
Inhibition or Stimulation of Autophagy Affects Early Formation of Lipofuscin-Like Autofluorescence in the Retinal Pigment Epithelium Cell

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
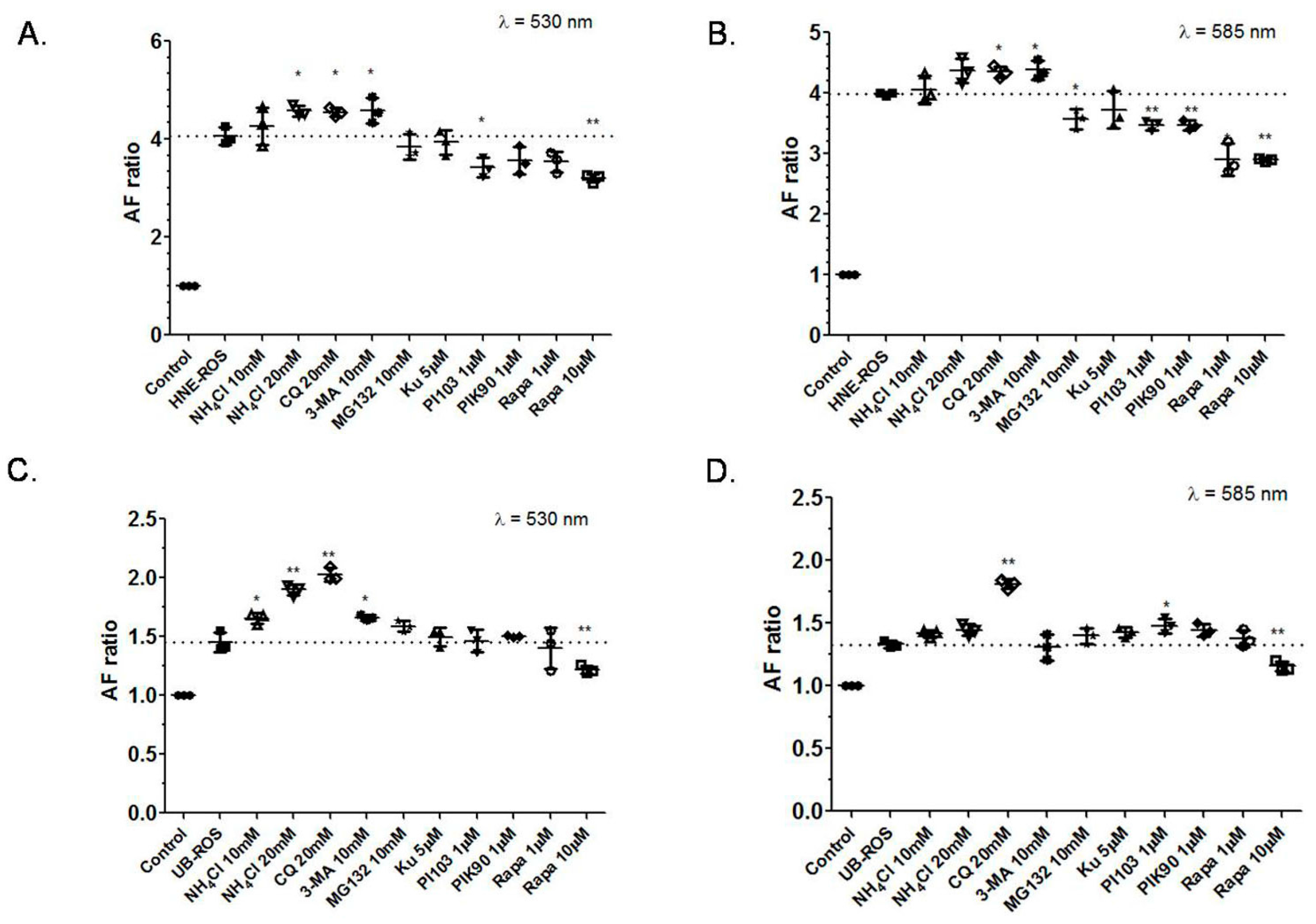
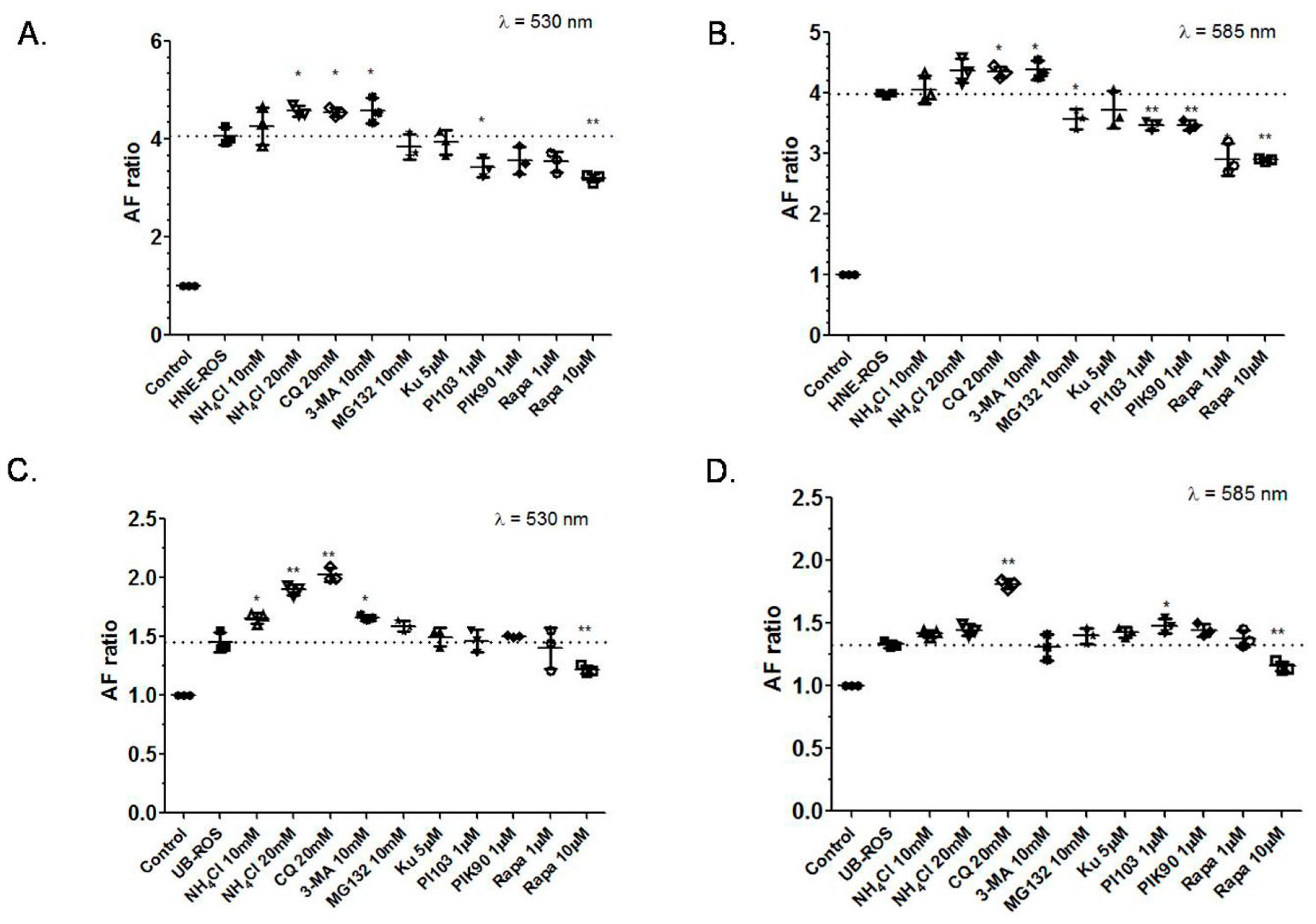
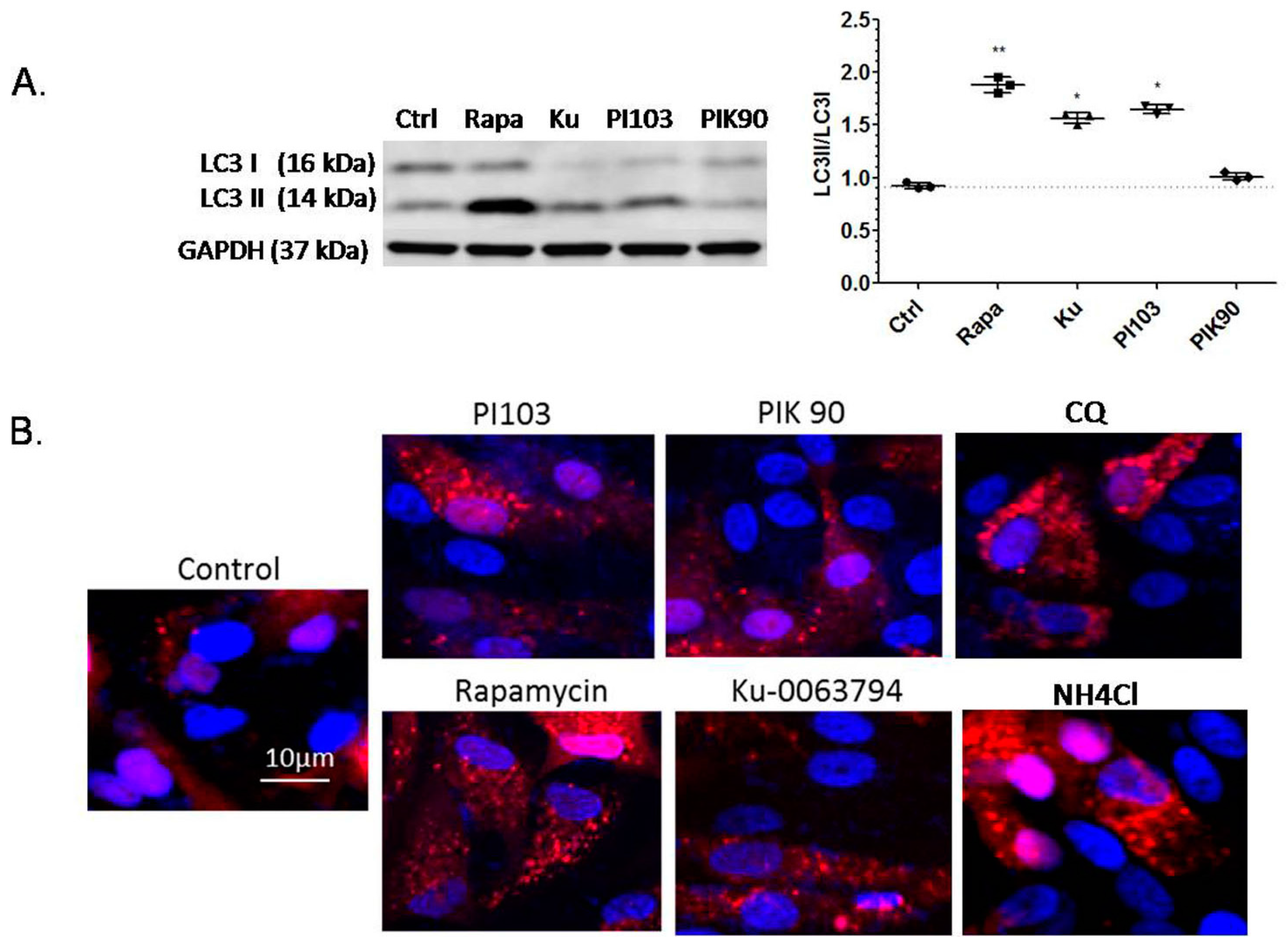
2.1. Effects of Autophagy Induction or Inhibition on RPE Cells Autofluorescence
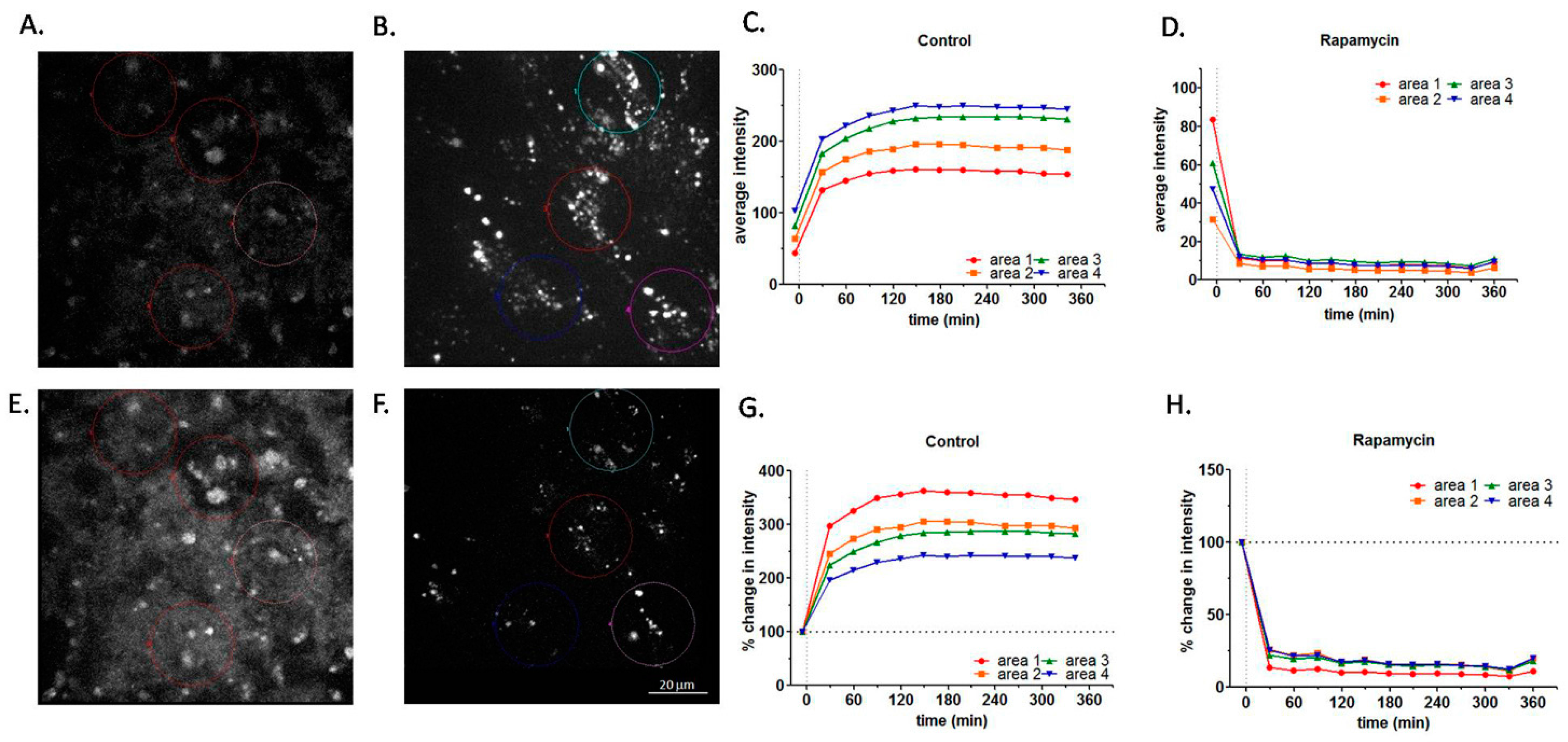
2.2. Effect of Rapamycin Treatment on RPE Autofluorescence by Live Cell Imaging
2.3. Effects of Autophagy Induction on Protein Expression and Lysosomal Activity by Confocal Microscopy
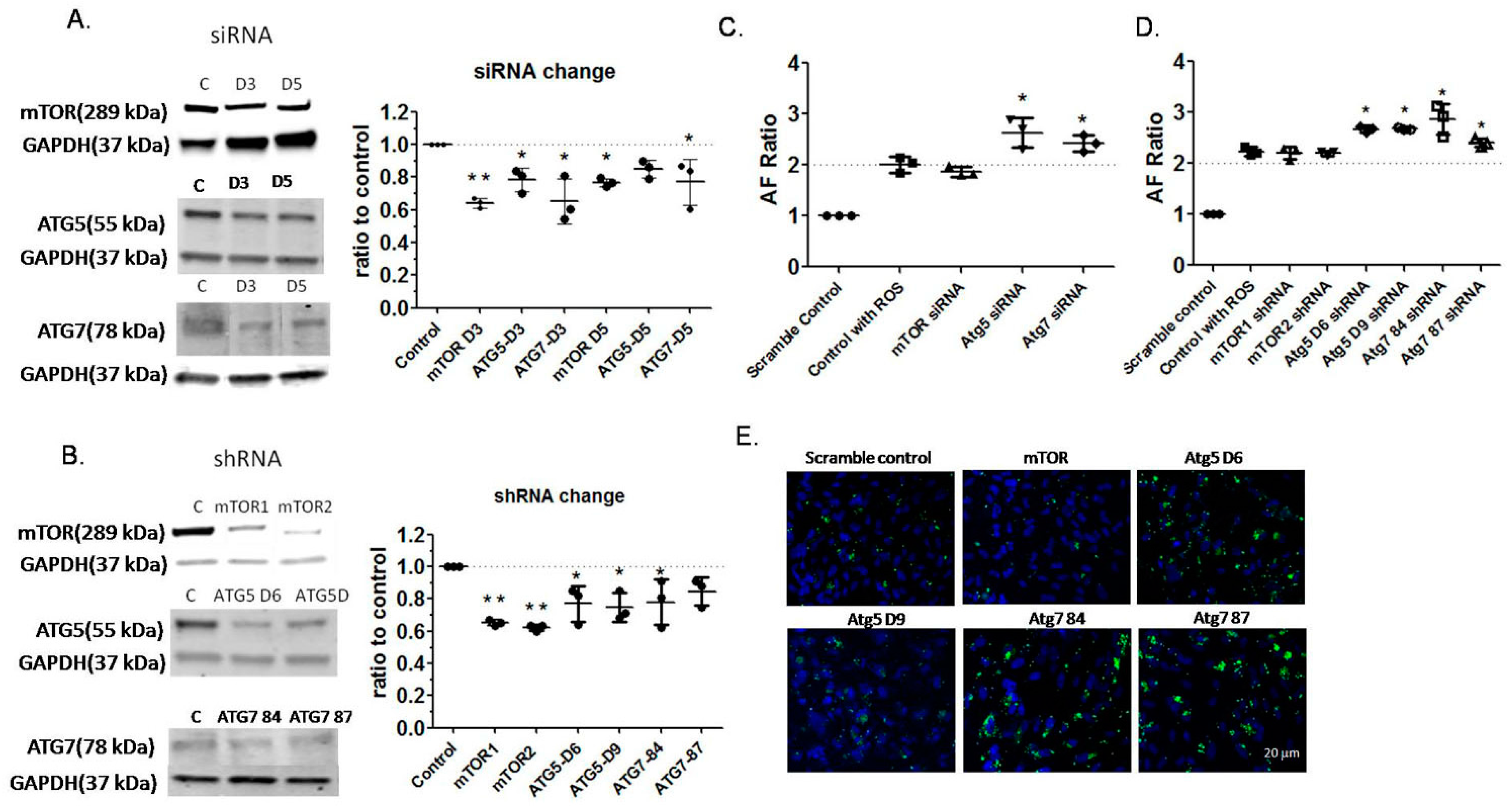
2.4. Effects of Autophagy Inhibition by RNA Interference on Autofluorescence of RPE Cells
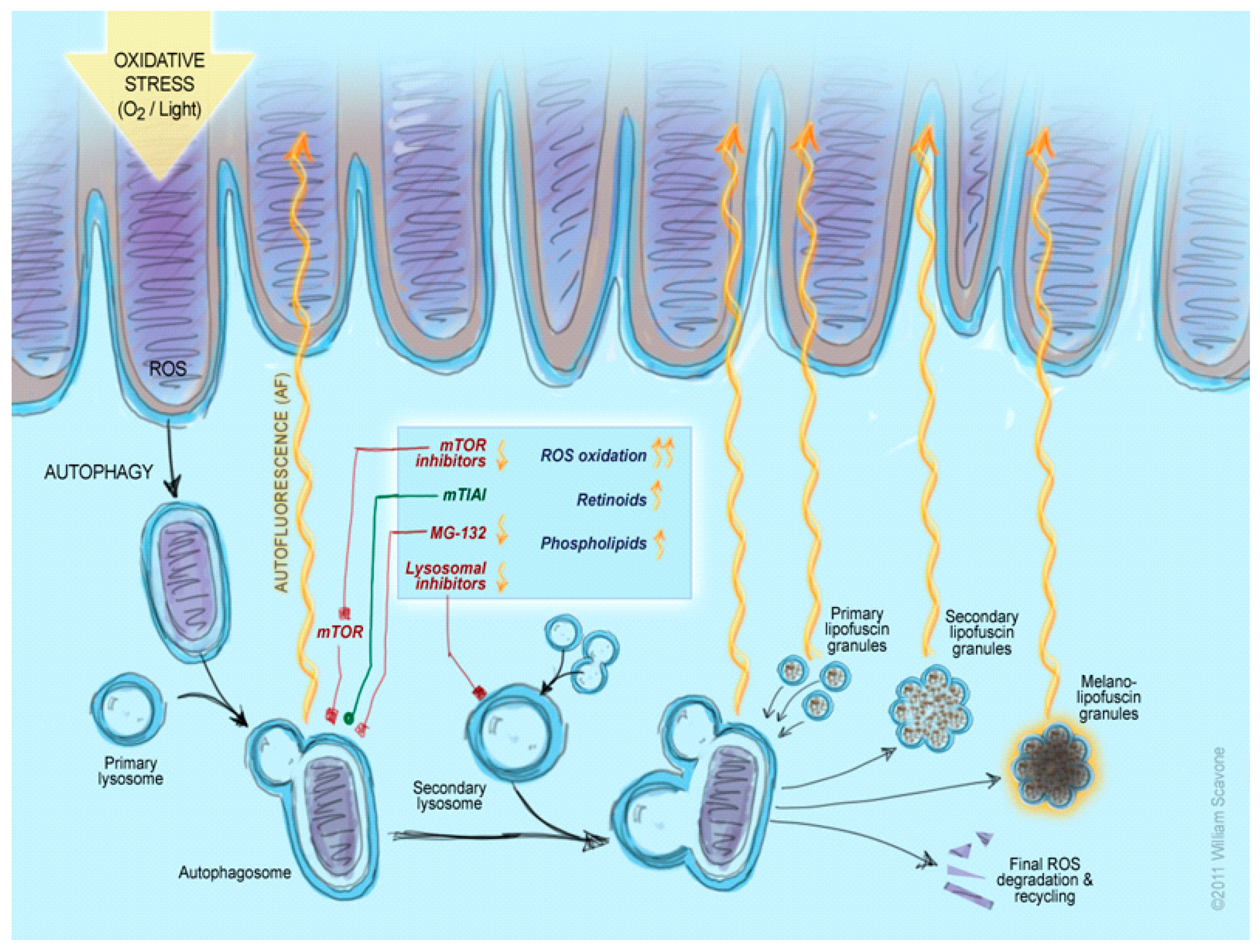
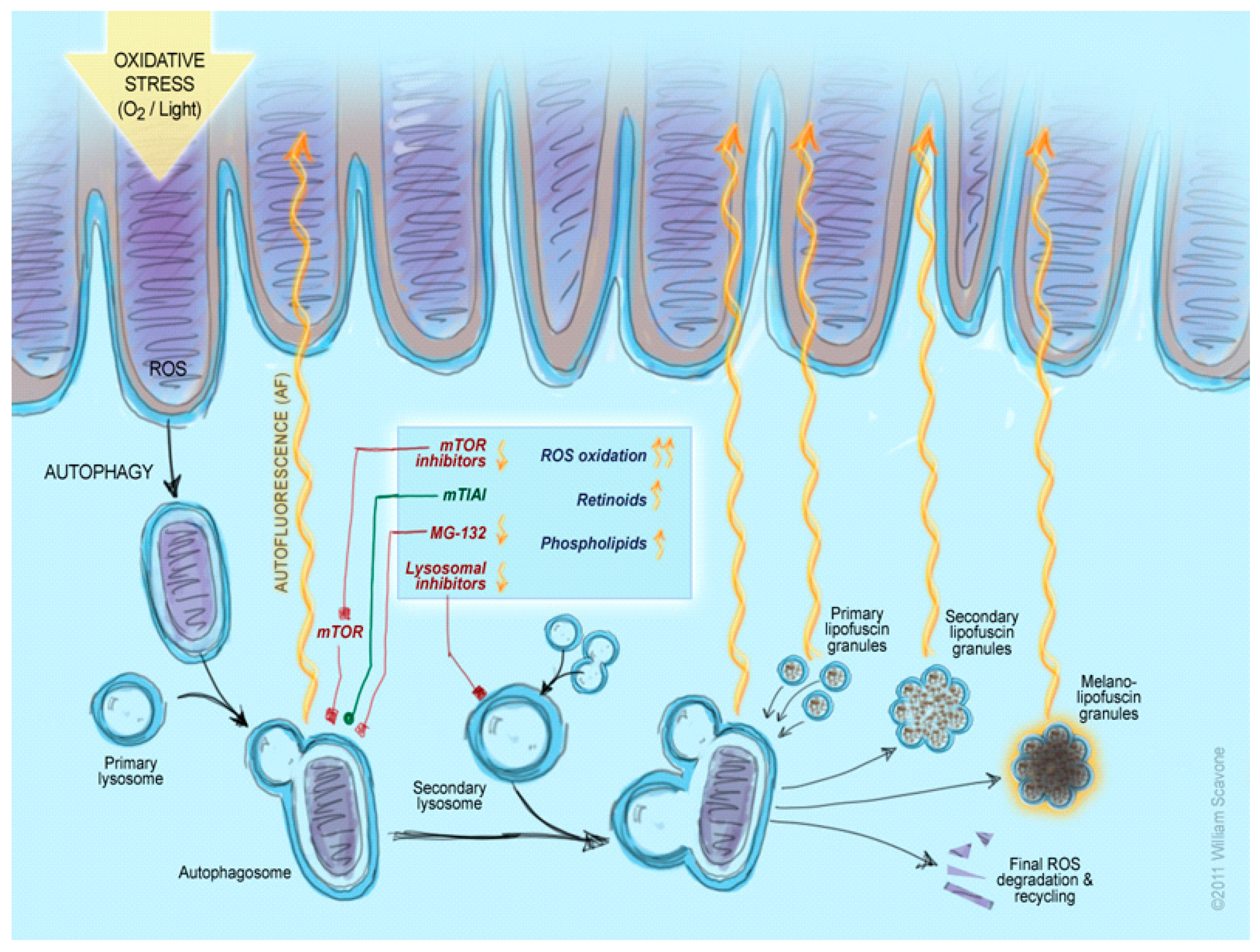
3. Discussion
3.1. Inhibition of the Lysosomal Complex and Autophagy Increases LLAF
3.2. Inhibition of mTOR and PIK3 α Pathways Decreases LLAF
4. Materials and Methods
4.1. RPE Cell Culture, ROS Isolation and Modification
4.2. RPE Cell Treatment
4.3. Flow Cytometry
4.4. RNA Interference
4.5. Lentivirus Vectors
- shATG5 D6 (TRCN0000151963): GGATGAGATAACTGAAAGG;
- shATG5 D9 (TRCN0000151474): GGCATTATCCAATTGGTTT;
- shATG7 84 (TRCN0000007584): GCCTGCTGAGGAGCTCTCCAT;
- shATG7 87 (TRCN0000007587): CCCAGCTATTGGAACACTGTA;
- mTOR1: TTCAGCGTCCCTACCTTCTTCT;
- mTOR 2: CCGCATTGTCTCTATCAAGTT.
4.6. Analysis of Punctate Cherry-LC3
4.7. Immunoblot Analysis
4.8. Confocal Microscopy
4.9. Live Cell Imaging and Fluorescence Quantification
4.10. Image Analysis of Live Cell Imaging
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Adler, L.T.; Boyer, N.P.; Chen, C.; Ablonczy, Z.; Crouch, R.K.; Koutalos, Y. The 11-cis Retinal Origins of Lipofuscin in the Retina. Prog. Mol. Biol. Transl. Sci. 2015, 134, e1–e12. [Google Scholar] [PubMed]
- Nandakumar, N.; Buzney, S.; Weiter, J.J. Lipofuscin and the principles of fundus autofluorescence: A review. Semin. Ophthalmol. 2012, 27, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Dorey, C.K.; Wu, G.; Ebenstein, D.; Garsd, A.; Weiter, J.J. Cell loss in the aging retina. Relationship to lipofuscin accumulation and macular degeneration. Investig. Ophthalmol. Vis. Sci. 1989, 30, 1691–1699. [Google Scholar]
- Sparrow, J.; Boulton, M. RPE lipofuscin and its role in retinal pathobiology. Exp. Eye Res. 2005, 80, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, J.R.; Hicks, D.; Hamel, C.P. The retinal pigment epithelium in health and disease. Curr. Mol. Med. 2010, 10, 802–823. [Google Scholar] [CrossRef] [PubMed]
- Brunk, U.T.; Terman, A. Lipofuscin: Mechanisms of age-related accumulation and influence on cell function. Free Radic. Biol. Med. 2002, 33, 611–619. [Google Scholar] [CrossRef]
- Kevany, B.M.; Palczewski, K. Phagocytosis of Retinal Rod and Cone Photoreceptors. Physiology 2010, 25, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Snodderly, D.M.; Sandstrom, M.M.; Leung, I.Y.; Zucker, C.L.; Neuringer, M. Retinal pigment epithelial cell distribution in central retina of rhesus monkeys. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2815–2818. [Google Scholar]
- Delori, F.C.; Goger, D.G.; Dorey, C.K. Age-related accumulation and spatial distribution of lipofuscin in RPE of normal subjects. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1855–1866. [Google Scholar]
- Kopitz, J.; Holz, F.; Kaemmerer, E.; Schutt, F. Lipids and lipid peroxidation products in the pathogenesis of age-related macular degeneration. Biochimie 2004, 86, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Valckenberg, S.; Fleckenstein, M.; Scholl, H.P.; Holz, F.G. Fundus autofluorescence and progression of age-related macular degeneration. Surv. Ophthalmol. 2009, 54, 96–117. [Google Scholar] [CrossRef] [PubMed]
- Terman, A.; Brunk, U.T. Lipofuscin. Int. J. Biochem. Cell Biol. 2004, 36, 1400–1404. [Google Scholar] [PubMed]
- Gerth, C.; Zawadzki, R.J.; Choi, S.S.; Keltner, J.L.; Park, S.S.; Werner, J.S. Visualization of lipofuscin accumulation in Stargardt macular dystrophy by high-resolution Fourier-domain optical coherence tomography. Arch. Ophthalmol. 2007, 125, 575. [Google Scholar] [CrossRef] [PubMed]
- Robson, A.G.; Michaelides, M.; Saihan, Z.; Bird, A.C.; Webster, A.R.; Moore, A.T.; Fitzke, F.W.; Holder, G.E. Functional characteristics of patients with retinal dystrophy that manifest abnormal parafoveal annuli of high density fundus autofluorescence: A review and update. Doc. Ophthalmol. 2008, 116, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Kellner, U.; Kellner, S.; Weber, B.H.; Fiebig, B.; Weinitz, S.; Ruether, K. Lipofuscin- and melanin-related fundus autofluorescence visualize different retinal pigment epithelial alterations in patients with retinitis pigmentosa. Eye 2009, 23, 1349–1359. [Google Scholar] [CrossRef] [PubMed]
- Seglen, P.O.; Bohley, P. Autophagy and other vacuolar protein degradation mechanisms. Experientia 1992, 48, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.; Schilling, J.D. Distinct lysosome phenotypes influence inflammatory function in peritoneal and bone marrow-derived macrophages. Int. J. Inflam. 2014, 2014, 154936. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C.J.; Rakoczy, P.E.; Constable, I.J. Lipofuscin of the retinal pigment epithelium: A review. Eye 1995, 9, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Hyttinen, J.; Ryhanen, T.; Viiri, J.; Paimela, T.; Toropainen, E.; Sorri, I.; Salminen, A. Mechanisms of protein aggregation in the retinal pigment epithelial cells. Front. Biosci. 2010, 2, 1374–1384. [Google Scholar] [CrossRef]
- Tzekov, R.; Stein, L.; Kaushal, S. Protein misfolding and retinal degeneration. Cold Spring Harb. Perspect. Biol. 2011, 3, a007492. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Sinha, D.; Blasiak, J.; Kauppinen, A.; Vereb, Z.; Salminen, A.; Boulton, M.E.; Petrovski, G. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy 2013, 9, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Mitter, S.K.; Rao, H.V.; Qi, X.; Cai, J.; Sugrue, A.; Dunn, W.A., Jr.; Grant, M.B.; Boulton, M.E. Autophagy in the retina: A potential role in age-related macular degeneration. Adv. Exp. Med. Biol. 2012, 723, 83–90. [Google Scholar] [PubMed]
- Frost, L.S.; Mitchell, C.H.; Boesze-Battaglia, K. Autophagy in the eye: Implications for ocular cell health. Exp. Eye Res. 2014, 124, 56–66. [Google Scholar] [PubMed]
- Li, C.P.; Yao, J.; Tao, Z.F.; Li, X.M.; Jiang, Q.; Yan, B. Epigallocatechin-gallate (EGCG) regulates autophagy in human retinal pigment epithelial cells: A potential role for reducing UVB light-induced retinal damage. Biochem. Biophys. Res. Commun. 2013, 438, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Krohne, T.U.; Stratmann, N.K.; Kopitz, J.; Holz, F.G. Effects of lipid peroxidation products on lipofuscinogenesis and autophagy in human retinal pigment epithelial cells. Exp. Eye Res. 2010, 90, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Tzekov, R.; McDowell, J.H.; Smith, W.C.; Tang, S.; Kaushal, S. Formation of lipofuscin-like material in the RPE Cell by different components of rod outer segments. Exp. Eye Res. 2013, 112, 57–67. [Google Scholar] [PubMed]
- Liu, J.; Lu, W.; Reigada, D.; Nguyen, J.; Laties, A.M.; Mitchell, C.H. Restoration of lysosomal pH in RPE cells from cultured human and ABCA4−/− mice: Pharmacologic approaches and functional recovery. Investig. Ophthalmol. Vis. Sci. 2008, 49, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.D.; Young, M.R. Ammonium chloride, an inhibitor of phagosome-lysosome fusion in macrophages, concurrently induces phagosome-endosome fusion, and opens a novel pathway: Studies of a pathogenic mycobacterium and a nonpathogenic yeast. J. Exp. Med. 1991, 174, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Sundelin, S.P.; Terman, A. Different effects of chloroquine and hydroxychloroquine on lysosomal function in cultured retinal pigment epithelial cells. APMIS 2002, 110, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.T.; Tan, H.L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.N.; Codogno, P.; Shen, H.M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Ravikumar, B.; Floto, R.A.; Rubinsztein, D.C. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009, 16, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martinez, J.M.; Moran, J.; Clarke, R.G.; Gray, A.; Cosulich, S.C.; Chresta, C.M.; Alessi, D.R. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR). Biochem. J. 2009, 421, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.W.; Knight, Z.A.; Goldenberg, D.D.; Yu, W.; Mostov, K.E.; Stokoe, D.; Shokat, K.M.; Weiss, W.A. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell 2006, 9, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.W.; Cheng, C.K.; Nicolaides, T.P.; Hackett, C.S.; Knight, Z.A.; Shokat, K.M.; Weiss, W.A. A dual phosphoinositide-3-kinase alpha/mTOR inhibitor cooperates with blockade of epidermal growth factor receptor in PTEN-mutant glioma. Cancer Res. 2007, 67, 7960–7965. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yamamoto, A.; Hatano, M.; Kobayashi, Y.; Kabeya, Y.; Suzuki, K.; Tokuhisa, T.; Ohsumi, Y.; Yoshimori, T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 2001, 152, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.H.; Cho, K.S.; Hwang, J.J.; Lee, S.J.; Choi, J.A.; Koh, J.Y. Induction of lysosomal dilatation, arrested autophagy, and cell death by chloroquine in cultured ARPE-19 cells. Investig. Ophthalmol. Vis. Sci. 2010, 51, 6030–6037. [Google Scholar] [CrossRef] [PubMed]
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The role of autophagy during the early neonatal starvation period. Nature 2004, 432, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, J.R.; Gregory-Roberts, E.; Yamamoto, K.; Blonska, A.; Ghosh, S.K.; Ueda, K.; Zhou, J. The bisretinoids of retinal pigment epithelium. Prog. Retin. Eye Res. 2012, 31, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yanase, E.; Feng, X.; Siegel, M.M.; Sparrow, J.R. Structural characterization of bisretinoid A2E photocleavage products and implications for age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 7275–7280. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, J.R. Bisretinoids of RPE lipofuscin: Trigger for complement activation in age-related macular degeneration. Adv. Exp. Med. Biol. 2010, 703, 63–74. [Google Scholar] [PubMed]
- Sparrow, J.R.; Yoon, K.D.; Wu, Y.; Yamamoto, K. Interpretations of fundus autofluorescence from studies of the bisretinoids of the retina. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4351–4357. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, M.; Enenkel, C. Intracellular Dynamics of the Ubiquitin-Proteasome-System. F1000Res 2015, 4, 367. [Google Scholar] [PubMed]
- Kenney, D.L.; Benarroch, E.E. The autophagy-lysosomal pathway: General concepts and clinical implications. Neurology 2015, 85, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Campello, L.; Esteve-Rudd, J.; Cuenca, N.; Martin-Nieto, J. The ubiquitin-proteasome system in retinal health and disease. Mol. Neurobiol. 2013, 47, 790–810. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.M.; Gombart, Z.J.; Chen, J.W. Chloroquine treatment of ARPE-19 cells leads to lysosome dilation and intracellular lipid accumulation: Possible implications of lysosomal dysfunction in macular degeneration. Cell Biosci. 2011, 1, 10. [Google Scholar] [PubMed]
- Tzekov, R. Ocular toxicity due to chloroquine and hydroxychloroquine: Electrophysiological and visual function correlates. Doc. Ophthalmol. 2005, 110, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, M.; Schutt, F.; Holz, F.G.; Kopitz, J. Inhibition of the ATP-driven proton pump in RPE lysosomes by the major lipofuscin fluorophore A2-E may contribute to the pathogenesis of age-related macular degeneration. FASEB J. 2004, 18, 562–564. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, C.J.; Rakoczy, P.E.; Constable, I.J. A simple flow cytometric technique to quantify rod outer segment phagocytosis in cultured retinal pigment epithelial cells. Curr. Eye Res. 1996, 15, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Haralampus-Grynaviski, N.M.; Lamb, L.E.; Clancy, C.M.; Skumatz, C.; Burke, J.M.; Sarna, T.; Simon, J.D. Spectroscopic and morphological studies of human retinal lipofuscin granules. Proc. Natl. Acad. Sci. USA 2003, 100, 179–3184. [Google Scholar] [CrossRef] [PubMed]
- Delori, F.; Greenberg, J.P.; Woods, R.L.; Fischer, J.; Duncker, T.; Sparrow, J.; Smith, R.T. Quantitative measurements of autofluorescence with the scanning laser ophthalmoscope. Investig. Ophthalmol. Vis. Sci. 2011, 52, 9379–9390. [Google Scholar] [CrossRef] [PubMed]
- Kaluzny, J.; Purta, P.; Poskin, Z.; Rogers, J.D.; Fawzi, A.A. Ex Vivo Confocal Spectroscopy of Autofluorescence in Age-Related Macular Degeneration. PLoS ONE 2016, 11, e0162869. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bai, Y.; Huang, L.; Qi, Y.; Zhang, Q.; Li, S.; Wu, Y.; Li, X. Protective effect of autophagy on human retinal pigment epithelial cells against lipofuscin fluorophore A2E: Implications for age-related macular degeneration. Cell Death Dis. 2015, 6, e1972. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.W.; Cheng, C.; Hackett, C.; Feldman, M.; Houseman, B.T.; Nicolaides, T.; Haas-Kogan, D.; James, C.D.; Oakes, S.A.; Debnath, J.; et al. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci. Signal. 2010, 3, ra81. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.L.; Lukas, T.J.; Yuan, M.; Du, N.; Tso, M.O.; Neufeld, A.H. Autophagy and exosomes in the aged retinal pigment epithelium: Possible relevance to drusen formation and age-related macular degeneration. PLoS ONE 2009, 4, e4160. [Google Scholar] [CrossRef] [PubMed]
- Ryhanen, T.; Hyttinen, J.M.; Kopitz, J.; Rilla, K.; Kuusisto, E.; Mannermaa, E.; Viiri, J.; Holmberg, C.I.; Immonen, I.; Meri, S.; et al. Crosstalk between Hsp70 molecular chaperone, lysosomes and proteasomes in autophagy-mediated proteolysis in human retinal pigment epithelial cells. J. Cell. Mol. Med. 2009, 13, 3616–3631. [Google Scholar] [CrossRef] [PubMed]
- Kazmin, R.; Rose, A.; Szczepek, M.; Elgeti, M.; Ritter, E.; Piechnick, R.; Hofmann, K.P.; Scheerer, P.; Hildebrand, P.W.; Bartl, F.J. The Activation Pathway of Human Rhodopsin in Comparison to Bovine Rhodopsin. J. Biol. Chem. 2015, 290, 20117–20127. [Google Scholar] [CrossRef] [PubMed]
- Andersson, H.; Baechi, T.; Hoechl, M.; Richter, C. Autofluorescence of living cells. J. Microsc. 1998, 191, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Tzekov, R.; Tang, S.B.; Kaushal, S. Accumulation and autofluorescence of phagocytized rod outer segment material in macrophages and microglial cells. Mol. Vis. 2012, 18, 103–113. [Google Scholar] [PubMed]
- Kaemmerer, E.; Schutt, F.; Krohne, T.U.; Holz, F.G.; Kopitz, J. Effects of Lipid Peroxidation-Related Protein Modifications on RPE Lysosomal Functions and POS Phagocytosis. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1342–1347. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, L.; Tzekov, R.; Li, H.; McDowell, J.H.; Gao, G.; Smith, W.C.; Tang, S.; Kaushal, S. Inhibition or Stimulation of Autophagy Affects Early Formation of Lipofuscin-Like Autofluorescence in the Retinal Pigment Epithelium Cell. Int. J. Mol. Sci. 2017, 18, 728. https://doi.org/10.3390/ijms18040728
Lei L, Tzekov R, Li H, McDowell JH, Gao G, Smith WC, Tang S, Kaushal S. Inhibition or Stimulation of Autophagy Affects Early Formation of Lipofuscin-Like Autofluorescence in the Retinal Pigment Epithelium Cell. International Journal of Molecular Sciences. 2017; 18(4):728. https://doi.org/10.3390/ijms18040728
Chicago/Turabian StyleLei, Lei, Radouil Tzekov, Huapeng Li, J. Hugh McDowell, Guangping Gao, W. Clay Smith, Shibo Tang, and Shalesh Kaushal. 2017. "Inhibition or Stimulation of Autophagy Affects Early Formation of Lipofuscin-Like Autofluorescence in the Retinal Pigment Epithelium Cell" International Journal of Molecular Sciences 18, no. 4: 728. https://doi.org/10.3390/ijms18040728
APA StyleLei, L., Tzekov, R., Li, H., McDowell, J. H., Gao, G., Smith, W. C., Tang, S., & Kaushal, S. (2017). Inhibition or Stimulation of Autophagy Affects Early Formation of Lipofuscin-Like Autofluorescence in the Retinal Pigment Epithelium Cell. International Journal of Molecular Sciences, 18(4), 728. https://doi.org/10.3390/ijms18040728