, K.; Sasaki, A.; Tsukamoto, T.; Okabayashi, K.; Hasegawa, H.; Kitagawa, Y.; Okada, Y. Epithelial cell-derived a disintegrin and metalloproteinase-17 confers resistance to colonic inflammation through EGFR activation. EBioMedicine 2016, 5, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Brynskov, J.; Foegh, P.; Pedersen, G.; Ellervik, C.; Kirkegaard, T.; Bingham, A.; Saermark, T. Tumour necrosis factor α converting enzyme (TACE) activity in the colonic mucosa of patients with inflammatory bowel disease. Gut 2002, 51, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Patel, I.R.; Attur, M.G.; Patel, R.N.; Stuchin, S.A.; Abagyan, R.A.; Abramson, S.B.; Amin, A.R. TNF-α convertase enzyme from human arthritis-affected cartilage: Isolation of cDNA by differential display, expression of the active enzyme, and regulation of TNF-α. J. Immunol. 1998, 160, 4570–4579. [Google Scholar] [PubMed]
- Zhang, Y.; Wang, X.; Loesch, K.; May, L.A.; Davis, G.E.; Jiang, J.; Frank, S.J. TIMP3 modulates GHR abundance and GH sensitivity. Mol. Endocrinol. 2016, 30, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Mohapatra, J.; Wagh, A.; Patel, H.M.; Pandey, D.; Kadam, S.; Argade, A.; Deshpande, S.S.; Shah, G.B.; Chatterjee, A.; et al. Involvement of TACE in colon inflammation: A novel mechanism of regulation via SIRT-1 activation. Cytokine 2014, 66, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jiang, J.; Warram, J.; Baumann, G.; Gan, Y.; Menon, R.K.; Denson, L.A.; Zinn, K.R.; Frank, S.J. Endotoxin-induced proteolytic reduction in hepatic growth hormone (GH) receptor: A novel mechanism for gh insensitivity. Mol. Endocrinol. 2008, 22, 1427–1437. [Google Scholar] [CrossRef] [PubMed]
- Schilbach, K.; Bidlingmaier, M. Growth hormone binding protein—Physiological and analytical aspects. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Haglund, K.; Sigismund, S.; Polo, S.; Szymkiewicz, I.; di Fiore, P.P.; Dikic, I. Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat. Cell Biol. 2003, 5, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Kazi, J.U.; Kabir, N.N.; Flores-Morales, A.; Ronnstrand, L. SOCS proteins in regulation of receptor tyrosine kinase signaling. Cell. Mol. Life Sci. 2014, 71, 3297–3310. [Google Scholar] [CrossRef] [PubMed]
- Davey, H.W.; McLachlan, M.J.; Wilkins, R.J.; Hilton, D.J.; Adams, T.E. STAT5b mediates the GH-induced expression of SOCS-2 and SOCS-3 mRNA in the liver. Mol. Cell. Endocrinol. 1999, 158, 111–116. [Google Scholar] [CrossRef]
- Alexander, W.S.; Starr, R.; Fenner, J.E.; Scott, C.L.; Handman, E.; Sprigg, N.S.; Corbin, J.E.; Cornish, A.L.; Darwiche, R.; Owczarek, C.M.; et al. SOCS1 is a critical inhibitor of interferon γ signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell 1999, 98, 597–608. [Google Scholar] [CrossRef]
- Metcalf, D.; Greenhalgh, C.J.; Viney, E.; Willson, T.A.; Starr, R.; Nicola, N.A.; Hilton, D.J.; Alexander, W.S. Gigantism in mice lacking suppressor of cytokine signalling-2. Nature 2000, 405, 1069–1073. [Google Scholar] [PubMed]
- Roberts, A.W.; Robb, L.; Rakar, S.; Hartley, L.; Cluse, L.; Nicola, N.A.; Metcalf, D.; Hilton, D.J.; Alexander, W.S. Placental defects and embryonic lethality in mice lacking suppressor of cytokine signaling 3. Proc. Natl. Acad. Sci. USA 2001, 98, 9324–9329. [Google Scholar] [CrossRef] [PubMed]
- Linossi, E.M.; Nicholson, S.E. Kinase inhibition, competitive binding and proteasomal degradation: Resolving the molecular function of the suppressor of cytokine signaling (SOCS) proteins. Immunol. Rev. 2015, 266, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Adams, T.E.; Hansen, J.A.; Starr, R.; Nicola, N.A.; Hilton, D.J.; Billestrup, N. Growth hormone preferentially induces the rapid, transient expression of SOCS-3, a novel inhibitor of cytokine receptor signaling. J. Biol. Chem. 1998, 273, 1285–1287. [Google Scholar] [CrossRef] [PubMed]
- Tollet-Egnell, P.; Flores-Morales, A.; Stavreus-Evers, A.; Sahlin, L.; Norstedt, G. Growth hormone regulation of SOCS-2, SOCS-3, and CIS messenger ribonucleic acid expression in the rat. Endocrinology 1999, 140, 3693–3704. [Google Scholar] [CrossRef] [PubMed]
- Ram, P.A.; Waxman, D.J. SOCS/CIS protein inhibition of growth hormone-stimulated STAT5 signaling by multiple mechanisms. J. Biol. Chem. 1999, 274, 35553–35561. [Google Scholar] [CrossRef] [PubMed]
- Wiejak, J.; Dunlop, J.; Gao, S.; Borland, G.; Yarwood, S.J. Extracellular signal-regulated kinase mitogen-activated protein kinase-dependent SOCS-3 gene induction requires c-Jun, signal transducer and activator of transcription 3, and specificity protein 3 transcription factors. Mol. Pharmacol. 2012, 81, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Wormald, S.; Zhang, J.G.; Krebs, D.L.; Mielke, L.A.; Silver, J.; Alexander, W.S.; Speed, T.P.; Nicola, N.A.; Hilton, D.J. The comparative roles of suppressor of cytokine signaling-1 and -3 in the inhibition and desensitization of cytokine signaling. J. Biol. Chem. 2006, 281, 11135–11143. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, H.; Ohishi, M.; Mori, H.; Murakami, M.; Chinen, T.; Aki, D.; Hanada, T.; Takeda, K.; Akira, S.; Hoshijima, M.; et al. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat. Immunol. 2003, 4, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Lang, R.; Pauleau, A.L.; Parganas, E.; Takahashi, Y.; Mages, J.; Ihle, J.N.; Rutschman, R.; Murray, P.J. SOCS3 regulates the plasticity of gp130 signaling. Nat. Immunol. 2003, 4, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Mihara, M.; Hashizume, M.; Yoshida, H.; Suzuki, M.; Shiina, M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin. Sci. Lond. 2012, 122, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Starr, R.; Hilton, D.J. SOCS: Suppressors of cytokine signalling. Int. J. Biochem. Cell Biol. 1998, 30, 1081–1085. [Google Scholar] [CrossRef]
- Yao, X.; Huang, J.; Zhong, H.; Shen, N.; Faggioni, R.; Fung, M.; Yao, Y. Targeting interleukin-6 in inflammatory autoimmune diseases and cancers. Pharmacol. Ther. 2014, 141, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, S.E.; de Souza, D.; Fabri, L.J.; Corbin, J.; Willson, T.A.; Zhang, J.G.; Silva, A.; Asimakis, M.; Farley, A.; Nash, A.D.; et al. Suppressor of cytokine signaling-3 preferentially binds to the SHP-2-binding site on the shared cytokine receptor subunit gp130. Proc. Natl. Acad. Sci. USA 2000, 97, 6493–6498. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.A.; Buzzelli, M.D.; Lang, C.H.; Capen, J.B.; Shumate, M.L.; Navaratnarajah, M.; Nagarajan, M.; Cooney, R.N. Interleukin-6 inhibits growth hormone-mediated gene expression in hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1793–G1803. [Google Scholar] [CrossRef] [PubMed]
- Atreya, R.; Neurath, M.F. New therapeutic strategies for treatment of inflammatory bowel disease. Mucosal Immunol. 2008, 1, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Ito, H. Treatment of Crohn’s disease with anti-IL-6 receptor antibody. J. Gastroenterol. 2005, 40 (Suppl. S16), 32–34. [Google Scholar] [CrossRef] [PubMed]
- Horino, J.; Fujimoto, M.; Terabe, F.; Serada, S.; Takahashi, T.; Soma, Y.; Tanaka, K.; Chinen, T.; Yoshimura, A.; Nomura, S.; et al. Suppressor of cytokine signaling-1 ameliorates dextran sulfate sodium-induced colitis in mice. Int. Immunol. 2008, 20, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Chinen, T.; Kobayashi, T.; Ogata, H.; Takaesu, G.; Takaki, H.; Hashimoto, M.; Yagita, H.; Nawata, H.; Yoshimura, A. Suppressor of cytokine signaling-1 regulates inflammatory bowel disease in which both ifngamma and IL-4 are involved. Gastroenterology 2006, 130, 373–388. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Hanada, T.; Mitsuyama, K.; Yoshida, T.; Kamizono, S.; Hoshino, T.; Kubo, M.; Yamashita, A.; Okabe, M.; Takeda, K.; et al. CIS3/SOCS3/SSI3 plays a negative regulatory role in STAT3 activation and intestinal inflammation. J. Exp. Med. 2001, 193, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, P.; MacSharry, J.; Darby, T.; Fanning, A.; Shanahan, F.; Houston, A.; Brint, E. Differential expression of key regulators of toll-like receptors in ulcerative colitis and Crohn’s disease: A role for tollip and peroxisome proliferator-activated receptor γ? Clin. Exp. Immunol. 2016, 183, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Miyanaka, Y.; Ueno, Y.; Tanaka, S.; Yoshioka, K.; Hatakeyama, T.; Shimamoto, M.; Sumii, M.; Chayama, K. Clinical significance of mucosal suppressors of cytokine signaling 3 expression in ulcerative colitis. World J. Gastroenterol. 2007, 13, 2939–2944. [Google Scholar] [CrossRef] [PubMed]
- Christophi, G.P.; Rong, R.; Holtzapple, P.G.; Massa, P.T.; Landas, S.K. Immune markers and differential signaling networks in ulcerative colitis and Crohn’s disease. Inflamm. Bowel Dis. 2012, 18, 2342–2356. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Shan, X.; Qian, J.; Ji, Q.; Wang, L.; Wang, X.; Li, M.; Ding, H.; Liu, Q.; Chen, L.; et al. The suppressor of cytokine signaling SOCS1 promotes apoptosis of intestinal epithelial cells via p53 signaling in Crohn’s disease. Exp. Mol. Pathol. 2016, 101, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.J.; Zhang, Y.; Lin, Y.; Jin, Y.; Zheng, C.Q. Identifying candidate genes for discrimination of ulcerative colitis and Crohn’s disease. Mol. Biol. Rep. 2014, 41, 6349–6355. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, S.; Zhang, R.; Denson, L.; Moriggl, R.; Steinbrecher, K.; Shroyer, N.; Lin, J.; Han, X. Enterocyte STAT5 promotes mucosal wound healing via suppression of myosin light chain kinase-mediated loss of barrier function and inflammation. EMBO Mol. Med. 2012, 4, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Ren, X.; Jurickova, I.; Groschwitz, K.; Pasternak, B.A.; Xu, H.; Wilson, T.A.; Hogan, S.P.; Denson, L.A. Regulation of intestinal barrier function by signal transducer and activator of transcription 5b. Gut 2009, 58, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Yue, C.; Wang, W.; Tian, W.L.; Huang, Q.; Zhao, R.S.; Zhao, Y.Z.; Li, Q.R.; Li, J.S. Lipopolysaccharide-induced failure of the gut barrier is site-specific and inhibitable by growth hormone. Inflamm. Res. 2013, 62, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Jiang, Z.; Wang, X.; Shu, H.; Cui, W.; Wilmore, D.W. Impact of perioperative treatment of recombinant human growth hormone on cell immune function and intestinal barrier function: Randomized, double-blind, controlled trial. World J. Surg. 2003, 27, 412–415. [Google Scholar] [CrossRef] [PubMed]
- Hattori, N.; Saito, T.; Yagyu, T.; Jiang, B.H.; Kitagawa, K.; Inagaki, C. Gh, GH receptor, GH secretagogue receptor, and ghrelin expression in human T cells, B cells, and neutrophils. J. Clin. Endocrinol. Metab. 2001, 86, 4284–4291. [Google Scholar] [CrossRef] [PubMed]
- Kermani, H.; Goffinet, L.; Mottet, M.; Bodart, G.; Morrhaye, G.; Dardenne, O.; Renard, C.; Overbergh, L.; Baron, F.; Beguin, Y.; et al. Expression of the growth hormone/insulin-like growth factor axis during Balb/c thymus ontogeny and effects of growth hormone upon ex vivo T cell differentiation. Neuroimmunomodulation 2012, 19, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Kimata, H.; Yoshida, A. Effect of growth hormone and insulin-like growth factor-I on immunoglobulin production by and growth of human B cells. J. Clin. Endocrinol. Metab. 1994, 78, 635–641. [Google Scholar] [PubMed]
- Matsuda, T.; Saito, H.; Inoue, T.; Fukatsu, K.; Han, I.; Furukawa, S.; Ikeda, S.; Muto, T. Growth hormone inhibits apoptosis and up-regulates reactive oxygen intermediates production by human polymorphonuclear neutrophils. J. Parenter. Enter. Nutr. 1998, 22, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Kumar, P.A.; Sun, J.; Aggarwal, A.; Fan, Y.; Sperling, M.A.; Lumeng, C.N.; Menon, R.K. Targeted deletion of growth hormone (GH) receptor in macrophage reveals novel osteopontin-mediated effects of GH on glucose homeostasis and insulin sensitivity in diet-induced obesity. J. Biol. Chem. 2013, 288, 15725–15735. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, M.; Frystyk, J.; Faber, J.; Kristensen, L.O. Gh activity and markers of inflammation: A crossover study in healthy volunteers treated with GH and a GH receptor antagonist. Eur. J. Endocrinol. 2012, 166, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Sosnowska, D.; Bonkowski, E.L.; Denson, L.A. Growth hormone inhibits signal transducer and activator of transcription 3 activation and reduces disease activity in murine colitis. Gastroenterology 2005, 129, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.L.; Fuller, C.R.; Dieleman, L.A.; DaCosta, C.M.; Haldeman, K.M.; Sartor, R.B.; Lund, P.K. Enhanced survival and mucosal repair after dextran sodium sulfate-induced colitis in transgenic mice that overexpress growth hormone. Gastroenterology 2001, 120, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Kara, E.; Sungurtekin, H.; Sungurtekin, U.; Alkanat, M.; Ilkgul, O. The effect of recombinant human growth hormone (rhGH) on trinitrobenzene sulfonic acid-induced colitis in rats: An experimental study. Inflamm. Bowel Dis. 2004, 10, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Christensen, H.; Flyvbjerg, A.; Orskov, H.; Laurberg, S. Effect of growth hormone on the inflammatory activity of experimental colitis in rats. Scand. J. Gastroenterol. 1993, 28, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Theiss, A.L.; Fuller, C.R.; Simmons, J.G.; Liu, B.; Sartor, R.B.; Lund, P.K. Growth hormone reduces the severity of fibrosis associated with chronic intestinal inflammation. Gastroenterology 2005, 129, 204–219. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; Cao, Y.; Wang, S.R.; Xu, Y.Z.; Huang, H.; Cui, Y.X.; Huang, Y. Beneficial effect of recombinant human growth hormone on the intestinal mucosa barrier of septic rats. Braz. J. Med. Biol. Res. 2007, 40, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, B.S.; Sutton, M.M. Somatomedin-C levels in growth-impaired children and adolescents with chronic inflammatory bowel disease. Gastroenterology 1986, 91, 830–836. [Google Scholar] [CrossRef]
- Thomas, A.G.; Holly, J.M.; Taylor, F.; Miller, V. Insulin like growth factor-I, insulin like growth factor binding protein-1, and insulin in childhood Crohn’s disease. Gut 1993, 34, 944–947. [Google Scholar] [CrossRef] [PubMed]
- Katsanos, K.H.; Tsatsoulis, A.; Christodoulou, D.; Challa, A.; Katsaraki, A.; Tsianos, E.V. Reduced serum insulin-like growth factor-1 (IGF-1) and igf-binding protein-3 levels in adults with inflammatory bowel disease. Growth Horm. IGF Res. 2001, 11, 364–367. [Google Scholar] [CrossRef] [PubMed]
- Gronbek, H.; Thogersen, T.; Frystyk, J.; Vilstrup, H.; Flyvbjerg, A.; Dahlerup, J.F. Low free and total insulinlike growth factor i (IGF-I) and IGF binding protein-3 levels in chronic inflammatory bowel disease: Partial normalization during prednisolone treatment. Am. J. Gastroenterol. 2002, 97, 673–678. [Google Scholar] [CrossRef]
- Eivindson, M.; Gronbaek, H.; Flyvbjerg, A.; Frystyk, J.; Zimmermann-Nielsen, E.; Dahlerup, J.F. The insulin-like growth factor (IGF)-system in active ulcerative colitis and Crohn’s disease: Relations to disease activity and corticosteroid treatment. Growth Horm. IGF Res. 2007, 17, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.C.; Smyth, A.; McNeill, E.; Galloway, P.J.; Hassan, K.; McGrogan, P.; Ahmed, S.F. The growth hormone insulin-like growth factor 1 axis in children and adolescents with inflammatory bowel disease and growth retardation. Clin. Endocrinol. Oxf. 2010, 73, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Tenore, A.; Berman, W.F.; Parks, J.S.; Bongiovanni, A.M. Basal and stimulated serum growth hormone concentrations in inflammatory bowel disease. J. Clin. Endocrinol. Metab. 1977, 44, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Ates, Y.; Degertekin, B.; Erdil, A.; Yaman, H.; Dagalp, K. Serum ghrelin levels in inflammatory bowel disease with relation to disease activity and nutritional status. Dig. Dis. Sci. 2008, 53, 2215–2221. [Google Scholar] [CrossRef] [PubMed]
- Reinecker, H.C.; Steffen, M.; Witthoeft, T.; Pflueger, I.; Schreiber, S.; MacDermott, R.P.; Raedler, A. Enhanced secretion of tumour necrosis factor-α, IL-6, and IL-1β by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn’s disease. Clin. Exp. Immunol. 1993, 94, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Peyrin-Biroulet, L.; Eisenhut, M.; Shin, J.I. IBD immunopathogenesis: A comprehensive review of inflammatory molecules. Autoimmun. Rev. 2017, 16, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Mauras, N.; George, D.; Evans, J.; Milov, D.; Abrams, S.; Rini, A.; Welch, S.; Haymond, M.W. Growth hormone has anabolic effects in glucocorticosteroid-dependent children with inflammatory bowel disease: A pilot study. Metabolism 2002, 51, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Henker, J. Therapy with recombinant growth hormone in children with crohn disease and growth failure. Eur. J. Pediatr. 1996, 155, 1066–1067. [Google Scholar] [CrossRef] [PubMed]
- Calenda, K.A.; Schornagel, I.L.; Sadeghi-Nejad, A.; Grand, R.J. Effect of recombinant growth hormone treatment on children with Crohn’s disease and short stature: A pilot study. Inflamm. Bowel Dis. 2005, 11, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Heyman, M.B.; Garnett, E.A.; Wojcicki, J.; Gupta, N.; Davis, C.; Cohen, S.A.; Gold, B.D.; Kirschner, B.S.; Baldassano, R.N.; Ferry, G.D.; et al. Growth hormone treatment for growth failure in pediatric patients with Crohn’s disease. J. Pediatr. 2008, 153, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Slonim, A.E.; Grovit, M.; Bulone, L. Effect of exclusion diet with nutraceutical therapy in juvenile Crohn’s disease. J. Am. Coll. Nutr. 2009, 28, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Denson, L.A.; Kim, M.O.; Bezold, R.; Carey, R.; Osuntokun, B.; Nylund, C.; Willson, T.; Bonkowski, E.; Li, D.; Ballard, E.; et al. A randomized controlled trial of growth hormone in active pediatric Crohn’s disease. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.C.; Kumar, P.; Galloway, P.J.; Blair, J.C.; Didi, M.; Dalzell, A.M.; Hassan, K.; McGrogan, P.; Ahmed, S.F. A preliminary trial of the effect of recombinant human growth hormone on short-term linear growth and glucose homeostasis in children with Crohn’s disease. Clin. Endocrinol. Oxf. 2011, 74, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.C.; Dalzell, A.M.; McGrogan, P.; Didi, M.; Laing, P.; Ahmed, S.F. The inflammatory milieu and the insulin like growth factor axis in children with inflammatory bowel disease following recombinant human growth hormone treatment. J. Biol. Regul. Homeost. Agents 2015, 29, 27–37. [Google Scholar] [PubMed]
- Slonim, A.E.; Bulone, L.; Damore, M.B.; Goldberg, T.; Wingertzahn, M.A.; McKinley, M.J. A preliminary study of growth hormone therapy for Crohn’s disease. N. Engl. J. Med. 2000, 342, 1633–1637. [Google Scholar] [CrossRef] [PubMed]
- Iannoli, P.; Miller, J.H.; Ryan, C.K.; Gu, L.H.; Ziegler, T.R.; Sax, H.C. Epidermal growth factor and human growth hormone accelerate adaptation after massive enterectomy in an additive, nutrient-dependent, and site-specific fashion. Surgery 1997, 122, 721–729. [Google Scholar] [CrossRef]
- Byrne, T.A.; Wilmore, D.W.; Iyer, K.; Dibaise, J.; Clancy, K.; Robinson, M.K.; Chang, P.; Gertner, J.M.; Lautz, D. Growth hormone, glutamine, and an optimal diet reduces parenteral nutrition in patients with short bowel syndrome: A prospective, randomized, placebo-controlled, double-blind clinical trial. Ann. Surg. 2005, 242, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Li, Y.; Wang, Z.; Wu, B.; Wang, J.; Li, J. Morphological adaptation in adult short bowel syndrome undergoing intestinal rehabilitation. J. Investig. Surg. 2013, 26, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Seguy, D.; Darmaun, D.; Duhamel, A.; Thuillier, F.; Cynober, L.; Cortot, A.; Gottrand, F.; Messing, B. Growth hormone enhances fat-free mass and glutamine availability in patients with short-bowel syndrome: An ancillary double-blind, randomized crossover study. Am. J. Clin. Nutr. 2014, 100, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Moller, J.; Jensen, M.B.; Frandsen, E.; Moller, N.; Kissmeyer, P.; Laurberg, S. Growth hormone treatment improves body fluid distribution in patients undergoing elective abdominal surgery. Clin. Endocrinol. Oxf. 1998, 49, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.B.; Kissmeyer-Nielsen, P.; Laurberg, S. Perioperative growth hormone treatment increases nitrogen and fluid balance and results in short-term and long-term conservation of lean tissue mass. Am. J. Clin. Nutr. 1998, 68, 840–846. [Google Scholar] [PubMed]
- Kissmeyer-Nielsen, P.; Jensen, M.B.; Laurberg, S. Perioperative growth hormone treatment and functional outcome after major abdominal surgery: A randomized, double-blind, controlled study. Ann. Surg. 1999, 229, 298–302. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soendergaard, C.; Young, J.A.; Kopchick, J.J. Growth Hormone Resistance—Special Focus on Inflammatory Bowel Disease. Int. J. Mol. Sci. 2017, 18, 1019. https://doi.org/10.3390/ijms18051019
Soendergaard C, Young JA, Kopchick JJ. Growth Hormone Resistance—Special Focus on Inflammatory Bowel Disease. International Journal of Molecular Sciences. 2017; 18(5):1019. https://doi.org/10.3390/ijms18051019
Chicago/Turabian StyleSoendergaard, Christoffer, Jonathan A. Young, and John J. Kopchick. 2017. "Growth Hormone Resistance—Special Focus on Inflammatory Bowel Disease" International Journal of Molecular Sciences 18, no. 5: 1019. https://doi.org/10.3390/ijms18051019
APA StyleSoendergaard, C., Young, J. A., & Kopchick, J. J. (2017). Growth Hormone Resistance—Special Focus on Inflammatory Bowel Disease. International Journal of Molecular Sciences, 18(5), 1019. https://doi.org/10.3390/ijms18051019