
Role of Genetics in the Etiology of Autistic Spectrum Disorder: Towards a Hierarchical Diagnostic Strategy

 ,
, 
Abstract
:1. Introduction
2. Known Genetic Syndromes Associated with Autism Spectrum Disorder
3. Recommended Investigations for the Identification of Genetic Disorders Associated with ASD
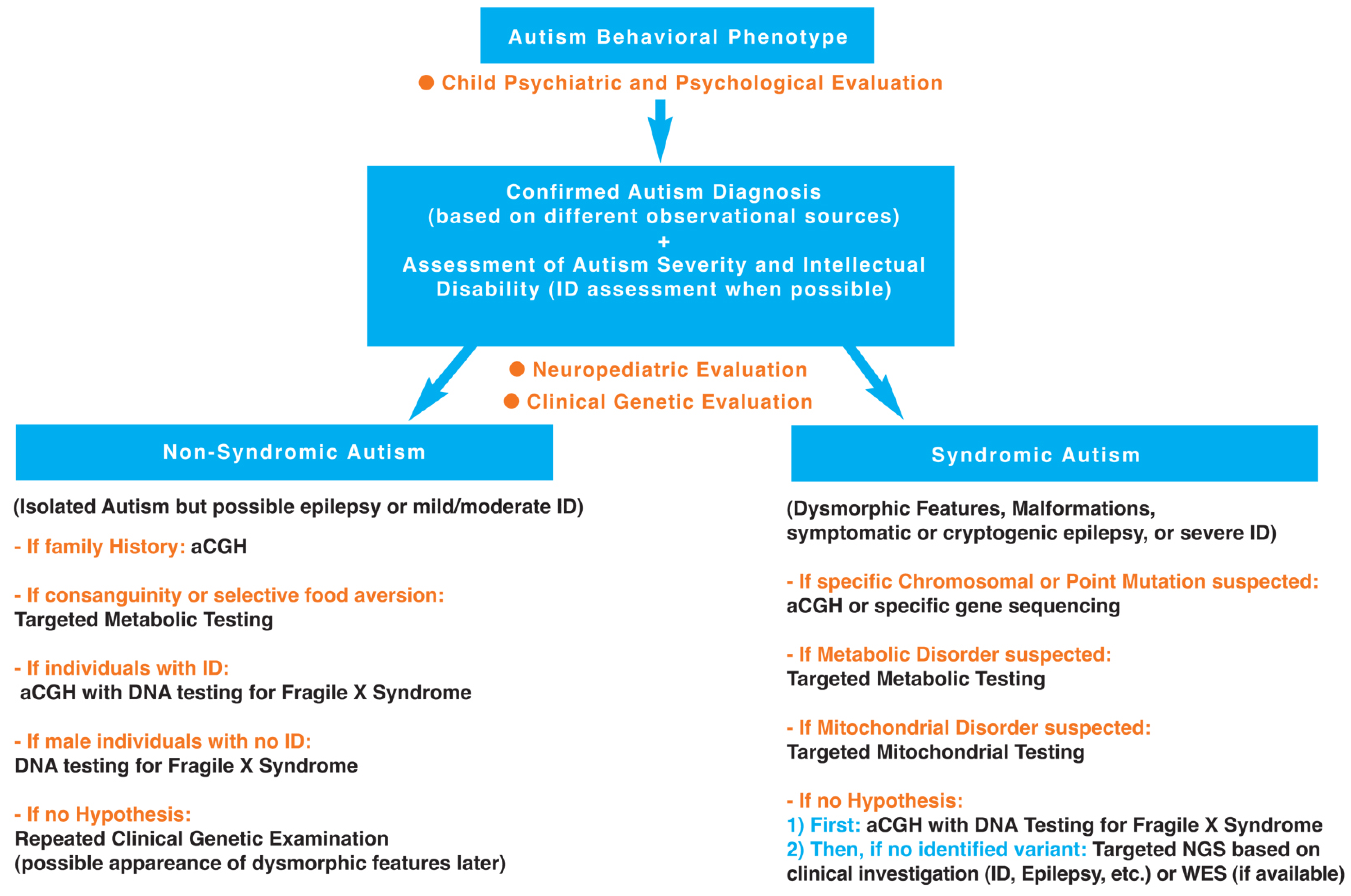
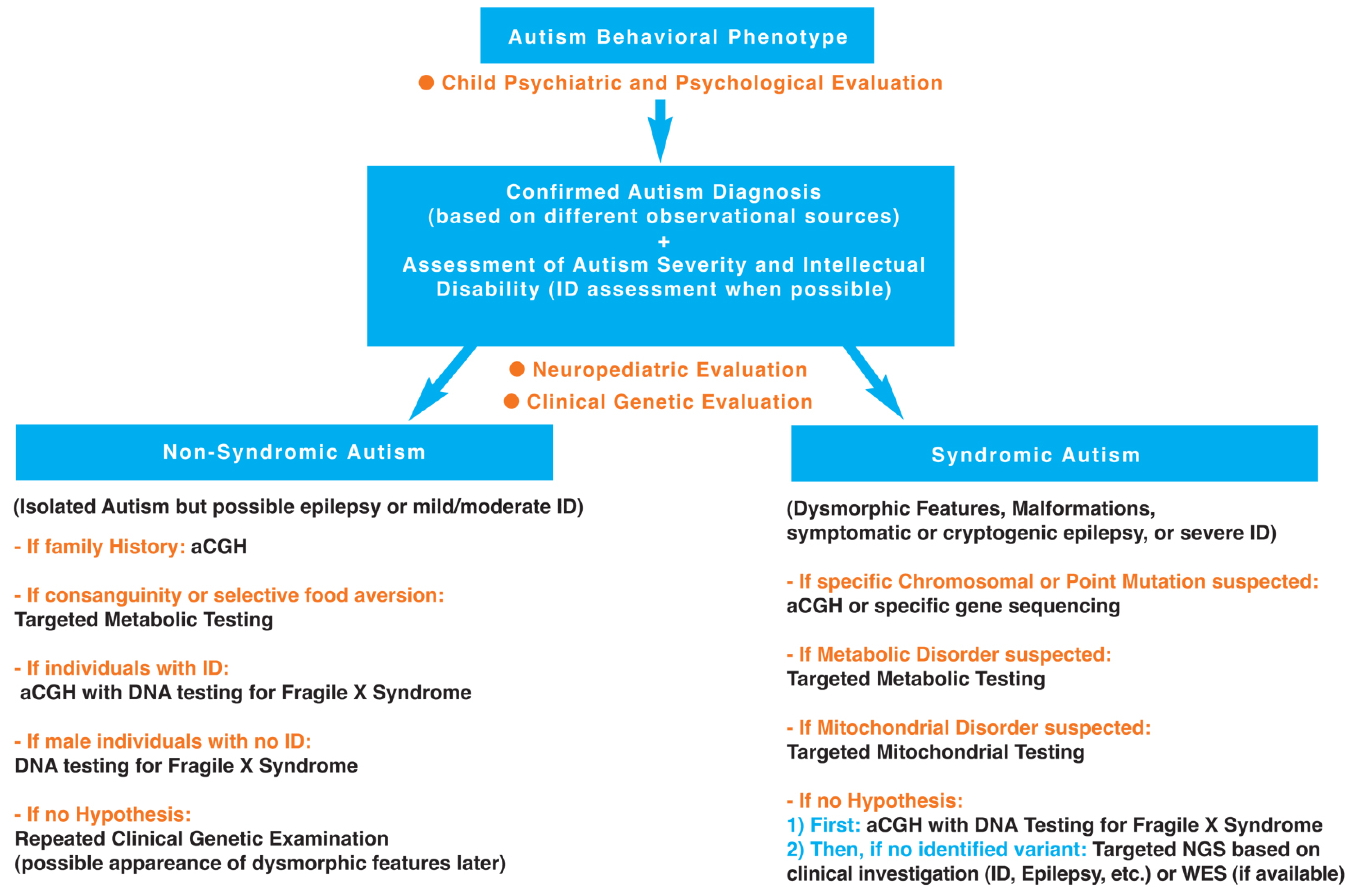
3.1. First Step: Clinical Investigation
3.1.1. Autism Diagnosis Using Different Observational Sources and Autism Severity Assessment (Child Psychiatric and Psychological Evaluation)
3.1.2. Developmental Milestones (Neuropediatric Evaluation)
3.1.3. Physical Examination (Genetic Evaluation)
3.2. Second Step: Complementary Investigations (Laboratory Studies)
3.2.1. Non-Syndromic Autism (Isolated ASD)
3.2.2. Syndromic Autism (ASD Associated with Dysmorphic Features and/or Malformations and/or Symptomatic or Cryptogenic Epilepsy, and/or Severe Intellectual Disability)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5), 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- Carrey, N.J. Itard’s 1828 Memoire on Mutism caused by a lesion of the intellectual functions: A historical analysis. J. Am. Acad. Child Adolesc. Psychiatry 1995, 34, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Kanner, L. Austistic disturbances of affective contact. Nervous Child 1943, 32, 217–253. [Google Scholar]
- Bleuler, E. Dementia Praecox oder Gruppe der Schizophrenien; Handbuch der Psychiatrie; Deuticke: Leipzig, Germany, 1911. [Google Scholar]
- Asperger, H. Die autistischen psychopathen im kindesalter. Arch. Psychiatry Nervenkrandeiten 1944, 117, 73–136. [Google Scholar] [CrossRef]
- Tordjman, S.; Somogyi, E.; Coulon, N.; Kermarrec, S.; Cohen, D.; Bronsard, G.; Bonnot, O.; Weismann-Arcache, C.; Botbol, M.; Lauth, B.; et al. Gene-environment interactions in autism spectrum disorders: Role of epigenetic mechanisms. Front. Psychiatry 2014, 5, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, B.S.; Geschwind, D.H. Advances in autism genetics: On the threshold of a new neurobiology. Nat. Rev. Genet. 2008, 9, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Bill, B.R.; Geschwind, D.H. Genetic advances in autism: Heterogeneity and convergence on shared pathways. Curr. Opin. Genet. Dev. 2009, 19, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Muhle, R.; Trentacoste, S.V.; Rapin, I. The genetics of autism. Pediatrics 2004, 113, 472–486. [Google Scholar] [CrossRef]
- Ronald, A.; Hoekstra, R.A. Autism spectrum disorders and autistic traits: A decade of new twin studies. Am. J. Med. Genet. 2011, 156, 255–274. [Google Scholar] [CrossRef] [PubMed]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Larsson, H.; Hultman, C.M.; Reichenberg, A. The familial risk of autism. JAMA 2014, 311, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Rosti, R.O.; Sadek, A.A.; Vaux, K.K.; Gleeson, J.G. The genetic landscape of autism spectrum disorders. Dev. Med. Child Neurol. 2014, 56, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Sebat, J.; Lakshmi, B.; Malhotra, D.; Troge, J.; Lese-Martin, C.; Walsh, T. Strong association of de novo copy number mutations with autism. Science 2007, 316, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Hegmann, J.P.; Possidente, B. Estimating genetic correlations from inbred strains. Behav. Genet. 1981, 11, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Miles, J.H. Autism spectrum disorders, a genetics review. Genet. Med. 2011, 13, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Bourgeron, T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat. Rev. Neurosc. 2015, 16, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Miles, J.H.; Hillman, R.E. Value of a clinical morphology examination in autism. Am. J. Med. Genet. 2000, 91, 245–253. [Google Scholar] [CrossRef]
- Jacquemont, M.L.; Sanlaville, D.; Redon, R.; Raoul, O.; Cormier-Daire, V.; Lyonnet, S.; Amiel, J.; Le Merrer, M.; Heron, D.; de Blois, M.C.; et al. Array-based comparative genomic hybridisation identifies high frequency of cryptic chromosomal rearrangements in patients with syndromic autism spectrum disorders. J. Med. Genet. 2006, 43, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Miles, J.H.; Takahashi, T.N.; Bagby, S.; Sahota, P.K.; Vaslow, D.F.; Wang, C.H.; Hillman, R.E.; Farmer, J.E. Essential versus complex autism: Definition of fundamental prognostic subtypes. Am. J. Med. Genet. 2005, 135, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.; Pichard, N.; Tordjman, S.; Baumann, C.; Burglen, L.; Excoffier, E.; Lazar, G.; Mazet, P.; Pinquier, C.; Verloes, A.; et al. Specific genetic disorders and autism: Clinical contribution towards their identification. J. Autism Dev. Disord. 2005, 35, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Amiet, C.; Gourfinkel-An, I.; Bouzamondo, A.; Tordjman, S.; Baulac, M.; Lechat, P.; Cohen, D.J. Epilepsy in autism is associated with intellectual disability and gender: Evidence from a meta-analysis. Biol. Psychiatry 2008, 64, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Amiet, C.; Gourfinkel-An, I.; Lauent, C.; Bodeneau, N.; Genin, B.; Leguern, E.; Tordjman, S.; Cohen, D.J. Does epilepsy in multiplex autism define a different subgroup of clinical characteristics and genetic risk? Mol. Autism 2013, 4, 47. [Google Scholar] [CrossRef] [PubMed]
- Amiet, C.; Gourfinkel-An, I.; Lauent, C.; Carayol, J.; Genin, B.; Leguern, E.; Tordjman, S.; Cohen, D.J. Epilepsy in simplex autism pedigrees is much lower than the rate in multiplex autism pedigrees. Biol. Psychiatry 2013, 74, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Canitano, R. Epilepsy in autism spectrum disorders. Eur. Child Adolesc. Psychiatry 2007, 16, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Tordjman, S.; Cohen, D.; Anderson, G.M.; Canitano, R.; Botbol, M.; Coulon, N.; Roubertoux, P.L. Reframing autism as a behavioral syndrome and not a specific mental disorder: Implications of genetic and phenotypic heterogeneity. Neurosci. Biobehav. Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Cook, E.H., Jr.; Lindgren, V.; Leventhal, B.L.; Courchesne, R.; Lincoln, A.; Shulman, C.; Lord, C.; Courchesne, E. Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am. J. Hum. Genet. 1997, 60, 928–934. [Google Scholar] [PubMed]
- Bolton, P.F.; Dennis, N.R.; Browne, C.E.; Thomas, N.S.; Veltman, M.W.; Thompson, R.J.; Jacobs, P. The phenotypic manifestations of interstitial duplications of proximal 15q with special reference to the autistic spectrum disorders. Am. J. Hum. Genet. 2001, 105, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.; Martel, C.; Wilson, A.; Déchambre, N.; Amy, C.; Duverger, L.; Guile, J.M.; Pipiras, E.; Benzacken, B.; Cavé, H.; et al. Brief report: visual-spatial deficit in a 16-year-old girl with maternally derived duplication of proximal 15q. J. Autism Dev. Disord. 2007, 37, 1585–1591. [Google Scholar] [CrossRef] [PubMed]
- Schroer, R.J.; Phelan, M.C.; Michaelis, R.C.; Crawford, E.C.; Skinner, S.A.; Cuccaro, M.; Simensen, R.J.; Bishop, J.; Skinner, C.; Fender, D.; et al. Autism and maternally derived aberrations of chromosome 15q. Am. J. Hum. Genet. 1998, 76, 327–336. [Google Scholar] [CrossRef]
- Wandstrat, A.E.; Leana-Cox, J.; Jenkins, L.; Schwartz, S. Molecular cytogenetic evidence for a common break-point in the largest inverted duplications for chromosomes 15. Am. J. Hum. Genet. 1998, 62, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, J.S.; Nurmi, E.L. Genetics of chilhood disorders: XLVII. Autism, Part 6: Duplication and inherited susceptibility of chromosome 15q11–q13 genes in autism. J. Am. Acad. Child Adolesc. Psychiatry 2003, 42, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, J.; King, B.H.; Leventhal, B.L.; Christian, S.L.; Ledbetter, D.H.; Cook, E.H., Jr. Molecular screening for proximal 15q abnormalities in a mentally retard population. J. Med. Genet. 1998, 35, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Steffenburg, S.; Gillberg, C.L.; Seffenburg, U.; Kyllerman, M. Autism in Angelman syndrome: A population-based study. Pediatr. Neurol. 1996, 14, 131–136. [Google Scholar] [CrossRef]
- Peters, S.U.; Beaudet, A.L.; Madduri, N.; Bacino, C.A. Autism in Angelman syndrome: Implications for autism research. Clin. Genet. 2004, 66, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, S.J.; Chen, P.F.; Ng, K.Y.; Bourgois-Rocha, F.; Lemtiri-Chlieh, F.; Levine, E.S.; Lalande, M. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proc. Natl. Acad. Sci. USA 2010, 107, 17668–17673. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, S.; D’Mello, S.R.; Narayanan, V. Epigenitics, autism spectrum, and neurodevelopmental disorders. Neurotherapeutics 2013, 10, 742–756. [Google Scholar] [CrossRef] [PubMed]
- Grafodatskaya, D.; Chung, B.; Szatmari, P.; Weksberg, R. Autism spectrum disorders and epigenetics. J. Am. Acad. Child Adolesc. Psychiatry 2010, 49, 794–809. [Google Scholar] [CrossRef] [PubMed]
- Dawson, A.J.; Cox, J.; Hovanes, K.; Spriggs, E. PWS/AS MS-MLPA confirms maternal origin of 15q11.2 microduplication. Case Rep. Genet. 2015, 2015, 474097. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.S.; Allen, J.A.; Mabb, A.M. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature 2011, 481, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Mabb, A.M.; Judson, M.C.; Zylka, M.J.; Philpot, B.D. Angelman syndrome: Insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 2011, 34, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Erickson, C.A.; Wink, L.K.; Baindu, B.; Ray, B.; Schaefer, T.L.; Pedapati, E.V.; Lahiri, D.K. Analysis of peripheral amyloid precursor protein in Angelman syndrome. Am. J. Med. Genet. 2016, 170, 2334–2337. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yao, A.; Zhi, H.; Kaur, K.; Zhu, Y.; Jia, M.; Wang, Q.; Jin, S.; Zhao, G.; et al. Angelman syndrome protein Ube3a regulates synaptic growth and endocytosis by inhibiting BMP signaling in Drosophila. PLoS Genet. 2016, 12, e1006062. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Person, R.E.; Beaudet, A.L. Ube3a-ATS is an atypical RNA polymerase II transcript that represses the paternal expression of Ube3a. Hum. Mol. Genet. 2012, 21, 3001–3012. [Google Scholar] [CrossRef] [PubMed]
- Kishino, T.; Lalande, M.; Wasgstaff, J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 1997, 15, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Petit, E.; Hérault, J.; Martineau, J.; Perrot, A.; Barthélémy, C.; Hameury, L.; Sauvage, D.; Lelord, G.; Müh, J.P. Association study with two markers of a human homeogene in infantile autism. J. Med. Genet. 1995, 32, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Clayton-Smith, J. Clinical research on Angelman syndrome in the United Kingdom: Observations on 82 affected individuals. Am. J. Med. Genet. 1993, 46, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Summers, J.A.; Allison, D.B.; Lynch, P.S.; Sandler, L. Behaviour problems in Angelman syndrome. J. Intellect. Disabil. Res. 1995, 39, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Walz, N.C. Parent report of stereotyped behaviors, social interaction, and developmental disturbances in individuals with Angelman syndrome. J. Autism Dev. Disord. 2007, 37, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Dykens, E.M.; Cassidy, S.B. Correlates of maladaptive behavior in children and adults with Prader-Willi syndrome. Am. J. Med. Genet. 1995, 60, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Demb, H.B.; Papola, P. PDD and Prader-Willi syndrome. J. Am. Acad. Child Adolesc. Psychiatry 1995, 34, 539–540. [Google Scholar] [CrossRef] [PubMed]
- Dykens, E.M.; Cassidy, S.B.; King, B.H. Maladaptive behavior differences in Prader-Willi syndrome due to paternal deletion versus maternal uniparental disomy. Am. J. Ment. Retard. 1995, 104, 67–77. [Google Scholar] [CrossRef]
- Veltman, M.W.; Thompson, R.J.; Roberts, S.E.; Thomas, N.S.; Whittington, J.; Bolton, P.F. Prader-Willi syndrome—A study comparing deletion and uniparental disomy cases with reference to autism spectrum disorders. Eur. Child Adolesc. Psychiatry 2004, 13, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Descheemaeker, M.J.; Govers, V.; Vermeulen, P.; Fryns, J.P. Pervasive developmental disorders in Prader-Willi syndrome: The Leuven experience in 59 subjects and controls. Am. J. Med. Genet. 2006, 140, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Hogart, A.; Wu, D.; LaSalle, J.M.; Schanen, N.C. The comorbidity of autism with the genomic disorders of chromosome 15q11.2–q13. Neurobiol. Dis. 2010, 38, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Horsthemke, B.; Wagstaff, J. Mechanisms of imprinting of the Prader-Willi/Angelman region. Am. J. Med. Genet. 2008, 146, 2041–2052. [Google Scholar] [CrossRef] [PubMed]
- Buiting, K. Prader-Willi and Angelman syndrome. Am. J. Med. Genet. 2010, 154, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, S.B.; Scwartz, S.; Miller, H.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.A.; Germani, T.; Haqq, A.M.; Zwaigenbaum, L. Autism spectrum disorder in Prader-Willi syndrome: A systematic review. Am. J. Med. Genet. 2015, 167, 2936–2944. [Google Scholar] [CrossRef] [PubMed]
- Veltman, M.W.; Thompson, R.J.; Craig, E.E.; Dennis, N.R.; Roberts, S.E.; Moore, V.; Brown, J.A.; Bolton, P.F. A paternally inherited duplication in the Prader-Willi/Angelman syndrome critical region: A case and family study. J. Autism Dev. Disord. 2015, 35, 117–127. [Google Scholar] [CrossRef]
- Bolton, P.F.; Roobol, M.; Allsopp, L.; Pickles, A. Association between idiopathic infantile macrocephaly and autism spectrum disorders. Lancet 2001, 358, 726–727. [Google Scholar] [CrossRef]
- Roberts, S.E.; Dennis, N.R.; Browne, C.E.; Willatt, L.; Woods, G.; Cross, I.; Jacobs, P.A.; Thomas, S. Characterisation of interstitial duplications and triplications of chromosome 15q11–q13. Hum. Genet. 2002, 110, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Goizet, C.; Excoffier, E.; Taine, L.; Taupiac, E.; El Moneim, A.A.; Arveiler, B.; Bouvard, M.; Lacombe, D. Case with autistic syndrome and chromosome 22q13.3 deletion detected by FISH. Am. J. Hum. Genet. 2000, 96, 839–844. [Google Scholar] [CrossRef]
- Manning, M.A.; Cassidy, S.B.; Clericuzio, C.; Cherry, A.M.; Schwartz, S.; Hudgins, L.; Enns, G.M.; Hoyme, H.E. Terminal 22q deletion syndrome: A newly recognized cause of speech and language disability in the autism spectrum. Pediatrics 2004, 114, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Prasad, C.; Prasad, A.N.; Chodirker, B.N.; Lee, C.; Dawson, A.K.; Jocelyn, L.J.; Chudley, A.E. Genetic evaluation of pervasive developmental disorders: The terminal 22q13 deletion syndrome may represent a recognizable phenotype. Clin.Genet. 2000, 57, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Phelan, M.C.; Rogers, R.C.; Saul, R.A.; Stapleton, G.A.; Sweet, K.; McDermid, H.; Shaw, S.R.; Claytor, J.; Willis, J.; Kelly, D.P. 22q13 Deletion Syndrome. Am. J. Hum. Genet. 2001, 101, 91–99. [Google Scholar] [CrossRef]
- Soorya, L.; Kolevzon, A.; Zweifach, J.; Lim, T.; Dobry, Y.; Schwartz, L.; Frank, Y.; Wang, A.T.; Cai, G.; Parkhomenko, E.; et al. Prospective investigation of autism and genotype-phenotype correlations in 22q13 deletion syndrome and SHANK3 deficiency. Mol. Autism 2013, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Shcheglovitov, A.; Shcheglovitova, O.; Yazawa, M.; Portmann, T.; Shu, R.; Sebastiano, V.; Krawisz, A.; Froehlich, W.; Bernstein, J.A.; Hallmayer, J.F.; et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 2013, 503, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Phelan, K.; McDermid, H.E. The 22q13.3 deletion syndrome (Phelan-McDermid syndrome). Mol. Syndromol. 2012, 2, 186–201. [Google Scholar] [CrossRef] [PubMed]
- Peca, J.; Feliciano, C.; Ting, J.T.; Wang, W.; Wells, M.F.; Venkatraman, T.N.; Lascola, C.D.; Fu, Z.; Feng, G. SHANK3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011, 472, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Jafri, F.; Fink, J.; Higgins, R.R.; Tervo, R. 22q13.32 deletion and duplication and inversion in the same family: A rare occurrence. ISRN Pediatr. 2011, 2011, 829825. [Google Scholar] [CrossRef] [PubMed]
- Zwanenburg, R.J.; Ruiter, S.A.J.; van Den Heuvel, E.R.; Flapper, B.C.T.; van Ravenswaaij-Arts, C.M.A. Developmental phenotype in Phelan-McDermid (22q13.3 deletion) syndrome: A systematic and prospective study in 34 children. J. Neurodev. Disord. 2016, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.R.; Noor, A.; Vincent, J.B.; Lionel, A.C.; Feuk, L.; Skaug, J.; Shago, M.; Moessner, R.; Pinto, D.; Ren, Y.; et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008, 82, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Hanson, E.; Nasir, R.H.; Fong, A.; Lian, A.; Hundley, R.; Shen, Y.; Wu, B.L.; Holm, I.A.; Miller, D.T. 16p11.2 Study group clinicians cognitive and behavioral characterization of 16p11.2 deletion syndrome. J. Dev. Behav. Pediatr. 2010, 31, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.A.; Shen, Y.; Korn, J.M.; Arking, D.E.; Miller, D.T.; Fossdal, R.; Saemundsen, E.; Stefansson, H.; Ferreira, M.A.; Green, T.; et al. Consortium association between microdeletion and microduplication at 16p11.2 and autism. New Engl. J. Med. 2008, 358, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.A.; KaraMohamed, S.; Sudi, J.; Conrad, D.F.; Brune, C.; Badner, J.A.; Gilliam, T.C.; Nowak, N.J.; Cook, E., Jr.; Dobyns, W.B.; et al. Recurrent 16p11.2 microdeletions in autism. Hum. Mol. Genet. 2008, 17, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Fombonne, E.; Du Mazaubrun, C.; Cans, C.; Grandjean, H. Autism and associated medical disorders in a French epidemiological survey. J. Am. Child Adolesc. Psychiatry 1997, 36, 1561–1569. [Google Scholar]
- Steinman, K.J.; Spence, S.J.; Ramocki, M.B.; Proud, M.B.; Kessler, S.K.; Marco, E.J.; Green Snyder, L.; D’Angelo, D.; Chen, Q.; Chung, W.K.; et al. 16p11.2 Deletion and duplication: Characterizing neurologic phenotypes in a large clinically ascertained cohort. Am. J. Med. Genet. 2016, 9999, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Green Snyder, L.; D’Angelo, D.; Chen, Q.; Bernier, R.; Goin-Kochel, R.P.; Wallace, A.S.; Gerdts, J.; Kanne, S.; Berry, L.; Blaskey, L.; et al. Autism spectrum disorder, developmental and psychiatric features in 16p11.2 duplication. J. Autism Dev. Disord. 2016, 46, 2734–2748. [Google Scholar] [CrossRef] [PubMed]
- Fisch, G.S.; Grossfeld, P.; Falk, R.; Battaglia, A.; Youngblom, J.; Simensen, R. Cognitive-behavioral features of Wolf-Hirschhorn syndrome and other subtelomeric microdeletions. Am. J. Med. Genet. 2010, 154, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Fisch, G.S.; Davis, R.; Youngblom, J.; Gregg, J. Genotype-phenotype association studies of chromosome 8p inverted duplication deletion syndrome. Behav. Genet. 2011, 41, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Bregmen, J.D.; Volkmar, F.R. Autism spectrum disorders in genetic syndromes: Implications for diagnosis, intervention and understanding the wider autism spectrum disorder population. J. Intellect. Disabil. Res. 1988, 53, 852–873. [Google Scholar]
- Mariner, R.; Jackson, A.W.; Levitas, A.; Hagerman, R.J.; Braden, M.; McBogg, P.M.; Smith, A.C.; Berry, R. Autism, mental retardation, and chromosomal abnormalities. J. Autism Dev. Disord. 1986, 16, 425–440. [Google Scholar] [CrossRef] [PubMed]
- Ghaziuddin, M. Autism in down syndrome: Family history correlates. J. Intellect. Disabil. Res. 1997, 41, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.; Howlin, P. Autism spectrum disorders in genetic syndromes: Implications for diagnosis, intervention and understanding the wider autism spectrum disorder population. J. Intellect. Disabil. Res. 2009, 53, 852–873. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Jing, Y.; Cost, G.J.; Chiang, J.C.; Kolpa, H.J.; Cotton, A.M.; Carone, D.M.; Carone, B.R.; Shivak, D.A.; Guschin, D.Y.; et al. Translating dosage compensation to trisomy 21. Nature 2013, 500, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.C.; McGavran, L.; Robinson, J.; Waldstein, G.; Macfarlane, J.; Zonona, J.; Reiss, J.; Lahr, M.; Allen, L.; Magenis, E. Interstitial deletion of (17) (P11.2p11.2) in nine patients. Am. J. Hum. Genet. 1986, 24, 393–414. [Google Scholar] [CrossRef] [PubMed]
- Vostanis, P.; Harrington, R.; Prendergast, M.; Farndon, P. Case reports of autism with interstitial deletion of chromosome 17 (p11.2 p11.2) and monosomy of chromosome 5 (5pter > 5p15.3). Psychiatr. Genet. 1994, 4, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Laje, G.; Morse, R.; Richter, W.; Ball, J.; Pao, M.; Smith, A.C.M. Autism spectrum features in smith-magenis syndrome. Am. J. Med. Genet. 2010, 154, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Potocki, L.; Bi, W.; Treadwell-Deering, D.; Carvalho, C.M.; Eifert, A.; Friedman, E.M.; Glaze, D.; Krull, K.; Lee, J.A.; Lewis, R.A.; et al. Characterization of Potocki-Lupski syndrome (dup(17) (p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am. J. Med. Genet. 2007, 80, 633–649. [Google Scholar] [CrossRef]
- Ghaziuddin, M.; Burmeister, M. Deletion of chromosome 2q37 and autism: A distinct subtype? J. Autism Dev. Disord. 1999, 29, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Devillard, F.; Guinchat, V.; Moreno-De-Luca, D.; Tabet, A.C.; Gruchy, N.; Guillem, P.; Nguyen Morel, M.A.; Leporrier, N.; Leboyer, M.; Jouk, P.S.; et al. Paracentric inversion of chromosome 2 associated with cryptic duplication of 2q14 and deletion of 2q37 in a patient with autism. Am. J. Med. Genet. 2010, 152, 2346–2354. [Google Scholar] [CrossRef] [PubMed]
- Galasso, C.; Lo-Castro, A.; Lalli, C.; Nardone, A.M.; Gullotta, F.; Curatolo, P. Deletion 2q37: An identifiable clinical syndrome with mental retardation and autism. J. Child Neurol. 2008, 23, 802–806. [Google Scholar] [CrossRef] [PubMed]
- Falk, R.E.; Casas, K.A. Chromosome 2q37 deletion: Clinical and molecular aspects. Am. J. Med. Genet. 2007, 145, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Fisch, G.S.; Falk, R.E.; Carey, J.C.; Imitola, J.; Sederberg, M.; Caravalho, K.S.; South, S. Deletion 2q37 syndrome: Cognitive-Behavioral trajectories and autistic features related to breakpoint and deletion size. Am. J. Med. Genet. 2016, 170, 2282–2291. [Google Scholar] [CrossRef] [PubMed]
- Lo-Castro, A.; Galasso, C.; Cerminara, C.; El-Malhany, N.; Benedetti, S.; Nardone, A.M.; Curatolo, P. Association of syndromic mental retardation and autism with 22q11.2 duplication. Neuropediatrics 2009, 40, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Lo-Castro, A.; Benvenuto, A.; Galasso, C.; Porfirio, C.; Curatolo, P. Autism spectrum disorders associated with chromosomal abnormalities. Res. Autism Spectr. Disord. 2010, 4, 319–327. [Google Scholar] [CrossRef]
- Portnoï, M.F. Microduplication 22q11.2: A new chromosomal syndrome. Eur. J. Med. Genet. 2009, 52, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.D.; Skuse, D.H. Adolescents and young adults with 22q11 deletion syndrome: Psychopathology in an at-risk group. Br. J. Psychiatry 2005, 186, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Wenger, T.L.; Miller, J.S.; DePolo, L.M.; de Marchena, A.B.; Clements, C.C.; Emanuel, B.S.; Zackai, E.H.; McDonald-McGinn, D.M.; Schultz, R.T. 22q11.2 duplication syndrome: Elevated rate of autism spectrum disorder and need for medical screening. Mol. Autism 2016, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Hidding, E.; Swaab, H.; de Sonneville, L.M.J.; van Engeland, H.; Vorstman, J.A.S. The role of COMT and plasma proline in the variable penetrance of autistic spectrum symptoms in 22q11.2 deletion syndrome. Clin. Genet. 2016, 90, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Mefford, H.C.; Sharp, A.J.; Baker, C.; Itsara, A.; Jiang, Z.; Buysse, K.; Huang, S.; Maloney, V.K.; Crolla, J.A.; Baralle, D.; et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N. Engl. J. Med. 2008, 359, 1685–1699. [Google Scholar] [CrossRef] [PubMed]
- Brunetti-Pierri, N.; Berg, J.S.; Scaglia, F.; Belmont, J.; Bacino, C.A.; Sahoo, T.; Lalani, S.R.; Graham, B.; Lee, B.; Shinawi, M.; et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat. Genet. 2008, 40, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Bernier, R.; Steinman, K.J.; Reilly, B.; Wallace, A.S.; Sherr, E.H.; Pojman, N.; Mefford, H.C.; Gerdts, J.; Earl, R.; Hanson, E.; et al. Clinical phenotype of the recurrent 1q21.1 copy-number variant. Gen. Med. 2016, 18, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Torres, F.; Barbosa, M.; Maciel, P. Recurrent copy number variations as risk factors for neurodevelopmental disorders: Critical overview and analysis of clinical implications. J. Med. Genet. 2016, 53, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.S.; Brunetti-Pierri, N.; Peters, S.U.; Kang, S.H.; Fong, C.T.; Salamone, J.; Freedenberg, D.; Hannig, V.L.; Prock, L.A.; Miller, D.T.; et al. Speech delay and autism spectrum behaviors are frequently associated with duplication of the 7q11.23 Williams-Beuren syndrome region. Genet. Med. 2007, 9, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Depienne, C.; Heron, D.; Betancur, C.; Benyahia, B.; Trouillard, O.; Bouteiller, D.; Verloes, A.; LeGuern, E.; Leboyer, M.; Brice, A. Autism, language delay and mental retardation in a patient with 7q11 duplication. J. Med. Genet. 2007, 44, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, L.; Prosnitz, A.; Pardo, S.; Bhatt, J.; Cohen, N.; Lauriat, T.; Ouchanov, L.; González, P.J.; Manghi, E.R.; Bondy, P.; et al. An atypical deletion of the Williams-Beuren syndrome interval implicates genes associated with defective visuospatial processing and autism. J. Med. Genet. 2007, 44, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, C.; Bonaglia, M.C.; Selicorni, A.; Borgatti, R.; Giorda, R. Unusual cognitive and behavioural profile in a Williams syndrome patient with atypical 7q11.23 deletion. J. Med. Genet. 2003, 40, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, A.J.; Searcy, Y.M.; Jones, W.; Lord, C. Social interaction behaviors discriminate young children with autism and Williams syndrome. J. Am. Acad. Child Adolesc. Psychiatry 2007, 46, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; Ercan-Sencicek, A.G.; Hus, V.; Luo, R.; Murtha, M.T.; Moreno-de-Luca, D.; Chu, S.H.; Moreau, M.P.; Gupta, A.R.; Thomson, S.A.; et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 2011, 70, 863–885. [Google Scholar] [CrossRef] [PubMed]
- Somerville, M.J.; Mervis, C.B.; Young, E.J.; Seo, E.J.; del Campo, M.; Bamforth, S.; Peregrine, E.; Loo, W.; Lilley, M.; Pérez-Jurado, L.A.; et al. Severe expressive-language delay related to duplication of the Williams-Beuren locus. N. Engl. J. Med. 2005, 353, 1694–1701. [Google Scholar] [CrossRef] [PubMed]
- Van der Aa, N.; Rooms, L.; Vandeweyer, G.; van den Ende, J.; Reyniers, E.; Fichera, M.; Romano, C.; Delle Chiaie, B.; Mortier, G.; Menten, B.; et al. Fourteen new cases contribute to the characterization of the 7q11.23 microduplication syndrome. Eur. J. Hum. Genet. 2009, 52, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Makeyev, A.V.; Enkhamandakh, B.; Hong, S.H.; Joshi, P.; Shin, D.G.; Bayarsaihan, D. Diversity and complexity in chromatin recognition by TFII-I transcription factors in pluripotent embryonic stem cells and embryonic tissues. PLoS ONE 2012, 7, e44443. [Google Scholar] [CrossRef] [PubMed]
- Tordjman, S.; Anderson, G.M.; Botbol, M.; Toutain, A.; Sarda, P.; Carlier, M.; Saugier-Veber, P.; Baumann, C.; Cohen, D.; Lagneaux, C.; et al. Autistic disorder in patients with Williams-Beuren syndrome: A reconsideration of the Williams-Beuren syndrome phenotype. PLoS ONE 2012, 7, e30778. [Google Scholar] [CrossRef] [PubMed]
- Tordjman, S.; Anderson, G.M.; Cohen, D.; Kermarrec, S.; Carlier, M.; Touitou, Y.; Saugier-Veber, P.; Lagneaux, C.; Chevreuil, C.; Verloes, A. Presence of autism, hyperserotonemia, and severe expressive language impairment in Williams-Beuren syndrome. Mol. Autism 2013, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Tordjman, S.; Najjar, I.; Bellissant, E.; Anderson, G.M.; Barburoth, M.; Cohen, D.; Jaafari, N.; Schischmanoff, O.; Fagard, R.; Lagdas, I.; et al. Advances in the research of melatonin in autism spectrum disorders: Literature review and new perspectives. Int. J. Mol. Sci. 2013, 14, 20508–20542. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.; Jones, C.; Groves, L.; Moss, J.; Oliver, C. Prevalence of autism spectrum disorder phenomenology in genetic disorders: A systematic review and meta-analysis. Lancet Psychiatry 2015, 2, 909–916. [Google Scholar] [CrossRef]
- Skuse, D.H.; James, R.S.; Bishop, D.V.; Coppin, B.; Dalton, P.; Aamodt-Leeper, G.; Bacarese-Hamilton, M.; Creswell, C.; McGurk, R.; Jacobs, P.A. Evidence from Turner’s syndrome of an imprinted X-linked locus affecting cognitive function. Nature 1997, 387, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Sybert, V.P.; McCauley, E. Turner’ syndrome. N. Engl. J. Med. 2004, 351, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Kent, L.; Bowdin, S.; Kirby, G.A.; Cooper, W.N.; Maher, E.R. Beckwith Wiedemann syndrome: A behavorial phenotype-genotype study. Am. Med. Genet. 2008, 147, 1295–1297. [Google Scholar] [CrossRef] [PubMed]
- DeBaun, M.R.; Niemitz, E.L.; Feinberg, A.P. Association of in vitro fertilization with Beckwith-Wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am. J. Hum. Genet. 2003, 72, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.C.; Choufani, S.; Ferreira, J.C.; Weksberg, R. Growth regulation, imprinted genes, and chromosome 11p15.5. Pediatr. Res. 2007, 61, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Weksberg, R.; Shuman, C.; Beckwith, J.B. Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 2010, 18, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Wolpert, C.M.; Menold, M.M.; Bass, M.P.; Qumsiyeh, M.B.; Donnelly, S.L.; Ravan, S.A.; Vance, J.M.; Gilbert, J.R.; Abramson, R.K.; Wright, H.H.; et al. Three probands with autistic disorder and isodicentric chromosome 15. Am. J. Med. Genet. 2000, 96, 365–372. [Google Scholar] [CrossRef]
- Wolpert, C.; Pericak-Vance, M.A.; Abramson, R.K.; Wright, H.H.; Cuccaro, M.L. Autistic symptoms among children and young adults with isodicentric chromosome 15. Am. J. Med. Genet. 2000, 96, 128–129. [Google Scholar] [CrossRef]
- Parisi, L.; di Filippo, T.; Roccella, M. Hypomelanosis of ito: Neurological and psychiatric pictures in developmental age. Minerva Pediatr. 2012, 64, 65–70. [Google Scholar] [PubMed]
- Åkefeldt, A.; Gillberg, C. Hypomelanosis of ito in three cases with autism and autistic-like conditions. Dev. Med. Child Neurol. 1991, 33, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.; Gillberg, C.; Råstam, M. Autism spectrum conditions in individuals with Möbius sequence, CHARGE syndrome and oculo-auriculo-vertebral spectrum: Diagnostic aspects. Res. Dev. Disabil. 2010, 31, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.; Råstam, M.; Billstedt, E.; Danielsson, S.; Strömland, K.; Miller, M.; Gillberg, C. Autism spectrum disorders and underlying brain pathology in CHARGE association. Dev. Med. Child Neurol. 2006, 48, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.M.; Nichols, S.L.; Issekutz, K.; Blake, K. Canadian paediatric surveillance program behavioral profiles and symptoms of autism in CHARGE syndrome: Preliminary Canadian epidemiological data. Am. J. Med. Genet. 2005, 133, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Hartshorne, T.S.; Grialou, T.L.; Parker, K.R. Autistic-like behavior in CHARGE syndrome. Am. J. Med. Genet. 2005, 133, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Patten, S.A.; Jacobs-McDaniels, N.L.; Zaouter, C.; Drapeau, P.; Albertson, R.C.; Moldovan, F. Role of Chd7 in zebrafish: A model for CHARGE syndrome. PLoS ONE 2012, 7, e31650. [Google Scholar] [CrossRef] [PubMed]
- Blake, K.D.; Prasad, C. CHARGE syndrome. Orphanet J. Rare Dis. 2006, 1, 34. [Google Scholar] [CrossRef] [PubMed]
- Bolton, P.F.; Park, R.J.; Higgins, J.N.; Griffiths, P.D.; Pickles, A. Neuro-epileptic determinants of autism spectrum disorders in tuberous sclerosis complex. Brain 2002, 125, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Curatolo, P.; Bombardieri, R.; Jozwiak, S. Tuberous sclerosis. Lancet 2008, 372, 657–668. [Google Scholar] [PubMed]
- Jeste, S.S.; Varcin, K.J.; Hellemann, G.S.; Gulsrud, A.C.; Bhatt, R.; Kasari, C.; Wu, J.Y.; Sahin, M.; Nelson, C.A. Symptom profiles of autism spectrum disorder in tuberous sclerosis complex. Neurology 2016. [Google Scholar] [CrossRef] [PubMed]
- McBride, K.L.; Varga, E.A.; Pastore, M.T.; Prior, T.W.; Manickam, K.; Atkin, J.F.; Herman, G.E. Confirmation study of PTEN mutations among individuals with autism or developmental delays/mental retardation and macrocephaly. Autism Res. 2010, 3, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Lintas, C.; Persico, A.M. Autistic phenotypes and genetic testing: State-of-the-art for the clinical geneticist. J. Med. Genet. 2009, 46, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.D.; Cai, G.; Chaste, P.; Nygren, G.; Goldsmith, J.; Reichert, J.; Anckarsäter, H.; Rastam, M.; Smith, C.J.; Silverman, J.M.; et al. Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am. J. Med. Genet. 2007, 144, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Herman, G.E.; Butter, E.; Enrile, B.; Pastore, M.; Prior, T.W.; Sommer, A. Increasing knowledge of PTEN germline mutations: Two additional patients with autism and macrocephaly. Am. J. Med. Genet. 2007, 143, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Goffin, A.; Hoefsloot, L.H.; Bosgoed, E.; Swillen, A.; Fryns, J.P. PTEN mutation in a family with Cowden syndrome and autism. Am. J. Med. Genet. 2001, 105, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, L.; Black, F.M.; Berg, J.N.; Eickholt, B.J.; Leslie, N.R. Functionally distinct groups of inherited PTEN mutations in autism and tumour syndromes. J. Med. Genet. 2015, 52, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Van Den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is cause by mutations in X-linked MECP2, encoding methil-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [PubMed]
- Olsonn, B.; Rett, A. A review of the Rett syndrome with a theory of autism. Brain Dev. 1990, 12, 11–15. [Google Scholar] [CrossRef]
- Young, D.J.; Bebbington, A.; Anderson, A.; Ravine, D.; Ellaway, C.; Kulkarni, A.; de Klerk, N.; Kaufmann, W.E.; Leonard, H. The diagnosis of autism in a female: Could it be Rett syndrome? Eur. J. Pediatr. 2008, 167, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L. The relationship of Rett syndrome and MECP2 disorders to autism. Dialogues Clin. Neurosci. 2012, 14, 253–262. [Google Scholar] [PubMed]
- Gadalla, K.; Bailey, M.E.; Cobb, S.R. MeCP2 and Rett syndrome: Reversibility and potential avenues for therapy. Biochem. J. 2011, 439, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Angus, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.S.; Schanen, C.; et al. Rett syndrome: Revised diagnostic criteria and nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Ramocki, M.B.; Peters, S.U.; Tavyev, Y.J.; Zhang, F.; Carvalho, C.M.; Schaaf, C.P.; Richman, R.; Fang, P.; Glaze, D.G.; Lupski, J.R.; et al. Autism and other neuropsychiatric symptoms are prevalent in individuals with MeCP2 duplication syndrome. Ann. Neurol. 2009, 66, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Nidiffer, F.D.; Kelly, T.E. Developmental and degenerative patterns associated with cognitive, behavioural and motor difficulties in the Sanfilippo syndrome: An epidemiological study. J. Ment. Defic. Res. 1983, 27, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Wraith, J.E. The mucopolysaccharidoses: A clinical review and guide to management. Arch. Dis. Child. 1995, 72, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Ritvo, E.R.; Mason-Brothers, A.; Freeman, B.J.; Pingree, C.; Jenson, W.R.; McMahon, W.M.; Petersen, P.B.; Jorde, L.B.; Mo, A.; Ritvo, A. The UCLA-university of Utah epidemiologic survey of autism: The etiologic role of rare diseases. Am. J. Psychiatry 1990, 147, 1614–1621. [Google Scholar] [PubMed]
- Shapiro, E.; King, K.; Ahmed, A.; Rudser, K.; Rumsey, R.; Yund, B.; Delaney, K.; Nestrasil, I.; Whitley, C.; Potegal, M. The neurobehavioral phenotype in mucopolysaccharidosis type IIIB: An exploratory study. Mol. Genet. Metab. Rep. 2016, 6, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Ramaekers, V.T.; Blau, N. Cerebral folate deficiency. Dev. Med. Child Neurol. 2004, 46, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Moretti, P.; Scaglia, F. Cerebral folate deficiency with developmental delay, autism, and response to folinic acid. Neurology 2005, 64, 1088–1090. [Google Scholar] [CrossRef] [PubMed]
- Gordon, N. Cerebral folate deficiency. Dev. Med. Child Neurol. 2009, 51, 180–182. [Google Scholar] [CrossRef] [PubMed]
- Raidah, S.A.; Mohammed, W.C. Diagnosis and management of cerebral folate deficiency. Neurosciences 2014, 19, 312–316. [Google Scholar]
- Ryan, A.K.; Bartlett, K.; Clayton, P.; Eaton, S.; Mills, L.; Donnai, D.; Winter, R.M.; Burn, J. Smith-Lemli-Opitz syndrome: A variable clinical and biochemical phenotype. J. Med. Genet. 1998, 35, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.I.; Hennekam, R.C.M. The Smith-Lemli-Opitz syndrome. J. Med. Genet. 2000, 37, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Tierny, E.; Nwokoro, N.A.; Porter, F.D.; Freund, L.S.; Ghuman, J.K.; Kelley, R.I. Behavior phenotype in the RSH/ Smith-Lemli-Opitz syndrome. Am. J. Med. Genet. 2001, 98, 191–200. [Google Scholar] [CrossRef]
- Tierny, E.; Nwokoro, N.A.; Kelley, R.I. Behavior phenotype of RSH/Smith-Lemli-Opitz syndrome. Ment. Retard. Dev. Disabil. Res. Rev. 2001, 98, 191–200. [Google Scholar]
- Tint, G.S.; Irons, M.; Elias, E.R.; Batta, A.K.; Frieden, R.; Chen, T.S.; Salen, G. Defective cholesterol biosynthesis associated with the Smith-Lemli-Opitz syndrome. N. Engl. J. Med. 1994, 330, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Thurm, A.; Tierney, E.; Farmer, C.; Albert, P.; Joseph, L.; Swedo, S.; Bianconi, S.; Bukelis, I.; Wheeler, C.; Sarphare, G.; et al. Development, behavior, and biomarker characterization of Smith-Lemli-Opitz syndrome: An update. J. Neurodev. Disord. 2016, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Baieli, S.; Pavone, L.; Meli, C.; Fiumara, A.; Coleman, M. Autism and phenylketonuria. J. Autism Dev. Disord. 2003, 33, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Miladi, N.; Larnaout, A.; Kaabachi, N.; Helayem, M.; Ben Hamida, M. Phenylketonuria: An underlying etiology of autistic syndrome report. J. Child Neurol. 1992, 7, 22–23. [Google Scholar] [CrossRef] [PubMed]
- Fon, E.A.; Sarrazin, J.; Meunier, C.; Alarcia, J.; Shevell, M.I.; Philippe, A.; Leboyer, M.; Rouleau, G.A. Adenylosuccinate lyase (ADSL) and infantile autism: Absence of previously reported point mutation. Am. J. Hum. Genet. 1995, 18, 554–557. [Google Scholar] [CrossRef] [PubMed]
- Jaeken, J.; van den Berghe, G. An infantile autistic syndrome characterized by the presence of succinylpurines in body fluids. Lancet 1984, 2, 1058–1061. [Google Scholar] [PubMed]
- Race, V.; Marie, S.; Vincent, M.F.; van der Berghe, G. Clinical, biochemical and molecular genetic correlations in adenylosuccinate lyase deficiency. Hum. Mol. Genet. 2000, 9, 2159–2163. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.L.; Aimi, J.; Barshop, B.A.; Jaeken, J.; van den Berghe, G.; Zalkin, H.; Dixon, J.E. A mutation in adenylosuccinate lyase associated with mental retardation autistic features. Nat. Genet. 1992, 1, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Jinnah, H.A.; Sabina, R.L.; Van Den Berghe, G. Metabolic disorders of purine metabolism affecting the nervous system. Handb. Clin. Neurol. 2013, 113, 1827–1836. [Google Scholar] [PubMed]
- Nasrallah, F.; Feki, M.; Kaabachi, N. Creatine and creatine deficiency syndromes: Biochemical and clinical aspects. Pediatr. Neurol. 2010, 42, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Póo-Argüelles, P.; Arias, A.; Vilaseca, M.A.; Ribes, A.; Artuch, R.; Sans-Fito, A.; Moreno, A.; Jakobs, C.; Salomons, G. X-linked creatine transporter deficiency in two patients with severe mental retardation and autism. J. Inherit. Metab. Dis. 2006, 29, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Bauman, M.; Tsai, A.C.; Reynolds, A.; Roberts, W.; Anagnostou, E.; Cameron, J.; Nozzolillo, A.A.; Chen, S.; Kyriakopoulou, L.; et al. Prevalence of creatine deficiency syndromes in children with nonsyndromic autism. Pediatrics 2016. [Google Scholar] [CrossRef] [PubMed]
- Campistol, J.; Díez-Juan, M.; Callejón, L.; Fernandez-De Miguel, A.; Casado, M.; Garcia Cazorla, A.; Lozano, R.; Artuch, R. Inborn error metabolic screening in individuals with nonsyndromic autism spectrum disorders. Dev. Med. Child Neurol. 2016, 58, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Durand, C.M.; Betancur, C.; Boeckers, T.M.; Bockmann, J.; Chaste, P.; Fauchereau, F.; Nygren, G.; Rastam, M.; Gillberg, I.C.; Anckarsäter, H.; et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2007, 39, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Moessner, R.; Marshall, C.R.; Sutcliffe, J.S.; Skaug, J.; Pinto, D.; Vincent, J.; Zwaigenbaum, L.; Fernandez, B.; Roberts, W.; Szatmari, P.; et al. Contribution of SHANK3 mutations to autism spectrum disorder. Am. J. Hum. Genet. 2007, 81, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Guiltmatre, A.; Huquet, G.; Delorme, R.; Bourgeron, T. The emerging role of SHANK genes in neuropsychiatric disorders. Dev. Neurobiol. 2014, 74, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Schroer, R.; Yan, J.; Song, W.; Yang, C.; Bockholt, A.; Cook, E.H., Jr.; Skinner, C.; Schwartz, C.E.; Sommer, S.S. High frequency of neurexin 1β signal peptide structural variants in patients with autism. Neurosci. Lett. 2006, 27, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Ching, M.S.; Shen, Y.; Tan, W.H.; Jeste, S.S.; Morrow, E.M.; Chen, X.; Mukaddes, N.M.; Yoo, S.Y.; Hanson, E.; Hundley, R.; et al. Deletions of NRXN1 (neurexin-1) predispose to a wide spectrum of developmental disorders. Am. J. Med. Genet. 2010, 153, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Autism Genome Project Consortium. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 2007, 39, 319–328. [Google Scholar]
- Viñas-Jornet, M.; Esteba-Castillo, S.; Gabau, E.; Ribas-Vidal, N.; Baena, N.; San, J.; Ruiz, A.; Coll, M.D.; Novell, R.; Guitart, M. A common cognitive, psychiatric and dysmorphic phenotype in carriers of NRXN1 deletion. Mol. Genet. Genom. Med. 2014, 2, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Dabell, M.P.; Rosenfeld, J.A.; Bader, P.; Escobar, L.F.; El-Khechen, D.; Vallee, S.E.; Dinulos, M.B.P.; Curry, C.; Fisher, J.; Tervo, R.; et al. Investigation of NRXN1 deletions: Clinical and molecular characterization. Am. J. Med. Genet. 2013, 161, 717–731. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, M.; Abrahams, B.S.; Stone, J.L. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am. J. Med. Genet. 2008, 82, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Werling, A.M.; Bobrowski, E.; Taurines, R.; Gundelfinger, R.; Romanos, M.; Grünblatt, E.; Walitza, S. CNTNAP2 gene in high functioning autism: No association according to family and meta-analysis approaches. J. Neural Transm. 2016, 123, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, J.D.; Gupta, A.R.; Sanders, S.J.; Walker, M.F.; Keaney, J.; Fernandez, T.V.; Murtha, M.T.; Anyanwu, S.; Ober, G.T.; Raubeson, M.J.; et al. No evidence for association of autism with rare heterozygous point mutations in contactin-associated protein-like 2 (CNTNAP2), or in other contactin-associated proteins or contactins. PLoS Genet. 2015, 11, e1004852. [Google Scholar] [CrossRef] [PubMed]
- Bakkaloglu, B.; O’Roak, B.J.; Louvi, A.; Gupta, A.R.; Abelson, J.F.; Morgan, T.M.; Chawarska, K.; Klin, A.; Ercan-Sencicek, A.G.; Stillman, A.A.; et al. Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am. J. Hum. Genet. 2008, 82, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, T.; Morgan, T.; Davis, N.; Klin, A.; Morris, A.; Farhi, A.; Lifton, R.P.; State, M.W. Disruption of contactin 4 (CNTN4) results in developmental delay and other features of 3p deletion syndrome. Am. J. Hum. Genet. 2004, 74, 1286–1293. [Google Scholar] [CrossRef] [PubMed]
- Roohi, J.; Montagna, C.; Tegay, D.H.; Palmer, L.E.; DeVincent, C.; Pomeroy, J.C.; Christian, S.L.; Nowak, N.; Hatchwell, E. Disruption of contactin 4 in three subjects with autism spectrum disorder. J. Med. Genet. 2008, 46, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Karayannis, T.; Au, E.; Patel, J.C.; Kruglikov, I.; Markx, S.; Delorme, R.; Héron, D.; Salomon, D.; Glessner, J.; Restituito, S.; et al. Cntnap4 differentially contributes to GABAergic and dopaminergic synaptic transmission. Nature 2014, 511, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Zhiling, Y.; Fujita, E.; Tanabe, Y. Mutations in the gene encoding CADM1 are associated with autism spectrum disorder. Biochem. Biophys. Res. Commun. 2008, 377, 926–929. [Google Scholar] [CrossRef] [PubMed]
- Fujita, E.; Tanabe, Y.; Imhof, B.A.; Mamsi, M.Y.; Momoi, T. A complex of synaptic adhesion molecule CADM1, a molecule related to autism spectrum disorder, with MUPP1 in the cerebellum. J. Neurochem. 2012, 123, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Kitagishi, Y.; Minami, A.; Nakanishi, A.; Ogura, Y.; Matsuda, S. Neuron membrane trafficking and protein kinases involved in autism and ADHD. Int. J. Mol. Sci. 2015, 16, 3095–3115. [Google Scholar] [CrossRef] [PubMed]
- Morrow, E.M.; Yoo, S.Y.; Flavell, S.W.; Kim, T.K.; Lin, Y.; Hill, R.S.; Mukaddes, N.M.; Balkhy, S.; Gascon, G.; Hashmi, A.; et al. Identifying autism loci and genes by tracing recent shared ancestry. Science 2008, 321, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Jamain, S.; Quach, H.; Betancur, C.; Rastam, M.; Colineaux, C.; Gillberg, I.C.; Soderstrom, H.; Giros, B.; Leboyer, M.; Gillberg, C.; et al. Mutations of the X-linked genes encodage neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003, 34, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Laumonnier, F.; Bonnet-Brilhault, F.; Gomot, M.; Blanc, R.; David, A.; Moizard, M.P.; Raynaud, M.; Ronce, N.; Lemonnier, E.; Calvas, P.; et al. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet. 2004, 74, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Oliveira, G.; Coutinho, A.; Yang, C.; Feng, J.; Katz, C.; Sram, J.; Bockholt, A.; Jones, I.R.; Craddock, N.; et al. Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol. Psychiatry 2005, 10, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Fisch, G.S. Is autism associated with the fragile X syndrome? Am. J. Med. Genet. 1992, 43, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Fisch, G.S. What is associated with the fragile X syndrome? Am. J. Med. Genet. 1993, 48, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.J.; Wehner, D.E.; Hagerman, R. The behavioral phenotype in fragile X: Symptoms of autism in very young children with fragile X syndrome, idiopathic autism, and other developmental disorders. J. Dev. Behav. Pediatr. 2001, 22, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.B., Jr.; Hatton, D.D.; Skinner, M.; Mesibov, G. Autistic behavior, FMR1 protein, and developmental trajectories in young males with fragile X syndrome. J. Autism Dev. Disord. 2001, 31, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Belmonte, M.K.; Bourgeron, T. Fragile X syndrome and autism at the intersection of genetic and neural netwoks. Nat. Neurosci. 2006, 9, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Kau, A.S.; Tierney, E.; Bukelis, I.; Stump, M.H.; Kates, W.R.; Trescher, W.H.; Kaufmann, W.E. Social behavior profile in young males with fragile X syndrome: Characteristics and specificity. Am. J. Med. Genet. 2004, 126, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Loesch, D.Z.; Bui, Q.M.; Dissanayake, C.; Clifford, S.; Gould, E.; Bulhak-Paterson, D.; Tassone, F.; Taylor, A.K.; Hessl, D.; Hagerman, R.; et al. Molecular and cognitive predictors of the continuum of autistic behaviours in fragile X. Neurosci. Biobehav. Rev. 2007, 31, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Clifford, S.; Dissanayake, C.; Bui, Q.M.; Huggins, R.; Taylor, A.K.; Loesch, D.Z. Autism spectrum phenotype in males and females with fragike X full mutation and premutation. J. Autism Dev. Disord. 2007, 337, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.W.; Hessl, D.; Goodlin-Jones, B.; Ferranti, J.; Bacalman, S.; Barbato, I.; Tassone, F.; Hagerman, P.J.; Herman, H.; Hagerman, R.J. Autism profiles of males with fragile X syndrome. Am. J. Ment. Retard. 2008, 113, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Dölen, G.; Carpenter, R.L.; Ocain, T.D.; Bear, M.F. Mechanism-based approaches to treating fragile X. Pharmacol. Ther. 2010, 127, 78–93. [Google Scholar] [CrossRef] [PubMed]
- McLennan, Y.; Polussa, J.; Tassone, F.; Hagerman, R. Fragile X syndrome. Curr. Genom. 2011, 12, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Bar-Nur, O.; Caspi, I.; Benvenisty, N. Molecular analysis of FMR 1 reactivation in fragile-X induced pluripotent stem cells and their neuronal derivatives. J. Mol. Cell. Biol. 2012, 4, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.; Lauterborn, J.; Au, J.; Berry-Kravis, E. Fragile X syndrome and targeted treatment trials. Results Probl. Cell Differ. 2012, 54, 297–335. [Google Scholar] [PubMed]
- Tabolacci, E.; Chiurazzi, P. Epignetics, fragile X syndrome and transcriptional therapy. Am. J. Med. Genet. 2013, 161, 2797–2808. [Google Scholar] [CrossRef] [PubMed]
- De Vries, B.B.; Halley, D.J.; Oostra, B.A.; Niermeijer, C. The fragile X syndrome. Am. J. Med. Genet. 1998, 35, 579–589. [Google Scholar] [CrossRef]
- Tolmie, J. The genetics of mental retardation. Curr. Opin. Psychiatry 1998, 11, 507–513. [Google Scholar] [CrossRef]
- Hagerman, R.J. Fragile X syndrome. Child Adolesc. Psychiatr. Clin. N. Am. 1996, 5, 895–911. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Chudley, A.E.; Knoll, J.H.; Jackson, A.W.; Kemper, M.; Ahmad, R. Autism in fragile X females. Am. J. Med. Genet. 1986, 23, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Hatton, D.D.; Sideris, J.; Skinner, M.; Mankowski, J.; Bailey, D.B., Jr.; Roberts, J.; Mirrett, P. Autistic behavior in children with fragile X syndrome: Prevalence, stability, and the impact of FMRP. Am. J. Med. Genet. 2006, 140, 1804–1813. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.E.; Tonnsen, B.L.; McCary, L.M.; Caravella, K.E.; Shinkareva, S.V. Brief report: Autism symptoms in infants with fragile X syndrome. J. Autism Dev. Disord. 2016, 46, 3830–3837. [Google Scholar] [CrossRef] [PubMed]
- Einfeld, S.; Molony, H.; Hall, W. Autism is not associated with the fragile X syndrome. Am. J. Med. Genet. 1989, 34, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Reiss, A.L.; Freund, L. Behavioral phenotype of fragile X syndrome; DSM-III-R autistic behavior in male children. Am. J. Med. Genet. 1992, 43, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Todd, R.D.; Hudziak, J.J. Genetics of autism. Curr. Opin. Psychiatry 1993, 6, 486–488. [Google Scholar] [CrossRef]
- Farzin, F.; Perry, H.; Hessl, D.; Loesch, D.; Cohen, J.; Bacalman, S.; Gane, L.; Tassone, F.; Hagerman, P.; Hagerman, R. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J. Dev. Behav. Pediatr. 2006, 27, 137–144. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Berry-Kravis, E.; Kaufmann, W.E.; Ono, M.Y.; Tartaglia, N.; Lachiewicz, A.; Kronk, R.; Delahunty, C.; Hessl, D.; Visootak, J.; et al. Advances in the treatment of fragile X syndrome. Pediatrics 2009, 123, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Hallmayer, J.; Hebert, J.M.; Spiker, D.; Lotspeich, L.; McMahon, W.M.; Petersen, P.B.; Nicholas, P.; Pingree, C.; Lin, A.A.; Cavalli-Sforza, L.L.; et al. Autism and the X chromosome. Multipoint sib-pair analysis. Arch. Gen. Psychiatry 1996, 53, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Constantino, J.N.; Zhang, Y.; Holzhauer, K.; Sant, S.; Long, K.; Vallorani, A.; Malik, L.; Gutmann, D.H. Distribution and Within-Family specificity of quantitative autistic traits in patients with neurofibromatosis type I. J. Pediatr. 2015, 167, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.; Plasschaert, E.; Descheemaeker, M.J.; Huson, S.; Borghgraef, M.; Vogels, A.; Evans, D.G.; Legius, E.; Green, J. Autism spectrum disorder profile in neurofibromatosis type I. J. Autism Dev. Disord. 2015, 45, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Plasschaert, E.; Descheemaeker, M.J.; Van Eylen, L.; Noens, I.; Steyaert, J.; Legius, E. Prevalence of autism spectrum disorder symptoms in children with neurofibromatosis type 1. Am. J. Med. Genet. 2014, 168, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Timonen-Soivio, L.; Vanhala, R.; Malm, H.; Hinkka-Yli-Salomäki, S.; Gissler, M.; Brown, A.; Sourander, A. Brief report: Syndromes in autistic children in a Finnish birth cohort. J. Autism Dev. Disord. 2016, 46, 2780–2784. [Google Scholar] [CrossRef] [PubMed]
- Sheth, K.; Moss, J.; Hyland, S.; Stinton, C.; Cole, T.; Oliver, C. The behavioral characteristics of Sotos syndrome. Am. J. Med. Genet. 2015, 167, 2945–2956. [Google Scholar] [CrossRef] [PubMed]
- Assumpcao, F.; Santos, R.C.; Rosario, M.; Mercadante, M. Brief report: Autism and Aarskog syndrome. J. Autism Dev. Disord. 1999, 29, 179–181. [Google Scholar] [CrossRef] [PubMed]
- De Wolf, V.; Crepel, A.; Schuit, F.; van Lommel, L.; Ceulemans, B.; Steyaert, J.; Seuntjens, E.; Peeters, H.; Devriendt, K. A complex Xp11.22 deletion in a patient with syndromic autism: Exploration of FAM120C as a positional candidate gene for autism. Am. J. Med. Genet. 2014, 164, 3035–3041. [Google Scholar] [CrossRef] [PubMed]
- Parisi, L.; Di Filippo, T.; Roccella, M. Behavioral phenotype and autism spectrum disorders in Cornelia de Lange syndrome. Ment. Illn. 2015, 7, 5988. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Landy-Schmitt, C.; Clark, B.; Kline, A.D.; Specht, M.; Grados, M.A. Autism traits in children and adolescents with Cornelia de Lange syndrome. Am. J. Med. Genet. 2014, 321, 1400–1410. [Google Scholar] [CrossRef] [PubMed]
- Alvarez Retuerto, A.I.; Cantor, R.M.; Gleeson, J.G.; Ustaszewska, A.; Schackwitz, W.S.; Pennacchio, L.A.; Geschwind, D.H. Association of common variants in the Joubert syndrome gene (AHI1) with autism. Hum. Mol. Genet. 2008, 17, 3887–3896. [Google Scholar] [CrossRef] [PubMed]
- Ionita-Laza, I.; Capanu, M.; De Rubeis, S.; McCallum, K.; Buxbaum, J.D. Identification of rare causal variants in sequence-based studies: Methods and applications to VPS13B, a gene involved in Cohen syndrome and Autism. PLoS Genet. 2014, 10, e1004729. [Google Scholar] [CrossRef] [PubMed]
- Howlin, P.; Karpf, J.; Turk, J. Behavioural characteristics and autistic features in individuals with Cohen syndrome. Eur. Child Adolesc. Psychiatry 2005, 14, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Chandler, K.E.; Moffett, M.; Clayton-Smith, J.; Baker, G.A. Neuropsychological assessment of a group of UK patients with Cohen syndrome. Neuropediatrics 2003, 34, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Lerma-Carrillo, I.; Molina, J.D.; Cuevas-Duran, T.; Julve-Correcher, C.; Espejo-Saavedra, J.M.; Andrade-Rosa, C.; Lopez-Muñoz, F. Psychopathology in the Lujan-Fryns syndrome: Report of two patients and review. Am. J. Med. Genet. 2006, 140, 2807–2811. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, P.; Piccini, G.; Caciolo, C.; Perrino, F.; Gambardella, M.L.; Mallardi, M.; Cesarini, L.; Leoni, C.; Leone, D.; Fossati, C.; et al. Behavioral profile in RASopathies. Am. J. Med. Genet. 2014, 164, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Adviento, B.; Corbin, I.L.; Widjaja, F.; Desachy, G.; Enrique, N.; Rosser, T.; Risi, S.; Marco, E.J.; Hendren, R.L.; Bearden, C.E.; et al. Autism traits in the RASopathies. J. Med. Genet. 2014, 51, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Ghaziuddin, M.; Bolyard, B.; Alessi, N. Autistic disorder in Noonan syndrome. J. Intellect. Disabil. Res. 1994, 38, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.; Wentz, E.; Fernell, E.; Strömland, K.; Miller, M.T.; Gillberg, C. Autistic spectrum disorders in Moëbius sequence: a comprehensive study of 25 cases. Dev. Med. Child Neurol. 2001, 43, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Briegel, W.; Schimek, M.; Kamp-Becker, I.; Hofmann, C.; Schwab, K.O. Autism spectrum disorders in children and adolescents with Moebius sequence. Eur. Child Adolesc. Psychiatry. 2009, 18, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Helsmoortel, C.; Vulto-van Silfhout, A.T.; Coe, B.P.; Vandeweyer, G.; Rooms, L.; van den Ende, J.; Schuurs-Hoeijmakers, J.H.; Marcelis, C.L.; Willemsen, M.H.; Vissers, L.E.; et al. A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat. Genet. 2014, 46, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Splawski, I.; Timothy, K.W.; Sharpe, L.M.; Decher, N.; Kumar, P.; Bloise, R.; Napolitano, C.; Schwartz, P.J.; Joseph, R.M.; Condouris, K.; et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004, 119, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Huguet, G.; Ey, E.; Bourgeron, T. The genetic landscapes of autism spectrum disorders. Annu. Rev. Genom. Hum. Genet. 2013, 14, 191–213. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, G.B. Clinical genetic aspects of autism spectrum sisorders. Int. J. Mol. Sci. 2016, 17, 180. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.; Rutter, M.; Le Couteur, A. Autism diagnostic interview-revised: A revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J. Autism Dev. Disord. 1994, 24, 659–685. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.; Risi, S.; Lambrecht, L.; Cook, E.; Leventhal, B.; DiValore, P.; Pickles, A.; Rutter, M. The autism diagnostic observation schedule-generic: A standard measures of social and communication deficits associated with the spectrum of autism. J. Autism Dev. Disord. 2000, 30, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Risi, S.; Lord, C.; Gotham, K.; Corsello, C.; Chrysler, C.; Szatmari, P.; Cook, E.H., Jr.; Leventhal, B.L.; Pickles, A. Combining information from multiple sources in the diagnosis of autism spectrum disorders. J. Am. Acad. Child Adolesc. Psychiatry 2006, 45, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, G.B.; Mendelson, N.J. Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions. Genet. Med. 2013, 15, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Ponsot, O.; Moutard, M.L.; Villeneuve, N. Orientation Diagnostique en Neurologie Pédiatrique. In Neurologie Pédiatrique, 2nd ed.; Flammarion Médecine-Sciences: Paris, France, 1998; pp. 75–87. [Google Scholar]
- Simonoff, E.; Bolton, P.; Rutter, M. Mental retardation: Genetics findings, clinical implications and research agenda. J. Child Psychol. Psychiatry 1996, 37, 259–280. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, T.J.; Windham, G.C.; Anderson, M.; Croen, L.A.; Grether, J.K.; Risch, N. Evidence of reproductive stoppage in families with autism spectrum disorder: A large, population-based cohort study. JAMA Psychiatry 2014, 71, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Ozonoff, S.; Young, G.S.; Carter, A.; Messinger, D.; Yirmiya, N.; Zwaigenbaum, L.; Bryson, S.; Carver, L.J.; Constantino, J.N.; Dobkins, K.; et al. Recurrence risk for autism spectrum disorders: A baby siblings research consortium study. Pediatrics 2011, 128, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Durkin, M.S.; Maenner, M.J.; Newschaffer, C.J.; Lee, L.C.; Cunniff, C.M.; Daniels, J.L.; Kirby, R.S.; Leavitt, L.; Miller, L.; Zahorodny, W.; et al. Advanced parental age and the risk of autism spectrum disorder. Am. J. Epidemiol. 2008, 168, 1268–1276. [Google Scholar] [CrossRef] [PubMed]
- Lord, C. Diagnostic Instruments in Autism Spectrum Disorders. In Handbook of Autism and Pervasive Developmental Disorders, 2nd ed.; Cohen, D.J., Volkmar, F.R., Eds.; John, Wiley and Sons Inc.: New York, NY, USA, 1997; pp. 460–483. [Google Scholar]
- Anastasi, A. Psychological Testing, 6th ed.; Macmillan Publishing Co Inc.: New York, NY, USA, 1988. [Google Scholar]
- Manual for the Raven’s Color Progressive Matrices (CPM); Les Editions Centre de Psychologie Appliquée (ECPA): Paris, France, 2016.
- Guinchat, V.; Thorsen, P.; Laurent, C.; Cans, C.; Bodeau, N.; Cohen, D. Pre-peri-and neonatal risk factors for autism. Acta Obstet. Gynecol. Scand. 2012, 91, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Gardener, H.; Spiegelman, D.; Buka, S.L. Perinatal and neonatal risk factors for autism: A comprehensive meta-analysis. Pediatrics 2011, 128, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Weissman, J.R.; Kelley, R.I.; Bauman, M.L.; Cohen, B.H.; Murray, K.F.; Mitchell, R.L.; Kern, R.L.; Natowicz, M.R. Mitochondrial disease in autism spectrum disorder patients: A cohort analysis. PLoS ONE 2008, 3, e3815. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Frye, R.E. Mitochondrial dysfunction in autism spectrum disorders: A systematic review and meta-analysis. Mol. Psychiatry 2012, 17, 290–314. [Google Scholar] [CrossRef] [PubMed]
- Demily, C.; Assouline, M.; Boddaert, N.; Barcia, G.; Besmond, C.; Poisson, A.; Sanlaville, D.; Munnich, A. Genetic approaches in autism spectrum disorders. Neuropsychiatr. Enfance Adolesc. 2016, 64, 395–401. [Google Scholar] [CrossRef]
- Lai, M.C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef]
- Tordjman, S.; Anderson, G.M.; Bellissant, E.; Botbol, M.; Charbuy, H.; Camus, F.; Graignic, R.; Kermarrec, S.; Fougerou, C.; Cohen, D.; et al. Day and nighttime excretion of 6-sulphatoxymelatonin in adolescents and young adults with autistic disorder. Psychoneuroendocrinology 2012, 37, 1990–1997. [Google Scholar] [CrossRef] [PubMed]
- Haag, G.; Tordjman, S.; Duprat, A.; Urwand, S.; Jardin, F.; Clement, M.C.; Cukierman, A.; Druon, C.; Maufras du Chatellier, A.; et al. Presentation of a diagnostic grid of the progressive stages of infantile autism as observed in treatment. Int. J. Psychoanal. 2005, 86, 335–352. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Genetic Disorder [References] | Estimated Rate (%) of the Disorder in Autism | Estimated Rate (%) of Autism in the Disorder | Degree of Intellectual Disability (ID) | Possible Autistic Behaviors | Other Possible Behaviors | Other Possible Symptoms |
|---|---|---|---|---|---|---|
| Chromosomal Disorders | ||||||
| Maternal * 15q11–q13 duplication [26,27,28,29,30,31,32,33,34,35,36,37,38] | 1–2 | 80–100 | Severe | Severe autistic syndrome with severe expressive language impairment | Hyperactivity and aggression | Seizures (75%), hypotonia, genitor/urinary abnormalities |
| Angelman syndrome * (maternal 15q11-q13 deletion, paternal uniparental disomy, mutations of UBE3A that encodes an ubiquitin E3 ligase) [35,39,40,41,42,43,44,45,46,47,48] | 1 | 48–80 | Severe | No language, stereotypies, sameness | Attention Deficit with Hyperactivity Disorder (ADHD), paroxysmal laughter, tantrums | Facial dysmorphism, microcephaly, seizures (>1 year), ataxia and walking disturbance |
| Prader–Willi syndrome * (maternal uniparental disomy at 15q11-q13, paternal deletions) [35,49,50,51,52,53,54,55,56,57,58] Paternal 15q11-q13 duplication (only few cases reported to be associated with autism): behavioral phenotype similar to Prader–Willi syndrome [58,59,60] | Not Available (NA) | 19–37 | Mild to moderate | Motor and verbal stereotypies, rituals | Hyperphagia, obsessive-compulsive traits, temper tantrums | Obesity, growth delay and hypogonadism, facial dysmorphism, Hypotonia |
| Phelan–McDermid * syndrome (Inherited, de novo deletions at 22q13.3 leading to loss of SHANK 3) 22q13.3 duplication [20,61,62,63,64,65,66,67,68,69,70] | NA | 75–84 | Severe | Variable autistic syndrome with social communication impairments, including delayed or absent verbal language | Global developmental delay, atatonia in adolescence and adulthood | Dysmorphic features, hypotonia, gait disturbance, recurring upper respiratory tract infections, gastroesophageal reflux and seizures |
| 16p11.2 duplication 16p11.2 deletion [71,72,73,74,75,76,77] | 1 | 33 | Severe | Severe autistic syndrome with speech impairment | Gross and fine coordination problems, SCH (Schizophrenia), anxiety, ADHD | Hypotonia (Multiple congenital anomalies are possible with more distal region) |
| Inverted duplication/deletion 8p21–23 [78,79] | NA | 30–57 | Variable | Mild to moderate autistic syndrome with absent or delayed verbal language | ADHD | Minor facial dysmorphism, hypotonia, agenesis of the corpus callosum, possible heart defect |
| Genetic disorder [References] | Estimated rate (%) of the disorder in autism | Estimated rate (%) of autism in the disorder | Degree of intellectual disability (ID) | Autistic behaviors | Other behaviors | Other symptoms |
| Down syndrome * (trisomy 21) [80,81,82,83,84] | 2 | 5–10 | Variable but usually severe when autism | Severe autistic syndrome | - | Facial dysmorphism, heart and intestine malformations |
| Smith–Magenis syndrome (17p11.2 deletion) [85,86,87,88] | <1 | 80–100 | Variable | Self-injurious and stereotyped behaviors, sameness | Tantrums, possible social contact, sleep disturbance | Facial dysmorphism, peripheral neuropathy, hypotonia |
| Potocki–Lupski syndrome (17p11.2 duplication) [89] | NA | 50–100 | Normal to moderate | Decreased eye contact, motor manierisms or posturing, sensory hypersensitivity or preoccupation, repetitive behaviors or interests, lack of appropriate functional or symbolic play and lack of joint attention | Developmental delay, language impairment, and cognitive impairment | hypotonia, poor feeding and failure to thrive in infancy, oral-pharyngeal dysphagia, obstructive and central sleep apnea, structural cardiovascular abnormalities, electroencephalography (EEG), abnormalities, and hypermetropia |
| 2q37 deletion [90,91,92,93,94] | <1 | 25–35 | Mild to moderate | Severe communication impairment, stereotypies | Hypotonia, hyperactivity, Obsessive-Compulsive Disorder (OCD), aggression, sleep disturbance | Facial dysmorphism, microcephaly, growth delay/short stature, intestine and heart malformations, seizure |
| 22q11.21 duplication and 22q11 deletion (DiGeorge/Vélocardio-facial syndrome) [95,96,97,98,99,100] | NA | <10 | Normal to severe ID | Autistic syndrome, Pervasive Developmental Disorder-Not Otherwise Specified PDD-NOS (ICD-10 criteria) | Learning disability, anxiety, ADHD, oppositional-defiant disorder, OCD, motor impairment | Facial dysmorphism, microcephaly, growth delay/short stature, craniofacial abnormalities/cleft palate, heart defect, hypotonia |
| 1q21.1 Copy-Number Variation (CNV) (1q21.1 deletion/duplication) [101,102,103,104] | NA | <30 | Normal to mild ID | Autistic syndrome, PDD-NOS (ICD-10 criteria) | Developmental delay, learning disability, anxiety, ADHD, aggression, SCZ and hallucination | Microcephaly (deletion) Macrocephaly (duplication) |
| Williams-Beuren syndrome * (7q11.23 deletion) and Reciprocal 7q11.23 duplication syndrome [105,106,107,108,109,110,111,112,113,114,115,116,117] | <1 | <10 | Mild to moderate | Autistic syndrome | Overfriendliness, over talkativeness, visual spatial deficit, hyperacusis, feeding and sleep problems | Facial dysmorphism, short stature, heart and endocrine malformations, hypercalcemia |
| Turner syndrome * (most common monosomy For X chromosome) [37,118,119] | NA | 3 | Usually normal IQ | Females monosomic for the maternal chromosome X score significantly worse on social adjustment and verbal skills | - | Short stature, skeletal abnormalities, absence of ovarian function, webbed neck, lymphedema in hands and feet, heart defects and kidney problems |
| Beckwith–Wiedemann * syndrome (abnormal expression of imprinted genes on chromosome 11p15.5 such as IGF2 and/or CDKN1C) [120,121,122,123] | NA | 6.8 (replication needed) | Usually normal IQ | Autistic syndrome | - | Pre- and postnatal overgrowth (hemihyperplasia, macroglossia, visceromegaly) and increased risk of embryonal tumors |
| Isodicentric chromosome 15 or duplication/inversion 15q11 [124,125] | NA | NA | Moderate to severe | Autistic behavior | Developmental delay and intellectual deficit, epilepsy | Early central hypotonia |
| Ito hypomelanosis [126,127] | NA | NA | Inconstant | Asperger syndrome(high functioning autism) or atypical autism | Psychomotor delay and cognitive deficit | hypopigmented skin lesions along the Blaschko lines, motor delay, seizures, microcephaly or macrocephaly, hypotonia, ophthalmological abnormalities |
| Single Gene Disorders | ||||||
| CHARGE syndrome * (CHD7, 8q21.1) [128,129,130,131,132,133] | <1 | 15–50 | Variable but often normal IQ | Variable autistic syndrome | Hyperactivity, obsessive-compulsive traits, tic disorders | Coloboma of the eye, Heart defects, Atresia of the nasal choane, Retardation of growth and/or development, Genital/urinary abnormalities, Ear abnormalities/deafness |
| Tuberous sclerosis (TSC1, 9q34) (TSC2, 16p13.3) [134,135,136] | 1–4 | 25–60 | Variable | Severe autistic syndrome | Learning disorder | Ectodermal anomalies, renal lesions, seizures |
| PTEN macrocephaly syndromes (PTEN, 10q23.31) [137,138,139,140,141,142] | 4 in ASD with macro-cephaly | NA | Severe | Autistic syndrome and language delay | = | Progressive macrocephaly, developmental delay, macrosomy, tumors in adulthood |
| Rett’s syndrome * (MECP2, Xq28) [117,143,144,145,146,147,148,149] | <1 in female | 61–100 | Severe | Stereotyped hand movements, absence of language, loss of social engagement | Stagnation stage (6–18 months) in girls, then regression stage (12–36 months), pseudostationary t stage (2–10 years), l and late motor t deterioration (>10 years) | Head growth deceleration, progressive motor neuron (gait and truncal apraxia, ataxia, decreasing mobility) and respiratory (hyperventilation, breath holding, apnea) symptoms |
| San Filippo syndrome # (SGSH, 17q25.3) [150,151,152,153] | 1 replication needed | 80–100 | Severe | Language impairment, autistic withdrawal, stereotyped behaviors impulsivity, inappropriate affects | Progressive loss of acquisitions | Motor regression, hepatomegaly |
| Cerebral folate deficiency # (FOLR1, 11q13.4) [154,155,156,157] | NA | NA | Variable | Autistic syndrome including especially social interaction and language impairment | irritability, movement (such as tremors) and gait disturbances with ataxia, sleep problems | Psychomotor regression, epilepsy, developmental delay, deceleration of head growth, dystonia/hypotonia, visual and hearing deficit |
| Smith–Lemli–Opitz syndrome # (DHCR7, 1q12–13) [158,159,160,161,162,163] | NA | 50 | Variable | Self-injurious behaviors, stereotypies (“opisthokinesis”) language impairment | Sensory hyper-reactivity, irritability, sleep disturbance | Facial dysmorphism, cleft palate, congenital heart disease, hypospadias, 2–3 toe syndactyly |
| Phenylketonuria # (PAH, 12q22-q24.1) [20,164,165] | NA | NA | Severe | Self-injurious behavior, lack of social responsiveness | Temper tantrums, hyperactivity | Eczema, hypertonia, seizures, hypo-pigmentation |
| Adenylosuccinate lyase deficiency # (ASL, 22q13.1–13.2) [166,167,168,169,170] | <1 | 80–100 | Variable | Severe autistic syndrome | - | Seizures |
| Creatine deficiency syndrome # (GAMT, 19p13.3) (CRTR, Xq28) [171,172,173,174] | <1 | 80–100 | Severe | Severe autistic syndrome with poor language | - | Seizures, hypotonia |
| SHANK 3 (22q13.3) [175,176,177] | <1 | NA | NA | Severe autistic syndrome with no language | - | - |
| Neurexin family: Neurexin 1 (NRX1, 2p16.3) [178,179,180,181,182] | 1 | NA | Variable | Autistic syndrome | Hyperactivity, depression, learning disability, but also normal behavior | Seizures, hypotonia, facial dysmorphism? |
| Contactin Associated Protein-like 2 (CNTNAP2, 7q35) [183,184,185] | NA | NA | Variable | Autistic syndrome including verbal language impairment | - | Seizures |
| Contactin 4 (CNTN4, 3p26.2–3p26.3) [186,187,188,189] | <1 | NA | Variable | Autistic syndrome, PDD-NOS (ICD-10 criteria) | Visual spatial impairment, regression | Facial dysmorphism, developmental delay, hypotonia, ptosis. |
| Cell adhesion molecule-1 (CADM1, 11q22.3–23.2) [190,191,192] | NA | NA | NA | Autistic syndrome with especially social communication impairment including verbal language deficit | - | - |
| Protocadherin 10 (PCDH10, 4q28) [193] | <1 | NA | NA | Autistic syndrome | - | - |
| Neuroligin family: Neuroligin 3 (NLG3, Xq13) Neuroligin 4 (NLG4, Xq22.33) [194,195,196] | <1 | NA | Variable | Severe autistic syndrome, PDD-NOS (ICD-10 criteria) | Regression | Tic |
| Fragile X * (FMR1, Xq27.3) [191,196,197,198,199,200,201,202,203,204,205,206,207,208,209,210,211,212,213,214,215,216,217,218,219,220,221,222] | 0–8 | 0–33 | Variable | Poor eye contact , social anxiety, language deficit and stereotypies | Hyperactivity with attention deficit, sensory hyper-reactivity | Facial dysmorphism, macro-orchidism |
| Neurofibromatosis type 1 [109,223,224,225] | NA | 21–40 | inconstant | Restrictive/repetitive behaviors and severe social-communicative impairments | Cognitive deficits and learning difficulties | Café-au-lait spots, iris Lisch nodules, axillary and inguinal freckling, multiple neurofibromas |
| Sotos syndrome [226,227] | NA | 70 (replication needed) | Mild to severe | Self-injurious behavior, physical aggression, and destruction | - | Facial dysmorphism, overgrowth of the body in early life with macrocephaly |
| Aarskog syndrome [228,229] | NA | NA | - | Variable autistic syndrome | Learning and behavioral disabilities | facial, limbs and genital features, acromelic short stature |
| Cornelia de Lange syndrome [117,230,231] | NA | 43 (replication needed) | Variable | Self-injury, excessive repetitive behaviors and expressive language deficits | Psychomotor delay, language acquisition difficulties | Facial dysmorphism, severe growth delay, abnormal hands and feet, and constant brachymetacarpia of the first metacarpus, various other malformations (heart, kidney, etc.) |
| Joubert syndrome [232] | NA | <40 | severe intellectual deficit to normal intelligence | Autistic syndrome | - | Facial dysmorphism, abnormal respiratory pattern, nystagmus, hypotonia, ataxia, and delay in achieving motor milestones |
| Cohen syndrome [117,233,234,235] | NA | 54 (replication needed) | variable | Atypical autism | Often sociable with a cheerful disposition, learning and behavioral problems | Microcephaly, facial dysmorphism, hypotonia, myopia, retinal dystrophy, neutropenia and truncal obesity |
| Lujan–Fryns syndrome [236] | NA | NA | Mild to moderate | Autistic-like disorder | Behavioral problems, emotional instability, hyperactivity and shyness, schizophrenia | Tall, marfanoid stature, facial dysmorphism, hypernasal voice and generalized hypotonia |
| Noonan syndrome [237,238,239] | NA | 15 (replication needed) | Mild | ASD | Poor feeding in infancy | Short stature, facial dysmorphism and congenital heart defects |
| Moebius syndrome [128,240,241] | NA | 0–45 | Inconstant, mild | ASD | Delayed walk development | Oculofacial paralysis, strabismus |
| Helsmoortel–Van der Aa syndrome (ADNP-related ID/ASD) [242] | 0.17 | 100 | Mild to severe | ASD, behavioral problems, sleep disturbance | Delayed developmental milestones (walking between 19 months and 4.5 years, and speech ranging from sentences to no words) | Facial dysmorphism, hypotonia, seizures, feeding difficulties, visual problems (hypermetropia, strabismus, cortical visual impairment), and cardiac defects |
| Timothy syndrome (CACNA1C) [243] | NA | 50–70 (replication needed) | Inconstant, mild to severe | ASD | Language, motor, and generalized cognitive impairment | Facial dysmorphism, rate-corrected QT (QTc) interval >480 ms, unilateral or bilateral cutaneous syndactyly, heart defects, seizures |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robert, C.; Pasquier, L.; Cohen, D.; Fradin, M.; Canitano, R.; Damaj, L.; Odent, S.; Tordjman, S. Role of Genetics in the Etiology of Autistic Spectrum Disorder: Towards a Hierarchical Diagnostic Strategy. Int. J. Mol. Sci. 2017, 18, 618. https://doi.org/10.3390/ijms18030618
Robert C, Pasquier L, Cohen D, Fradin M, Canitano R, Damaj L, Odent S, Tordjman S. Role of Genetics in the Etiology of Autistic Spectrum Disorder: Towards a Hierarchical Diagnostic Strategy. International Journal of Molecular Sciences. 2017; 18(3):618. https://doi.org/10.3390/ijms18030618
Chicago/Turabian StyleRobert, Cyrille, Laurent Pasquier, David Cohen, Mélanie Fradin, Roberto Canitano, Léna Damaj, Sylvie Odent, and Sylvie Tordjman. 2017. "Role of Genetics in the Etiology of Autistic Spectrum Disorder: Towards a Hierarchical Diagnostic Strategy" International Journal of Molecular Sciences 18, no. 3: 618. https://doi.org/10.3390/ijms18030618
APA StyleRobert, C., Pasquier, L., Cohen, D., Fradin, M., Canitano, R., Damaj, L., Odent, S., & Tordjman, S. (2017). Role of Genetics in the Etiology of Autistic Spectrum Disorder: Towards a Hierarchical Diagnostic Strategy. International Journal of Molecular Sciences, 18(3), 618. https://doi.org/10.3390/ijms18030618