
Function and Clinical Implications of Long Non-Coding RNAs in Melanoma

 ,
,
Abstract
1. Introduction
1.1. Therapeutic Modalities
1.1.1. Targeted Therapy
BRAF Mutated Melanoma
NRAS Mutated Melanoma
NF1 Mutated Melanoma
KIT Mutated Melanoma
1.1.2. Immunotherapy
2. Long Non-Coding RNAs
2.1. Signal LncRNA
2.2. Decoy LncRNA
2.3. Guide LncRNA
2.4. Scaffold LncRNA
2.5. Multifunctionality and Novel Mechanisms
3. Long Non-Coding RNAs in Melanoma
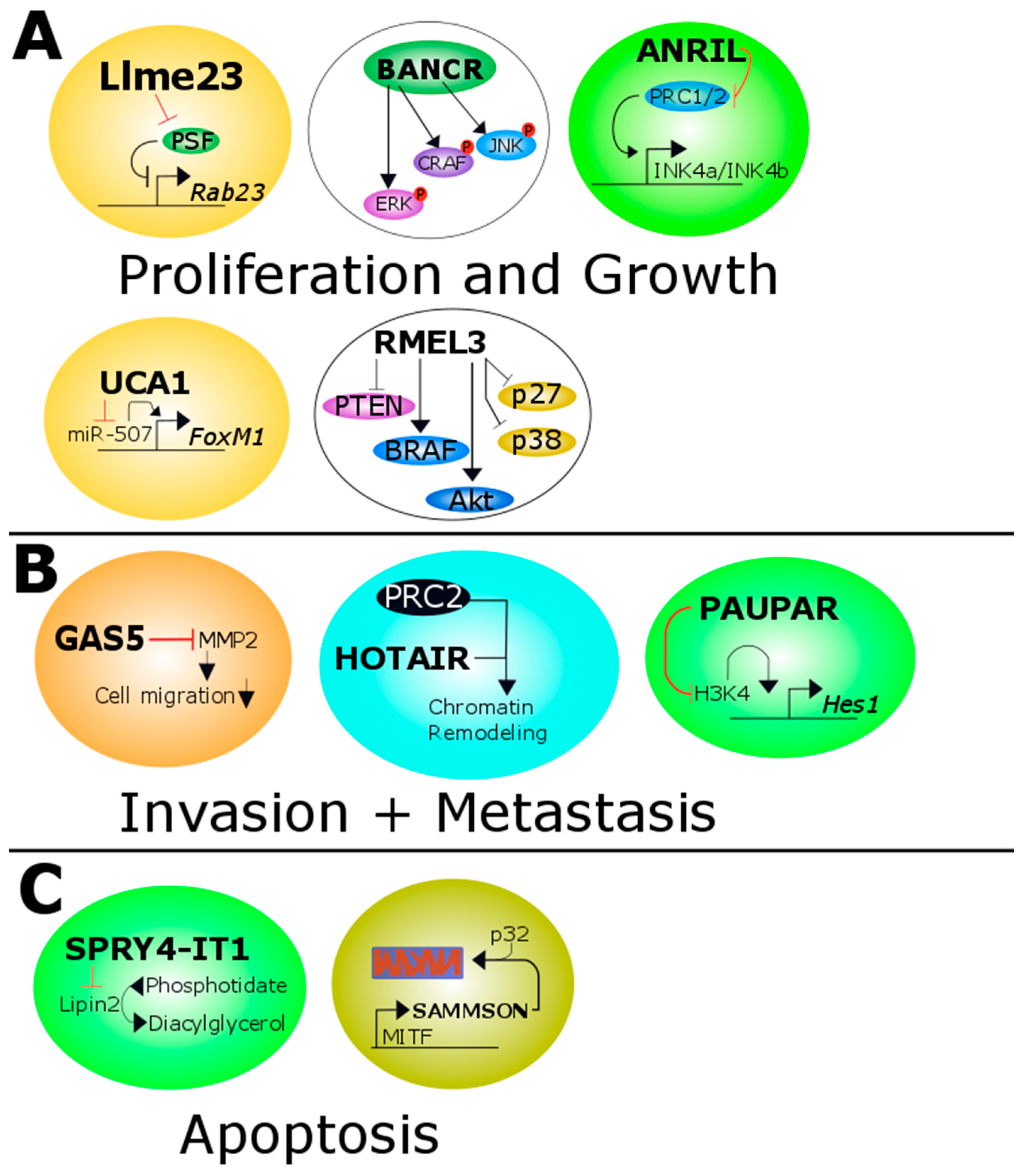
3.1. ANRIL
3.2. BANCR
3.3. CASC15
3.4. GAS5
3.5. HOTAIR
3.6. Llme23
3.7. MALAT1
3.8. PAUPAR
3.9. RMEL3
3.10. SAMMSON
3.11. SNHG5
3.12. SPRY4-IT1
3.13. UCA1
4. Conclusions and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AMPK | adenosine monophosphate-activated protein kinase |
| ANRIL | antisense lncRNA in INK4 locus |
| BANCR | BRAF-activated non-coding RNA |
| BRAF | B-Raf proto-oncogene, serine/threonine kinase |
| BRIM-3 | BRAF Inhibitor in Melanoma 3 |
| CASC15 | cancer susceptibility candidate 15 |
| CDKN2A | cyclin dependent kinase inhibitor 2A |
| ceRNA | competing endogenous RNA |
| COOLAIR | cold induced long antisense intragenic RNA |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein 4 |
| DTIC | dacarbazine |
| ENCODE | ENCyclopedia of DNA Elements |
| ERK | extracellular signal-regulated kinase |
| GAS5 | growth arrest-specific transcript 5 |
| HES1 | hairy and enhancer of split-1 |
| HOTAIR | HOX transcript antisense RNA |
| HOTTIP | HOXA distal transcript antisense RNA |
| HOX | homebox |
| HOXA | homebox A |
| KIT | KIT proto-oncogene receptor tyrosine kinase |
| lncRNA | long non-coding RNA |
| LSD1 | lysine-specific histone demethylase 1 |
| mAb | monoclonal antibody |
| MALAT1 | metastasis-associated lung adenocarcinoma transcript 1 |
| MAPK | mitogen-activated protein kinase |
| MEK | MAPK/ERK Kinase |
| miRNA | microRNA |
| MITF | melanogenesis associated transcription factor |
| MMP | matrix metalloproteinase |
| NBR2 | lncRNA neighbor of BRCA1 gene 2 |
| ncRNA | non-coding RNA |
| NEAT | nuclear-enriched transcript |
| NF1 | neurofibromin 1 |
| NRAS | neuroblastoma RAS viral (v-ras) oncogene homolog |
| nt | nucleotides |
| PAUPAR | PAX6 upstream antisense RNA |
| PD-1 | programmed cell death 1 |
| PD-L1 | programmed cell death ligand-1 |
| piRNA | PIWI-interacting RNA |
| PRC | polycomb repressive complexe |
| PSF | protein associated splicing factor |
| RAF | rapidly Accelerated Fibrosarcoma |
| RAS | rat sarcoma |
| RTK | receptor tyrosine kinase |
| SAMMSON | survival associated mitochondrial melanoma-specific oncogenic non-coding RNA |
| siRNA | small interfering RNA |
| SNHG5 | SnoRNA host gene 5 |
| SPRY4-IT1 | SPRY4 intronic transcript 1 |
| TERT | telomerase reverse transcriptase |
| TIM-3 | T-cell immunoglobulin and mucin-domain containing-3 |
| TP53 | tumor protein p53 |
| UCA1 | urothelial carcinoma-associated 1 |
| UV | ultraviolet |
References
- Deeks, E.D. Nivolumab: A review of its use in patients with malignant melanoma. Drugs 2014, 74, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Richtig, G.; Richtig, E.; Massone, C.; Hofmann-Wellenhof, R. Analysis of clinical, dermoscopic and histopathological features of primary melanomas of patients with metastatic disease—A retrospective study at the Department of Dermatology, Medical University of Graz, 2000–2010. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 1776–1781. [Google Scholar] [CrossRef] [PubMed]
- Balch, C.M.; Gershenwald, J.E.; Soong, S.J.; Thompson, J.F.; Atkins, M.B.; Byrd, D.R.; Buzaid, A.C.; Cochran, A.J.; Coit, D.G.; Ding, S.; et al. Final version of 2009 AJCC melanoma staging and classification. J. Clin. Oncol. 2009, 27, 6199–6206. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Masliah-Planchon, J.; Garinet, S.; Pasmant, E. RAS-MAPK pathway epigenetic activation in cancer: miRNAs in action. Oncotarget 2015, 7, 38892–38907. [Google Scholar]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Colombino, M.; Capone, M.; Lissia, A.; Cossu, A.; Rubino, C.; De Giorgi, V.; Massi, D.; Fonsatti, E.; Staibano, S.; Nappi, O.; et al. BRAF/NRAS mutation frequencies among primary tumors and metastases in patients with melanoma. J. Clin. Oncol. 2012, 30, 2522–2529. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Menzies, A.M.; Nagrial, A.M.; Haydu, L.E.; Hamilton, A.L.; Mann, G.J.; Hughes, T.M.; Thompson, J.F.; Scolyer, R.A.; Kefford, R.F. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J. Clin. Oncol. 2011, 29, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Richtig, G.; Aigelsreiter, A.; Kashofer, K.; Talakic, E.; Kupsa, R.; Schaider, H.; Richtig, E. Two case reports of rare BRAF mutations in exon 11 and exon 15 with discussion of potential treatment options. Case Rep. Oncol. 2016, 9, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Richtig, G.; Hoeller, C.; Kashofer, K.; Aigelsreiter, A.; Heinemann, A.; Kwong, L.N.; Pichler, M.; Richtig, E. Beyond the BRAFV600E hotspot-biology and clinical implications of rare BRAF gene mutations in melanoma patients. Br. J. Dermatol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ellerhorst, J.A.; Greene, V.R.; Ekmekcioglu, S.; Warneke, C.L.; Johnson, M.M.; Cooke, C.P.; Wang, L.E.; Prieto, V.G.; Gershenwald, J.E.; Wei, Q.; et al. Clinical correlates of NRAS and BRAF mutations in primary human melanoma. Clin. Cancer Res. 2011, 17, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Rafnar, T.; Sulem, P.; Stacey, S.N.; Geller, F.; Gudmundsson, J.; Sigurdsson, A.; Jakobsdottir, M.; Helgadottir, H.; Thorlacius, S.; Aben, K.K.H.; et al. Sequence variants at the TERT-CLPTM1L locus associate with many cancer types. Nat. Genet. 2009, 41, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in uveal melanoma. N. Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The genetic evolution of melanoma from precursor lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.; Watson, I.R. Molecular characterisation of cutaneous melanoma: Creating a framework for targeted and immune therapies. Br. J. Cancer 2016, 115, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H.; Lao, C.D.; et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2014, 372, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.B.; Kefford, R.; Pavlick, A.C.; Infante, J.R.; Ribas, A.; Sosman, J.A.; Fecher, L.A.; Millward, M.; McArthur, G.A.; Hwu, P.; et al. Phase II study of the MEK1/MEK2 inhibitor trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J. Clin. Oncol. 2013, 31, 482–489. [Google Scholar] [CrossRef] [PubMed]
- McGettigan, S. Dabrafenib: A new therapy for use in BRAF-mutated metastatic melanoma. J. Adv. Pract. Oncol. 2014, 5, 211–215. [Google Scholar] [PubMed]
- Livingstone, E.; Zimmer, L.; Piel, S.; Schadendorf, D. PLX4032: Does it keep its promise for metastatic melanoma treatment? Expert Opin. Investig. Drugs 2010, 19, 1439–1449. [Google Scholar] [CrossRef] [PubMed]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, K.; Zhu, X.; Lin, G.; Song, F.; Zhao, Y.; Piao, Y.; Liu, J.; Cheng, W.; Bi, X.; et al. Encorafenib (LGX818), a potent BRAF inhibitor, induces senescence accompanied by autophagy in BRAFV600E melanoma cells. Cancer Lett. 2016, 370, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.-K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAFV600E inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, L.; Hillen, U.; Livingstone, E.; Lacouture, M.E.; Busam, K.; Carvajal, R.D.; Egberts, F.; Hauschild, A.; Kashani-Sabet, M.; Goldinger, S.M.; et al. Atypical melanocytic proliferations and new primary melanomas in patients with advanced melanoma undergoing selective BRAF Inhibition. J. Clin. Oncol. 2012, 30, 2375–2383. [Google Scholar] [CrossRef] [PubMed]
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.; Stokoe, D.; Gloor, S.L.; Vigers, G.; et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010, 464, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAFV600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Marincola, F.M.; Atkins, M.B. What’s new in melanoma? Combination! J. Transl. Med. 2015, 13, 213. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Gonzalez, R.; Pavlick, A.; Hamid, O.; Gajewski, T.F.; Daud, A.; Flaherty, L.; Logan, T.; Chmielowski, B.; Lewis, K.; et al. Combination of vemurafenib and cobimetinib in patients with advanced BRAFV600-mutated melanoma: A phase 1B study. Lancet Oncol. 2014, 15, 954–965. [Google Scholar] [CrossRef]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Puzanov, I. Treatment of NRAS-mutant melanoma. Curr. Treat. Options Oncol. 2015, 16, 15. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Schadendorf, D.; Berking, C.; Agarwala, S.S.; van Herpen, C.M.L.; Queirolo, P.; Blank, C.U.; Hauschild, A.; Beck, J.T.; St-Pierre, A.; et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: A non-randomised, open-label phase 2 study. Lancet Oncol. 2013, 14, 249–256. [Google Scholar] [CrossRef]
- Richtig, G.; Richtig, E.; Kashofer, K.; Koch, L.; Winter, G.; Hoefler, G.; Pichler, M.; Ehall, B.; Grübler, M.R.; Heinemann, A.; et al. Testing and clinical implications for non-V600 BRAF mutations in metastatic NRAS(mt) melanoma. Br. J. Dermatol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Nissan, M.H.; Pratilas, C.A.; Jones, A.M.; Ramirez, R.; Won, H.; Liu, C.; Tiwari, S.; Kong, L.; Hanrahan, A.J.; Yao, Z.; Merghoub, T.; et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 2014, 74, 2340–2350. [Google Scholar] [CrossRef] [PubMed]
- Ranzani, M.; Alifrangis, C.; Perna, D.; Dutton-Regester, K.; Pritchard, A.; Wong, K.; Rashid, M.; Robles-Espinoza, C.D.; Hayward, N.K.; Mcdermott, U.; et al. BRAF/NRAS wild-type melanoma, NF1 status and sensitivity to trametinib. Pigment Cell Melanoma Res. 2015, 28, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, S.R.; Theurillat, J.-P.; van Allen, E.; Wagle, N.; Hsiao, J.; Cowley, G.S.; Schadendorf, D.; Root, D.E.; Garraway, L.A. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013, 3, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Krauthammer, M.; Kong, Y.; Bacchiocchi, A.; Evans, P.; Pornputtapong, N.; Wu, C.; McCusker, J.P.; Ma, S.; Cheng, E.; Straub, R.; et al. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat. Genet. 2015, 47, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.A.; Busam, K.; Pinkel, D.; Bastian, B.C. Somatic activation of KIT in distinct subtypes of melanoma. J. Clin. Oncol. 2006, 24, 4340–4346. [Google Scholar] [CrossRef] [PubMed]
- Bastian, B.C.; Esteve-Puig, R. Targeting activated KIT signaling for melanoma therapy. J. Clin. Oncol. 2013, 31, 3288–3290. [Google Scholar] [CrossRef] [PubMed]
- Posch, C.; Moslehi, H.; Sanlorenzo, M.; Green, G.; Panzer-Grümayer, R.; Rappersberger, K.; Ortiz-Urda, S. Pharmacological inhibitors of c-KIT block mutant c-KIT mediated migration of melanocytes and melanoma cells in vitro and in vivo. Oncotarget 2016, 7, 45916–45925. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Corless, C.L.; Giobbie-Hurder, A.; Fletcher, J.A.; Zhu, M.; Marino-Enriquez, A.; Friedlander, P.; Gonzalez, R.; Weber, J.S.; Gajewski, T.F.; et al. Imatinib for melanomas harboring mutationally activated or amplified kit arising on mucosal, acral, and chronically sun-damaged skin. J. Clin. Oncol. 2013, 31, 3182–3190. [Google Scholar] [CrossRef] [PubMed]
- Minor, D.R.; Kashani-Sabet, M.; Garrido, M.; O’Day, S.J.; Hamid, O.; Bastian, B.C. Sunitinib therapy for melanoma patients with KIT mutations. Clin. Cancer Res. 2012, 18, 1457–1463. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Lawrence, D.P.; Weber, J.S.; Gajewski, T.F.; Gonzalez, R.; Lutzky, J.; O’Day, S.J.; Hamid, O.; Wolchok, J.D.; Chapman, P.B.; et al. Phase II study of nilotinib in melanoma harboring KIT alterations following progression to prior KIT inhibition. Clin. Cancer Res. 2015, 21, 2289–2296. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Kunkel, L.; Sznol, M.; Rosenberg, S.A. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: Long-term survival update. Cancer J. Sci. Am. 2000, 6, 11–14. [Google Scholar]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.-F.; Testori, A.; Grob, J.-J.; et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.-T.; Berman, D.M.; Wolchok, J.D. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.; Kuball, J.; Voelkl, S.; Wiendl, H.; Becker, B.; Walter, B.; Majdic, O.; Gajewski, T.F.; Theobald, M.; Andreesen, R.; et al. Blockade of PD-L1 (B7-H1) augments human tumor-specific T cell responses in vitro. Int. J. Cancer 2006, 119, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Kluger, H.; Sznol, M.; Carvajal, R.; Lawrence, D.; Atkins, M.; Powderly, J.; Sharfman, W.; Puzanov, I.; Smith, D.; et al. Durable, Long-Term Survival in Previously Treated Patients with Advanced Melanoma (MEL) Who Received Nivolumab (NIVO) Monotherapy in a Phase I Trial. Available online: http://www.abstractsonline.com/Plan/ViewAbstract.aspx?mID=4017&sKey=371fa616-a0cf-4bf-993d-ce424853b52c&cKey=616f965e-a236-4bd2-9f7a-6399bd6f3f6c&mKey=1d10d749-4b6a-4ab3-bcd4-f80fb1922267 (accessed on 22 January 2017).
- Galluzzi, L.; Eggermont, A.; Kroemer, G. Doubling the blockade for melanoma immunotherapy. Oncoimmunology 2016, 5, e1106127. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.-A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Romero, D. Immunotherapy: PD-1 says goodbye, TIM-3 says hello. Nat. Rev. Clin. Oncol. 2016, 13, 202–203. [Google Scholar] [CrossRef] [PubMed]
- Mittal, D.; Sinha, D.; Barkauskas, D.; Young, A.; Kalimutho, M.; Stannard, K.; Caramia, F.; Haibe-Kains, B.; Stagg, J.; Khanna, K.K.; et al. Adenosine 2B receptor expression on cancer cells promotes metastasis. Cancer Res. 2016, 76, 4372–4382. [Google Scholar] [CrossRef] [PubMed]
- Knowling, S.; Morris, K.V. Non-coding RNA and antisense RNA. Nature’s trash or treasure? Biochimie 2011, 93, 1922–1927. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project; Bernstein, B.E.; Birney, E.; Dunham, I.; Green, E.D.; Gunter, C.; Snyder, M. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Smolle, M.A.; Calin, H.N.; Pichler, M.; Calin, G.A. Noncoding RNAs and immune checkpoints: Clinical implications as cancer therapeutics. FEBS J. 2017. [Google Scholar] [CrossRef] [PubMed]
- Pichler, M.; Stiegelbauer, V.; Vychytilova-Faltejskova, P.; Ivan, C.; Ling, H.; Winter, E.; Zhang, X.; Goblirsch, M.; Wulf-Goldenberg, A.; Ohtsuka, M.; et al. Genome-wide miRNA analysis identifies miR-188-3p as a novel prognostic marker and molecular factor involved in colorectal carcinogenesis. Clin. Cancer Res. 2016, 23, 1323–1333. [Google Scholar] [CrossRef] [PubMed]
- Pichler, M.; Calin, G.A. MicroRNAs in cancer: From developmental genes in worms to their clinical application in patients. Br. J. Cancer 2015, 113, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Vincent, K.; Pichler, M.; Fodde, R.; Berindan-Neagoe, I.; Slack, F.J.; Calin, G.A. Junk DNA and the long non-coding RNA twist in cancer genetics. Oncogene 2015. [Google Scholar] [CrossRef] [PubMed]
- Dinger, M.E.; Pang, K.C.; Mercer, T.R.; Mattick, J.S. Differentiating protein-coding and noncoding RNA: Challenges and ambiguities. PLoS Comput. Biol. 2008, 4, e1000176. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; Carmichael, G.G. Long noncoding RNAs in mammalian cells: What, where, and why? Wiley Interdiscip. Rev. RNA 2010, 1, 2–21. [Google Scholar] [CrossRef] [PubMed]
- Lipovich, L.; Johnson, R.; Lin, C.Y. MacroRNA underdogs in a microRNA world: Evolutionary, regulatory, and biomedical significance of mammalian long non-protein-coding RNA. Biochim. Biophys. Acta Gene Regul. Mech. 2010, 1799, 597–615. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Cerk, S.; Schwarzenbacher, D.; Adiprasito, J.B.; Stotz, M.; Hutterer, G.C.; Gerger, A.; Ling, H.; Calin, G.A.; Pichler, M. Current status of long non-coding RNAs in human breast cancer. Int. J. Mol. Sci. 2016, 17, 1485. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, M.; Ling, H.; Ivan, C.; Pichler, M.; Matsushita, D.; Goblirsch, M.; Stiegelbauer, V.; Shigeyasu, K.; Zhang, X.; Chen, M.; et al. H19 noncoding RNA, an independent prognostic factor, regulates essential Rb-E2F and CDK8-β-catenin signaling in colorectal cancer. EBioMedicine 2016, 13, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Seles, M.; Hutterer, G.; Kiesslich, T.; Pummer, K.; Berindan-Neagoe, I.; Perakis, S.; Schwarzenbacher, D.; Stotz, M.; Gerger, A.; Pichler, M. Current insights into long non-coding RNAs in renal cell carcinoma. Int. J. Mol. Sci. 2016, 17, 573. [Google Scholar] [CrossRef] [PubMed]
- Smolle, M.A.; Bullock, M.D.; Ling, H.; Pichler, M.; Haybaeck, J. Long non-coding RNAs in endometrial carcinoma. Int. J. Mol. Sci. 2015, 16, 26463–26472. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.F. Non-coding RNAs: Meet thy masters. BioEssays 2010, 32, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Huarte, M.; Guttman, M.; Feldser, D.; Garber, M.; Koziol, M.J.; Kenzelmann-Broz, D.; Khalil, A.M.; Zuk, O.; Amit, I.; Rabani, M.; et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 2010, 142, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Guenther, M.G.; Levine, S.S.; Boyer, L.A.; Jaenisch, R.; Young, R.A. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 2007, 130, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, X.; Wu, H.; Ni, P.; Gu, Z.; Qiao, Y.; Chen, N.; Sun, F.; Fan, Q. CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucl. Acids Res. 2010, 38, 5366–5383. [Google Scholar] [CrossRef] [PubMed]
- Martianov, I.; Ramadass, A.; Serra Barros, A.; Chow, N.; Akoulitchev, A. Repression of the human dihydrofolate reductase gene by a non-coding interfering transcript. Nature 2007, 445, 666–670. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Hämmerle, M.; Eißmann, M.; Hsu, J.; Kim, Y.; Hung, G.; Revenko, A.; Arun, G.; Stentrup, M.; Groß, M.; et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013, 73, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Shen, Z.; Chakraborty, A.; Giri, S.; Freier, S.M.; Wu, X.; Zhang, Y.; Gorospe, M.; Prasanth, S.G.; Lal, A.; et al. Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B-MYB. PLoS Genet. 2013, 9, e1003368. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.; Chang, H.Y. Long noncoding RNA in genome regulation: Prospects and mechanisms. RNA Biol. 2011, 7, 582–585. [Google Scholar] [CrossRef]
- Good, M.C.; Zalatan, J.G.; Lim, W.A. Scaffold proteins: Hubs for controlling the flow of cellular information. Science 2011, 332, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, X.; Zhou, J.; Hu, J.; Zhang, D.; Liu, J.; Qiao, Y.; Zhan, Q. LncRNA HULC modulates the phosphorylation of YB-1 through serving as a scaffold of ERK and YB-1 to enhance hepatocarcinogenesis. Hepatology 2016. [Google Scholar] [CrossRef]
- Sun, C.-C.; Li, S.-J.; Li, G.; Hua, R.-X.; Zhou, X.-H.; Li, D.-J. Long intergenic noncoding RNA 00511 acts as an oncogene in non-small-cell lung cancer by binding to EZH2 and suppressing p57. Mol. Ther. Acids 2016, 5, e385. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Nakagawa, T.; Kitagawa, K.; Suzuki, S.; Liu, N.; Kitagawa, M.; Xiong, Y. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15 (INK4B) tumor suppressor gene. Oncogene 2011, 30, 1956–1962. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.-C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xiao, Z.-D.; Han, L.; Zhang, J.; Lee, S.-W.; Wang, W.; Lee, H.; Zhuang, L.; Chen, J.; Lin, H.-K.; et al. LncRNA NBR2 engages a metabolic checkpoint by regulating AMPK under energy stress. Nat. Cell Biol. 2016, 18, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xiao, Z.D.; Gan, B. An lncRNA switch for AMPK activation. Cell Cycle 2016, 15, 1948–1949. [Google Scholar] [CrossRef] [PubMed]
- Pasmant, E.; Laurendeau, I.; Héron, D.; Vidaud, M.; Vidaud, D.; Bièche, I. Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: Identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res. 2007, 67, 3963–3969. [Google Scholar] [CrossRef] [PubMed]
- Bishop, D.T.; Demenais, F.; Iles, M.M.; Harland, M.; Taylor, J.C.; Corda, E.; Randerson-Moor, J.; Aitken, J.F.; Avril, M.F.; Azizi, E.; et al. Genome-wide association study identifies three loci associated with melanoma risk. Nat. Genet. 2009, 41, 920–925. [Google Scholar] [CrossRef] [PubMed]
- Vanneste, R.; Smith, E.; Graham, G. Multiple neurofibromas as the presenting feature of familial atypical multiple malignant melanoma (FAMMM) syndrome. Am. J. Med. Genet. Part A 2013, 161, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Yap, K.L.; Li, S.; Muñoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M.M. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell 2010, 38, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Cunnington, M.S.; Koref, M.S.; Mayosi, B.M.; Burn, J.; Keavney, B. Chromosome 9p21 SNPs associated with multiple disease phenotypes correlate with ANRIL expression. PLoS Genet. 2010, 6, e1000899. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Wang, H.; Pan, H.; Shi, Y.; Li, T.; Ge, S.; Jia, R.; Zhang, H.; Fan, X. ANRIL lncRNA triggers efficient therapeutic efficacy by reprogramming the aberrant INK4-hub in melanoma. Cancer Lett. 2016, 381, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Flockhart, R.J.; Webster, D.E.; Qu, K.; Mascarenhas, N.; Kovalski, J.; Kretz, M.; Khavari, P.A. BRAFV600E remodels the melanocyte transcriptome and induces BANCR to regulate melanoma cell migration. Genome Res. 2012, 22, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhang, L.; Jia, L.; Duan, Y.; Li, Y.; Bao, L.; Sha, N. Long non-coding RNA BANCR promotes proliferation in malignant melanoma by regulating MAPK pathway activation. PLoS ONE 2014, 9, e100893. [Google Scholar] [CrossRef] [PubMed]
- Lessard, L.; Liu, M.; Marzese, D.M.; Wang, H.; Chong, K.; Kawas, N.; Donovan, N.C.; Kiyohara, E.; Hsu, S.; Nelson, N.; et al. The CASC15 long intergenic noncoding RNA locus is involved in melanoma progression and phenotype switching. J. Investig. Dermatol. 2015, 135, 2464–2474. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.M.; Steitz, J.A. Classificatiocelln of gas5 as a multi-small-nucleolar-RNA (snoRNA) host gene and a member of the 5′-terminal oligopyrimidine gene family reveals common features of snoRNA host genes. Mol. Cell. Biol. 1998, 18, 6897–6909. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, H.; Xiao, Y.; Tang, X.; Li, Y.; Han, Q.; Fu, J.; Yang, Y.; Zhu, Y. Lentiviral-mediated overexpression of long non-coding RNA GAS5 reduces invasion by mediating MMP2 expression and activity in human melanoma cells. Int. J. Oncol. 2016, 48, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Zhang, W.; Su, B.; Yu, B. Long noncoding RNA HOTAIR is associated with motility, invasion, and metastatic potential of metastatic melanoma. BioMed Res. Int. 2013, 2013, 251098. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhou, L.; Wu, L.-M.; Lai, M.-C.; Xie, H.-Y.; Zhang, F.; Zheng, S.-S. Overexpression of long non-coding RNA HOTAIR predicts tumor recurrence in hepatocellular carcinoma patients following liver transplantation. Ann. Surg. Oncol. 2011, 18, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Murat, P.; Matak-Vinkovic, D.; Murrell, A.; Balasubramanian, S. Binding interactions between long noncoding RNA HOTAIR and PRC2 proteins. Biochemistry 2013, 52, 9519–9527. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Sui, A.; Garen, A. Binding of mouse VL30 retrotransposon RNA to PSF protein induces genes repressed by PSF: Effects on steroidogenesis and oncogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.F.; Tan, G.H.; Ma, C.C.; Li, L. The non-coding RNA llme23 drives the malignant property of human melanoma cells. J. Genet. Genom. 2013, 40, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Diederichs, S.; Wang, W.; Böing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a novel noncoding RNA, and thymosin β4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Hämmerle, M.; Diederichs, S. MALAT1—A paradigm for long noncoding RNA function in cancer. J. Mol. Med. 2013, 91, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Zhang, X.; Hao, Y.; Fang, Z.; He, Y. Potential roles of abnormally expressed long noncoding RNA UCA1 and Malat-1 in metastasis of melanoma. Melanoma Res. 2014, 24, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Engreitz, J.M.; Sirokman, K.; McDonel, P.; Shishkin, A.A.; Surka, C.; Russell, P.; Grossman, S.R.; Chow, A.Y.; Guttman, M.; Lander, E.S. RNA-RNA interactions enable specific targeting of noncoding RNAs to nascent pre-mRNAs and chromatin sites. Cell 2014, 159, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Arancio, W.; Pizzolanti, G.; Genovese, S.I.; Baiamonte, C.; Giordano, C. Competing endogenous RNA and interactome bioinformatic analyses on human telomerase. Rejuvenation Res. 2014, 17, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Luan, W.; Li, L.; Shi, Y.; Bu, X.; Xia, Y.; Wang, J.; Djangmah, H.S.; Liu, X.; You, Y.; Xu, B.; et al. Long non-coding RNA MALAT1 acts as a competing endogenous RNA to promote malignant melanoma growth and metastasis by sponging miR-22. Oncotarget 2016, 7, 63901–63912. [Google Scholar] [CrossRef] [PubMed]
- Silayoi, P.; Speece, M. The importance of packaging attributes: A conjoint analysis approach. Eur. J. Mark 2007, 41, 1495–1517. [Google Scholar] [CrossRef]
- Ding, X.; Wang, X.; Lin, M.; Xing, Y.; Ge, S.; Jia, R.; Zhang, H.; Fan, X.; Li, J. PAUPAR lncRNA suppresses tumourigenesis by H3K4 demethylation in uveal melanoma. FEBS Lett. 2016, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Sousa, J.F.; Torrieri, R.; Silva, R.R.; Pereira, C.G.; Valente, V.; Torrieri, E.; Peronni, K.C.; Martins, W.; Muto, N.; Francisco, G.; et al. Novel primate-specific genes, RMEL 1, 2 and 3, with highly restricted expression in melanoma, assessed by new data mining tool. PLoS ONE 2010, 5, e13510. [Google Scholar] [CrossRef] [PubMed]
- Goedert, L.; Pereira, C.G.; Roszik, J.; Plaça, J.R.; Cardoso, C.; Chen, G.; Deng, W.; Yennu-Nanda, V.G.; Silva, W.A., Jr.; Davies, M.A.; et al. RMEL3, a novel BRAFV600E-associated long noncoding RNA, is required for MAPK and PI3K signaling in melanoma. Oncotarget 2016, 7, 36711–36718. [Google Scholar] [PubMed]
- Leucci, E.; Vendramin, R.; Spinazzi, M.; Laurette, P.; Fiers, M.; Wouters, J.; Radaelli, E.; Eyckerman, S.; Leonelli, C.; Vanderheyden, K.; et al. Melanoma addiction to the long non-coding RNA SAMMSON. Nature 2016, 531, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, R.; Satoh, H.; Moriyama, M.; Satoh, K.; Morishita, Y.; Yoshida, S.; Watanabe, T.; Nakamura, Y.; Mori, S. Intronic U50 small-nucleolar-RNA (snoRNA) host gene of no protein-coding potential is mapped at the chromosome breakpoint t(3;6)(q27;q15) of human B-cell lymphoma. Genes Cells 2000, 5, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Ichigozaki, Y.; Fukushima, S.; Jinnin, M.; Miyashita, A.; Nakahara, S.; Tokuzumi, A.; Yamashita, J.; Kajihara, I.; Aoi, J.; Masuguchi, S.; et al. Serum long non-coding RNA, snoRNA host gene 5 level as a new tumor marker of malignant melanoma. Exp. Dermatol. 2016, 25, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Mazar, J.; Zhao, W.; Khalil, A.M.; Lee, B.; Shelley, J.; Govindarajan, S.S.; Yamamoto, F.; Ratnam, M.; Aftab, M.N.; Collins, S.; et al. The functional characterization of long noncoding RNA SPRY4-IT1 in human melanoma cells. Oncotarget 2014, 5, 8959–8969. [Google Scholar] [CrossRef] [PubMed]
- Khaitan, D.; Dinger, M.E.; Mazar, J.; Crawford, J.; Smith, M.A.; Mattick, J.S.; Perera, R.J. The melanoma-upregulated long noncoding RNA SPRY4-IT1 modulates apoptosis and invasion. Cancer Res. 2011, 71, 3852–3862. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Mazar, J.; Lee, B.; Sawada, J.; Li, J.L.; Shelley, J.; Govindarajan, S.; Towler, D.; Mattick, J.S.; Komatsu, M.; et al. The long noncoding RNA SPRIGHTLY regulates cell proliferation in primary human melanocytes. J. Investig. Dermatol. 2016, 136, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Shen, S.-K.; Xiong, J.-G.; Xu, Y.; Zhang, H.-Q.; Liu, H.-J.; Lu, Z.-G. Clinical significance of long noncoding RNA SPRY4-IT1 in melanoma patients. FEBS Open Bio 2016, 6, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, W.; Yang, C.; Wu, W.; Wu, S.; Qin, X.; Li, X. Long non-coding RNA UCA1a(CUDR) promotes proliferation and tumorigenesis of bladder cancer. Int. J. Oncol. 2012, 41, 276–284. [Google Scholar] [PubMed]
- Wei, Y.; Sun, Q.; Zhao, L.; Wu, J.; Chen, X.; Wang, Y.; Zang, W.; Zhao, G. LncRNA UCA1-miR-507-FOXM1 axis is involved in cell proliferation, invasion and G0/G1 cell cycle arrest in melanoma. Med. Oncol. 2016, 33, 88. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.-D.; Zhuang, L.; Gan, B. Long non-coding RNAs in cancer metabolism. BioEssays 2016, 38, 991–996. [Google Scholar] [CrossRef] [PubMed]
- Diederichs, S. The four dimensions of noncoding RNA conservation. Trends Genet. 2014, 30, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, M.; Gutschner, T. Long non-coding RNAs in cancer and development: Where do we go from here? Int. J. Mol. Sci. 2015, 16, 1395–1405. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| LncRNA Name | Function | References |
|---|---|---|
| ANRIL | Represses the transcription of CDKN2A/B which leads to perturbation in the cell cycle, increased migration and colony formation. | [94,95,96] |
| BANCR | High levels of BANCR lead to increased migration (by targeting CXCL11) and proliferation. High levels of BANCR directly correlated with tumor stage and indirectly with survival. | [97,98] |
| CASC15 | Promotes melanoma progression and invasiveness. Direct correlation between tumor stage and expression levels. | [99] |
| GAS5 | Indirectly correlates with melanoma migration and invasiveness over reduced levels of MMP2. | [101] |
| HOTAIR | HOTAIR is up-regulated in metastases compared to the primary tumor, favoring a pro-metastatic role. | [102] |
| Llme23 | Llme23 promotes the expression of the proto-oncogenic RAS-related small GTPase Rab23. | [106] |
| MALAT1 | Possibly involved in cell proliferation and invasion. It does this by targeting miR-22 in cutaneous melanoma and miR-140 in uveal melanoma. | [109,112,113] |
| PAUPAR | It is a tumor suppressor lncRNA and reduces cell migration and metastases. | [114] |
| RMEL3 | Depletion led to decreased cell survival and proliferation in BRAFV600E melanoma cell lines. | [116] |
| SAMMSON | Promotes cell viability and growth irrespective of melanomas mutational status. | [117] |
| SNGH5 | Increased serum levels in patients with melanoma. | [119] |
| SPRTY4-IT1 (SPRIGHTLY) | Associated with melanoma-genesis; Associated with higher tumor stage and worse prognosis. | [122,123] |
| UCA1 | Promotes invasion and cell proliferation. | [109,125] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richtig, G.; Ehall, B.; Richtig, E.; Aigelsreiter, A.; Gutschner, T.; Pichler, M. Function and Clinical Implications of Long Non-Coding RNAs in Melanoma. Int. J. Mol. Sci. 2017, 18, 715. https://doi.org/10.3390/ijms18040715
Richtig G, Ehall B, Richtig E, Aigelsreiter A, Gutschner T, Pichler M. Function and Clinical Implications of Long Non-Coding RNAs in Melanoma. International Journal of Molecular Sciences. 2017; 18(4):715. https://doi.org/10.3390/ijms18040715
Chicago/Turabian StyleRichtig, Georg, Barbara Ehall, Erika Richtig, Ariane Aigelsreiter, Tony Gutschner, and Martin Pichler. 2017. "Function and Clinical Implications of Long Non-Coding RNAs in Melanoma" International Journal of Molecular Sciences 18, no. 4: 715. https://doi.org/10.3390/ijms18040715
APA StyleRichtig, G., Ehall, B., Richtig, E., Aigelsreiter, A., Gutschner, T., & Pichler, M. (2017). Function and Clinical Implications of Long Non-Coding RNAs in Melanoma. International Journal of Molecular Sciences, 18(4), 715. https://doi.org/10.3390/ijms18040715