
The Impacts of Cellular Senescence in Elderly Pneumonia and in Age-Related Lung Diseases That Increase the Risk of Respiratory Infections

{kind=link}
Abstract
:1. Introduction
2. Cellular Senescence, SASP, and Aging
2.1. Cellular Senescence
2.2. SASP
2.3. The Role of Cellular Senescence in Aging and Aging-Related Diseases
3. The Impacts of Aging in Respiratory Tract Antimicrobial Defense System
4. The Impacts of Cellular Senescence in Age-Related Lung Diseases
4.1. The Role of Cellular Senescence in COPD
4.2. The Role of Cellular Senescence in IPF
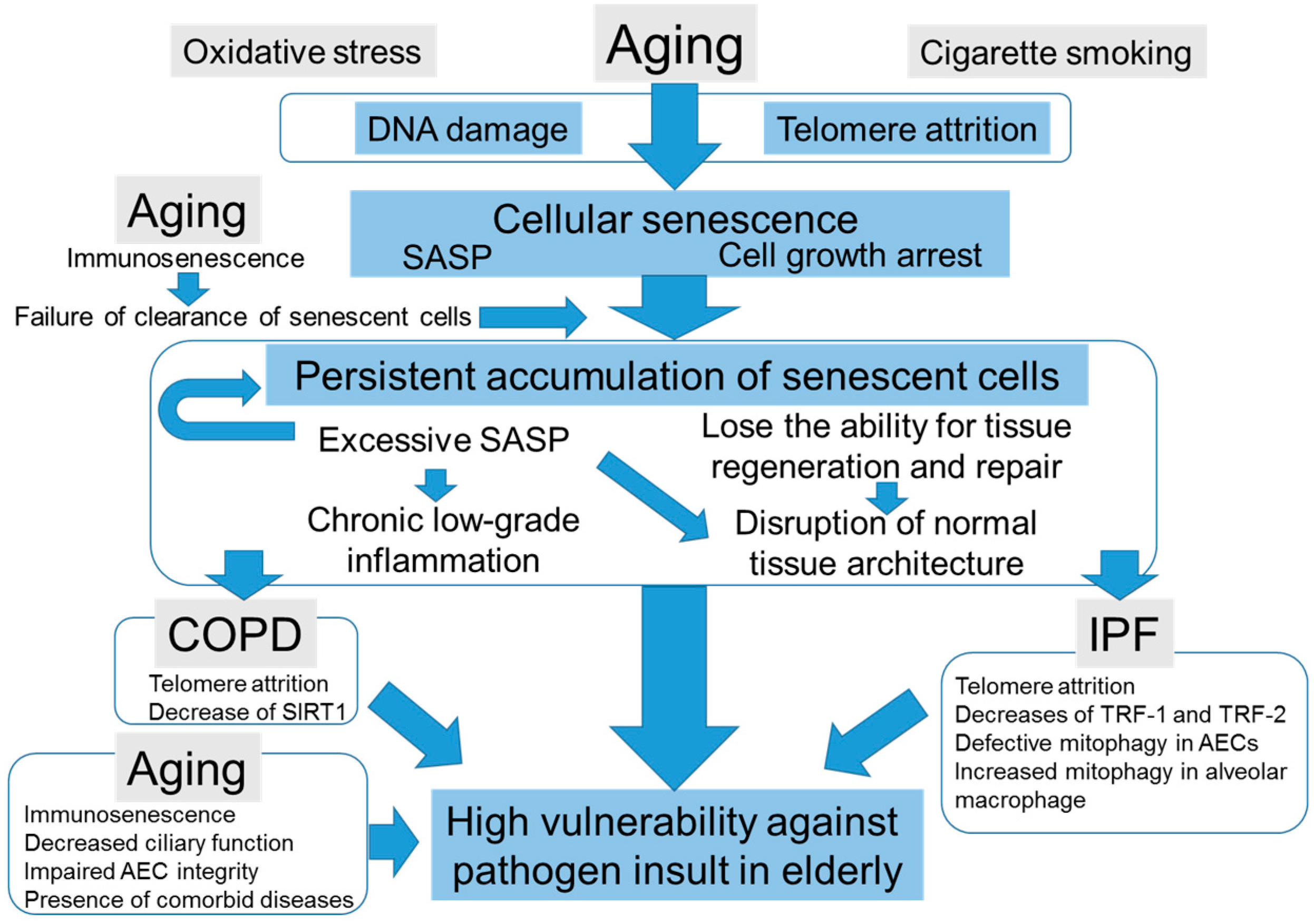
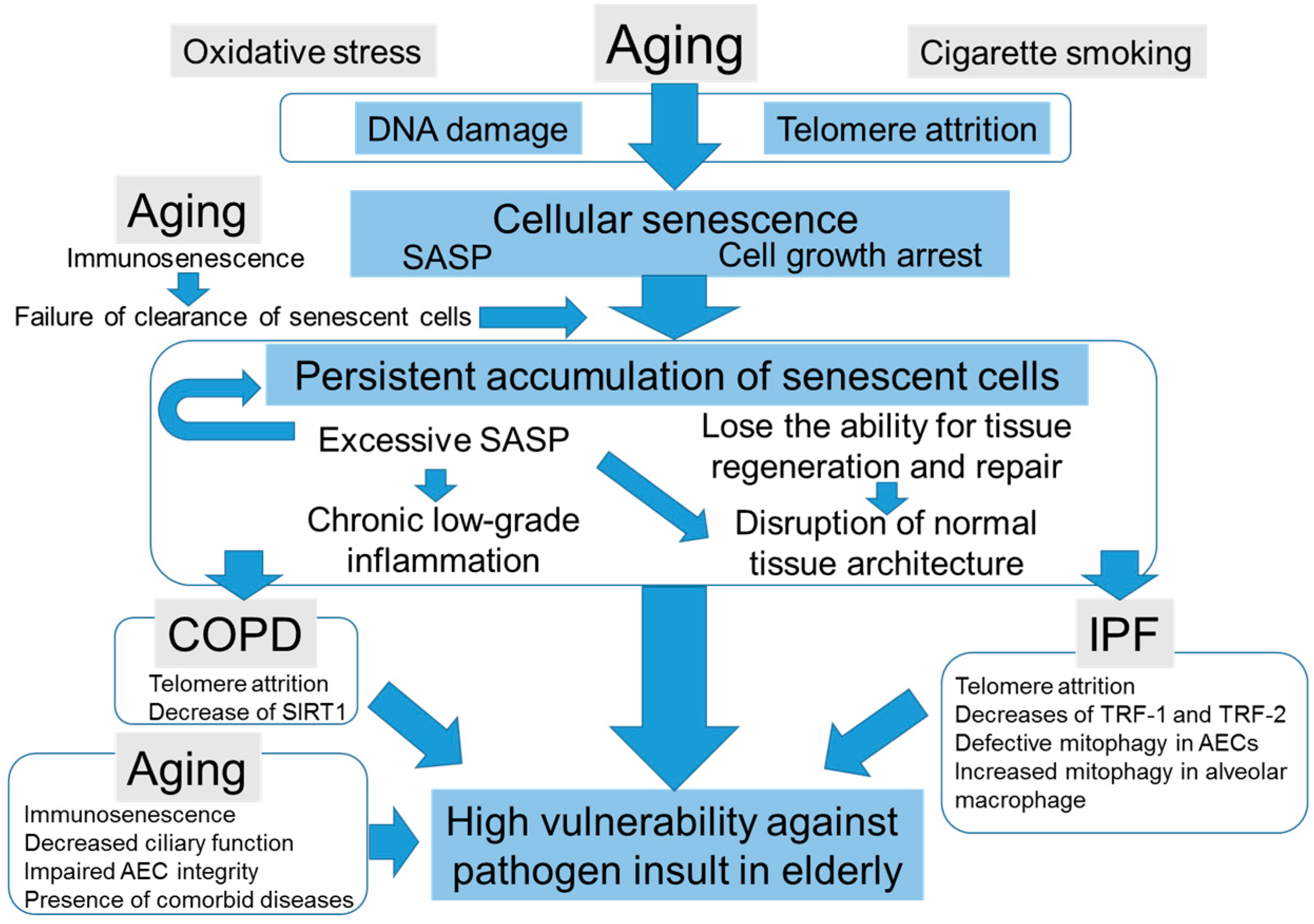
4.3. The Potential Scenarios of How Cellular Senescence Causes Distinct Age-Related Lung Pathologies
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| TNF | Tumor necrosis factor |
| IL | Interleukin |
| SASP | Senescence-associated secretory phenotype |
| ECM | Extracellular matrix |
| COPD | Chronic obstructive pulmonary disease |
| IPF | Idiopathic pulmonary fibrosis |
| SAβGAL | Senescence-associated β-galactosidase |
| TGF-β1 | Transforming growth factor-β1 |
| IGFBP | Insulin-like growth factor 1 binding protein |
| CDK | Cyclin-dependent kinase |
| NF-κB | Nuclear factor-κB |
| MMP | Matrix metalloproteinase |
| AEC | Alveolar epithelial cell |
| TLR | Toll like receptor |
| SIRT1 | Sirtuin 1 |
| TRF | Telomere repeat binding factor |
| PINK1 | PTEN-induced putative kinase 1 |
References
- El-Solh, A.A.; Sikka, P.; Ramadan, F.; Davies, J. Etiology of severe pneumonia in the very elderly. Am. J. Respir. Crit. Care Med. 2001, 163, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Fry, A.M.; Shay, D.K.; Holman, R.C.; Curns, A.T.; Anderson, L.J. Trends in hospitalizations for pneumonia among persons aged 65 years or older in the United States, 1988–2002. JAMA 2005, 294, 2712–2719. [Google Scholar] [CrossRef] [PubMed]
- Stupka, J.E.; Mortensen, E.M.; Anzueto, A.; Restrepo, M.I. Community-acquired pneumonia in elderly patients. Aging Health 2009, 5, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, L.; Conn, L.A.; Pinner, R.W.; Teutsch, S. Trends in infectious disease hospitalizations in the United States, 1980–1994. Arch. Intern. Med. 1998, 158, 1923–1928. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.L.; Neuzil, K.M.; Thompson, W.W.; Shay, D.K.; Yu, O.; Hanson, C.A.; Jackson, L.A. The burden of community-acquired pneumonia in seniors: Results of a population-based study. Clin. Infect. Dis. 2004, 39, 1642–1650. [Google Scholar] [CrossRef] [PubMed]
- Janssens, J.P.; Krause, K.H. Pneumonia in the very old. Lancet Infect. Dis. 2004, 4, 112–124. [Google Scholar] [CrossRef]
- Donowitz, G.R.; Cox, H.L. Bacterial community-acquired pneumonia in older patients. Clin. Geriatr. Med. 2007, 23, 515–534. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Adeyemi, A.O.; Rascati, K.L. Direct Medical Costs and Utilization of Health Care Services to Treat Pneumonia in the United States: An Analysis of the 2007–2011 Medical Expenditure Panel Survey. Clin. Ther. 2015, 37, 1466.e1–1476.e1. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Goodwin, J. Effect of aging on respiratory system physiology and immunology. Clin. Interv. Aging 2006, 1, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.C.; Chan, K.N.; Hu, W.H.; Lam, W.K.; Zheng, L.; Tipoe, G.L.; Sun, J.; Leung, R.; Tsang, K.W. The effect of aging on nasal mucociliary clearance, beat frequency, and ultrastructure of respiratory cilia. Am. J. Respir. Crit. Care Med. 2001, 163, 983–988. [Google Scholar] [CrossRef] [PubMed]
- McClaran, S.R.; Babcock, M.A.; Pegelow, D.F.; Reddan, W.G.; Dempsey, J.A. Longitudinal effects of aging on lung function at rest and exercise in healthy active fit elderly adults. J. Appl. Physiol. 1995, 78, 1957–1968. [Google Scholar] [PubMed]
- Gillooly, M.; Lamb, D. Airspace size in lungs of lifelong non-smokers: Effect of age and sex. Thorax 1993, 48, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Koivula, I.; Sten, M.; Makela, P.H. Risk factors for pneumonia in the elderly. Am. J. Med. 1994, 96, 313–320. [Google Scholar] [CrossRef]
- Farr, B.M.; Bartlett, C.L.; Wadsworth, J.; Miller, D.L. Risk factors for community-acquired pneumonia diagnosed upon hospital admission. British Thoracic Society Pneumonia Study Group. Respir. Med. 2000, 94, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Kothe, H.; Bauer, T.; Marre, R.; Suttorp, N.; Welte, T.; Dalhoff, K. Outcome of community-acquired pneumonia: Influence of age, residence status and antimicrobial treatment. Eur. Respir. J. 2008, 32, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Yende, S.; Tuomanen, E.I.; Wunderink, R.; Kanaya, A.; Newman, A.B.; Harris, T.; de Rekeneire, N.; Kritchevsky, S.B. Preinfection systemic inflammatory markers and risk of hospitalization due to pneumonia. Am. J. Respir. Crit. Care Med. 2005, 172, 1440–1446. [Google Scholar] [CrossRef] [PubMed]
- Shivshankar, P.; Boyd, A.R.; Le Saux, C.J.; Yeh, I.T.; Orihuela, C.J. Cellular senescence increases expression of bacterial ligands in the lungs and is positively correlated with increased susceptibility to pneumococcal pneumonia. Aging Cell 2011, 10, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Hinojosa, E.; Boyd, A.R.; Orihuela, C.J. Age-associated inflammation and toll-like receptor dysfunction prime the lungs for pneumococcal pneumonia. J. Infect. Dis. 2009, 200, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Hayflick, L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.I.; Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Long, E.O. Cellular senescence induced by CD158d reprograms natural killer cells to promote vascular remodeling. Proc. Natl. Acad. Sci. USA 2012, 109, 20596–20601. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Espin, D.; Canamero, M.; Maraver, A.; Gomez-Lopez, G.; Contreras, J.; Murillo-Cuesta, S.; Rodriguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [PubMed]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Bavik, C.; Coleman, I.; Dean, J.P.; Knudsen, B.; Plymate, S.; Nelson, P.S. The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res. 2006, 66, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L.; Logsdon, N.J.; Kurundkar, D.; Kurundkar, A.; Bernard, K.; Hock, T.; Meldrum, E.; Sanders, Y.Y.; Thannickal, V.J. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med. 2014, 6, 231ra247. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.; Peetermans, W.E.; Viegi, G.; Blasi, F. Risk factors for community-acquired pneumonia in adults in Europe: A literature review. Thorax 2013, 68, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Burney, P.G.; Silverman, E.K.; Celli, B.R.; Vestbo, J.; Wedzicha, J.A.; Wouters, E.F. Chronic obstructive pulmonary disease. Nat. Rev. Dis. Prim. 2015, 1, 15076. [Google Scholar] [CrossRef] [PubMed]
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Vestbo, J.; Hurd, S.S.; Agusti, A.G.; Jones, P.W.; Vogelmeier, C.; Anzueto, A.; Barnes, P.J.; Fabbri, L.M.; Martinez, F.J.; Nishimura, M.; et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am. J. Respir. Crit. Care Med. 2013, 187, 347–365. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Kurz, D.J.; Decary, S.; Hong, Y.; Erusalimsky, J.D. Senescence-associated (β)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J. Cell Sci. 2000, 113, 3613–3622. [Google Scholar] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-associated β-galactosidase is lysosomal β-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Marino, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Del Roso, A.; Vittorini, S.; Cavallini, G.; Donati, A.; Gori, Z.; Masini, M.; Pollera, M.; Bergamini, E. Ageing-related changes in the in vivo function of rat liver macroautophagy and proteolysis. Exp. Gerontol. 2003, 38, 519–527. [Google Scholar] [CrossRef]
- Tomaru, U.; Takahashi, S.; Ishizu, A.; Miyatake, Y.; Gohda, A.; Suzuki, S.; Ono, A.; Ohara, J.; Baba, T.; Murata, S.; et al. Decreased proteasomal activity causes age-related phenotypes and promotes the development of metabolic abnormalities. Am. J. Pathol. 2012, 180, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M.; et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007, 13, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Elledge, S.J. How autophagy both activates and inhibits cellular senescence. Autophagy 2016, 12, 898–899. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Hara, H.; Araya, J.; Takasaka, N.; Kojima, J.; Ito, S.; Minagawa, S.; Yumino, Y.; Ishikawa, T.; Numata, T.; et al. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. Oncoimmunology 2012, 1, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.T.; Lee, K.B.; Kim, S.Y.; Choi, H.R.; Park, S.C. Autophagy impairment induces premature senescence in primary human fibroblasts. PLoS ONE 2011, 6, e23367. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavare, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Xu, C.; Donahue, G.; Shimi, T.; Pan, J.A.; Zhu, J.; Ivanov, A.; Capell, B.C.; Drake, A.M.; Shah, P.P.; et al. Autophagy mediates degradation of nuclear lamina. Nature 2015, 527, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Serrano, M. Senescence in tumours: Evidence from mice and humans. Nat. Rev. Cancer 2010, 10, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, B.D.; Gilbert, P.M.; Porpiglia, E.; Mourkioti, F.; Lee, S.P.; Corbel, S.Y.; Llewellyn, M.E.; Delp, S.L.; Blau, H.M. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat. Med. 2014, 20, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Bernet, J.D.; Doles, J.D.; Hall, J.K.; Kelly Tanaka, K.; Carter, T.A.; Olwin, B.B. p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat. Med. 2014, 20, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Rodier, F.; Patil, C.K.; Freund, A.; Desprez, P.Y.; Campisi, J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J. Biol. Chem. 2011, 286, 36396–36403. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Hubackova, S.; Krejcikova, K.; Bartek, J.; Hodny, Z. IL1- and TGFβ-Nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine ‘bystander senescence’. Aging 2012, 4, 932–951. [Google Scholar] [CrossRef] [PubMed]
- Pribluda, A.; Elyada, E.; Wiener, Z.; Hamza, H.; Goldstein, R.E.; Biton, M.; Burstain, I.; Morgenstern, Y.; Brachya, G.; Billauer, H.; et al. A senescence-inflammatory switch from cancer-inhibitory to cancer-promoting mechanism. Cancer Cell 2013, 24, 242–256. [Google Scholar] [CrossRef] [PubMed]
- Erusalimsky, J.D.; Kurz, D.J. Cellular senescence in vivo: Its relevance in ageing and cardiovascular disease. Exp. Gerontol. 2005, 40, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Jeyapalan, J.C.; Ferreira, M.; Sedivy, J.M.; Herbig, U. Accumulation of senescent cells in mitotic tissue of aging primates. Mech. Ageing Dev. 2007, 128, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Melk, A.; Kittikowit, W.; Sandhu, I.; Halloran, K.M.; Grimm, P.; Schmidt, B.M.; Halloran, P.F. Cell senescence in rat kidneys in vivo increases with growth and age despite lack of telomere shortening. Kidney Int. 2003, 63, 2134–2143. [Google Scholar] [CrossRef] [PubMed]
- Paradis, V.; Youssef, N.; Dargere, D.; Ba, N.; Bonvoust, F.; Deschatrette, J.; Bedossa, P. Replicative senescence in normal liver, chronic hepatitis C, and hepatocellular carcinomas. Hum. Pathol. 2001, 32, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jurk, D.; Maddick, M.; Nelson, G.; Martin-Ruiz, C.; von Zglinicki, T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 2009, 8, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Perez-Terzic, C.; Jin, F.; Pitel, K.S.; Niederlander, N.J.; Jeganathan, K.; Yamada, S.; Reyes, S.; Rowe, L.; Hiddinga, H.J.; et al. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat. Cell Biol. 2008, 10, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Bhaumik, D.; Scott, G.K.; Schokrpur, S.; Patil, C.K.; Orjalo, A.V.; Rodier, F.; Lithgow, G.J.; Campisi, J. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging 2009, 1, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Cellular Senescence and Lung Function during Aging. Yin and Yang. Ann. Am. Thorac. Soc. 2016, 13, S402–S406. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; van Steeg, H.; Dolle, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.E.; Sorrentino, J.A.; Clark, K.S.; Darr, D.B.; Krishnamurthy, J.; Deal, A.M.; Bardeesy, N.; Castrillon, D.H.; Beach, D.H.; Sharpless, N.E. Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell 2013, 152, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, N.; Imamura, Y.; Yamakoshi, K.; Hirota, F.; Nakayama, R.; Kubo, Y.; Ishimaru, N.; Takahashi, A.; Hirao, A.; Shimizu, T.; et al. Visualizing the dynamics of p21(Waf1/Cip1) cyclin-dependent kinase inhibitor expression in living animals. Proc. Natl. Acad. Sci. USA 2007, 104, 15034–15039. [Google Scholar] [CrossRef] [PubMed]
- Banito, A.; Rashid, S.T.; Acosta, J.C.; Li, S.; Pereira, C.F.; Geti, I.; Pinho, S.; Silva, J.C.; Azuara, V.; Walsh, M.; et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009, 23, 2134–2139. [Google Scholar] [CrossRef] [PubMed]
- Utikal, J.; Polo, J.M.; Stadtfeld, M.; Maherali, N.; Kulalert, W.; Walsh, R.M.; Khalil, A.; Rheinwald, J.G.; Hochedlinger, K. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature 2009, 460, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Boe, D.M.; Boule, L.A.; Kovacs, E.J. Innate immune responses in the ageing lung. Clin. Exp. Immunol. 2017, 187, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Lowery, E.M.; Brubaker, A.L.; Kuhlmann, E.; Kovacs, E.J. The aging lung. Clin. Interv. Aging 2013, 8, 1489–1496. [Google Scholar] [PubMed]
- Shaw, A.C.; Goldstein, D.R.; Montgomery, R.R. Age-dependent dysregulation of innate immunity. Nat. Rev. Immunol. 2013, 13, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Castañeda-Delgado, J.E.; Miranda-Castro, N.Y.; Gonzalez-Amaro, R.; Gonzalez-Curiel, I.; Montoya-Rosales, A.; Rivas-Calderon, B.; Rivas-Santiago, B. Production of antimicrobial peptides is preserved in aging. Clin. Immunol. 2013, 148, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Bailey, K.L.; Bonasera, S.J.; Wilderdyke, M.; Hanisch, B.W.; Pavlik, J.A.; deVasure, J.; Robinson, J.E.; Sisson, J.H.; Wyatt, T.A. Aging causes a slowing in ciliary beat frequency, mediated by PKCepsilon. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L584–L589. [Google Scholar] [CrossRef] [PubMed]
- Kling, K.M.; Lopez-Rodriguez, E.; Pfarrer, C.; Muhlfeld, C.; Brandenberger, C. Aging exacerbates acute lung injury-induced changes of the air-blood barrier, lung function, and inflammation in the mouse. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L1–L12. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Zheng, D.; Limmon, G.V.; Leung, N.H.; Xu, S.; Rajapakse, J.C.; Yu, H.; Chow, V.T.; Chen, J. Aging exacerbates damage and delays repair of alveolar epithelia following influenza viral pneumonia. Respir. Res. 2014, 15, 116. [Google Scholar] [CrossRef] [PubMed]
- Renshaw, M.; Rockwell, J.; Engleman, C.; Gewirtz, A.; Katz, J.; Sambhara, S. Cutting edge: Impaired Toll-like receptor expression and function in aging. J. Immunol. 2002, 169, 4697–4701. [Google Scholar] [CrossRef] [PubMed]
- Chelvarajan, R.L.; Collins, S.M.; van Willigen, J.M.; Bondada, S. The unresponsiveness of aged mice to polysaccharide antigens is a result of a defect in macrophage function. J. Leukoc. Biol. 2005, 77, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Boehmer, E.D.; Meehan, M.J.; Cutro, B.T.; Kovacs, E.J. Aging negatively skews macrophage TLR2- and TLR4-mediated pro-inflammatory responses without affecting the IL-2-stimulated pathway. Mech. Ageing Dev. 2005, 126, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Rutgers, S.R.; Postma, D.S.; ten Hacken, N.H.; Kauffman, H.F.; van Der Mark, T.W.; Koeter, G.H.; Timens, W. Ongoing airway inflammation in patients with COPD who do not currently smoke. Thorax 2000, 55, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Aoshiba, K.; Nagai, A. Alveolar cell senescence in patients with pulmonary emphysema. Am. J. Respir. Crit. Care Med. 2006, 174, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.C.; Welker, L.; Paasch, K.; Feindt, B.; Erpenbeck, V.J.; Hohlfeld, J.M.; Krug, N.; Nakashima, M.; Branscheid, D.; Magnussen, H.; et al. Lung fibroblasts from patients with emphysema show markers of senescence in vitro. Respir. Res. 2006, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Morla, M.; Busquets, X.; Pons, J.; Sauleda, J.; MacNee, W.; Agusti, A.G. Telomere shortening in smokers with and without COPD. Eur. Respir. J. 2006, 27, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Savale, L.; Chaouat, A.; Bastuji-Garin, S.; Marcos, E.; Boyer, L.; Maitre, B.; Sarni, M.; Housset, B.; Weitzenblum, E.; Matrat, M.; et al. Shortened telomeres in circulating leukocytes of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2009, 179, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Aoshiba, K.; Nagai, A. Cigarette smoke induces senescence in alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 2004, 31, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Nyunoya, T.; Monick, M.M.; Klingelhutz, A.; Yarovinsky, T.O.; Cagley, J.R.; Hunninghake, G.W. Cigarette smoke induces cellular senescence. Am. J. Respir. Cell Mol. Biol. 2006, 35, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Rajendrasozhan, S.; Yang, S.R.; Kinnula, V.L.; Rahman, I. SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2008, 177, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Chung, S.; Hwang, J.W.; Rajendrasozhan, S.; Sundar, I.K.; Dean, D.A.; McBurney, M.W.; Guarente, L.; Gu, W.; Ronty, M.; et al. SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice. J. Clin. Investig. 2012, 122, 2032–2045. [Google Scholar] [CrossRef] [PubMed]
- Richmond, B.W.; Brucker, R.M.; Han, W.; Du, R.H.; Zhang, Y.; Cheng, D.S.; Gleaves, L.; Abdolrasulnia, R.; Polosukhina, D.; Clark, P.E.; et al. Airway bacteria drive a progressive COPD-like phenotype in mice with polymeric immunoglobulin receptor deficiency. Nat. Commun. 2016, 7, 11240. [Google Scholar] [CrossRef] [PubMed]
- Polosukhin, V.V.; Cates, J.M.; Lawson, W.E.; Zaynagetdinov, R.; Milstone, A.P.; Massion, P.P.; Ocak, S.; Ware, L.B.; Lee, J.W.; Bowler, R.P.; et al. Bronchial secretory immunoglobulin a deficiency correlates with airway inflammation and progression of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2011, 184, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Weycker, D.; Edelsberg, J.; Bradford, W.Z.; Oster, G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2006, 174, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Gribbin, J.; Hubbard, R.B.; Le Jeune, I.; Smith, C.J.; West, J.; Tata, L.J. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax 2006, 61, 980–985. [Google Scholar] [CrossRef] [PubMed]
- Natsuizaka, M.; Chiba, H.; Kuronuma, K.; Otsuka, M.; Kudo, K.; Mori, M.; Bando, M.; Sugiyama, Y.; Takahashi, H. Epidemiologic survey of Japanese patients with idiopathic pulmonary fibrosis and investigation of ethnic differences. Am. J. Respir. Crit. Care Med. 2014, 190, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Araya, J.; Kojima, J.; Takasaka, N.; Ito, S.; Fujii, S.; Hara, H.; Yanagisawa, H.; Kobayashi, K.; Tsurushige, C.; Kawaishi, M.; et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L56–L69. [Google Scholar] [CrossRef] [PubMed]
- Minagawa, S.; Araya, J.; Numata, T.; Nojiri, S.; Hara, H.; Yumino, Y.; Kawaishi, M.; Odaka, M.; Morikawa, T.; Nishimura, S.L.; et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-beta-induced senescence of human bronchial epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L391–L401. [Google Scholar] [CrossRef] [PubMed]
- Disayabutr, S.; Kim, E.K.; Cha, S.I.; Green, G.; Naikawadi, R.P.; Jones, K.D.; Golden, J.A.; Schroeder, A.; Matthay, M.A.; Kukreja, J.; et al. miR-34 miRNAs Regulate Cellular Senescence in Type II Alveolar Epithelial Cells of Patients with Idiopathic Pulmonary Fibrosis. PLoS ONE 2016, 11, e0158367. [Google Scholar] [CrossRef] [PubMed]
- Alder, J.K.; Chen, J.J.; Lancaster, L.; Danoff, S.; Su, S.C.; Cogan, J.D.; Vulto, I.; Xie, M.; Qi, X.; Tuder, R.M.; et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13051–13056. [Google Scholar] [CrossRef] [PubMed]
- Naikawadi, R.P.; Disayabutr, S.; Mallavia, B.; Donne, M.L.; Green, G.; La, J.L.; Rock, J.R.; Looney, M.R.; Wolters, P.J. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight 2016, 1, e86704. [Google Scholar] [CrossRef] [PubMed]
- Povedano, J.M.; Martinez, P.; Flores, J.M.; Mulero, F.; Blasco, M.A. Mice with Pulmonary Fibrosis Driven by Telomere Dysfunction. Cell Rep. 2015, 12, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Alder, J.K.; Barkauskas, C.E.; Limjunyawong, N.; Stanley, S.E.; Kembou, F.; Tuder, R.M.; Hogan, B.L.; Mitzner, W.; Armanios, M. Telomere dysfunction causes alveolar stem cell failure. Proc. Natl. Acad. Sci. USA 2015, 112, 5099–5104. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Shteinberg, A.; Porat, Z.; Budovsky, A.; Braiman, A.; Ziesche, R.; Fraifeld, V.E. Cellular senescence-like features of lung fibroblasts derived from idiopathic pulmonary fibrosis patients. Aging 2015, 7, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Im, J.; Hergert, P.; Nho, R.S. Reduced FoxO3a expression causes low autophagy in idiopathic pulmonary fibrosis fibroblasts on collagen matrices. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L552–L561. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.; Lai, Y.C.; Romero, Y.; Brands, J.; St Croix, C.M.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.S.; et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015, 125, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Larson-Casey, J.L.; Deshane, J.S.; Ryan, A.J.; Thannickal, V.J.; Carter, A.B. Macrophage Akt1 Kinase-Mediated Mitophagy Modulates Apoptosis Resistance and Pulmonary Fibrosis. Immunity 2016, 44, 582–596. [Google Scholar] [CrossRef] [PubMed]
- Chilosi, M.; Poletti, V.; Zamo, A.; Lestani, M.; Montagna, L.; Piccoli, P.; Pedron, S.; Bertaso, M.; Scarpa, A.; Murer, B.; et al. Aberrant Wnt/β-catenin pathway activation in idiopathic pulmonary fibrosis. Am. J. Pathol. 2003, 162, 1495–1502. [Google Scholar] [CrossRef]
- Konigshoff, M.; Kramer, M.; Balsara, N.; Wilhelm, J.; Amarie, O.V.; Jahn, A.; Rose, F.; Fink, L.; Seeger, W.; Schaefer, L.; et al. WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J. Clin. Investig. 2009, 119, 772–787. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Wei, Y.; Szekeres, C.; Kugler, M.C.; Wolters, P.J.; Hill, M.L.; Frank, J.A.; Brumwell, A.N.; Wheeler, S.E.; Kreidberg, J.A.; et al. Epithelial cell α3β1 integrin links β-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J. Clin. Investig. 2009, 119, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Bolanos, A.L.; Milla, C.M.; Lira, J.C.; Ramirez, R.; Checa, M.; Barrera, L.; Garcia-Alvarez, J.; Carbajal, V.; Becerril, C.; Gaxiola, M.; et al. Role of Sonic Hedgehog in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L978–L990. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Pardo, A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. An integral model. Am. J. Respir. Crit. Care Med. 2014, 189, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, K.; Yanagi, S.; Kawahara, K.; Nishio, M.; Tsubouchi, H.; Imazu, Y.; Koshida, R.; Matsumoto, N.; Taguchi, A.; Yamashita, S.; et al. Epithelial Pten controls acute lung injury and fibrosis by regulating alveolar epithelial cell integrity. Am. J. Respir. Crit. Care Med. 2013, 187, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Tsujino, K.; Takeda, Y.; Arai, T.; Shintani, Y.; Inagaki, R.; Saiga, H.; Iwasaki, T.; Tetsumoto, S.; Jin, Y.; Ihara, S.; et al. Tetraspanin CD151 protects against pulmonary fibrosis by maintaining epithelial integrity. Am. J. Respir. Crit. Care Med. 2012, 186, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Chilosi, M.; Poletti, V.; Rossi, A. The pathogenesis of COPD and IPF: Distinct horns of the same devil? Respir. Res. 2012, 13, 3. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Seeger, W.; Voswinckel, R. Senescence-associated secretory phenotype and its possible role in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2014, 51, 323–333. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yanagi, S.; Tsubouchi, H.; Miura, A.; Matsuo, A.; Matsumoto, N.; Nakazato, M. The Impacts of Cellular Senescence in Elderly Pneumonia and in Age-Related Lung Diseases That Increase the Risk of Respiratory Infections. Int. J. Mol. Sci. 2017, 18, 503. https://doi.org/10.3390/ijms18030503
Yanagi S, Tsubouchi H, Miura A, Matsuo A, Matsumoto N, Nakazato M. The Impacts of Cellular Senescence in Elderly Pneumonia and in Age-Related Lung Diseases That Increase the Risk of Respiratory Infections. International Journal of Molecular Sciences. 2017; 18(3):503. https://doi.org/10.3390/ijms18030503
Chicago/Turabian StyleYanagi, Shigehisa, Hironobu Tsubouchi, Ayako Miura, Ayako Matsuo, Nobuhiro Matsumoto, and Masamitsu Nakazato. 2017. "The Impacts of Cellular Senescence in Elderly Pneumonia and in Age-Related Lung Diseases That Increase the Risk of Respiratory Infections" International Journal of Molecular Sciences 18, no. 3: 503. https://doi.org/10.3390/ijms18030503
APA StyleYanagi, S., Tsubouchi, H., Miura, A., Matsuo, A., Matsumoto, N., & Nakazato, M. (2017). The Impacts of Cellular Senescence in Elderly Pneumonia and in Age-Related Lung Diseases That Increase the Risk of Respiratory Infections. International Journal of Molecular Sciences, 18(3), 503. https://doi.org/10.3390/ijms18030503