
A Review on Ubiquitination of Neurotrophin Receptors: Facts and Perspectives


Abstract
:1. Neurotrophins and Their Receptors
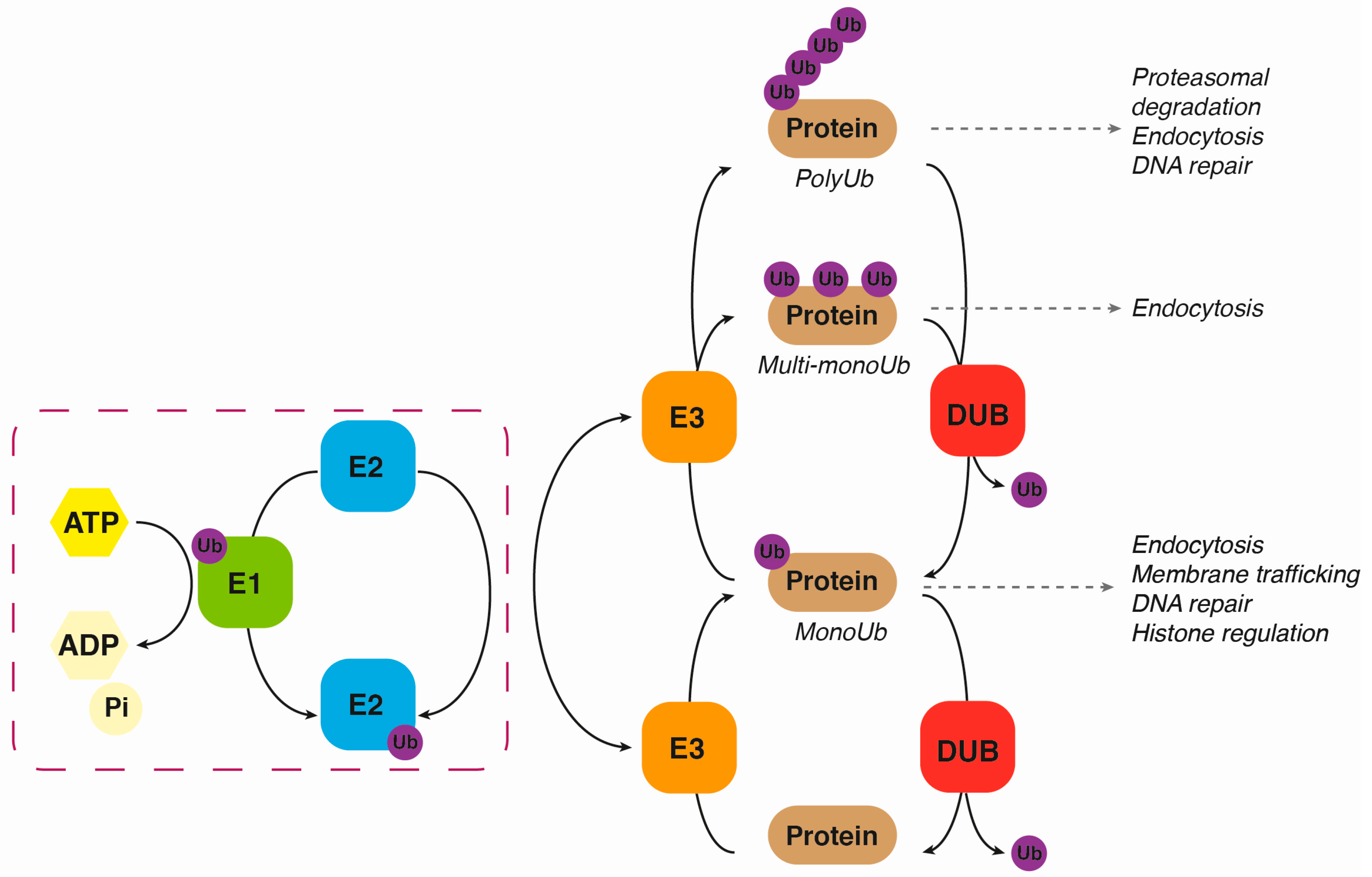
2. Ubiquitination
3. Deubiquitination
4. Neurotrophin Receptors Ubiquitination
4.1. p75NTR Ubiquitination
4.2. Ubiquitination of TrkA
4.3. TrkB and TrkC Ubiquitination
5. Deubiquitination of Trk Neurotrophin Receptors
6. Clinical Relevance and Future Approaches
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chao, M.V. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat. Rev. 2003, 4, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Reichardt, L.F. Trk receptors: Roles in neuronal signal transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Levi-Montalcini, R.; Hamburger, V. A nerve growth-stimulating factor isolated from sarcomas 37 and 180. Proc. Natl. Acad. Sci. USA 1954, 40, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Barde, Y.A.; Edgar, D.; Thoenen, H. Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1982, 1, 549–553. [Google Scholar] [PubMed]
- Maisonpierre, P.C.; Belluscio, L.; Friedman, B.; Alderson, R.F.; Wiegand, S.J.; Furth, M.E.; Lindsay, R.M.; Yancopoulos, G.D. NT-3, BDNF, and NGF in the developing rat nervous system: Parallel as well as reciprocal patterns of expression. Neuron 1990, 5, 501–509. [Google Scholar] [CrossRef]
- Berkemeier, L.R.; Winslow, J.W.; Kaplan, D.R.; Nikolics, K.; Goeddel, D.V.; Rosenthal, A. Neurotrophin-5: A novel neurotrophic factor that activates Trk and TrkB. Neuron 1991, 7, 857–866. [Google Scholar] [CrossRef]
- Ip, N.Y.; Ibanez, C.F.; Nye, S.H.; McClain, J.; Jones, P.F.; Gies, D.R.; Belluscio, L.; Le Beau, M.M.; Espinosa, R., 3rd; Squinto, S.P.; et al. Mammalian neurotrophin-4: Structure, chromosomal localization, tissue distribution, and receptor specificity. Proc. Natl. Acad. Sci. USA 1992, 89, 3060–3064. [Google Scholar] [CrossRef] [PubMed]
- Esposito, D.; Patel, P.; Stephens, R.M.; Perez, P.; Chao, M.V.; Kaplan, D.R.; Hempstead, B.L. The cytoplasmic and transmembrane domains of the p75 and Trk a receptors regulate high affinity binding to nerve growth factor. J. Biol. Chem. 2001, 276, 32687–32695. [Google Scholar] [CrossRef] [PubMed]
- Delcroix, J.D.; Valletta, J.S.; Wu, C.; Hunt, S.J.; Kowal, A.S.; Mobley, W.C. NGF signaling in sensory neurons: Evidence that early endosomes carry NGF retrograde signals. Neuron 2003, 39, 69–84. [Google Scholar] [CrossRef]
- Ye, H.; Kuruvilla, R.; Zweifel, L.S.; Ginty, D.D. Evidence in support of signaling endosome-based retrograde survival of sympathetic neurons. Neuron 2003, 39, 57–68. [Google Scholar] [CrossRef]
- Arevalo, J.C.; Waite, J.; Rajagopal, R.; Beyna, M.; Chen, Z.Y.; Lee, F.S.; Chao, M.V. Cell survival through Trk neurotrophin receptors is differentially regulated by ubiquitination. Neuron 2006, 50, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Geetha, T.; Jiang, J.; Wooten, M.W. Lysine 63 polyubiquitination of the nerve growth factor receptor TrkA directs internalization and signaling. Mol. Cell 2005, 20, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Makkerh, J.P.; Ceni, C.; Auld, D.S.; Vaillancourt, F.; Dorval, G.; Barker, P.A. P75 neurotrophin receptor reduces ligand-induced Trk receptor ubiquitination and delays Trk receptor internalization and degradation. EMBO Rep. 2005, 6, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Arévalo, J.C.; Gu, S.H. Neurotrophin signaling: Many exciting surprises! Cell. Mol. Life Sci. 2006, 63, 1523–1537. [Google Scholar] [CrossRef] [PubMed]
- Ciehanover, A.; Hod, Y.; Hershko, A. A heat-stable polypeptide component of an ATP-dependent proteolytic system from reticulocytes. Biochem. Biophys. Res. Commun. 1978, 81, 1100–1105. [Google Scholar] [CrossRef]
- Sun, L.; Chen, Z.J. The novel functions of ubiquitination in signaling. Curr. Opin. Cell Biol. 2004, 16, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J.; Joazeiro, C.A. Ring domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, S.; Nakayama, K.I. U-box proteins as a new family of ubiquitin ligases. Biochem. Biophys. Res. Commun. 2003, 302, 635–645. [Google Scholar] [CrossRef]
- Rotin, D.; Kumar, S. Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2009, 10, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.J.; Rape, M. Enhanced protein degradation by branched ubiquitin chains. Cell 2014, 157, 910–921. [Google Scholar] [CrossRef] [PubMed]
- Kirisako, T.; Kamei, K.; Murata, S.; Kato, M.; Fukumoto, H.; Kanie, M.; Sano, S.; Tokunaga, F.; Tanaka, K.; Iwai, K. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 2006, 25, 4877–4887. [Google Scholar] [CrossRef] [PubMed]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [PubMed]
- Fraile, J.M.; Quesada, V.; Rodríguez, D.; Freije, J.M.; López-Otín, C. Deubiquitinases in cancer: New functions and therapeutic options. Oncogene 2012, 31, 2373–2388. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Urbe, S. Integration of cellular ubiquitin and membrane traffic systems: Focus on deubiquitylases. FEBS J. 2017. [Google Scholar] [CrossRef] [PubMed]
- Heride, C.; Urbe, S.; Clague, M.J. Ubiquitin code assembly and disassembly. Curr. Biol. 2014, 24, R215–R220. [Google Scholar] [CrossRef] [PubMed]
- Huang, O.W.; Cochran, A.G. Regulation of deubiquitinase proteolytic activity. Curr. Opin. Struct. Biol. 2013, 23, 806–811. [Google Scholar] [CrossRef] [PubMed]
- McCann, A.P.; Scott, C.J.; van Schaeybroeck, S.; Burrows, J.F. Deubiquitylating enzymes in receptor endocytosis and trafficking. Biochem. J. 2016, 473, 4507–4525. [Google Scholar] [CrossRef] [PubMed]
- Brummelkamp, T.R.; Nijman, S.M.; Dirac, A.M.; Bernards, R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-κB. Nature 2003, 424, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Tan, X.; Shi, Y.; Xu, G.; Mao, R.; Gu, X.; Fan, Y.; Yu, Y.; Burlingame, S.; Zhang, H.; et al. Usp11 negatively regulates TNFα-induced NF-κB activation by targeting on Iκ-α. Cell Signal. 2010, 22, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Sacco, J.J.; Coulson, J.M.; Clague, M.J.; Urbe, S. Emerging roles of deubiquitinases in cancer-associated pathways. IUBMB Life 2010, 62, 140–157. [Google Scholar] [CrossRef] [PubMed]
- Ohrt, T.; Mancini, A.; Tamura, T.; Niedenthal, R. c-Cbl binds to tyrosine-phosphorylated neurotrophin receptor p75 and induces its ubiquitination. Cell Signal. 2004, 16, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Khursigara, G.; Orlinick, J.R.; Chao, M.V. Association of the p75 neurotrophin receptor with TRAF6. J. Biol. Chem. 1999, 274, 2597–2600. [Google Scholar] [CrossRef] [PubMed]
- Yeiser, E.C.; Rutkoski, N.J.; Naito, A.; Inoue, J.; Carter, B.D. Neurotrophin signaling through the p75 receptor is deficient in TRAF6−/− mice. J. Neurosci. 2004, 24, 10521–10529. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.C.; Twomey, C.; Jain, R.; McCarthy, J.V. Association between presenilin-1 and TRAF6 modulates regulated intramembrane proteolysis of the p75NTR neurotrophin receptor. J. Neurochem. 2009, 108, 216–230. [Google Scholar] [CrossRef] [PubMed]
- Geetha, T.; Zheng, C.; Unroe, B.; Sycheva, M.; Kluess, H.; Babu, J.R. Polyubiquitination of the neurotrophin receptor p75 directs neuronal cell survival. Biochem. Biophys. Res. Commun. 2012, 421, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Cell biology of protein misfolding: The examples of Alzheimer’s and Parkinson’s diseases. Nat. Cell Biol. 2004, 6, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Fahnestock, M.; Michalski, B.; Xu, B.; Coughlin, M.D. The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in Alzheimer’s disease. Mol. Cell. Neurosci. 2001, 18, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Pedraza, C.E.; Podlesniy, P.; Vidal, N.; Arevalo, J.C.; Lee, R.; Hempstead, B.; Ferrer, I.; Iglesias, M.; Espinet, C. Pro-NGF isolated from the human brain affected by Alzheimer’s disease induces neuronal apoptosis mediated by p75NTR. Am. J. Pathol. 2005, 166, 533–543. [Google Scholar] [CrossRef]
- Geetha, T.; Zheng, C.; McGregor, W.C.; Douglas White, B.; Diaz-Meco, M.T.; Moscat, J.; Babu, J.R. TRAF6 and p62 inhibit amyloid β-induced neuronal death through p75 neurotrophin receptor. Neurochem. Int. 2012, 61, 1289–1293. [Google Scholar] [CrossRef] [PubMed]
- Tessarollo, L.; Tsoulfas, P.; Martin-Zanca, D.; Gilbert, D.J.; Jenkins, N.A.; Copeland, N.G.; Parada, L.F. TrkC, a receptor for neurotrophin-3, is widely expressed in the developing nervous system and in non-neuronal tissues. Development 1993, 118, 463–475. [Google Scholar] [PubMed]
- Jadhav, T.; Geetha, T.; Jiang, J.; Wooten, M.W. Identification of a consensus site for TRAF6/p62 polyubiquitination. Biochem. Biophys. Res. Commun. 2008, 371, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, T.S.; Wooten, M.W.; Wooten, M.C. Mining the TRAF6/p62 interactome for a selective ubiquitination motif. BMC Proc. 2011, 5 (Suppl. S2), S4. [Google Scholar] [CrossRef] [PubMed]
- Geetha, T.; Seibenhener, M.L.; Chen, L.; Madura, K.; Wooten, M.W. P62 serves as a shuttling factor for TrkA interaction with the proteasome. Biochem. Biophys. Res. Commun. 2008, 374, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Thien, C.B.; Langdon, W.Y. C-CBL and CBL-B ubiquitin ligases: Substrate diversity and the negative regulation of signalling responses. Biochem. J. 2005, 391, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Emdal, K.B.; Pedersen, A.K.; Bekker-Jensen, D.B.; Tsafou, K.P.; Horn, H.; Lindner, S.; Schulte, J.H.; Eggert, A.; Jensen, L.J.; Francavilla, C.; et al. Temporal proteomics of NGF-TrkA signaling identifies an inhibitory role for the E3 ligase Cbl-b in neuroblastoma cell differentiation. Sci Signal. 2015, 8, ra40. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Shimokawa, N.; Esmaeili-Mahani, S.; Morita, A.; Masuda, H.; Iwasaki, T.; Tamura, J.; Haglund, K.; Koibuchi, N. Ligand-induced downregulation of TrkA is partly regulated through ubiquitination by Cbl. FEBS Lett. 2011, 585, 1741–1747. [Google Scholar] [CrossRef] [PubMed]
- Georgieva, M.V.; de Pablo, Y.; Sanchis, D.; Comella, J.X.; Llovera, M. Ubiquitination of TrkA by Nedd4-2 regulates receptor lysosomal targeting and mediates receptor signaling. J. Neurochem. 2011, 117, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Calvo, L.; Anta, B.; Lopez-Benito, S.; Southon, E.; Chao, M.V.; Tessarollo, L.; Arevalo, J.C. Regulation of trafficking of activated TrkA is critical for NGF-mediated functions. Traffic 2011, 12, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Calvo, L.; Anta, B.; Lopez-Benito, S.; Lopez-Bellido, R.; Vicente-Garcia, C.; Tessarollo, L.; Rodriguez, R.E.; Arevalo, J.C. In vivo regulation of NGF-mediated functions by Nedd4-2 ubiquitination of TrkA. J. Neurosci. 2014, 34, 6098–6106. [Google Scholar] [CrossRef] [PubMed]
- Kiris, E.; Wang, T.; Yanpallewar, S.; Dorsey, S.G.; Becker, J.; Bavari, S.; Palko, M.E.; Coppola, V.; Tessarollo, L. TrkA in vivo function is negatively regulated by ubiquitination. J. Neurosci. 2014, 34, 4090–4098. [Google Scholar] [CrossRef] [PubMed]
- Pandya, C.; Kutiyanawalla, A.; Turecki, G.; Pillai, A. Glucocorticoid regulates TrkB protein levels via c-Cbl dependent ubiquitination: A decrease in c-Cbl mRNA in the prefrontal cortex of suicide subjects. Psychoneuroendocrinology 2014, 45, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Monteggia, L.M. A neurotrophic model for stress-related mood disorders. Biol. Psychiatry 2006, 59, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Proenca, C.C.; Song, M.; Lee, F.S. Differential effects of BDNF and neurotrophin 4 (NT-4) on endocytic sorting of TrkB receptors. J. Neurochem. 2016, 138, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Geetha, T.; Wooten, M.W. TrkA receptor endolysosomal degradation is both ubiquitin and proteasome dependent. Traffic 2008, 9, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Sommerfeld, M.T.; Schweigreiter, R.; Barde, Y.A.; Hoppe, E. Down-regulation of the neurotrophin receptor TrkB following ligand binding. Evidence for an involvement of the proteasome and differential regulation of TrkA and TrkB. J. Biol. Chem. 2000, 275, 8982–8990. [Google Scholar] [CrossRef] [PubMed]
- Moises, T.; Wuller, S.; Saxena, S.; Senderek, J.; Weis, J.; Kruttgen, A. Proteasomal inhibition alters the trafficking of the neurotrophin receptor TrkA. Biochem. Biophys. Res. Commun. 2009, 387, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Bignell, G.R.; Warren, W.; Seal, S.; Takahashi, M.; Rapley, E.; Barfoot, R.; Green, H.; Brown, C.; Biggs, P.J.; Lakhani, S.R.; et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat. Genet. 2000, 25, 160–165. [Google Scholar] [PubMed]
- Wooten, M.W.; Geetha, T.; Babu, J.R.; Seibenhener, M.L.; Peng, J.; Cox, N.; Diaz-Meco, M.T.; Moscat, J. Essential role of sequestosome 1/p62 in regulating accumulation of lys63-ubiquitinated proteins. J. Biol. Chem. 2008, 283, 6783–6789. [Google Scholar] [CrossRef] [PubMed]
- Ceriani, M.; Amigoni, L.; D’Aloia, A.; Berruti, G.; Martegani, E. The deubiquitinating enzyme UBPY/USP8 interacts with TrkA and inhibits neuronal differentiation in PC12 cells. Exp. Cell Res. 2015, 333, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Anta, B.; Martin-Rodriguez, C.; Gomis-Perez, C.; Calvo, L.; Lopez-Benito, S.; Calderon-Garcia, A.A.; Vicente-Garcia, C.; Villarroel, A.; Arevalo, J.C. Ubiquitin-specific protease 36 (USP36) controls neuronal precursor cell-expressed developmentally down-regulated 4-2 (Nedd4-2) actions over the neurotrophin receptor TrkA and potassium voltage-gated channels 7.2/3 (Kv7.2/3). J. Biol. Chem. 2016, 291, 19132–19145. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, V. Deciphering Parkinson’s disease—PARK8. Lancet Neurol. 2002, 1, 83. [Google Scholar] [CrossRef]
- Bonifati, V.; Dekker, M.C.; Vanacore, N.; Fabbrini, G.; Squitieri, F.; Marconi, R.; Antonini, A.; Brustenghi, P.; dalla Libera, A.; De Mari, M.; et al. Autosomal recessive early onset parkinsonism is linked to three loci: PARK2, PARK6, and PARK7. Neurol. Sci. 2002, 23 (Suppl. S2), S59–S60. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Lin, D.C.; Yin, D.; Koeffler, H.P. An emerging role of PARK2 in cancer. J. Mol. Med. 2014, 92, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Pfoh, R.; Lacdao, I.K.; Saridakis, V. Deubiquitinases and the new therapeutic opportunities offered to cancer. Endocr. Relat. Cancer 2015, 22, T35–T54. [Google Scholar] [CrossRef] [PubMed]
- Laedermann, C.J.; Cachemaille, M.; Kirschmann, G.; Pertin, M.; Gosselin, R.D.; Chang, I.; Albesa, M.; Towne, C.; Schneider, B.L.; Kellenberger, S.; et al. Dysregulation of voltage-gated sodium channels by ubiquitin ligase Nedd4-2 in neuropathic pain. J. Clin. Investig. 2013, 123, 3002–3013. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; Binder, A.; Wasner, G. Neuropathic pain: Diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurol. 2010, 9, 807–819. [Google Scholar] [CrossRef]
- Arevalo, J.C. Nedd4-2 regulation of voltage-gated ion channels: An update on structure-function relationships and the pathophysiological consequences of dysfunction. J. Recept. Ligand Channel Res. 2014, 8, 53–63. [Google Scholar] [CrossRef]
- Anand, P. Neurotrophic factors and their receptors in human sensory neuropathies. Prog. Brain Res. 2004, 146, 477–492. [Google Scholar] [PubMed]
- Chang, D.S.; Hsu, E.; Hottinger, D.G.; Cohen, S.P. Anti-nerve growth factor in pain management: Current evidence. J. Pain Res. 2016, 9, 373–383. [Google Scholar] [PubMed]
- Ligon, C.O.; Moloney, R.D.; Greenwood-Van Meerveld, B. Targeting epigenetic mechanisms for chronic pain: A valid approach for the development of novel therapeutics. J. Pharmacol. Exp. Ther. 2016, 357, 84–93. [Google Scholar] [CrossRef] [PubMed]
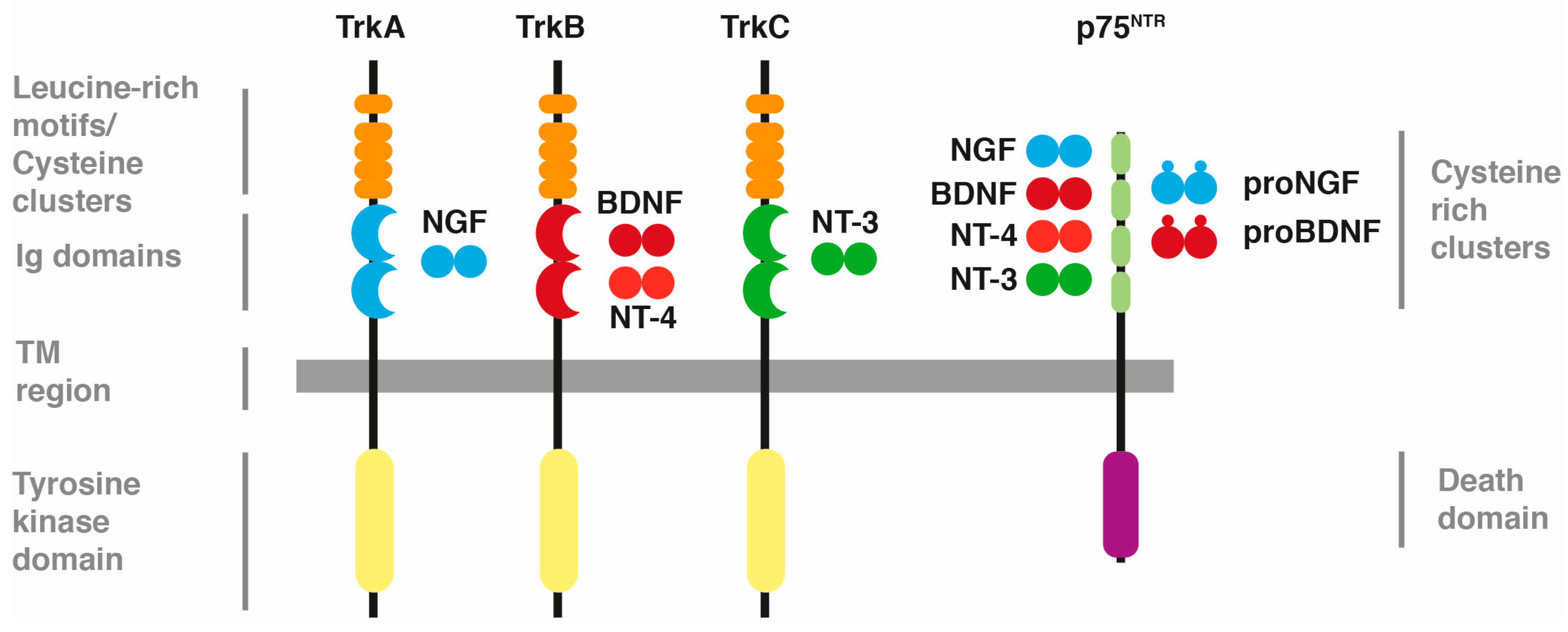

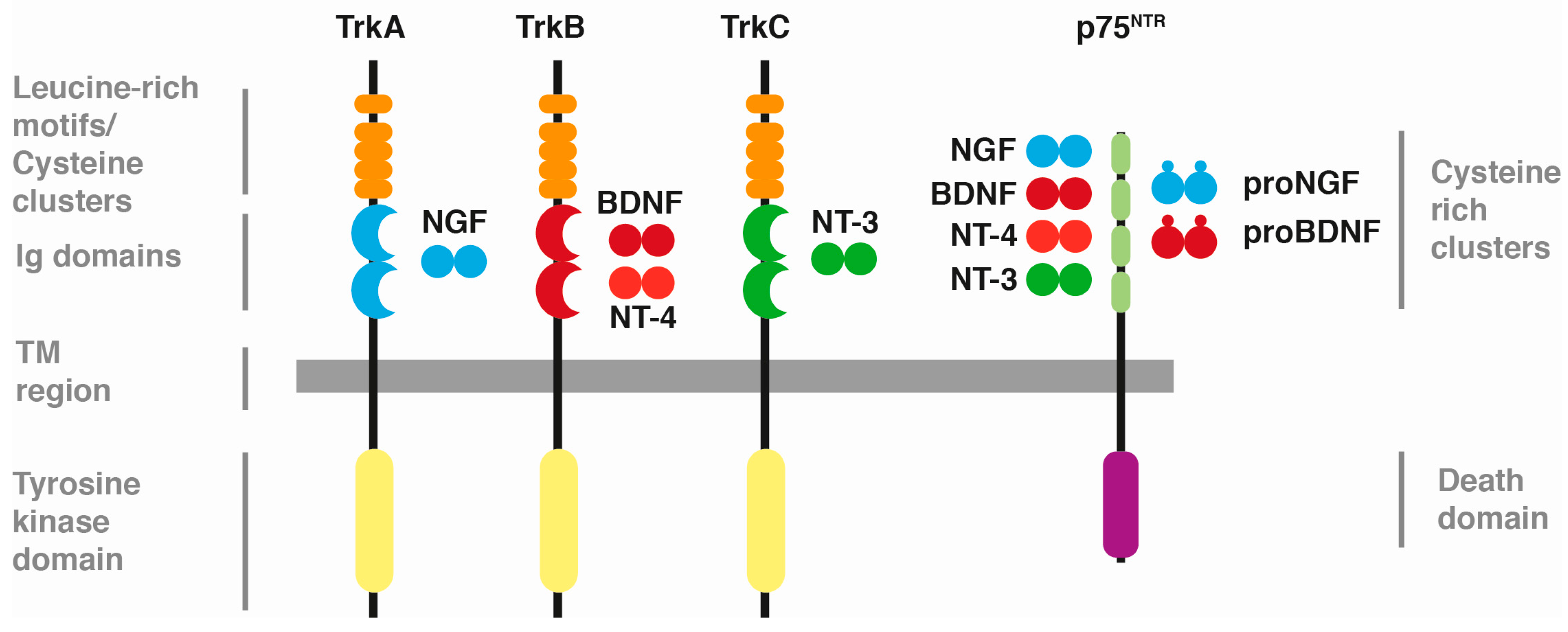{kind=link}
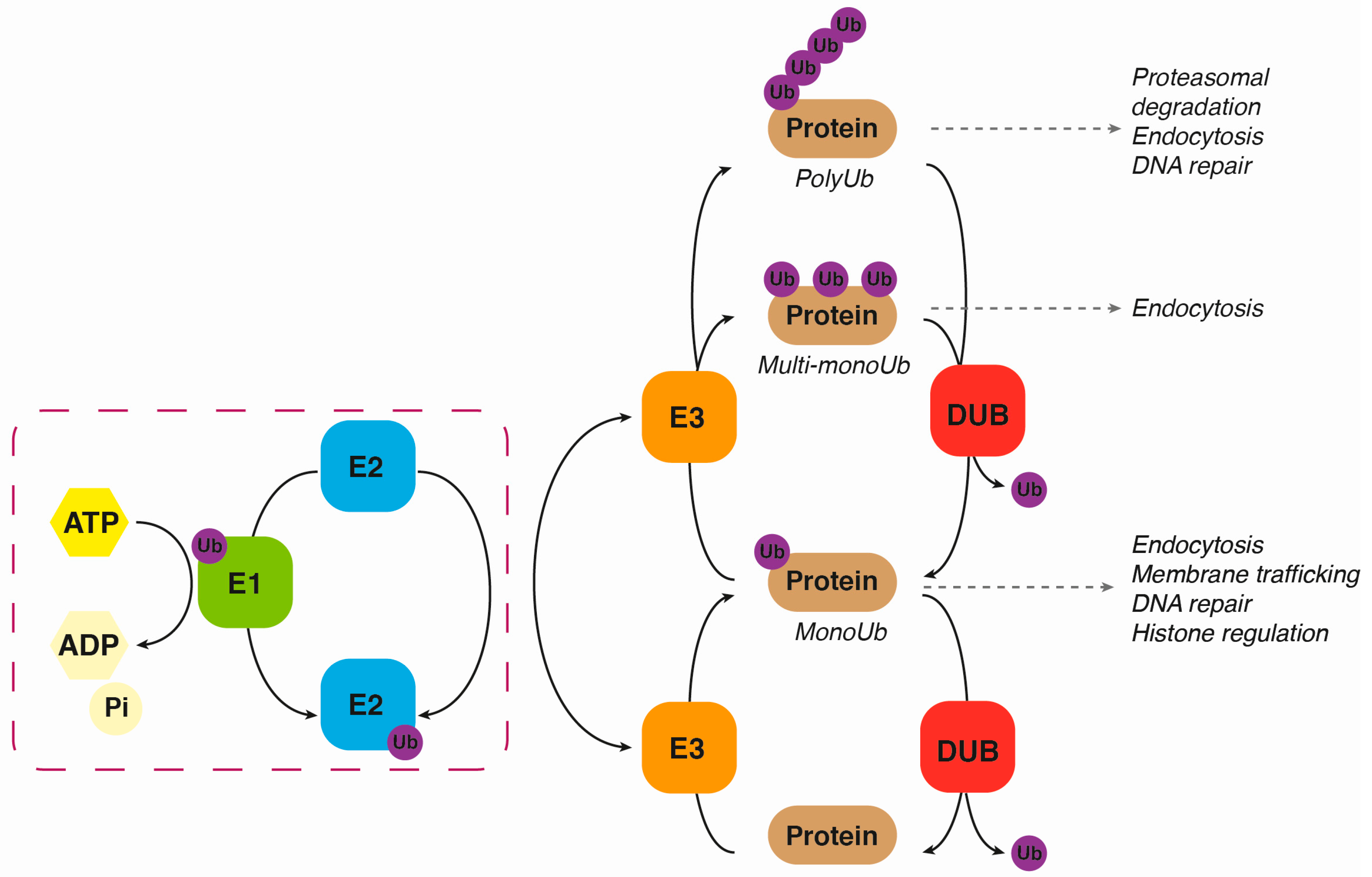{kind=link}
| Neurotrophin Receptor | Ub-Ligase | Ub-Ligase Family | References |
|---|---|---|---|
| p75NTR | c-Cbl | RING-family | [32] |
| TRAF6 | RING-family | [33,34,35,36] | |
| TrkA | TRAF6 | RING-family | [12,13,42,43] |
| c-Cbl | RING-family | [47] | |
| Cbl-b | RING-family | [46] | |
| Nedd4-2 | HECT-family | [11,48,49,50] | |
| TrkB | TRAF6 | RING-family | [42,43] |
| c-Cbl | RING-family | [52] | |
| TrkC | c-Cbl * | RING-family | [52] |
| TRAF6 * | RING-family | [42,43] |
| Neurotrophin Receptor | Deubiquitinase | DUB Family | References |
|---|---|---|---|
| TrkA | CYLD | USP-family | [12,59] |
| USP8 | USP-family | [60,61] | |
| USP36 * | USP-family | [61] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Sánchez, J.; Arévalo, J.C. A Review on Ubiquitination of Neurotrophin Receptors: Facts and Perspectives. Int. J. Mol. Sci. 2017, 18, 630. https://doi.org/10.3390/ijms18030630
Sánchez-Sánchez J, Arévalo JC. A Review on Ubiquitination of Neurotrophin Receptors: Facts and Perspectives. International Journal of Molecular Sciences. 2017; 18(3):630. https://doi.org/10.3390/ijms18030630
Chicago/Turabian StyleSánchez-Sánchez, Julia, and Juan Carlos Arévalo. 2017. "A Review on Ubiquitination of Neurotrophin Receptors: Facts and Perspectives" International Journal of Molecular Sciences 18, no. 3: 630. https://doi.org/10.3390/ijms18030630
APA StyleSánchez-Sánchez, J., & Arévalo, J. C. (2017). A Review on Ubiquitination of Neurotrophin Receptors: Facts and Perspectives. International Journal of Molecular Sciences, 18(3), 630. https://doi.org/10.3390/ijms18030630