Maillard Proteomics: Opening New Pages


Abstract

1. Introduction
2. Part 1. Probing the Structure of Glycated Proteins by Mass Spectrometry
2.1. Analysis of Intact Proteins
2.1.1. MALDI-TOF-MS of Intact Glycated Proteins
2.1.2. ESI-MS of Intact Glycated Proteins
2.2. Proteomics Approach in Glycation Research
2.2.1. Top-Down Proteomic Strategy
2.2.2. Bottom-Up Proteomic Strategy
Limited Enzymatic Proteolysis
Application of Gel-Based Proteomics in Maillard Research
Application of LC-Based Proteomics in Maillard Research
3. Part 2. New Prospectives in Maillard Proteomics
3.1. Synthetic Peptides as Model Systems in Maillard Proteomics
3.2. Individual Glycation Sites in Human Proteins as the Markers of Diabetes Mellitus
3.3. Proteomics in the Study of Anti-Glycative Defense
3.4. Glycation of Plant Proteins as the Marker of Ageing and Environmental Stress
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 2D-GE | two-dimensional gel electrophoresis |
| 2-HNP | 2(hydroxymethyl)pyrylium |
| 3-DG | 3-deoxyglucasone |
| 3-DG-H | 3-deoxyglucosone-derived hydroimidazolone |
| AALS | anionic acid labile surfactant |
| α-La | α-lactalbumin |
| ABC | ammonium bicarbonate buffer |
| ACN | acetonitrile |
| AGEs | advanced glycation end-products |
| ALEs | advanced lipoxidation end products |
| Apo-I | apolipoprotein I |
| β-Lg | β-lactoglobulin |
| BAC | boronic acid affinity chromatography |
| BSA | bovine serum albumin |
| BUP | bottom-up proteomic |
| CAD | collision-activated dissociation |
| CE | capillary electrophoresis; |
| CEA | Nδ-(carboxyethyl)arginine |
| CEL | Nε-(carboxyethyl)lysine |
| CMA | Nδ-(carboxymethyl)arginine |
| CML | Nε-(carboxymethyl)lysine |
| CZE | capillary zone electrophoresis |
| DCP | dicarbonyl proteome |
| DDA | data-dependent acquisition |
| DHX | deuterium–hydrogen exchange |
| DIA | data-independent acquisition |
| DIGE | difference gel electrophoresis |
| DM | diabetes mellitus |
| DR | double resonance |
| DTE | dithioeritritol |
| DTT | dithiothreitol |
| ECD | electron capture dissociation |
| EI | electron (impact) ionization |
| ESI | electrospray ionization |
| ETD | electron transfer dissociation |
| EXC | cation exchange chromatography |
| FA | formic acid |
| FIA | flow injection analysis |
| Fmoc | 9-fluorenylmethoxycarbonyl |
| FT-ICR | Fourier transform-ion cyclotron resonance |
| Fru-Lys | Nε-(fructosyl)lysine, Amadori compound |
| GC | gas chromatography |
| G-DHI | glyoxal-derived dihydroxyimidazolidine |
| GELFrEE | gel-eluted liquid fraction entrapment electrophoresis |
| Glarg | 1-(4-amino-4-carboxybutyl)-2-imino-5-oxo-imidazolidine, glyoxal-derived hydroimidazolone |
| Glo1 | glyoxalase 1 |
| Glo2 | glyoxalase 2 |
| GO | glyoxal |
| GOLD | glyoxal-derived lysine dimer |
| GPF | gas phase fractionation |
| GSH | glutathione |
| HbA | hemoglobin A |
| HbA1C | glycated hemoglobin |
| HIF1α | hypoxia-inducible factor 1α |
| HILIC | hydrophilic interaction liquid chromatography |
| HOAc | acetic acid |
| HPD | high pressure denaturation |
| HPLC | high-performance liquid chromatography |
| HR | high resolution |
| HR-MS | high resolution mass spectrometry |
| HSA | human serum albumin |
| IA | iodoacetamide |
| IgG | immunoglobulin G |
| IGT | impaired glucose tolerance |
| IMAC | immobilized metal affinity chromatography |
| IP-RP-HPLC | ion-pair reversed-phase high performance liquid chromatography |
| IT | ion trap |
| LC | liquid chromatography |
| LC-MS/MS | liquid chromatography-tandem mass spectrometry |
| LC-UV | liquid chromatography with ultraviolet detection |
| LIT | linear ion trap |
| MALDI | matrix assisted laser desorption/ionization |
| MG-DHI | methylglyoxal-derived dihydroxyimidazolidine |
| MG-H1 | Nδ-(5-methyl-4-oxo-5-hydroimidazo-linone-2-yl)ornithine, methylglyoxal-derived hydroimidazolone 1 |
| MGO | methylglyoxal |
| MOA | methylhydroxylamine hydrochloride |
| MRM | multiple reaction monitoring |
| MS | mass spectrometry |
| MS/MS | tandem mass spectrometry |
| MSA | multi-stage activation |
| MSTFA | N-methyl-N-(trimethylsylil)trifluoroacetamide |
| nanoLC | nano-scaled liquid chromatography |
| NLMS3 | neutral loss triggered MS3 |
| oPDA | o-phenylenediamine |
| PEG | polyethylene glycol |
| PTMs | post-translational modifications |
| q | RF-only quadrupole |
| Q | quadrupole mass analyzer |
| QIT | quadrupole ion trap |
| QqQ | triple quadrupole |
| QqTOF | quadrupole-time of flight |
| RAGEs | receptors to advanced glycation end products |
| RCCs | reactive dicarbonyl compounds |
| RI | ribonuclease inhibitor |
| RNase A | ribonuclease A |
| ROS | reactive oxygen species |
| RP-HPLC | reversed-phase high performance liquid chromatography |
| RP-SPE | reversed phase-solid phase extraction |
| SDS | sodium dodecyl sulfate |
| SDS-PAGE | polyacrylamide gel electrophoresis is sodium dodecyl sulfate |
| SELDI | surface-enhanced laser desorption/ionization |
| SPE | solid phase extraction. |
| SPPS | solid phase peptide synthesis |
| SWATH | Sequential Window Acquisition of all Theoretical Mass Spectra |
| T2DM | type 2 diabetes mellitus |
| TCEP | tris-(2-carboxyethyl)-phosphine hydrochloride |
| TDP | top-down proteomics |
| THP | tetrahydropyrimidine |
| TOF | time of flight |
| UHPLC | ultra-high performance liquid chromatography |
| v/v | volume/volume |
References
- Brings, S.; Fleming, T.; Freichel, M.; Muckenthaler, M.U.; Herzig, S.; Nawroth, P.P. Dicarbonyls and advanced glycation end-products in the development of diabetic complications and targets for intervention. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ames, J.M.; Smith, R.D.; Baynes, J.W.; Metz, T.O. A perspective on the Maillard reaction and the analysis of protein glycation by mass spectrometry: Probing the pathogenesis of chronic disease. J. Proteome Res. 2009, 8, 754–769. [Google Scholar] [CrossRef] [PubMed]
- Amadori, M. The product of the condensation of glucose and p-phenetidine. Atti Della R. Accad. Naz. Dei Lincei 1929, 9, 68–73. [Google Scholar]
- Heyns, K.; Noack, H. Die Umsetzung von d-Fructose mit l-Lysin and l-Arginin und dered Beziehung zu nichtenzymatischen Bräunungsreaktion. Chem. Ber. 1962, 95, 720–727. [Google Scholar] [CrossRef]
- Hodge, J.E. The Amadori rearrangement. Adv. Carbohydr. Chem. 1955, 10, 169–205. [Google Scholar] [PubMed]
- Wells-Knecht, K.J.; Zyzak, D.V.; Litchfield, J.E.; Thorpe, S.R.; Baynes, J.W. Mechanism of autoxidative glycosylation: Identification of glyoxal and arabinose as intermediates in the autoxidative modification of proteins by glucose. Biochemistry 1995, 34, 3702–3709. [Google Scholar] [CrossRef] [PubMed]
- Vistoli, G.; de Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): An overview of their mechanisms of formation. Free Radic. Res. 2013, 47, 3–27. [Google Scholar] [CrossRef] [PubMed]
- Wolff, S.P.; Dean, R.T. Glucose autoxidation and protein modification. The potential role of “autoxidative glycosylation” in diabetes. Biochem. J. 1987, 245, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Dhar, A.; Desai, K.; Kazachmov, M.; Yu, P.; Wu, L. Methylglyoxal production in vascular smooth muscle cells from different metabolic precursors. Metabolism 2008, 57, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef] [PubMed]
- Kalapos, M.P. Methylglyoxal in living organisms: Chemistry, biochemistry, toxicology and biological implications. Toxicol. Lett. 1999, 110, 145–175. [Google Scholar] [CrossRef]
- Araki, A. Oxidative stress and diabetes mellitus: A possible role of alpha-dicarbonyl compounds in free radical formation. Nihon Ronen Igakkai Zasshi Jpn. J. Geriatr. 1997, 34, 716–720. [Google Scholar]
- Ott, C.; Jacobs, K.; Haucke, E.; Navarrete Santos, A.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Henning, C.; Smuda, M.; Girndt, M.; Ulrich, C.; Glomb, M.A. Molecular basis of maillard amide-advanced glycation end product (AGE) formation in vivo. J. Biol. Chem. 2011, 286, 44350–44356. [Google Scholar] [CrossRef] [PubMed]
- Gkogkolou, P.; Böhm, M. Advanced glycation end products. Dermatoendocrinology 2012, 4, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.P.; Bali, A.; Singh, N.; Jaggi, A.S. Advanced glycation end products and diabetic complications. Korean J. Physiol. Pharmacol. 2014, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Comazzi, S.; Bertazzolo, W.; Bonfanti, U.; Spagnolo, V.; Sartorelli, P. Advanced glycation end products and sorbitol in blood from differently compensated diabetic dogs. Res. Vet. Sci. 2008, 84, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Sajithlal, G.B.; Chandrakasan, G. Role of lipid peroxidation products in the formation of advanced glycation end products: An in vitro study on collagen. Proc. Indian Acad. Sci. Chem. Sci. 1999, 111, 215–229. [Google Scholar] [CrossRef]
- Greifenhagen, U.; Frolov, A.; Blüher, M.; Hoffmann, R. Plasma proteins modified by advanced glycation end products (AGEs) reveal site-specific susceptibilities to glycemic control in patients with type 2 diabetes. J. Biol. Chem. 2016, 291, 9610–9616. [Google Scholar] [CrossRef] [PubMed]
- Haucke, E.; Navarrete Santos, A.; Simm, A.; Henning, C.; Glomb, M.A.; Gürke, J.; Schindler, M.; Fischer, B.; Navarrete Santos, A. Accumulation of advanced glycation end products in the rabbit blastocyst under maternal diabetes. Reprod. Camb. Engl. 2014, 148, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Jaisson, S.; Souchon, P.-F.; Desmons, A.; Salmon, A.-S.; Delemer, B.; Gillery, P. Early formation of serum advanced glycation end-products in children with type 1 diabetes mellitus: Relationship with glycemic control. J. Pediatr. 2016, 172, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, C.T.; Smuda, M.; Smith, A.J.O.; Howell, S.; Smith, D.G.; Singh, A.; Gupta, P.; Glomb, M.A.; Wormstone, I.M.; Nagaraj, R.H. AGEs in human lens capsule promote the TGFβ2-mediated EMT of lens epithelial cells: Implications for age-associated fibrosis. Aging Cell 2016, 15, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Kartika, R.; Spiegel, D.A. Exploring post-translational arginine modification using chemically synthesized methylglyoxal hydroimidazolones. J. Am. Chem. Soc. 2012, 134, 8958–8967. [Google Scholar] [CrossRef] [PubMed]
- Younessi, P.; Yoonessi, A. Advanced glycation end-products and their receptor-mediated roles: Inflammation and oxidative stress. Iran. J. Med. Sci. 2011, 36, 154–166. [Google Scholar] [PubMed]
- Huang, S.-M.; Wu, C.-H.; Yen, G.-C. Effects of flavonoids on the expression of the pro-inflammatory response in human monocytes induced by ligation of the receptor for AGEs. Mol. Nutr. Food Res. 2006, 50, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, R.; Yan, S.F.; Schmidt, A.M. Receptor for AGE (RAGE): Signaling mechanisms in the pathogenesis of diabetes and its complications. Ann. N. Y. Acad. Sci. 2011, 1243, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Goh, S.-Y.; Cooper, M.E. Clinical review: The role of advanced glycation end products in progression and complications of diabetes. J. Clin. Endocrinol. Metab. 2008, 93, 1143–1152. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, M.W.; Hedegaard, R.V.; Andersen, J.M.; de Courten, B.; Bügel, S.; Nielsen, J.; Skibsted, L.H.; Dragsted, L.O. Advanced glycation endproducts in food and their effects on health. Food Chem. Toxicol. 2013, 60, 10–37. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M.; Stern, D.M. RAGE: A new target for the prevention and treatment of the vascular and inflammatory complications of diabetes. Trends Endocrinol. Metab. 2000, 11, 368–375. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Fages, C.; Rauvala, H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-κB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J. Biol. Chem. 1999, 274, 19919–19924. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Shaheen, F.; Anwar, A.; Masania, J.; Thornalley, P.J. Assay of methylglyoxal-derived protein and nucleotide AGEs. Biochem. Soc. Trans. 2014, 42, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Bilova, T.; Lukasheva, E.; Brauch, D.; Greifenhagen, U.; Paudel, G.; Tarakhovskaya, E.; Frolova, N.; Mittasch, J.; Balcke, G.U.; Tissier, A.; et al. A snapshot of the plant glycated proteome: Structural, functional, and mechanistic aspects. J. Biol. Chem. 2016, 291, 7621–7636. [Google Scholar] [CrossRef] [PubMed]
- Scheijen, J.L.J.M.; Clevers, E.; Engelen, L.; Dagnelie, P.C.; Brouns, F.; Stehouwer, C.D.A.; Schalkwijk, C.G. Analysis of advanced glycation endproducts in selected food items by ultra-performance liquid chromatography tandem mass spectrometry: Presentation of a dietary AGE database. Food Chem. 2016, 190, 1145–1150. [Google Scholar] [CrossRef] [PubMed]
- Makita, Z.; Vlassara, H.; Cerami, A.; Bucala, R. Immunochemical detection of advanced glycosylation end products in vivo. J. Biol. Chem. 1992, 267, 5133–5138. [Google Scholar] [PubMed]
- Mitsuhashi, T.; Vlassara, H.; Founds, H.W.; Li, Y.M. Standardizing the immunological measurement of advanced glycation endproducts using normal human serum. J. Immunol. Methods 1997, 207, 79–88. [Google Scholar] [CrossRef]
- Smuda, M.; Henning, C.; Raghavan, C.T.; Johar, K.; Vasavada, A.R.; Nagaraj, R.H.; Glomb, M.A. Comprehensive analysis of maillard protein modifications in human lenses: Effect of age and cataract. Biochemistry 2015, 54, 2500–2507. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J.; Rabbani, N. Detection of oxidized and glycated proteins in clinical samples using mass spectrometry—A user’s perspective. Biochim. Biophys. Acta 2014, 1840, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Wellner, A.; Huettl, C.; Henle, T. Formation of Maillard reaction products during heat treatment of carrots. J. Agric. Food Chem. 2011, 59, 7992–7998. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, H.; Hanke, T.; Frolov, A.; Langrock, T.; Hoffmann, R.; Fischer, C.; Schwarzenbolz, U.; Henle, T.; Born, R.; Worch, H. Modification of collagen in vitro with respect to formation of Nε-carboxymethyllysine. Int. J. Biol. Macromol. 2009, 44, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Slight, S.H.; Prabhakaram, M.; Shin, D.B.; Feather, M.S.; Ortwerth, B.J. The extent of N ε-(carboxymethyl)lysine formation in lens proteins and polylysine by the autoxidation products of ascorbic acid. Biochim. Biophys. Acta 1992, 1117, 199–206. [Google Scholar] [CrossRef]
- Glomb, M.A.; Pfahler, C. Amides are novel protein modifications formed by physiological sugars. J. Biol. Chem. 2001, 276, 41638–41647. [Google Scholar] [CrossRef] [PubMed]
- Badoud, R.; Fay, L.B. Mass spectrometric analysis of N-carboxymethylamino acids as periodate oxidation derivatives of Amadori compounds application to glycosylated haemoglobin. Amino Acids 1993, 5, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Thornalley, P.J. Quantitative screening of protein biomarkers of early glycation, advanced glycation, oxidation and nitrosation in cellular and extracellular proteins by tandem mass spectrometry multiple reaction monitoring. Biochem. Soc. Trans. 2003, 31, 1417–1422. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, H.; Hanke, T.; Simon, P.; Born, R.; Fischer, C.; Frolov, A.; Langrock, T.; Hoffmann, R.; Schwarzenbolz, U.; Henle, T.; et al. Carboxymethylation of the fibrillar collagen with respect to formation of hydroxyapatite. J. Biomed. Mater. Res. B Appl. Biomater. 2010, 92, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Penndorf, I.; Li, C.; Schwarzenbolz, U.; Henle, T. N-terminal glycation of proteins and peptides in foods and in vivo: Evaluation of N-(2-furoylmethyl)valine in acid hydrolyzates of human hemoglobin. Ann. N. Y. Acad. Sci. 2008, 1126, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Troise, A.D.; Fiore, A.; Wiltafsky, M.; Fogliano, V. Quantification of Nε-(2-Furoylmethyl)-l-lysine (furosine), Nε-(Carboxymethyl)-l-lysine (CML), Nε-(Carboxyethyl)-l-lysine (CEL) and total lysine through stable isotope dilution assay and tandem mass spectrometry. Food Chem. 2015, 188, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Milkovska-Stamenova, S.; Schmidt, R.; Frolov, A.; Birkemeyer, C. GC-MS method for the quantitation of carbohydrate intermediates in glycation systems. J. Agric. Food Chem. 2015, 63, 5911–5919. [Google Scholar] [CrossRef] [PubMed]
- Navarro, M.; Atzenbeck, L.; Pischetsrieder, M.; Morales, F.J. Investigations on the Reaction of C3 and C6 α-dicarbonyl compounds with hydroxytyrosol and related compounds under competitive conditions. J. Agric. Food Chem. 2016, 64, 6327–6332. [Google Scholar] [CrossRef] [PubMed]
- Henning, C.; Liehr, K.; Girndt, M.; Ulrich, C.; Glomb, M.A. Extending the spectrum of α-dicarbonyl compounds in vivo. J. Biol. Chem. 2014, 289, 28676–28688. [Google Scholar] [CrossRef] [PubMed]
- Weigel, K.U.; Opitz, T.; Henle, T. Studies on the occurrence and formation of 1,2-dicarbonyls in honey. Eur. Food Res. Technol. 2004, 218, 147–151. [Google Scholar] [CrossRef]
- Venkatraman, J.; Aggarwal, K.; Balaram, P. Helical peptide models for protein glycation: Proximity effects in catalysis of the Amadori rearrangement. Chem. Biol. 2001, 8, 611–625. [Google Scholar] [CrossRef]
- Zhang, Q.; Monroe, M.E.; Schepmoes, A.A.; Clauss, T.R.W.; Gritsenko, M.A.; Meng, D.; Petyuk, V.A.; Smith, R.D.; Metz, T.O. Comprehensive identification of glycated peptides and their glycation motifs in plasma and erythrocytes of control and diabetic subjects. J. Proteome Res. 2011, 10, 3076–3088. [Google Scholar] [CrossRef] [PubMed]
- Bilova, T.; Paudel, G.; Shilyaev, N.; Schmidt, R.; Brauch, D.; Tarakhovskaya, E.; Milrud, S.; Smolikova, G.; Tissier, A.; Vogt, T.; et al. Global proteomic analysis of advanced glycation end products in the Arabidopsis proteome provides evidence for age-related glycation Hotspots. J. Biol. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.V.; Mann, M. Status of large-scale analysis of post-translational modifications by mass spectrometry. Mol. Cell. Proteom. 2013, 12, 3444–3452. [Google Scholar] [CrossRef] [PubMed]
- Priego Capote, F.; Sanchez, J.-C. Strategies for proteomic analysis of non-enzymatically glycated proteins. Mass Spectrom. Rev. 2009, 28, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Wrodnigg, T.M.; Eder, B. The Amadori and Heyns Rearrangements: Landmarks in the History of Carbohydrate Chemistry or Unrecognized Synthetic Opportunities? In Glycoscience; Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2001; pp. 115–152. ISBN 978-3-540-41383-7. [Google Scholar]
- Mossine, V.V.; Linetsky, M.; Glinsky, G.V.; Ortwerth, B.J.; Feather, M.S. Superoxide free radical generation by Amadori compounds: The role of acyclic forms and metal ions. Chem. Res. Toxicol. 1999, 12, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Hinton, D.J.S.; Ames, J.M. Site specificity of glycation and carboxymethylation of bovine serum albumin by fructose. Amino Acids 2006, 30, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Barnaby, O.; Wa, C.; Cerny, R.L.; Clarke, W.; Hage, D.S. Quantitative analysis of glycation sites on human serum albumin using 16O/18O-Labeling and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Clin. Chim. Acta Int. J. Clin. Chem. 2010, 411, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Anguizola, J.; Matsuda, R.; Barnaby, O.S.; Hoy, K.S.; Wa, C.; DeBolt, E.; Koke, M.; Hage, D.S. Review: Glycation of human serum albumin. Clin. Chim. Acta Int. J. Clin. Chem. 2013, 425, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Akıllıoğlu, H.G.; Çelikbıçak, Ö.; Salih, B.; Gökmen, V. Monitoring protein glycation by electrospray ionization (ESI) quadrupole time-of-flight (Q-TOF) mass spectrometer. Food Chem. 2017, 217, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Lössl, P.; van de Waterbeemd, M.; Heck, A.J. The diverse and expanding role of mass spectrometry in structural and molecular biology. EMBO J. 2016, 35, 2634–2657. [Google Scholar] [CrossRef] [PubMed]
- Signor, L.; Boeri Erba, E. Matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) mass spectrometric analysis of intact proteins larger than 100 kDa. J. Vis. Exp. 2013, 79, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lapolla, A.; Gerhardinger, C.; Baldo, L.; Fedele, D.; Keane, A.; Seraglia, R.; Catinella, S.; Traldi, P. A study on in vitro glycation processes by matrix-assisted laser desorption ionization mass spectrometry. Biochim. Biophys. Acta 1993, 1225, 33–38. [Google Scholar] [CrossRef]
- Kańska, U.; Boratyński, J. Thermal glycation of proteins by d-glucose and d-fructose. Arch. Immunol. Ther. Exp. 2002, 50, 61–66. [Google Scholar]
- Lapolla, A.; Fedele, D.; Seraglia, R.; Catinella, S.; Baldo, L.; Aronica, R.; Traldi, P. A new effective method for the evaluation of glycated intact plasma proteins in diabetic subjects. Diabetologia 1995, 38, 1076–1081. [Google Scholar] [CrossRef] [PubMed]
- Meltretter, J.; Pischetsrieder, M. Application of mass spectrometry for the detection of glycation and oxidation products in milk proteins. Ann. N. Y. Acad. Sci. 2008, 1126, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Lapolla, A.; Fedele, D.; Seraglia, R.; Catinella, S.; Traldi, P. Matrix-assisted laser desorption/ionization capabilities in the study of non-enzymatic protein glycation. Rapid Commun. Mass Spectrom. 1994, 8, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Cohenford, M.A.; Frost, L.; Seneviratne, C.; Dain, J.A. Inhibitory effect of gold nanoparticles on the d-ribose glycation of bovine serum albumin. Int. J. Nanomed. 2014, 9, 5461–5469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Tu, Z.; Wang, H.; Huang, X.; Shi, Y.; Sha, X.; Xiao, H. Improved glycation after ultrasonic pretreatment revealed by high-performance liquid chromatography-linear ion trap/Orbitrap high-resolution mass spectrometry. J. Agric. Food Chem. 2014, 62, 2522–2530. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Liu, W.; Frost, L.; Kirschenbaum, L.J.; Dain, J.A.; Seeram, N.P. Glucitol-core containing gallotannins inhibit the formation of advanced glycation end-products mediated by their antioxidant potential. Food Funct. 2016, 7, 2213–2222. [Google Scholar] [CrossRef] [PubMed]
- Lapolla, A.; Fedele, D.; Aronica, R.; Baldo, L.; D’Alpaos, M.; Seraglia, R.; Traldi, P. The in vitro glycation of lysozyme and the influence of buffer concentration investigated by mass spectrometry. Rapid Commun. Mass Spectrom. 1996, 10, 1512–1518. [Google Scholar] [CrossRef]
- Lee, B.-S.; Jayathilaka, G.D.; Huang, J.-S.; Vida, L.N.; Honig, G.R.; Gupta, S. Analyses of in vitro nonenzymatic glycation of normal and variant hemoglobins by MALDI-TOF mass spectrometry. J. Biomol. Tech. 2011, 22, 90–94. [Google Scholar] [PubMed]
- Carulli, S.; Calvano, C.D.; Palmisano, F.; Pischetsrieder, M. MALDI-TOF MS characterization of glycation products of whey proteins in a glucose/galactose model system and lactose-free milk. J. Agric. Food Chem. 2011, 59, 1793–1803. [Google Scholar] [CrossRef] [PubMed]
- Paradela-Dobarro, B.; Rodiño-Janeiro, B.K.; Alonso, J.; Raposeiras-Roubín, S.; González-Peteiro, M.; González-Juanatey, J.R.; Álvarez, E. Key structural and functional differences between early and advanced glycation products. J. Mol. Endocrinol. 2016, 56, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, S.; Ghosh, P.; Parveen, S.; Dasgupta, S. Glycation of human γB-crystallin: A biophysical investigation. Int. J. Biol. Macromol. 2017, 96, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Dinda, A.K.; Tripathy, D.R.; Dasgupta, S. Glycation of Ribonuclease A affects its enzymatic activity and DNA binding ability. Biochimie 2015, 118, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Hrynets, Y.; Ndagijimana, M.; Betti, M. Rapid myoglobin aggregation through glucosamine-induced α-dicarbonyl formation. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Hattan, S.J.; Parker, K.C.; Vestal, M.L.; Yang, J.Y.; Herold, D.A.; Duncan, M.W. Analysis and quantitation of glycated hemoglobin by matrix assisted laser desorption/ionization time of flight mass spectrometry. J. Am. Soc. Mass Spectrom. 2016, 27, 532–541. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, A.; Mirasole, C.; Zolla, L. Haemoglobin glycation (Hb1Ac) increases during red blood cell storage: A MALDI-TOF mass-spectrometry-based investigation. Vox Sang. 2013, 105, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Traldi, P.; Castilho, G.; Sartori, C.H.; Machado-Lima, A.; Nakandakare, E.R.; Corrêa-Giannella, M.L.C.; Roverso, M.; Porcu, S.; Lapolla, A.; Passarelli, M. Glycated human serum albumin isolated from poorly controlled diabetic patients impairs cholesterol efflux from macrophages: An investigation by mass spectrometry. Eur. J. Mass Spectrom. Chichester Engl. 2015, 21, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Machado-Lima, A.; Iborra, R.T.; Pinto, R.S.; Castilho, G.; Sartori, C.H.; Oliveira, E.R.; Okuda, L.S.; Nakandakare, E.R.; Giannella-Neto, D.; Machado, U.F.; et al. In type 2 diabetes mellitus glycated albumin alters macrophage gene expression impairing ABCA1-mediated cholesterol efflux. J. Cell. Physiol. 2015, 230, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Wang, Z.; Huang, C.; Wang, Z.; Chi, L. Investigation of non-enzymatic glycosylation of human serum albumin using ion trap-time of flight mass spectrometry. Molecules 2012, 17, 8782–8794. [Google Scholar] [CrossRef] [PubMed]
- Lapolla, A.; Fedele, D.; Aronica, R.; Garbeglio, M.; D’Alpaos, M.; Seraglia, R.; Traldi, P. The in vivo glyco-oxidation of α- and β-globins investigated by matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun. Mass Spectrom. 1996, 10, 1133–1135. [Google Scholar] [CrossRef]
- Biroccio, A.; Urbani, A.; Massoud, R.; di Ilio, C.; Sacchetta, P.; Bernardini, S.; Cortese, C.; Federici, G. A quantitative method for the analysis of glycated and glutathionylated hemoglobin by matrix-assisted laser desorption ionization-time of flight mass spectrometry. Anal. Biochem. 2005, 336, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Porcu, S.; Lapolla, A.; Biasutto, L.; Zoratti, M.; Piarulli, F.; Eliana, G.; Basso, D.; Roverso, M.; Seraglia, R. A preliminary fastview of mitochondrial protein profile from healthy and type 2 diabetic subjects. Eur. J. Mass Spectrom. Chichester Engl. 2014, 20, 307–315. [Google Scholar] [CrossRef]
- Lapolla, A.; Porcu, S.; Roverso, M.; Desoye, G.; Cosma, C.; Nardelli, G.B.; Bogana, G.; Carrozzini, M.; Traldi, P. A preliminary investigation on placenta protein profile reveals only modest changes in well controlled gestational diabetes mellitus. Eur. J. Mass Spectrom. Chichester Engl. 2013, 19, 211–223. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Argirova, M.; Ahmed, N.; Mann, V.M.; Argirov, O.; Dawnay, A. Mass spectrometric monitoring of albumin in uremia. Kidney Int. 2000, 58, 2228–2234. [Google Scholar] [CrossRef] [PubMed]
- Goswami, J. Different separation or experimental techniques for clinical chromatography: Small review. J. Chromatogr. Sep. Tech. 2015. [Google Scholar] [CrossRef]
- De Seny, D.; Cobraiville, G.; Leprince, P.; Fillet, M.; Collin, C.; Mathieu, M.; Hauzeur, J.-P.; Gangji, V.; Malaise, M.G. Biomarkers of inflammation and innate immunity in atrophic nonunion fracture. J. Transl. Med. 2016, 14, 258. [Google Scholar] [CrossRef] [PubMed]
- Nedić, O.; Lagundžin, D.; Masnikosa, R. Posttranslational modifications of the insulin-like growth factor-binding protein 3 in patients with type 2 diabetes mellitus assessed by affinity chromatography. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2012, 904, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Jabeen, R.; Saleemuddin, M.; Petersen, J.; Mohammad, A. Inactivation and modification of superoxide dismutase by glyoxal: Prevention by antibodies. Biochimie 2007, 89, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Seibert, V.; Wiesner, A.; Buschmann, T.; Meuer, J. Surface-enhanced laser desorption ionization time-of-flight mass spectrometry (SELDI TOF-MS) and ProteinChip technology in proteomics research. Pathol. Res. Pract. 2004, 200, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Mazumdar, S. Electrospray ionization mass spectrometry: A technique to access the information beyond the molecular weight of the analyte. Int. J. Anal. Chem. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Krusemark, C.J.; Frey, B.L.; Belshaw, P.J.; Smith, L.M. Modifying the charge state distribution of proteins in electrospray ionization mass spectrometry by chemical derivatization. J. Am. Soc. Mass Spectrom. 2009, 20, 1617–1625. [Google Scholar] [CrossRef] [PubMed]
- Redman, E.A.; Ramos-Payan, M.; Mellors, J.S.; Ramsey, J.M. Analysis of hemoglobin glycation using microfluidic CE-MS: A rapid, mass spectrometry compatible method for assessing diabetes management. Anal. Chem. 2016, 88, 5324–5330. [Google Scholar] [CrossRef] [PubMed]
- Kijewska, M.; Radziszewska, K.; Cal, M.; Waliczek, M.; Stefanowicz, P.; Szewczuk, Z. The influence of glycation on a high pressure denaturation of ubiquitin. Biosci. Rep. 2016, 36. [Google Scholar] [CrossRef] [PubMed]
- Bertoletti, L.; Regazzoni, L.; Altomare, A.; Colombo, R.; Colzani, M.; Vistoli, G.; Marchese, L.; Carini, M.; de Lorenzi, E.; Aldini, G. Advanced glycation end products of β2-microglobulin in uremic patients as determined by high resolution mass spectrometry. J. Pharm. Biomed. Anal. 2014, 91, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Roberts, N.B.; Green, B.N.; Morris, M. Potential of electrospray mass spectrometry for quantifying glycohemoglobin. Clin. Chem. 1997, 43, 771–778. [Google Scholar] [PubMed]
- Stefanowicz, P.; Boratyński, J.; Kańska, U.; Petry, I.; Szewczuk, Z. Evaluation of high temperature glycation of proteins and peptides by electrospray ionization mass spectrometry. Acta Biochim. Pol. 2001, 48, 1137–1141. [Google Scholar] [PubMed]
- Marie, A.-L.; Przybylski, C.; Gonnet, F.; Daniel, R.; Urbain, R.; Chevreux, G.; Jorieux, S.; Taverna, M. Capillary zone electrophoresis and capillary electrophoresis-mass spectrometry for analyzing qualitative and quantitative variations in therapeutic albumin. Anal. Chim. Acta 2013, 800, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Peterson, K.P.; Pavlovich, J.G.; Goldstein, D.; Little, R.; England, J.; Peterson, C.M. What is hemoglobin A1c? An analysis of glycated hemoglobins by electrospray ionization mass spectrometry. Clin. Chem. 1998, 44, 1951–1958. [Google Scholar] [PubMed]
- McKillop, A.M.; McCluskey, J.T.; Boyd, A.C.; Mooney, M.H.; Flatt, P.R.; O’Harte, F.P. Production and characterization of specific antibodies for evaluation of glycated insulin in plasma and biological tissues. J. Endocrinol. 2000, 167, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.J.; Boyd, A.C.; O’Harte, F.P.M.; McKillop, A.M.; Wiggam, M.I.; Mooney, M.H.; McCluskey, J.T.; Lindsay, J.R.; Ennis, C.N.; Gamble, R.; et al. Demonstration of glycated insulin in human diabetic plasma and decreased biological activity assessed by euglycemic-hyperinsulinemic clamp technique in humans. Diabetes 2003, 52, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.P.; Jackson, H.; Green, B.N. Hb Belleville [β10(A7)Ala→Thr] affects the determination of Hb A1c by routine cation exchange high performance liquid chromatography. Hemoglobin 2009, 33, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Yeboah, F.K.; Alli, I.; Yaylayan, V.A.; Konishi, Y.; Stefanowicz, P. Monitoring glycation of lysozyme by electrospray ionization mass spectrometry. J. Agric. Food Chem. 2000, 48, 2766–2774. [Google Scholar] [CrossRef] [PubMed]
- Al-Abed, Y.; Mitsuhashi, T.; Li, H.; Lawson, J.A.; FitzGerald, G.A.; Founds, H.; Donnelly, T.; Cerami, A.; Ulrich, P.; Bucala, R. Inhibition of advanced glycation endproduct formation by acetaldehyde: Role in the cardioprotective effect of ethanol. Proc. Natl. Acad. Sci. USA 1999, 96, 2385–2390. [Google Scholar] [CrossRef] [PubMed]
- Panuwet, P.; Hunter, R.E.; D’Souza, P.E.; Chen, X.; Radford, S.A.; Cohen, J.R.; Marder, M.E.; Kartavenka, K.; Ryan, P.B.; Barr, D.B. Biological matrix effects in quantitative tandem mass spectrometry-based analytical methods: Advancing biomonitoring. Crit. Rev. Anal. Chem. 2016, 46, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Bozhinov, A.S.; Boyanova, M.; Niwa, T.; Ivanov, I. Evidence for the presence of glycation adducts in protein therapeutics. Biotechnol. Biotechnol. Equip. 2010, 24, 1904–1909. [Google Scholar] [CrossRef]
- Borges, C.R.; Oran, P.E.; Buddi, S.; Jarvis, J.W.; Schaab, M.R.; Rehder, D.S.; Rogers, S.P.; Taylor, T.; Nelson, R.W. Building multidimensional biomarker views of type 2 diabetes on the basis of protein microheterogeneity. Clin. Chem. 2011, 57, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Catherman, A.D.; Skinner, O.S.; Kelleher, N.L. Top down proteomics: Facts and perspectives. Biochem. Biophys. Res. Commun. 2014, 445, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Blein-Nicolas, M.; Zivy, M. Thousand and one ways to quantify and compare protein abundances in label-free bottom-up proteomics. Biochim. Biophys. Acta 2016, 1864, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Uygun, B.E.; Geerts, S.; Ozer, S.; Scalf, M.; Gilpin, S.E.; Ott, H.C.; Yarmush, M.L.; Smith, L.M.; Welham, N.V.; Frey, B.L. Proteomic analysis of naturally-sourced biological scaffolds. Biomaterials 2016, 75, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.-C.; Yates, J.R. Protein analysis by Shotgun/Bottom-up proteomics. Chem. Rev. 2013, 113, 2343–2394. [Google Scholar] [CrossRef] [PubMed]
- Frolov, A.; Blüher, M.; Hoffmann, R. Glycation sites of human plasma proteins are affected to different extents by hyperglycemic conditions in type 2 diabetes mellitus. Anal. Bioanal. Chem. 2014, 406, 5755–5763. [Google Scholar] [CrossRef] [PubMed]
- Lohnes, K.; Quebbemann, N.R.; Liu, K.; Kobzeff, F.; Loo, J.A.; Rachel, R.; Loo, O. Combining high-throughput MALDI-TOF mass spectrometry and isoelectric focusing gel electrophoresis for virtual 2D gel-based proteomics. Methods 2016, 104, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Bogdanov, B.; Smith, R.D. Proteomics by FTICR mass spectrometry: Top down and bottom up. Mass Spectrom. Rev. 2005, 24, 168–200. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Wang, D. Top-down proteomics of a drop of blood for diabetes monitoring. J. Proteome Res. 2014, 13, 1560–1569. [Google Scholar] [CrossRef] [PubMed]
- Serra, A.; Gallart-Palau, X.; See-Toh, R.S.-E.; Hemu, X.; Tam, J.P.; Sze, S.K. Commercial processed soy-based food product contains glycated and glycoxidated lunasin proteoforms. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Brede, C.; Hop, B.; Jørgensen, K.; Skadberg, Ø. Measurement of glycated albumin in serum and plasma by LC-MS/MS. Scand. J. Clin. Lab. Investig. 2016, 76, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, X.; Liu, Y.-H.; Richardson, D.; Li, H.; Shameem, M.; Yang, X. Simultaneous monitoring of oxidation, deamidation, isomerization, and glycosylation of monoclonal antibodies by liquid chromatography-mass spectrometry method with ultrafast tryptic digestion. Mabs 2016, 8, 1477–1486. [Google Scholar] [CrossRef] [PubMed]
- Sze, S.K.; Ge, Y.; Oh, H.; McLafferty, F.W. Top-down mass spectrometry of a 29-kDa protein for characterization of any posttranslational modification to within one residue. Proc. Natl. Acad. Sci. USA 2002, 99, 1774–1779. [Google Scholar] [CrossRef] [PubMed]
- Chmelik, J.; Zidkova, J.; Rehulka, P.; Petry-Podgorska, I.; Bobalova, J. Influence of different proteomic protocols on degree of high-coverage identification of nonspecific lipid transfer protein 1 modified during malting. Electrophoresis 2009, 30, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Catherman, A.D.; Li, M.; Tran, J.C.; Durbin, K.R.; Compton, P.D.; Early, B.P.; Thomas, P.M.; Kelleher, N.L. Top down proteomics of human membrane proteins from enriched mitochondrial fractions. Anal. Chem. 2013, 85, 1880–1888. [Google Scholar] [CrossRef] [PubMed]
- Perkel, J.M. Tearing the top off ‘Top-Down’ proteomics. BioTechniques 2012, 53, 75–78. [Google Scholar] [PubMed]
- Gregorich, Z.R.; Ge, Y. Top-down proteomics in health and disease: Challenges and opportunities. Proteomics 2014, 14, 1195–1210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ge, Y. Comprehensive analysis of protein modifications by top-down mass spectrometry. Circ. Cardiovasc. Genet. 2011, 4. [Google Scholar] [CrossRef] [PubMed]
- Whitelegge, J.; Halgand, F.; Souda, P.; Zabrouskov, V. Top-down mass spectrometry of integral membrane proteins. Expert Rev. Proteom. 2006, 3, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, N.L. Top-down proteomics. Anal. Chem. 2004, 76, 197A–203A. [Google Scholar] [CrossRef] [PubMed]
- Tran, J.C.; Zamdborg, L.; Ahlf, D.R.; Lee, J.E.; Catherman, A.D.; Durbin, K.R.; Tipton, J.D.; Vellaichamy, A.; Kellie, J.F.; Li, M.; et al. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature 2011, 480, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Priego-Capote, F.; Ramírez-Boo, M.; Finamore, F.; Gluck, F.; Sanchez, J.-C. Quantitative analysis of glycated proteins. J. Proteome Res. 2014, 13, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Verrastro, I.; Pasha, S.; Jensen, K.T.; Pitt, A.R.; Spickett, C.M. Mass spectrometry-based methods for identifying oxidized proteins in disease: Advances and challenges. Biomolecules 2015, 5, 378–411. [Google Scholar] [CrossRef] [PubMed]
- Arena, S.; Salzano, A.M.; Renzone, G.; D’Ambrosio, C.; Scaloni, A. Non-enzymatic glycation and glycoxidation protein products in foods and diseases: An interconnected, complex scenario fully open to innovative proteomic studies. Mass Spectrom. Rev. 2014, 33, 49–77. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Chen, R.; Aebersold, R.; Brentnall, T.A. Mass spectrometry based glycoproteomics—From a proteomics perspective. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.R.; Ruse, C.I.; Nakorchevsky, A. Proteomics by mass cpectrometry: Approaches, advances, and applications. Annu. Rev. Biomed. Eng. 2009, 11, 49–79. [Google Scholar] [CrossRef] [PubMed]
- Garcia, B.A. What does the future hold for Top Down mass spectrometry? J. Am. Soc. Mass Spectrom. 2010, 21, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Tang, N.; Schepmoes, A.A.; Phillips, L.S.; Smith, R.D.; Metz, T.O. Proteomic profiling of nonenzymatically glycated proteins in human plasma and erythrocyte membranes. J. Proteome Res. 2008, 7, 2025–2032. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, N.L.; Lin, H.Y.; Valaskovic, G.A.; Aaserud, D.J.; Fridriksson, E.K.; McLafferty, F.W. Top Down versus Bottom Up protein characterization by tandem high-resolution mass spectrometry. J. Am. Chem. Soc. 1999, 121, 806–812. [Google Scholar] [CrossRef]
- Marotta, E.; Lapolla, A.; Fedele, D.; Senesi, A.; Reitano, R.; Witt, M.; Seraglia, R.; Traldi, P. Accurate mass measurements by Fourier transform mass spectrometry in the study of advanced glycation end products/peptides. J. Mass Spectrom. 2003, 38, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, P.; Müller, M.; Appel, R.D. Automated protein identification by tandem mass spectrometry: Issues and strategies. Mass Spectrom. Rev. 2006, 25, 235–254. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.W.; Aebersold, R.; Caprioli, R.M. The pros and cons of peptide-centric proteomics. Nat. Biotechnol. 2010, 28, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Grant, R.P.; Hoofnagle, A.N. From lost in translation to paradise found: Enabling protein biomarker method transfer by mass spectrometry. Clin. Chem. 2014, 60, 941–944. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Liu, T.; Qian, W.-J.; Petyuk, V.A.; Smith, R.D. Liquid chromatography-mass spectrometry-based quantitative proteomics. J. Biol. Chem. 2011, 286, 25443–25449. [Google Scholar] [CrossRef] [PubMed]
- Kielmas, M.; Szewczuk, Z.; Stefanowicz, P. A study on human serum albumin influence on glycation of fibrinogen. Biochem. Biophys. Res. Commun. 2013, 439, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Gorisse, L.; Pietrement, C.; Vuiblet, V.; Schmelzer, C.E.H.; Köhler, M.; Duca, L.; Debelle, L.; Fornès, P.; Jaisson, S.; Gillery, P. Protein carbamylation is a hallmark of aging. Proc. Natl. Acad. Sci. USA 2016, 113, 1191–1196. [Google Scholar] [CrossRef] [PubMed]
- Frahm, G.E.; Smith, D.G.S.; Kane, A.; Lorbetskie, B.; Cyr, T.D.; Girard, M.; Johnston, M.J.W. Determination of supplier-to-supplier and lot-to-lot variability in glycation of recombinant human serum albumin expressed in Oryza sativa. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-H.; Wang, T.-F.; Wu, C.-H.; Chen, S.-H. In-depth comparative characterization of hemoglobin glycation in normal and diabetic bloods by LC-MSMS. J. Am. Soc. Mass Spectrom. 2014, 25, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Wa, C.; Cerny, R.L.; Clarke, W.A.; Hage, D.S. Characterization of glycation adducts on human serum albumin by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Clin. Chim. Acta Int. J. Clin. Chem. 2007, 385, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Meltretter, J.; Wüst, J.; Pischetsrieder, M. Comprehensive analysis of nonenzymatic post-translational β-lactoglobulin modifications in processed milk by ultrahigh-performance liquid chromatography-tandem mass spectrometry. J. Agric. Food Chem. 2013, 61, 6971–6981. [Google Scholar] [CrossRef] [PubMed]
- Humeny, A.; Kislinger, T.; Becker, C.-M.; Pischetsrieder, M. Qualitative determination of specific protein glycation products by matrix-assisted laser desorption/ionization mass spectrometry Peptide mapping. J. Agric. Food Chem. 2002, 50, 2153–2160. [Google Scholar] [CrossRef] [PubMed]
- Stefanowicz, P.; Kijewska, M.; Kluczyk, A.; Szewczuk, Z. Detection of glycation sites in proteins by high-resolution mass spectrometry combined with isotopic labeling. Anal. Biochem. 2010, 400, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Lapolla, A.; Fedele, D.; Senesi, A.; Arico, N.C.; Reitano, R.; Seraglia, R.; Astner, H.; Traldi, P. Advanced glycation end-products/peptides: A preliminary investigation by LC and LC/MS. Il Farmaco 2002, 57, 845–852. [Google Scholar] [CrossRef]
- Lapolla, A.; Fedele, D.; Garbeglio, M.; Martano, L.; Tonani, R.; Seraglia, R.; Favretto, D.; Fedrigo, M.A.; Traldi, P. Matrix-assisted laser desorption/ionization mass spectrometry, enzymatic digestion, and molecular modeling in the study of nonenzymatic glycation of IgG. J. Am. Soc. Mass Spectrom. 2000, 11, 153–159. [Google Scholar] [CrossRef]
- Tsiatsiani, L.; Heck, A.J.R. Proteomics beyond trypsin. FEBS J. 2015, 282, 2612–2626. [Google Scholar] [CrossRef] [PubMed]
- Loziuk, P.L.; Wang, J.; Li, Q.; Sederoff, R.R.; Chiang, V.L.; Muddiman, D.C. Understanding the role of proteolytic digestion on discovery and targeted proteomic measurements using liquid chromatography tandem mass spectrometry and design of experiments. J. Proteome Res. 2013, 12, 5820–5829. [Google Scholar] [CrossRef] [PubMed]
- Lapolla, A.; Fedele, D.; Reitano, R.; Aricò, N.C.; Seraglia, R.; Traldi, P.; Marotta, E.; Tonani, R. Enzymatic digestion and mass spectrometry in the study of advanced glycation end products/peptides. J. Am. Soc. Mass Spectrom. 2004, 15, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Cotham, W.E.; Metz, T.O.; Ferguson, P.L.; Brock, J.W.C.; Hinton, D.J.S.; Thorpe, S.R.; Baynes, J.W.; Ames, J.M. Proteomic analysis of arginine adducts on glyoxal-modified ribonuclease. Mol. Cell. Proteom. 2004, 3, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Farah, M.A.; Bose, S.; Lee, J.-H.; Jung, H.-C.; Kim, Y. Analysis of glycated insulin by MALDI-TOF mass spectrometry. Biochim. Biophys. Acta 2005, 1725, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Meltretter, J.; Seeber, S.; Humeny, A.; Becker, C.-M.; Pischetsrieder, M. Site-specific formation of Maillard, oxidation, and condensation products from whey proteins during reaction with lactose. J. Agric. Food Chem. 2007, 55, 6096–6103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Schepmoes, A.A.; Brock, J.W.C.; Wu, S.; Moore, R.J.; Purvine, S.O.; Baynes, J.W.; Smith, R.D.; Metz, T.O. Improved methods for the enrichment and analysis of glycated peptides. Anal. Chem. 2008, 80, 9822–9829. [Google Scholar] [CrossRef] [PubMed]
- Ajjan, R.A.; Gamlen, T.; Standeven, K.F.; Mughal, S.; Hess, K.; Smith, K.A.; Dunn, E.J.; Anwar, M.M.; Rabbani, N.; Thornalley, P.J.; et al. Diabetes is associated with posttranslational modifications in plasminogen resulting in reduced plasmin generation and enzyme-specific activity. Blood 2013, 122, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Proc, J.L.; Kuzyk, M.A.; Hardie, D.B.; Yang, J.; Smith, D.S.; Jackson, A.M.; Parker, C.E.; Borchers, C.H. A quantitative study of the effects of chaotropic agents, surfactants, and solvents on the digestion efficiency of human plasma proteins by trypsin. J. Proteome Res. 2010, 9, 5422–5437. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Liu, N.; Long, J.; Tang, C.; Li, J.; Huo, L.; Wang, X.; Chen, P.; Liang, S. Blue native/SDS-PAGE combined with iTRAQ analysis reveals advanced glycation end-product-induced changes of synaptosome proteins in C57 BL/6 mice. Electrophoresis 2011, 32, 2194–2205. [Google Scholar] [CrossRef] [PubMed]
- Griesser, E.; Vemula, V.; Raulien, N.; Wagner, U.; Reeg, S.; Grune, T.; Fedorova, M. Cross-talk between lipid and protein carbonylation in a dynamic cardiomyocyte model of mild nitroxidative stress. Redox Biol. 2017, 11, 438–455. [Google Scholar] [CrossRef] [PubMed]
- Bollineni, R.C.; Hoffmann, R.; Fedorova, M. Proteome-wide profiling of carbonylated proteins and carbonylation sites in HeLa cells under mild oxidative stress conditions. Free Radic. Biol. Med. 2014, 68, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Karpievitch, Y.V.; Polpitiya, A.D.; Anderson, G.A.; Smith, R.D.; Dabney, A.R. Liquid chromatography mass spectrometry-based proteomics: Biological and technological aspects. Ann. Appl. Stat. 2010, 4, 1797–1823. [Google Scholar] [CrossRef] [PubMed]
- Meltretter, J.; Becker, C.-M.; Pischetsrieder, M. Identification and site-specific relative quantification of β-lactoglobulin modifications in heated milk and dairy products. J. Agric. Food Chem. 2008, 56, 5165–5171. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, M.; He, D.; Zhao, X.; Zhang, W.; Wei, L.; Huang, E.; Ji, L.; Zhang, M.; Willard, B.; et al. HDL in diabetic nephropathy has less effect in endothelial repairing than diabetes without complications. Lipids Health Dis. 2016, 15, 76. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.W.; Gupta, R.; Deeth, H.C.; Alewood, P.F. Proteomic analysis of temperature-dependent changes in stored UHT milk. J. Agric. Food Chem. 2011, 59, 1837–1846. [Google Scholar] [CrossRef] [PubMed]
- Le, T.T.; Deeth, H.C.; Bhandari, B.; Alewood, P.F.; Holland, J.W. A proteomic approach to detect lactosylation and other chemical changes in stored milk protein concentrate. Food Chem. 2012, 132, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Calvano, C.D.; Monopoli, A.; Loizzo, P.; Faccia, M.; Zambonin, C. Proteomic approach based on MALDI-TOF MS to detect powdered milk in fresh cow’s milk. J. Agric. Food Chem. 2013, 61, 1609–1617. [Google Scholar] [CrossRef] [PubMed]
- Marvin, L.F.; Parisod, V.; Fay, L.B.; Guy, P.A. Characterization of lactosylated proteins of infant formula powders using two-dimensional gel electrophoresis and nanoelectrospray mass spectrometry. Electrophoresis 2002, 23, 2505–2512. [Google Scholar] [CrossRef]
- Kueper, T.; Grune, T.; Prahl, S.; Lenz, H.; Welge, V.; Biernoth, T.; Vogt, Y.; Muhr, G.-M.; Gaemlich, A.; Jung, T.; et al. Vimentin is the specific target in skin glycation. Structural prerequisites, functional consequences, and role in skin aging. J. Biol. Chem. 2007, 282, 23427–23436. [Google Scholar] [CrossRef] [PubMed]
- Soboleva, A.; Modzel, M.; Didio, A.; Płóciennik, H.; Kijewska, M.; Grischina, T.; Karonova, T.; Bilova, T.; Stefanov, V.; Stefanowicz, P.; et al. Quantification of prospective type 2 diabetes mellitus biomarkers by stable isotope dilution with bi-labeled standard glycated peptides. Anal. Methods 2017, 9, 409–418. [Google Scholar] [CrossRef]
- Frolov, A.; Bilova, T.; Paudel, G.; Berger, R.; Balcke, G.U.; Birkemeyer, C.; Wessjohann, L.A. Early responses of mature Arabidopsis thaliana plants to reduced water potential in the agar-based polyethylene glycol infusion drought model. J. Plant Physiol. 2017, 208, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Hinton, D.J.S.; Ames, J.M. Analysis of glycated protein by capillary electrophoresis. Int. Congr. Ser. 2002, 471–474. [Google Scholar] [CrossRef]
- Lacinová, K.; Eckhardt, A.; Pataridis, S.; Sedláková, P.; Mikšík, I. Glycation of proteins—Their analysis and physiological aspects. In Advances in Molecular Mechanisms and Pharmacology of Diabetic Complications; Transworld Research Network: Kerala, India, 2010; pp. 17–38. ISBN 978-81-7895-499-8. [Google Scholar]
- Singh, N.R.; Bourdon, E.; Rondeau, P. Identification of up-regulated low molecular weight proteins in human adipocytes treated with glycoxidized albumin. Open Obes. J. 2009, 2, 1–6. [Google Scholar]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.V.; Lu, X.; Standley, M.; Pattee, P.; Neelima, G.; Girisesh, G.; Dakshinamurthy, K.V.; Roberts, C.T.; Nagalla, S.R. Proteomic identification of urinary biomarkers of diabetic nephropathy. Diabetes Care 2007, 30, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Nesatyy, V.J.; Ross, N.W. Recovery of intact proteins from silver stained gels. Analyst 2002, 127, 1180–1187. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Jayaramaiah, R.H.; Agawane, S.B.; Vannuruswamy, G.; Korwar, A.M.; Anand, A.; Dhaygude, V.S.; Shaikh, M.L.; Joshi, R.S.; Boppana, R.; et al. Potential dual role of eugenol in inhibiting advanced glycation end products in diabetes: Proteomic and mechanistic insights. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.I.; Cociorva, D.; Norris, J.L.; Yates, J.R. Optimization of mass spectrometry-compatible surfactants for shotgun proteomics. J. Proteome Res. 2007, 6, 2529–2538. [Google Scholar] [CrossRef] [PubMed]
- Jagadeeshaprasad, M.G.; Batkulwar, K.B.; Meshram, N.N.; Tiwari, S.; Korwar, A.M.; Unnikrishnan, A.G.; Kulkarni, M.J. Targeted quantification of N-1-(carboxymethyl) valine and N-1-(carboxyethyl) valine peptides of β-hemoglobin for better diagnostics in diabetes. Clin. Proteom. 2016, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Vannuruswamy, G.; Jagadeeshaprasad, M.G.; Kashinath, K.; Kesavan, S.K.; Bhat, S.; Korwar, A.M.; Chougale, A.D.; Boppana, R.; Reddy, D.S.; Kulkarni, M.J. Molecules with O-acetyl group protect protein glycation by acetylating lysine residues. RSC Adv. 2016, 6, 65572–65578. [Google Scholar] [CrossRef]
- Nagai-Okatani, C.; Minamino, N. Aberrant glycosylation in the left ventricle and plasma of rats with cardiac hypertrophy and heart failure. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Eng, W.S.; Colquhoun, D.R.; Dinglasan, R.R.; Graham, D.R.; Mahal, L.K. Complex N-linked glycans serve as a determinant for exosome/microvesicle cargo recruitment. J. Biol. Chem. 2014, 289, 32526–32537. [Google Scholar] [CrossRef] [PubMed]
- Laurie, D.E.; Splan, R.K.; Green, K.; Still, K.M.; McKown, R.L.; Laurie, G.W. Detection of prosecretory mitogen lacritin in nonprimate tears primarily as a C-terminal-like fragment. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6130–6136. [Google Scholar] [CrossRef] [PubMed]
- Spiller, S.; Frolov, A.; Hoffmann, R. Quantification of specific glycation sites in human serum albumin as prospective type 2 diabetes mellitus biomarkers. Protein Pept. Lett. 2017. [Google Scholar] [CrossRef] [PubMed]
- Luche, S.; Diemer, H.; Tastet, C.; Chevallet, M.; Van Dorsselaer, A.; Leize-Wagner, E.; Rabilloud, T. About thiol derivatization and resolution of basic proteins in two-dimensional electrophoresis. Proteomics 2004, 4, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Boyatzis, A.E.; Bringans, S.D.; Piggott, M.J.; Duong, M.N.; Lipscombe, R.J.; Arthur, P.G. Limiting the hydrolysis and oxidation of maleimide-peptide adducts improves detection of protein thiol oxidation. J. Proteome Res. 2017, 16, 2004–2015. [Google Scholar] [CrossRef] [PubMed]
- Yokono, T.; Mineki, R.; Taka, H.; Kotaniguchi, H.; Murayama, K. Improvement of automatic in-gel digestion by in situ alkylation of proteins. J. Biomol. Tech. 2003, 14, 191–196. [Google Scholar] [PubMed]
- Moritz, R.L.; Eddes, J.S.; Reid, G.E.; Simpson, R.J. S-pyridylethylation of intact polyacrylamide gels and in situ digestion of electrophoretically separated proteins: A rapid mass spectrometric method for identifying cysteine-containing peptides. Electrophoresis 1996, 17, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Feckler, C.; Muster, G.; Feser, W.; Römer, A.; Palme, K. Mass spectrometric analysis reveals a cysteine bridge between residues 2 and 61 of the auxin-binding protein 1 from Zea mays L. FEBS Lett. 2001, 509, 446–450. [Google Scholar] [CrossRef]
- Amoresano, A.; Andolfo, A.; Siciliano, R.A.; Cozzolino, R.; Minchiotti, L.; Galliano, M.; Pucci, P. Analysis of human serum albumin variants by mass spectrometric procedures. Biochim. Biophys. Acta 1998, 1384, 79–92. [Google Scholar] [CrossRef]
- Wang, S.; Kaltashov, I.A. A new strategy of using O18-labeled iodoacetic acid for mass spectrometry-based protein quantitation. J. Am. Soc. Mass Spectrom. 2012, 23, 1293–1297. [Google Scholar] [CrossRef] [PubMed]
- Starita, L.M.; Lo, R.S.; Eng, J.K.; von Haller, P.D.; Fields, S. Sites of ubiquitin attachment in Saccharomyces cerevisiae. Proteomics 2012, 12, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Hains, P.G.; Robinson, P.J. The impact of commonly used alkylating agents on artifactual peptide modification. J. Proteome Res. 2017, 16, 3443–3447. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Su, P.; Yang, Y.; Wang, T.; Yang, Y. DNA directed immobilization enzyme on polyamidoamine tethered magnetic composites with high reusability and stability. J. Mater. Chem. B 2016, 4, 5873–5882. [Google Scholar] [CrossRef]
- Banerjee, S.; Chakraborti, A.S. Structural alterations of hemoglobin and myoglobin by glyoxal: A comparative study. Int. J. Biol. Macromol. 2014, 66, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.R. Mass spectrometry and the age of the proteome. J. Mass Spectrom. 1998, 33, 1–19. [Google Scholar] [CrossRef]
- Keilhauer, E.C.; Geyer, P.E.; Mann, M. HCD fragmentation of glycated peptides. J. Proteome Res. 2016, 15, 2881–2890. [Google Scholar] [CrossRef] [PubMed]
- Petry-Podgórska, I.; Zídková, J.; Flodrová, D.; Bobálová, J. 2D-HPLC and MALDI-TOF/TOF analysis of barley proteins glycated during brewing. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2010, 878, 3143–3148. [Google Scholar] [CrossRef] [PubMed]
- Gygi, S.P.; Corthals, G.L.; Zhang, Y.; Rochon, Y.; Aebersold, R. Evaluation of two-dimensional gel electrophoresis-based proteome analysis technology. Proc. Natl. Acad. Sci. USA 2000, 97, 9390–9395. [Google Scholar] [CrossRef] [PubMed]
- Köcher, T.; Pichler, P.; Swart, R.; Mechtler, K. Analysis of protein mixtures from whole-cell extracts by single-run nanoLC-MS/MS using ultralong gradients. Nat. Protoc. 2012, 7, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Chernushevich, I.V.; Loboda, A.V.; Thomson, B.A. An introduction to quadrupole-time-of-flight mass spectrometry. J. Mass Spectrom. 2001, 36, 849–865. [Google Scholar] [CrossRef] [PubMed]
- Pozo, O.J.; Van Eenoo, P.; Deventer, K.; Elbardissy, H.; Grimalt, S.; Sancho, J.V.; Hernandez, F.; Ventura, R.; Delbeke, F.T. Comparison between triple quadrupole, time of flight and hybrid quadrupole time of flight analysers coupled to liquid chromatography for the detection of anabolic steroids in doping control analysis. Anal. Chim. Acta 2011, 684, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Yost, R.A.; Enke, C.G. Triple quadrupole mass spectrometry for direct mixture analysis and structure elucidation. Anal. Chem. 1979, 51, 1251–1264. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-Y.; Pace, N.; Kerns, E.H.; Kleintop, T.; Kagan, N.; Sakuma, T. Hybrid triple quadrupole-linear ion trap mass spectrometry in fragmentation mechanism studies: Application to structure elucidation of buspirone and one of its metabolites. J. Mass Spectrom. 2005, 40, 1017–1029. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-F.; Wu, H.-T.; Tan, G.-G.; Zhu, Z.-Y.; Chai, Y.-F. Liquid chromatography coupled with time-of-flight and ion trap mass spectrometry for qualitative analysis of herbal medicines. J. Pharm. Anal. 2011, 1, 235–245. [Google Scholar] [CrossRef]
- Douglas, D.J.; Frank, A.J.; Mao, D. Linear ion traps in mass spectrometry. Mass Spectrom. Rev. 2005, 24, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Scigelova, M.; Hornshaw, M.; Giannakopulos, A.; Makarov, A. Fourier Transform Mass Spectrometry. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Scigelova, M.; Makarov, A. Orbitrap mass analyzer-overview and applications in proteomics. Proteomics 2006, 6, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Gallien, S.; Domon, B. Quantitative proteomics using the high resolution accurate mass capabilities of the quadrupole-orbitrap mass spectrometer. Bioanalysis 2014, 6, 2159–2170. [Google Scholar] [CrossRef] [PubMed]
- Senko, M.W.; Remes, P.M.; Canterbury, J.D.; Mathur, R.; Song, Q.; Eliuk, S.M.; Mullen, C.; Earley, L.; Hardman, M.; Blethrow, J.D.; et al. Novel parallelized quadrupole/linear ion trap/Orbitrap tribrid mass spectrometer improving proteome coverage and peptide identification rates. Anal. Chem. 2013, 85, 11710–11714. [Google Scholar] [CrossRef] [PubMed]
- Krokhin, O.V.; Ens, W.; Standing, K.G. MALDI QqTOF MS combined with off-line HPLC for characterization of protein primary structure and post-translational modifications. J. Biomol. Tech. 2005, 16, 429–440. [Google Scholar] [PubMed]
- Da Costa, G.; Gomes, R.A.; Guerreiro, A.; Mateus, É.; Monteiro, E.; Barroso, E.; Coelho, A.V.; Freire, A.P.; Cordeiro, C. Beyond genetic factors in familial amyloidotic polyneuropathy: Protein glycation and the loss of fibrinogen’s chaperone activity. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Moreno, F.J.; Quintanilla-López, J.E.; Lebrón-Aguilar, R.; Olano, A.; Sanz, M.L. Mass spectrometric characterization of glycated β-lactoglobulin peptides derived from galacto-oligosaccharides surviving the in vitro gastrointestinal digestion. J. Am. Soc. Mass Spectrom. 2008, 19, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Frolov, A.; Hoffmann, R. Analysis of amadori peptides enriched by boronic acid affinity chromatography. Ann. N. Y. Acad. Sci. 2008, 1126, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, E.J.; Bereman, M.S.; Durand, S.; Valaskovic, G.A.; MacCoss, M.J. Effects of column and gradient lengths on peak capacity and peptide identification in nanoflow LC-MS/MS of complex proteomic samples. J. Am. Soc. Mass Spectrom. 2013, 24, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Priego-Capote, F.; Scherl, A.; Müller, M.; Waridel, P.; Lisacek, F.; Sanchez, J.-C. Glycation isotopic labeling with 13C-reducing sugars for quantitative analysis of glycated proteins in human plasma. Mol. Cell. Proteom. 2010, 9, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Finamore, F.; Priego-Capote, F.; Nolli, S.; Fontana, P.; Sanchez, J.-C. Aspirin-mediated acetylation of haemoglobin increases in presence of high glucose concentration and decreases protein glycation. EuPA Open Proteom. 2015, 8, 116–127. [Google Scholar] [CrossRef]
- Paudel, G.; Bilova, T.; Schmidt, R.; Greifenhagen, U.; Berger, R.; Tarakhovskaya, E.; Stöckhardt, S.; Balcke, G.U.; Humbeck, K.; Brandt, W.; et al. Osmotic stress is accompanied by protein glycation in Arabidopsis thaliana. J. Exp. Bot. 2016, 67, 6283–6295. [Google Scholar] [CrossRef] [PubMed]
- Kalli, A.; Smith, G.T.; Sweredoski, M.J.; Hess, S. Evaluation and optimization of mass spectrometric settings during data-dependent acquisition mode: Focus on LTQ-Orbitrap mass analyzers. J. Proteome Res. 2013, 12, 3071–3086. [Google Scholar] [CrossRef] [PubMed]
- Canterbury, J.D.; Merrihew, G.E.; Goodlett, D.R.; MacCoss, M.J.; Shaffer, S.A. Comparison of data acquisition strategies on quadrupole ion trap instrumentation for shotgun proteomics. J. Am. Soc. Mass Spectrom. 2014, 25, 2048–2059. [Google Scholar] [CrossRef] [PubMed]
- Herold, D.A.; Boyd, J.C.; Bruns, D.E.; Emerson, J.C.; Burns, K.G.; Bray, R.E.; Vandenhoff, G.E.; Freedlender, A.E.; Fortier, G.A.; Pohl, S.L. Measurement of glycosylated hemoglobins using boronate affinity chromatography. Ann. Clin. Lab. Sci. 1983, 13, 482–488. [Google Scholar] [PubMed]
- Frantzen, F.; Grimsrud, K.; Heggli, D.E.; Faaren, A.L.; Løvli, T.; Sundrehagen, E. Glycohemoglobin filter assay for doctors’ offices based on boronic acid affinity principle. Clin. Chem. 1997, 43, 2390–2396. [Google Scholar] [PubMed]
- Pepaj, M.; Thorsby, P.M. Analysis of glycated albumin by on-line two-dimensional liquid chromatography mass spectrometry. J. Liq. Chromatogr. Relat. Technol. 2015, 38, 20–28. [Google Scholar] [CrossRef]
- Mitsuhashi, T.; Li, Y.M.; Fishbane, S.; Vlassara, H. Depletion of reactive advanced glycation endproducts from diabetic uremic sera using a lysozyme-linked matrix. J. Clin. Investig. 1997, 100, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Degani, G.; Altomare, A.A.; Colzani, M.; Martino, C.; Mazzolari, A.; Fritz, G.; Vistoli, G.; Popolo, L.; Aldini, G. A capture method based on the VC1 domain reveals new binding properties of the human receptor for advanced glycation end products (RAGE). Redox Biol. 2017, 11, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, R.R.; Venkatraman, K.; Vijayalakshmi, M.A. Pseudoaffinity chromatography enrichment of glycated peptides for monitoring advanced glycation end products (AGEs) in metabolic disorders. J. Proteins Proteom. 2016, 7, 167–176. [Google Scholar]
- Bollineni, R.C.; Hoffmann, R.; Fedorova, M. Identification of protein carbonylation sites by two-dimensional liquid chromatography in combination with MALDI- and ESI-MS. J. Proteom. 2011, 74, 2338–2350. [Google Scholar] [CrossRef] [PubMed]
- Pernemalm, M.; Lehtiö, J. A novel prefractionation method combining protein and peptide isoelectric focusing in immobilized pH gradient strips. J. Proteome Res. 2013, 12, 1014–1019. [Google Scholar] [CrossRef] [PubMed]
- Greifenhagen, U.; Frolov, A.; Blüher, M.; Hoffmann, R. Site-specific analysis of advanced glycation end products in plasma proteins of type 2 diabetes mellitus patients. Anal. Bioanal. Chem. 2016, 408, 5557–5566. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.; Yi, E.C. Use of gas-phase fractionation to increase protein identifications: Application to the peroxisome. Methods Mol. Biol. 2008, 432, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Hu, A.; Noble, W.S.; Wolf-Yadlin, A. Technical advances in proteomics: New developments in data-independent acquisition. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.-J.; Cho, K.; Kim, J.-Y.; Kim, Y.-H.; Oh, H.-B. Protein analysis using a combination of an online monolithic trypsin immobilized enzyme reactor and collisionally-activated dissociation/electron transfer dissociation dual tandem mass spectrometry. Bull. Korean Chem. Soc. 2012, 33, 3233–3240. [Google Scholar] [CrossRef]
- Gadgil, H.S.; Bondarenko, P.V.; Treuheit, M.J.; Ren, D. Screening and sequencing of glycated proteins by neutral loss scan LC/MS/MS method. Anal. Chem. 2007, 79, 5991–5999. [Google Scholar] [CrossRef] [PubMed]
- Frolov, A.; Hoffmann, P.; Hoffmann, R. Fragmentation behavior of glycated peptides derived from d-glucose, d-fructose and d-ribose in tandem mass spectrometry. J. Mass Spectrom. 2006, 41, 1459–1469. [Google Scholar] [CrossRef] [PubMed]
- Milkovska-Stamenova, S.; Hoffmann, R. Hexose-derived glycation sites in processed bovine milk. J. Proteom. 2016, 134, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Kijewska, M.; Kuc, A.; Kluczyk, A.; Waliczek, M.; Man-Kupisinska, A.; Lukasiewicz, J.; Stefanowicz, P.; Szewczuk, Z. Selective detection of carbohydrates and their peptide conjugates by ESI-MS using synthetic quaternary ammonium salt derivatives of phenylboronic acids. J. Am. Soc. Mass Spectrom. 2014, 25, 966–976. [Google Scholar] [CrossRef] [PubMed]
- Greifenhagen, U.; Nguyen, V.D.; Moschner, J.; Giannis, A.; Frolov, A.; Hoffmann, R. Sensitive and site-specific identification of carboxymethylated and carboxyethylated peptides in tryptic digests of proteins and human plasma. J. Proteome Res. 2015, 14, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Böhme, D.; Singer, D.; Frolov, A. Specific tandem mass spectrometric detection of AGE-modified arginine residues in peptides. J. Mass Spectrom. 2015, 50, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Petyuk, V.A.; Schepmoes, A.A.; Orton, D.J.; Monroe, M.E.; Yang, F.; Smith, R.D.; Metz, T.O. Analysis of non-enzymatically glycated peptides: Neutral-loss-triggered MS(3) versus multi-stage activation tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2008, 22, 3027–3034. [Google Scholar] [CrossRef] [PubMed]
- Stefanowicz, P.; Kijewska, M.; Szewczuk, Z. Does electron capture dissociation (ECD) provide quantitative information on the chemical modification of lysine side chains in proteins? The glycation of ubiquitin. Anal. Chem. 2014, 86, 7247–7251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Tang, N.; Brock, J.W.C.; Mottaz, H.M.; Ames, J.M.; Baynes, J.W.; Smith, R.D.; Metz, T.O. Enrichment and analysis of nonenzymatically glycated peptides: Boronate affinity chromatography coupled with electron-transfer dissociation mass spectrometry. J. Proteome Res. 2007, 6, 2323–2330. [Google Scholar] [CrossRef] [PubMed]
- Swaney, D.L.; McAlister, G.C.; Wirtala, M.; Schwartz, J.C.; Syka, J.E.P.; Coon, J.J. Supplemental activation method for high-efficiency electron-transfer dissociation of doubly protonated peptide precursors. Anal. Chem. 2007, 79, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Barnaby, O.S.; Cerny, R.L.; Clarke, W.; Hage, D.S. Quantitative analysis of glycation patterns in human serum albumin using 16O/18O-labeling and MALDI-TOF MS. Clin. Chim. Acta Int. J. Clin. Chem. 2011, 412, 1606–1615. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Xu, W.; Deng, Y. A new strategy for early diagnosis of type 2 diabetes by standard-free, label-free LC-MS/MS quantification of glycated peptides. Diabetes 2013, 62, 3936–3942. [Google Scholar] [CrossRef] [PubMed]
- Kielmas, M.; Kijewska, M.; Kluczyk, A.; Oficjalska, J.; Gołębiewska, B.; Stefanowicz, P.; Szewczuk, Z. Comparison of modification sites in glycated crystallin in vitro and in vivo. Anal. Bioanal. Chem. 2015, 407, 2557–2567. [Google Scholar] [CrossRef] [PubMed]
- Glomb, M.A.; Lang, G. Isolation and characterization of glyoxal-arginine modifications. J. Agric. Food Chem. 2001, 49, 1493–1501. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbolz, U.; Henle, T.; Haeßner, R.; Klostermeyer, H. On the reaction of glyoxal with proteins. Z. Für Lebensm. Forsch. A 1997, 205, 121–124. [Google Scholar] [CrossRef]
- Henle, T.; Walter, A.W.; Haessner, R.; Klostermeyer, H. Isolation to AGEs formed endogenously. Corresponding studies, and identification of a protein-bound imidazolone resulting from the reaction of arginine residues and methylglyoxal. Z. Für Lebensm. Unters. Forsch. 1997, 204, 95–98. [Google Scholar] [CrossRef]
- Hellwig, M.; Henle, T. Formyline, a new glycation compound from the reaction of lysine and 3-deoxypentosone. Eur. Food Res. Technol. 2010, 230, 903–914. [Google Scholar] [CrossRef]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J. Am. Diet. Assoc. 2010, 110, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J.; Battah, S.; Ahmed, N.; Karachalias, N.; Agalou, S.; Babaei-Jadidi, R.; Dawnay, A. Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem. J. 2003, 375, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liang, L.; Liu, X.; Labuza, T.P.; Zhou, P. Effect of fructose and glucose on glycation of β-lactoglobulin in an intermediate-moisture food model system: Analysis by liquid chromatography–mass spectrometry (LC–MS) and data-independent acquisition LC–MS (LC–MSE). J. Agric. Food Chem. 2012, 60, 10674–10682. [Google Scholar] [CrossRef] [PubMed]
- Korwar, A.M.; Vannuruswamy, G.; Jagadeeshaprasad, M.G.; Jayaramaiah, R.H.; Bhat, S.; Regin, B.S.; Ramaswamy, S.; Giri, A.P.; Mohan, V.; Balasubramanyam, M.; et al. Development of diagnostic fragment ion library for glycated peptides of human serum albumin: Targeted quantification in prediabetic, diabetic, and microalbuminuria plasma by parallel reaction monitoring, SWATH, and MSE. Mol. Cell. Proteom. 2015, 14, 2150–2159. [Google Scholar] [CrossRef] [PubMed]
- Baraka-Vidot, J.; Planesse, C.; Meilhac, O.; Militello, V.; van den Elsen, J.; Bourdon, E.; Rondeau, P. Glycation alters ligand binding, enzymatic, and pharmacological properties of human albumin. Biochemistry 2015, 54, 3051–3062. [Google Scholar] [CrossRef] [PubMed]
- Klöpfer, A.; Spanneberg, R.; Glomb, M.A. Formation of arginine modifications in a model system of Nα-tert-butoxycarbonyl (Boc)-arginine with methylglyoxal. J. Agric. Food Chem. 2011, 59, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.U.; Thorpe, S.R.; Baynes, J.W. Identification of Nε-carboxymethyllysine as a degradation product of fructoselysine in glycated protein. J. Biol. Chem. 1986, 261, 4889–4894. [Google Scholar] [PubMed]
- Modzel, M.; Płóciennik, H.; Kielmas, M.; Szewczuk, Z.; Stefanowicz, P. A synthesis of new, bi-labeled peptides for quantitative proteomics. J. Proteom. 2015, 115, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.R.; Thornalley, P.J. Influence of pH and phosphate ions on the kinetics of enolisation and degradation of fructosamines. Studies with the model fructosamine, Nε-1-deoxy-d-fructos-1-yl-hippuryl-lysine. Biochem. Int. 1992, 28, 429–439. [Google Scholar] [PubMed]
- Smith, P.R.; Thornalley, P.J. Mechanism of the degradation of non-enzymatically glycated proteins under physiological conditions. Studies with the model fructosamine, Nε-(1-deoxy-d-fructos-1-yl)hippuryl-lysine. Eur. J. Biochem. 1992, 210, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Krause, R.; Kühn, J.; Penndorf, I.; Knoll, K.; Henle, T. N-Terminal pyrazinones: A new class of peptide-bound advanced glycation end-products. Amino Acids 2004, 27, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Mittelmaier, S.; Pischetsrieder, M. Multistep ultrahigh performance liquid chromatography/tandem mass spectrometry analysis for untargeted quantification of glycating activity and identification of most relevant glycation products. Anal. Chem. 2011, 83, 9660–9668. [Google Scholar] [CrossRef] [PubMed]
- Greifenhagen, U.; Frolov, A.; Hoffmann, R. Oxidative degradation of Nε-fructosylamine-substituted peptides in heated aqueous systems. Amino Acids 2015, 47, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Frolov, A.; Schmidt, R.; Spiller, S.; Greifenhagen, U.; Hoffmann, R. Arginine-derived advanced glycation end products generated in peptide-glucose mixtures during boiling. J. Agric. Food Chem. 2014, 62, 3626–3635. [Google Scholar] [CrossRef] [PubMed]
- Jakas, A.; Horvat, S. Study of degradation pathways of Amadori compounds obtained by glycation of opioid pentapeptide and related smaller fragments: Stability, reactions, and spectroscopic properties. Biopolymers 2003, 69, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Shakkottai, V.G.; Sudha, R.; Balaram, P. Gramicidin S: A peptide model for protein glycation and reversal of glycation using nucleophilic amines. J. Pept. Res. 2002, 60, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Sasaki, T.; Niwa, S.; Oishi, T.; Murata, M.; Kawakami, T.; Aimoto, S. Intact glycation end products containing carboxymethyl-lysine and glyoxal lysine dimer obtained from synthetic collagen model peptide. Bioorg. Med. Chem. Lett. 2004, 14, 5677–5680. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, A.; Matsui, K.; Konoki, K.; Matsumori, N.; Murata, M.; Kawakami, T.; Aimoto, S. Lysine proximity significantly affects glycation of lysine-containing collagen model peptides. Bioorg. Med. Chem. 2011, 19, 2125–2129. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Li, L.; Qi, H.; Zhang, Z.X.X.; Li, B. Kinetic study on peptide-bound pyrraline formation and elimination in the Maillard reaction using single- and multiple-response models. J. Food Sci. 2016, 81, C2405–C2424. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Li, L.; Qi, H.; Wan, L.; Cai, P.; Xu, Z.; Li, B. Formation of peptide bound pyrraline in the Maillard model systems with different Lys-containing dipeptides and tripeptides. Molecules 2016, 21, 463. [Google Scholar] [CrossRef] [PubMed]
- Kapurniotu, A.; Bernhagen, J.; Greenfield, N.; Al-Abed, Y.; Teichberg, S.; Frank, R.W.; Voelter, W.; Bucala, R. Contribution of advanced glycosylation to the amyloidogenicity of islet amyloid polypeptide. Eur. J. Biochem. 1998, 251, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Schalkwijk, C.G.; Posthuma, N.; ten Brink, H.J.; ter Wee, P.M.; Teerlink, T. Induction of 1,2-dicarbonyl compounds, intermediates in the formation of advanced glycation end-products, during heat-sterilization of glucose-based peritoneal dialysis fluids. Perit. Dial. Int. J. Int. Soc. Perit. Dial. 1999, 19, 325–333. [Google Scholar]
- Gensberger-Reigl, S.; Huppert, J.; Pischetsrieder, M. Quantification of reactive carbonyl compounds in icodextrin-based peritoneal dialysis fluids by combined UHPLC-DAD and -MS/MS detection. J. Pharm. Biomed. Anal. 2016, 118, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Horvat, S.; Jakas, A. Peptide and amino acid glycation: New insights into the Maillard reaction. J. Pept. Sci. 2004, 10, 119–137. [Google Scholar] [CrossRef] [PubMed]
- Vinale, F.; Fogliano, V.; Schieberle, P.; Hofmann, T. Development of a stable isotope dilution assay for an accurate quantification of protein-bound Nε-(1-deoxy-d-fructos-1-yl)-l-lysine using a 13C-labeled internal standard. J. Agric. Food Chem. 1999, 47, 5084–5092. [Google Scholar] [CrossRef] [PubMed]
- Stefanowicz, P.; Kijewska, M.; Kapczyńska, K.; Szewczuk, Z. Methods of the site-selective solid phase synthesis of peptide-derived Amadori products. Amino Acids 2010, 38, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Frolov, A.; Singer, D.; Hoffmann, R. Site-specific synthesis of Amadori-modified peptides on solid phase. J. Pept. Sci. 2006, 12, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Frolov, A.; Singer, D.; Hoffmann, R. Solid-phase synthesis of glucose-derived Amadori peptides. J. Pept. Sci. 2007, 13, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Stefanowicz, P.; Kapczyńska, K.; Kluczyk, A.; Szewczuk, Z. A new procedure for the synthesis of peptide-derived Amadori products on a solid support. Tetrahedron Lett. 2007, 48, 967–969. [Google Scholar] [CrossRef]
- Frolov, A.; Hoffmann, R. Separation of Amadori peptides from their unmodified analogs by ion-pairing RP-HPLC with heptafluorobutyric acid as ion-pair reagent. Anal. Bioanal. Chem. 2008, 392, 1209–1214. [Google Scholar] [CrossRef] [PubMed]
- Gruber, P.; Hofmann, T. Chemoselective synthesis of peptides containing major advanced glycation end-products of lysine and arginine. J. Pept. Res. 2005, 66, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Iberg, N.; Flückiger, R. Nonenzymatic glycosylation of albumin in vivo. Identification of multiple glycosylated sites. J. Biol. Chem. 1986, 261, 13542–13545. [Google Scholar] [PubMed]
- Howard, M.J.; Smales, C.M. NMR analysis of synthetic human serum albumin α-helix 28 identifies structural distortion upon amadori modification. J. Biol. Chem. 2005, 280, 22582–22589. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-H.; Yen, G.-C. Inhibitory effect of naturally occurring flavonoids on the formation of advanced glycation endproducts. J. Agric. Food Chem. 2005, 53, 3167–3173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Frolov, A.; Tang, N.; Hoffmann, R.; van de Goor, T.; Metz, T.O.; Smith, R.D. Application of electron transfer dissociation mass spectrometry in analyses of non-enzymatically glycated peptides. Rapid Commun. Mass Spectrom. 2007, 21, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Yaylayan, V.; Sporns, P. Diagnostic ion series for the identification of Amadori rearrangement products by MS techniques based on electron-impact ionization. J. Agric. Food Chem. 1989, 37, 978–981. [Google Scholar] [CrossRef]
- Frolov, A.; Hoffmann, R. Identification and relative quantification of specific glycation sites in human serum albumin. Anal. Bioanal. Chem. 2010, 397, 2349–2356. [Google Scholar] [CrossRef] [PubMed]
- Milkovska-Stamenova, S.; Hoffmann, R. Identification and quantification of bovine protein lactosylation sites in different milk products. J. Proteom. 2016, 134, 112–126. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, M.; Frolov, A.; Hoffmann, R. Fragmentation behavior of Amadori-peptides obtained by non-enzymatic glycosylation of lysine residues with ADP-ribose in tandem mass spectrometry. J. Mass Spectrom. 2010, 45, 664–669. [Google Scholar] [CrossRef] [PubMed]
- Ruan, E.D.; Wang, H.; Ruan, Y.; Juárez, M. Study of fragmentation behavior of amadori rearrangement products in lysine-containing peptide model by tandem mass spectrometry. Eur. J. Mass Spectrom. Chichester Engl. 2013, 19, 295–303. [Google Scholar] [CrossRef]
- Stefanowicz, P.; Kapczynska, K.; Jaremko, M.; Jaremko, Ł.; Szewczuk, Z. A mechanistic study on the fragmentation of peptide-derived Amadori products. J. Mass Spectrom. 2009, 44, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Stefanowicz, P.; Kijewska, M.; Szewczuk, Z. Sequencing of peptide-derived Amadori products by the electron capture dissociation method. J. Mass Spectrom. 2009, 44, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Gehrig, P.M.; Hunziker, P.E.; Zahariev, S.; Pongor, S. Fragmentation pathways of NG-methylated and unmodified arginine residues in peptides studied by ESI-MS/MS and MALDI-MS. J. Am. Soc. Mass Spectrom. 2004, 15, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Suh, M.-S.; Thangadurai, T.D.; Kim, J.; Rhee, Y.H.; Yoon, H.-J.; Shin, S.K. Mass-balanced 1H/2H isotope dipeptide tag for simultaneous protein quantitation and identification. Anal. Chem. 2008, 80, 6145–6153. [Google Scholar] [CrossRef] [PubMed]
- Snow, L.M.; Fugere, N.A.; Thompson, L.V. Advanced glycation end-product accumulation and associated protein modification in type II skeletal muscle with aging. J. Gerontol. A. Biol. Sci. Med. Sci. 2007, 62, 1204–1210. [Google Scholar] [CrossRef] [PubMed]
- Rahbar, S.; Blumenfeld, O.; Ranney, H.M. Studies of an unusual hemoglobin in patients with diabetes mellitus. Biochem. Biophys. Res. Commun. 1969, 36, 838–843. [Google Scholar] [CrossRef]
- Hellwig, M.; Henle, T. Baking, ageing, diabetes: A short history of the Maillard reaction. Angew. Chem. Int. Ed. Engl. 2014, 53, 10316–10329. [Google Scholar] [CrossRef] [PubMed]
- Bozkaya, G.; Ozgu, E.; Karaca, B. The association between estimated average glucose levels and fasting plasma glucose levels. Clinics 2010, 65, 1077–1080. [Google Scholar] [CrossRef] [PubMed]
- Neelofar; Ahmad, J.; Alam, K. Impact of in vitro non-enzymatic glycation on biophysical and biochemical regimes of human serum albumin: Relevance in diabetes associated complications. RSC Adv. 2015, 5, 63605–63614. [Google Scholar] [CrossRef]
- Ikeda, K.; Sakamoto, Y.; Kawasaki, Y.; Miyake, T.; Tanaka, K.; Urata, T.; Katayama, Y.; Ueda, S.; Horiuchi, S. Determination of glycated albumin by enzyme-linked boronate immunoassay (ELBIA). Clin. Chem. 1998, 44, 256–263. [Google Scholar] [PubMed]
- Ohe, Y.; Matsuura, M.; Nakajima, Y.; Shin, S.; Hashimoto, F.; Hirota, M.; Shima, K. Radioimmunoassay of glycosylated albumin with monoclonal antibody to glucitol-lysine. Clin. Chim. Acta Int. J. Clin. Chem. 1987, 169, 229–238. [Google Scholar] [CrossRef]
- Barnaby, O.S.; Cerny, R.L.; Clarke, W.; Hage, D.S. Comparison of modification sites formed on human serum albumin at various stages of glycation. Clin. Chim. Acta Int. J. Clin. Chem. 2011, 412, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Saenz-Suarez, H.; Poutou-Pinales, R.A.; Gonzalez-Santos, J.; Barreto, G.E.; Rieto-Navarrera, L.P.; Saenz-Moreno, J.A.; Landazuri, P.; Barrera-Avellanda, L.A. Prediction of glycation sites: New insights from protein structural analysis. Turk. J. Biol. 2016, 40, 12–25. [Google Scholar] [CrossRef]
- Demirag, B.; Sarisozen, B.; Durak, K.; Bilgen, O.F.; Ozturk, C. The effect of α-2 macroglobulin on the healing of ruptured anterior cruciate ligament in rabbits. Connect. Tissue Res. 2004, 45, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, C.B.; Ruddy, S.; Shehadeh, I.H.; Müller-Eberhard, H.J.; Merrill, J.P.; Austen, K.F. Complement metabolism in man: Hypercatabolism of the fourth (C4) and third (C3) components in patients with renal allograft rejection and hereditary angioedema (HAE). J. Clin. Investig. 1969, 48, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, S.J.; Stolzenberg-Solomon, R.Z.; Kopp, W.; Rager, H.; Virtamo, J.; Albanes, D. Impact of circulating vitamin D binding protein levels on the association between 25-hydroxyvitamin D and pancreatic cancer risk: A nested case-control study. Cancer Res. 2012, 72, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, S.; Conti, A.; Iannaccone, S.; Cannistraci, C.V.; Campanella, A.; Barbariga, M.; Codazzi, F.; Pelizzoni, I.; Magnani, G.; Pesca, M.; et al. Ceruloplasmin oxidation, a feature of Parkinson’s disease CSF, inhibits ferroxidase activity and promotes cellular iron retention. J. Neurosci. 2011, 31, 18568–18577. [Google Scholar] [CrossRef] [PubMed]
- Nanjee, M.N.; Crouse, J.R.; King, J.M.; Hovorka, R.; Rees, S.E.; Carson, E.R.; Morgenthaler, J.-J.; Lerch, P.; Miller, N.E. Effects of intravenous infusion of lipid-free Apo A-I in humans. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.C.-M. Structure and function of transferrin. Biochem. Educ. 1984, 12, 146–154. [Google Scholar] [CrossRef]
- Sand, K.M.K.; Bern, M.; Nilsen, J.; Noordzij, H.T.; Sandlie, I.; Andersen, J.T. Unraveling the interaction between FcRn and albumin: Opportunities for design of albumin-based therapeutics. Front. Immunol. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, I.B.; Brownlee, M. Should minimal blood glucose variability become the gold standard of glycemic control? J. Diabetes Complicat. 2005, 19, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-E. Alternative biomarkers for assessing glycemic control in diabetes: Fructosamine, glycated albumin, and 1,5-anhydroglucitol. Ann. Pediatr. Endocrinol. Metab. 2015, 20, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Spiller, S.; Li, Y.; Blüher, M.; Welch, L.; Hoffmann, R. Glycated lysine-141 in haptoglobin improves the diagnostic accuracy for type 2 diabetes mellitus in combination with glycated hemoglobin HbA1c and fasting plasma glucose. Clin. Proteom. 2017, 14. [Google Scholar] [CrossRef] [PubMed]
- Wondrak, G.T.; Cervantes-Laurean, D.; Roberts, M.J.; Qasem, J.G.; Kim, M.; Jacobson, E.L.; Jacobson, M.K. Identification of α-dicarbonyl scavengers for cellular protection against carbonyl stress. Biochem. Pharmacol. 2002, 63, 361–373. [Google Scholar] [CrossRef]
- McLellan, A.C.; Thornalley, P.J.; Benn, J.; Sonksen, P.H. Glyoxalase system in clinical diabetes mellitus and correlation with diabetic complications. Clin. Sci. 1994, 87, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Dicarbonyls (glyoxal, methylglyoxal, and 3-deoxyglucosone). In Uremic Toxins; Niwa, T., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 177–192. ISBN 978-1-118-42403-2. [Google Scholar]
- Ahmed, N.; Thornalley, P.J.; Dawczynski, J.; Franke, S.; Strobel, J.; Stein, G.; Haik, G.M. Methylglyoxal-derived hydroimidazolone advanced glycation end-products of human lens proteins. Investig. Ophthalmol. Vis. Sci. 2003, 44, 5287–5292. [Google Scholar] [CrossRef]
- Xue, M.; Rabbani, N.; Thornalley, P.J. Glyoxalase in ageing. Semin. Cell Dev. Biol. 2011, 22, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Glyoxalase in diabetes, obesity and related disorders. Semin. Cell Dev. Biol. 2011, 22, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Jack, M.; Wright, D. Role of advanced glycation endproducts and glyoxalase I in diabetic peripheral sensory neuropathy. Transl. Res. J. Lab. Clin. Med. 2012, 159, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Thornalley, P.J.; Giardino, I.; Beisswenger, P.; Thorpe, S.R.; Onorato, J.; Brownlee, M. Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. J. Clin. Investig. 1998, 101, 1142–1147. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Methylglyoxal, glyoxalase 1 and the dicarbonyl proteome. Amino Acids 2012, 42, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Kaur, C.; Ghosh, A.; Pareek, A.; Sopory, S.K.; Singla-Pareek, S.L. Glyoxalases and stress tolerance in plants. Biochem. Soc. Trans. 2014, 42, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Auburger, G.; Kurz, A. The role of glyoxalases for sugar stress and aging, with relevance for dyskinesia, anxiety, dementia and Parkinson’s disease. Aging 2011, 3, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. The dicarbonyl proteome: Proteins susceptible to dicarbonyl glycation at functional sites in health, aging, and disease. Ann. N. Y. Acad. Sci. 2008, 1126, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Dicarbonyl proteome and genome damage in metabolic and vascular disease. Biochem. Soc. Trans. 2014, 42, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Kaur, C.; Singla-Pareek, S.L.; Sopory, S.K. Glyoxalase and methylglyoxal as biomarkers for plant stress tolerance. Crit. Rev. Plant Sci. 2014, 33, 429–456. [Google Scholar] [CrossRef]
- Sobhanian, H.; Motamed, N.; Jazii, F.R.; Nakamura, T.; Komatsu, S. Salt stress induced differential proteome and metabolome response in the shoots of Aeluropus lagopoides (Poaceae), a halophyte C(4) plant. J. Proteome Res. 2010, 9, 2882–2897. [Google Scholar] [CrossRef] [PubMed]
- Zang, X.; Komatsu, S. A proteomics approach for identifying osmotic-stress-related proteins in rice. Phytochemistry 2007, 68, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Castillejo, Á.M.; Maldonado, A.M.; Ogueta, S.; Jorrín, J.V. Proteomic analysis of responses to drought stress in sunflower (Helianthus annuus) leaves by 2DE gel electrophoresis and mass spectrometry. Open Proteom. J. 2008, 1, 59–71. [Google Scholar] [CrossRef]
- Akashi, K.; Yoshida, K.; Kuwano, M.; Kajikawa, M.; Yoshimura, K.; Hoshiyasu, S.; Inagaki, N.; Yokota, A. Dynamic changes in the leaf proteome of a C3 xerophyte, Citrullus lanatus (wild watermelon), in response to water deficit. Planta 2011, 233, 947–960. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, N.; Lee, D.-G.; Lee, S.-H.; Kang, K.Y.; Lee, J.J.; Kim, P.J.; Yoon, H.-S.; Kim, J.-S.; Lee, B.-H. Excess copper induced physiological and proteomic changes in germinating rice seeds. Chemosphere 2007, 67, 1182–1193. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Cui, J.; Zhang, H.; Wang, G.; Zhao, F.-J.; Shen, Z. Proteomic analysis of copper stress responses in the roots of two rice (Oryza sativa L.) varieties differing in Cu tolerance. Plant Soil 2013, 366, 647–658. [Google Scholar] [CrossRef]
- Ahsan, N.; Lee, S.-H.; Lee, D.-G.; Lee, H.; Lee, S.W.; Bahk, J.D.; Lee, B.-H. Physiological and protein profiles alternation of germinating rice seedlings exposed to acute cadmium toxicity. C. R. Biol. 2007, 330, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Hasanuzzaman, M.; Hossain, M.A.; Fujita, M. Exogenous selenium pretreatment protects rapeseed seedlings from cadmium-induced oxidative stress by upregulating antioxidant defense and methylglyoxal detoxification systems. Biol. Trace Elem. Res. 2012, 149, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H. Advanced glycation end-products and atherosclerosis. Ann. Med. 1996, 28, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H. Protein glycation in the kidney: Role in diabetes and aging. Kidney Int. 1996, 49, 1795–1804. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H.; Bucala, R. Recent progress in advanced glycation and diabetic vascular disease: Role of advanced glycation end product receptors. Diabetes 1996, 45, S65–S66. [Google Scholar] [CrossRef] [PubMed]
- Sebeková, K.; Krajcoviová-Kudlácková, M.; Schinzel, R.; Faist, V.; Klvanová, J.; Heidland, A. Plasma levels of advanced glycation end products in healthy, long-term vegetarians and subjects on a western mixed diet. Eur. J. Nutr. 2001, 40, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Huang, G.; Xiao, L.; Mitchell, A.E. Determination of advanced glycation endproducts by LC-MS/MS in raw and roasted almonds (Prunus dulcis). J. Agric. Food Chem. 2011, 59, 12037–12046. [Google Scholar] [CrossRef] [PubMed]
- Bechtold, U.; Rabbani, N.; Mullineaux, P.M.; Thornalley, P.J. Quantitative measurement of specific biomarkers for protein oxidation, nitration and glycation in Arabidopsis leaves. Plant J. Cell Mol. Biol. 2009, 59, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Luevano-Contreras, C.; Chapman-Novakofski, K. Dietary advanced glycation end products and aging. Nutrients 2010, 2, 1247–1265. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Yamamoto, H.; Makino, A.; Sugimoto, T.; Miyake, C. Methylglyoxal functions as Hill oxidant and stimulates the photoreduction of O2 at photosystem I: A symptom of plant diabetes. Plant Cell Environ. 2011, 34, 1454–1464. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Shimakawa, G.; Nishi, A.; Iwamoto, T.; Sakamoto, K.; Yamamoto, H.; Amako, K.; Makino, A.; Miyake, C. Functional analysis of the AKR4C subfamily of Arabidopsis thaliana: Model structures, substrate specificity, acrolein toxicity, and responses to light and [CO2]. Biosci. Biotechnol. Biochem. 2013, 77, 2038–2045. [Google Scholar] [CrossRef] [PubMed]
- Takagi, D.; Inoue, H.; Odawara, M.; Shimakawa, G.; Miyake, C. The Calvin cycle inevitably produces sugar-derived reactive carbonyl methylglyoxal during photosynthesis: A potential cause of plant diabetes. Plant Cell Physiol. 2014, 55, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, B.C.; Oelmüller, R. Reactive oxygen species generation and signaling in plants. Plant Signal. Behav. 2012, 7, 1621–1633. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Chan, Z. ROS regulation during abiotic stress responses in crop plants. Front. Plant Sci. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Slesak, I.; Libik, M.; Karpinska, B.; Karpinski, S.; Miszalski, Z. The role of hydrogen peroxide in regulation of plant metabolism and cellular signalling in response to environmental stresses. Acta Biochim. Pol. 2007, 54, 39–50. [Google Scholar] [PubMed]
- Mirzaee, M.; Moieni, A.; Ghanati, F. Effects of drought stress on the lipid peroxidation and antioxidant enzyme activities in two canola (Brassica napus L.) cultivars. J. Agric. Sci. Technol. 2013, 15, 593–602. [Google Scholar]
- Rabbani, N.; Thornalley, P.J. Dicarbonyl stress in cell and tissue dysfunction contributing to ageing and disease. Biochem. Biophys. Res. Commun. 2015, 458, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.A.; Wani, S.H.; Bhattacharjee, S.; Burritt, D.J.; Tran, L.-S.P. MG-mediated disruption in cellular functioning. In Drought Stress Tolerance in Plants, Vol 1: Physiology and Biochemistry; Mohammad Anwar, H., Wani, S.H., Bhattacharjee, S., Burritt, D.J., Tran, L.-S.P., Eds.; Springer: Berlin, Germany, 2016; Volume 1, p. 387. ISBN 978-3-319-28899-4. [Google Scholar]
- Shipanova, I.N.; Glomb, M.A.; Nagaraj, R.H. Protein modification by methylglyoxal: Chemical nature and synthetic mechanism of a major fluorescent adduct. Arch. Biochem. Biophys. 1997, 344, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Oya, T.; Hattori, N.; Mizuno, Y.; Miyata, S.; Maeda, S.; Osawa, T.; Uchida, K. Methylglyoxal modification of protein. Chemical and immunochemical characterization of methylglyoxal-arginine adducts. J. Biol. Chem. 1999, 274, 18492–18502. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Shimohira, K.; Watanabe, H.; Hayase, F. Detection and determination of glyceraldehyde-derived pyridinium-type advanced glycation end product in streptozotocin-induced diabetic rats. Biosci. Biotechnol. Biochem. 2007, 71, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.U.; Brinkmann Frye, E.; Degenhardt, T.P.; Thorpe, S.R.; Baynes, J.W. Nε-(carboxyethyl)lysine, a product of the chemical modification of proteins by methylglyoxal, increases with age in human lens proteins. Biochem. J. 1997, 324(pt 8), 565–570. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Yang, Y.; Yuk, I.; Pai, R.; McKay, P.; Eigenbrot, C.; Dennis, M.; Katta, V.; Francissen, K.C. Unveiling a glycation hot spot in a recombinant humanized monoclonal antibody. Anal. Chem. 2008, 80, 2379–2390. [Google Scholar] [CrossRef] [PubMed]
- Ottum, M.S.; Mistry, A.M. Advanced glycation end-products: Modifiable environmental factors profoundly mediate insulin resistance. J. Clin. Biochem. Nutr. 2015, 57, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mildner-Szkudlarz, S.; Siger, A.; Szwengiel, A.; Przygoński, K.; Wojtowicz, E.; Zawirska-Wojtasiak, R. Phenolic compounds reduce formation of Nε-(carboxymethyl)lysine and pyrazines formed by Maillard reactions in a model bread system. Food Chem. 2017, 231, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Hoque, T.S.; Uraji, M.; Hoque, M.A.; Nakamura, Y.; Murata, Y. Methylglyoxal induces inhibition of growth, accumulation of anthocyanin, and activation of glyoxalase I and II in Arabidopsis thaliana. J. Biochem. Mol. Toxicol. 2017, 31. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
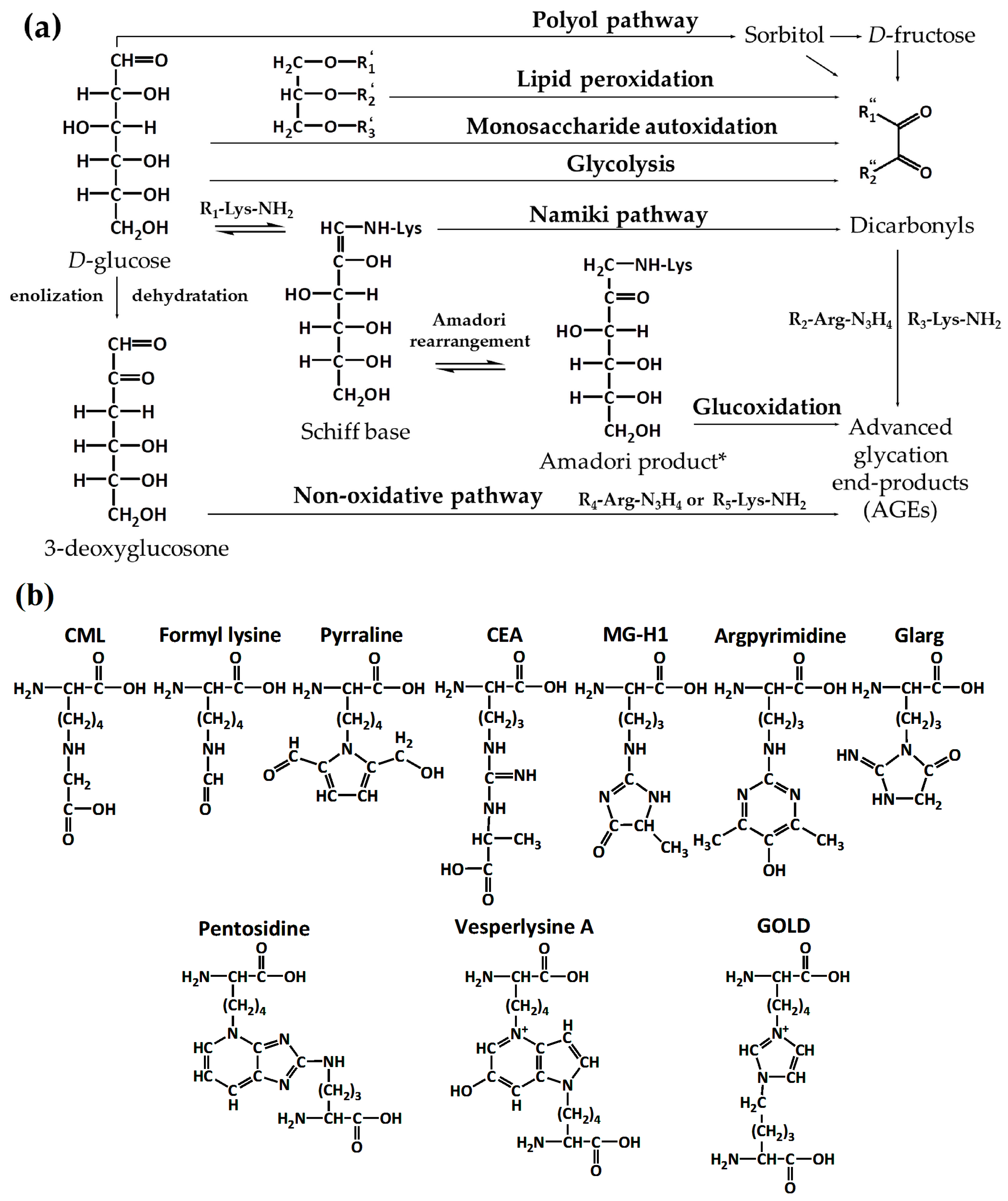{kind=link}
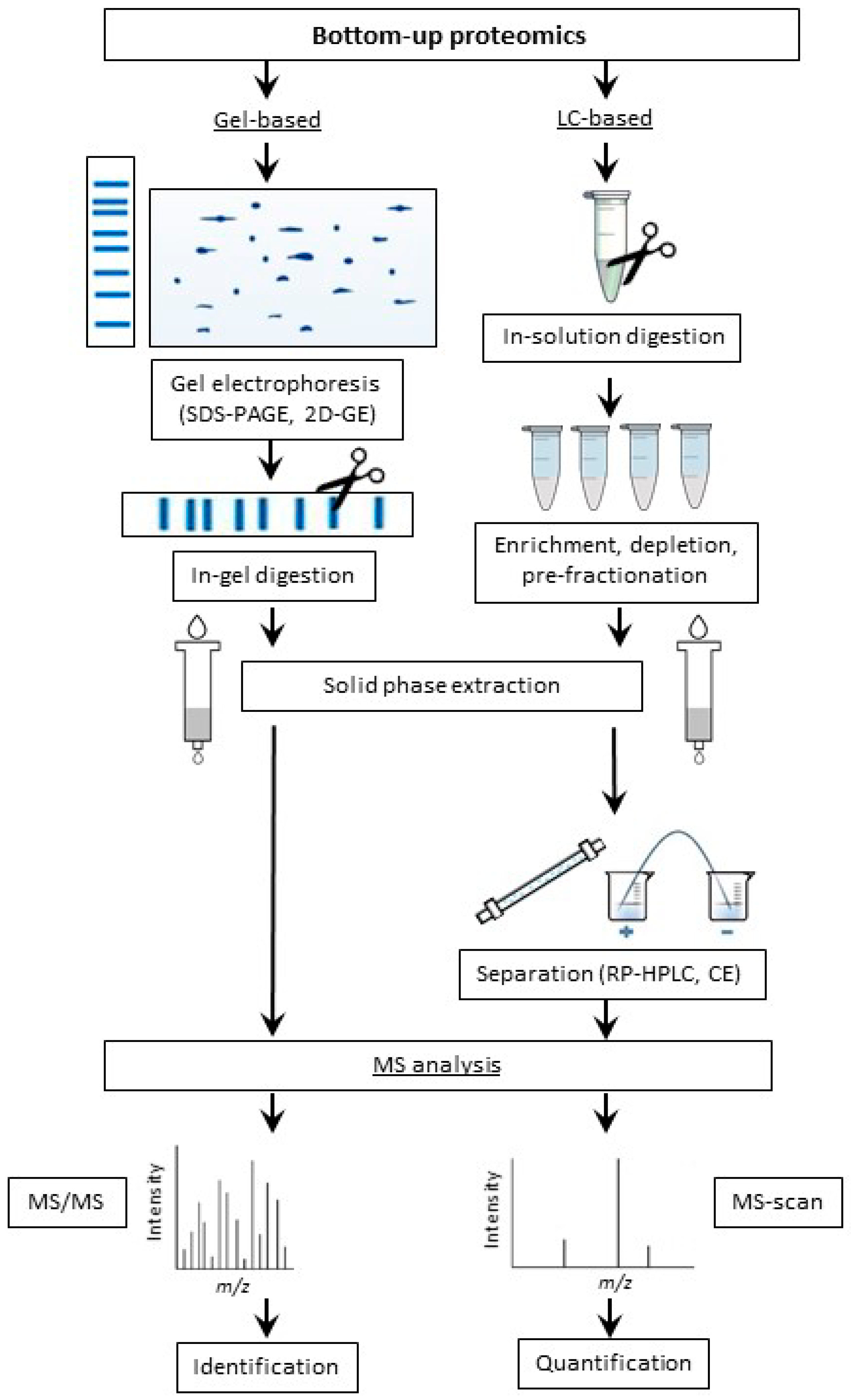{kind=link}
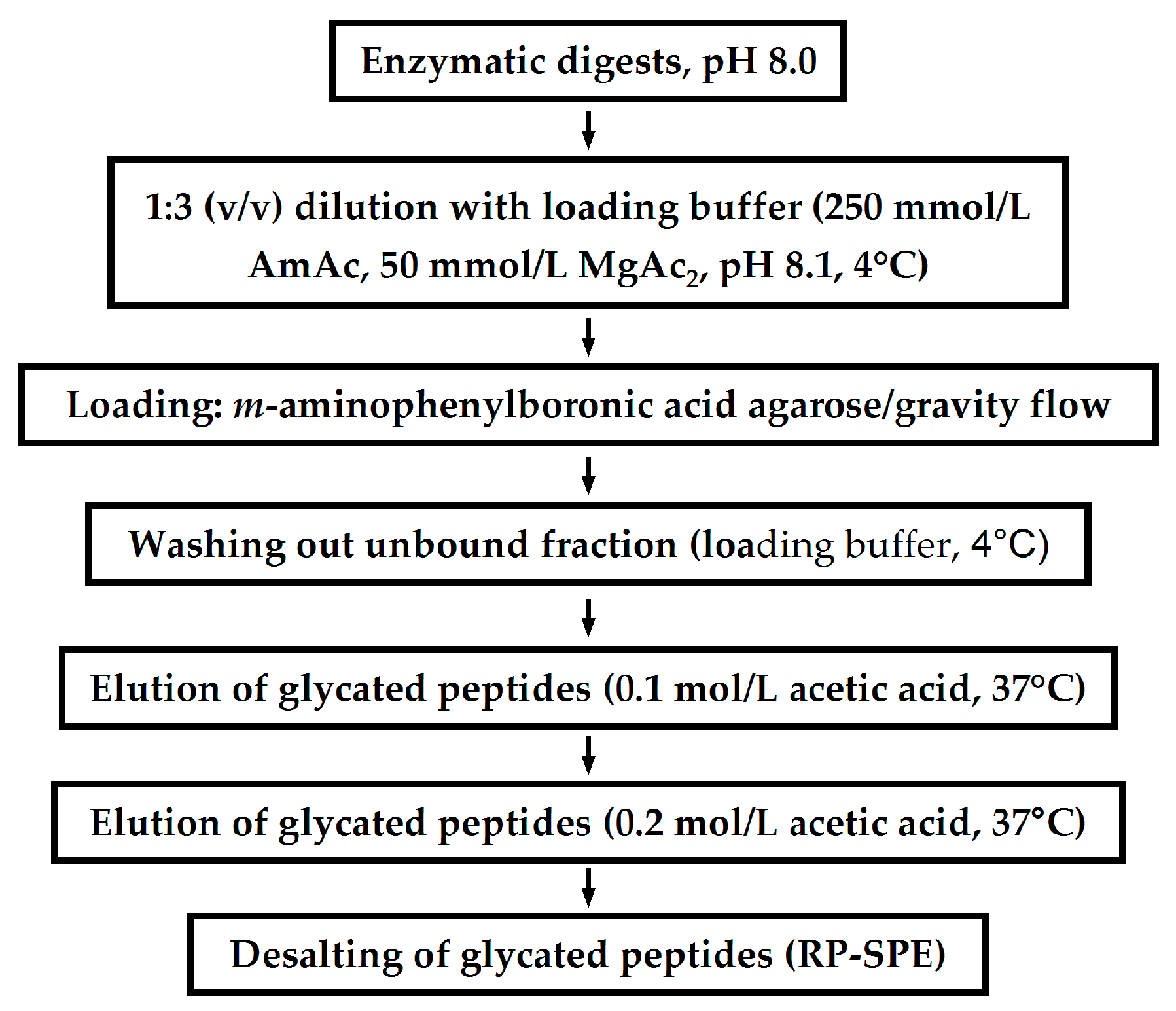{kind=link}
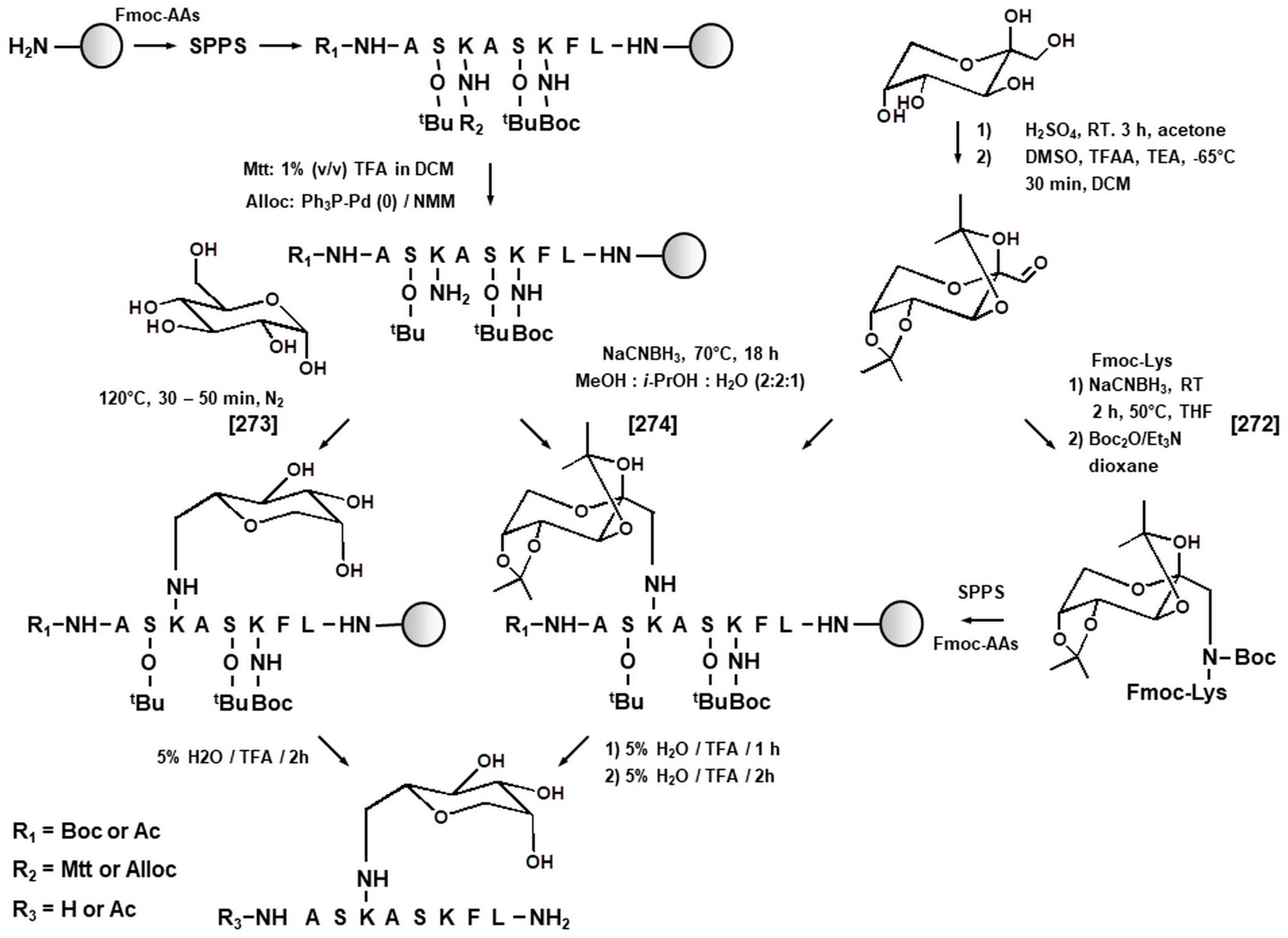{kind=link}
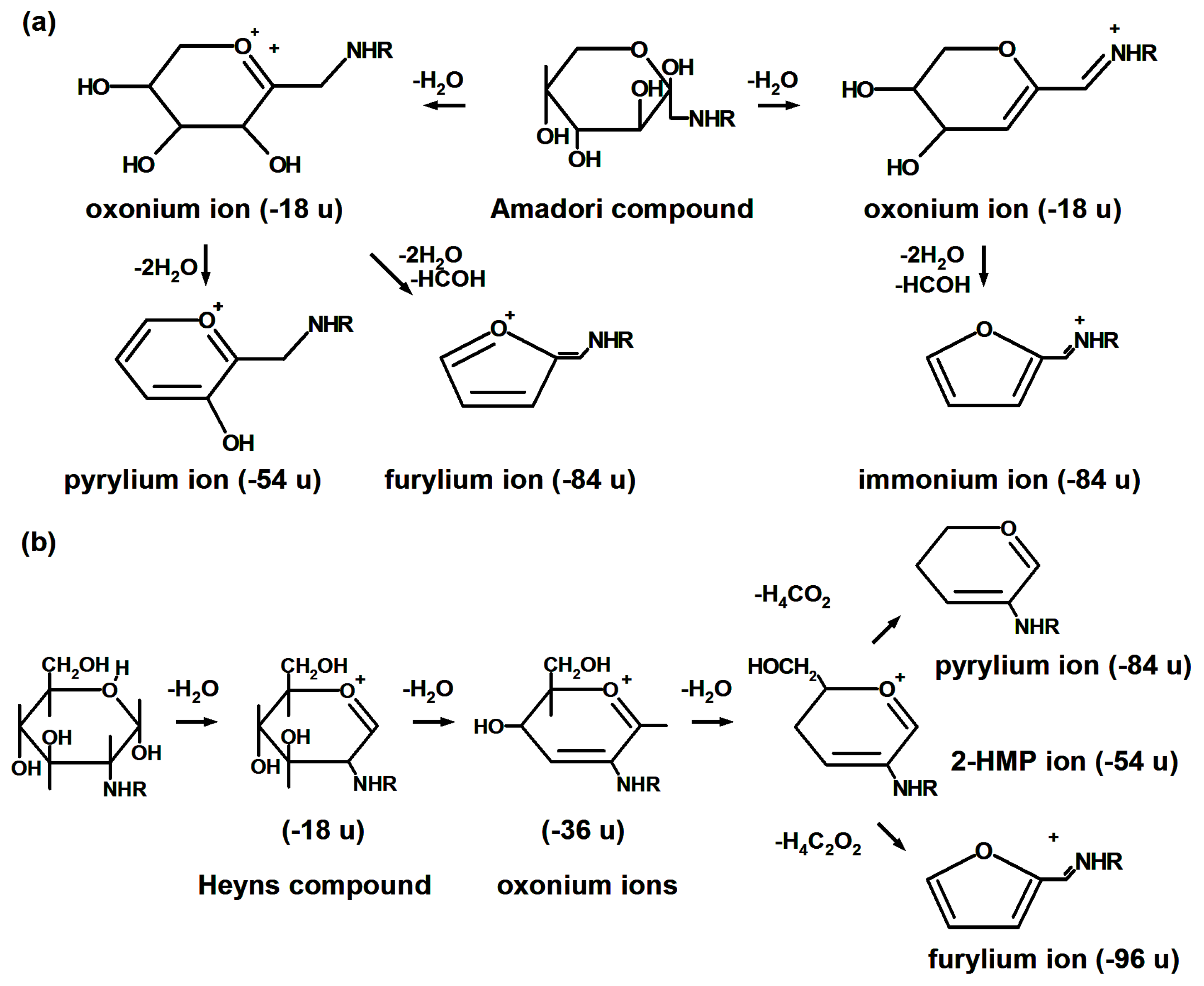{kind=link}
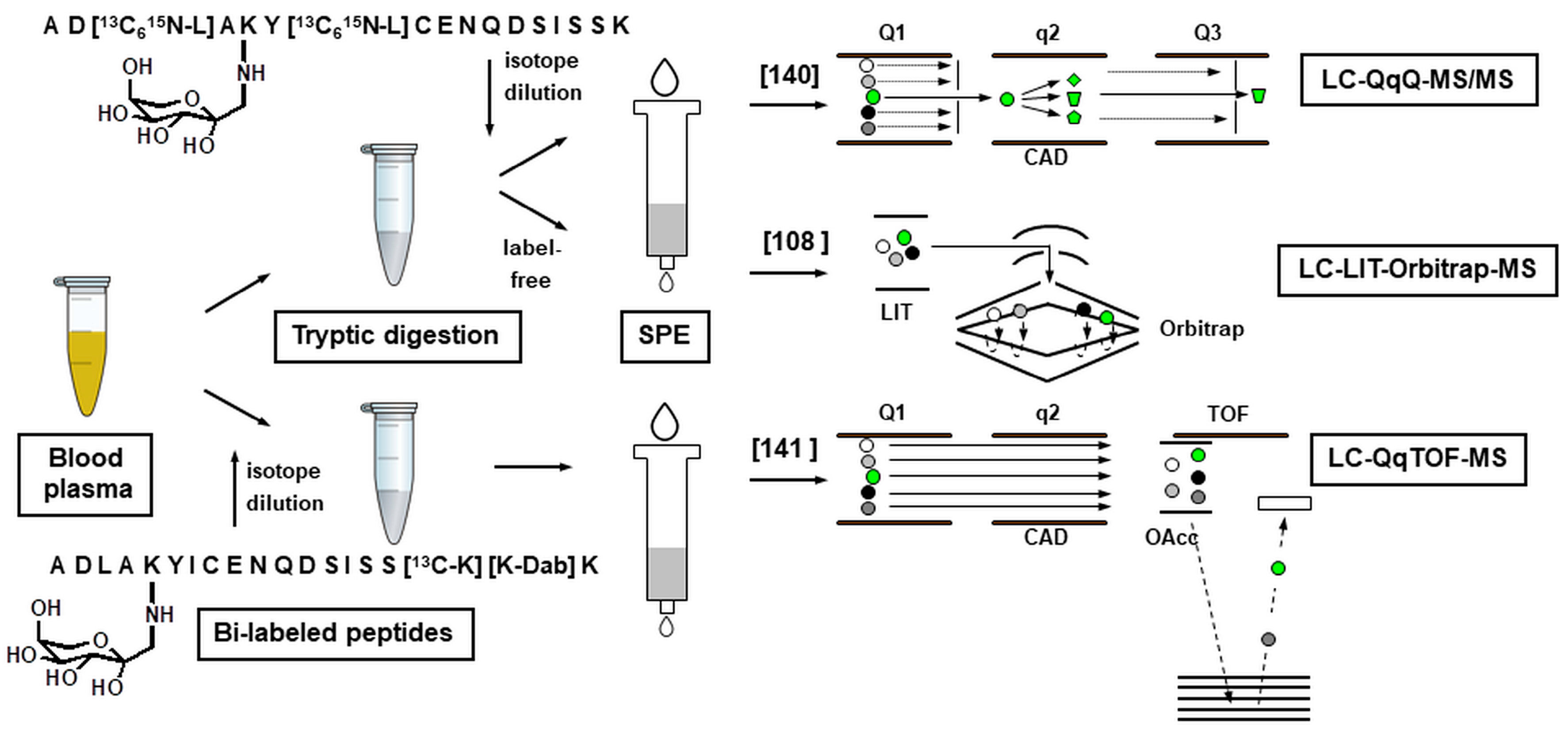{kind=link}
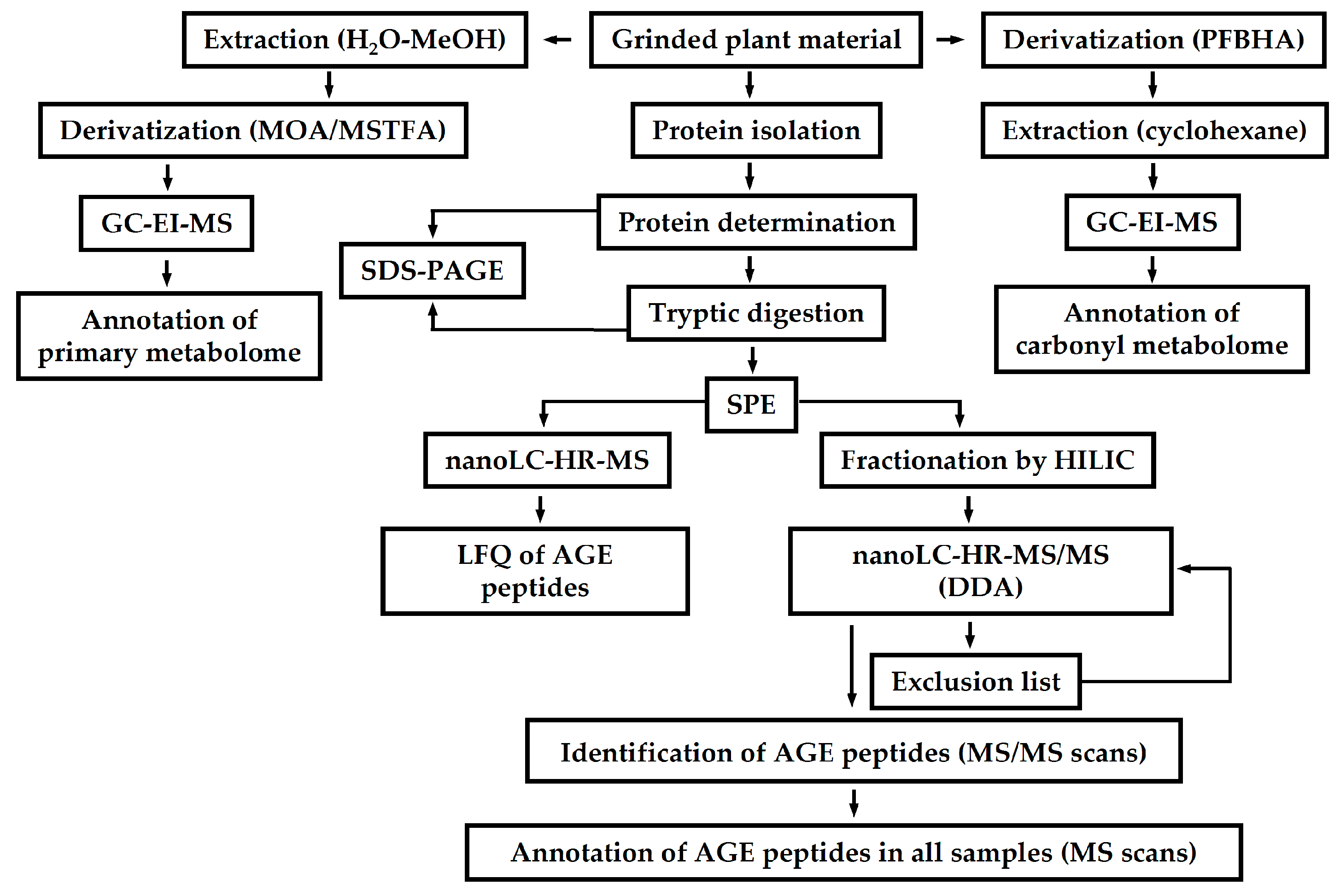{kind=link}
| # | Object | Analyzed Adducts | Methodology | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Technique | Protein Isolation | Denaturing Buffer or Detergent | Reduction Alkylation | Protease | Chromatographic System | MS | ||||
| 1 | HSA | Fru-Lys | cap-HPLC-MS | PEG 6000, affinity chromato-graphy | 0.5 M tris-HCl, 2.75 mmol/L EDTA, 6 mol/L guanidine-HCl, pH 8.1 | DTT/IA | trypsin | C18 A: 2% ACN, 0.1% aq. FA B: 98% aq. ACN, 0.1% FA | ESI-IT-TOF-MS | [83] |
| 2 | plasma proteins | Fru-Lys | nano-UHPLC-MS | - | 1% (w/v) SDS | TCEP/IA | trypsin | RP, nanoAcquity UPLC BEH130 A: 0.1% aq. FA B: 0.1% FA in ACN | ESI-LTQ-Orbitrap | [115] |
| 3 | HSA | Fru-Lys | nano-HPLC-MS | - | 76% acetonitrile | - | trypsin | RP, C18 A: 0.2% aq. FA B: 0.2% FA in ACN | ESI-QqTOF, ESI-QqQ | [120] |
| 4 | HSA | AGEs | HPLC-MS, MALDI-MS | - | - | DTT | trypsin | RP, C18 A: 0.1% aq. TFA B: 0.1% TFA in ACN | ESI-IT, MALDI-TOF | [156] |
| 5 | RNase | AGEs | HPLC-MS | - | 0.1 mol/L MOPS buffer, 6 mol/L urea, 1 mmol/L EDTA | DTT | trypsin | RP, C18 A: 0.1% aq. TFA/FA B: 0.1% TFA/FA in ACN | ESI-QqTOF, ESI-QqQ | [157] |
| 6 | HSA | Fru-Lys | nano-HPLC-MS | centrifugal conc. | - | DTT/IA | chymo-trypsin | RP, C18 A: 0.1% aq. FA B: 0.1% FA in ACN | ESI-QqTOF | [146] |
| 7 | HSA | Fru-Lys | 2D-nano-HPLC-MS | PEG 6000, affinity chromato-graphy | 0.5 mol/L tris-HCl, 2.75 mol/L EDTA, 6 mol/L guanidine-HCl, pH 8.1 | DTT/IA | Glu-C | RP, BetaBasic C18 A: 0.1% aq. FA/ 2% ACN, B: 98% ACN, 0.1% FA | ESI-IT-TOF | [83] |
| 8 | β-Lg | AGEs | UHPLC-MS | - | - | DTT (after hydrolysis) | Glu-C | RP, C18 A: 0.1% aq. FA B: ACN | ESI-QqLIT | [149] |
| 9 | insulin | Fru-Lys | MALDI-MS | - | 4 mol/L urea | DTT/ IA | Glu-C | - | MALDI-TOF | [158] |
| 10 | HSA | Fru-Lys | MALDI-MS | - | 6 mol/L guanidine-HCl, pH 8.5, 100 mmol/L ABC | DTT/IA | Glu-C | - | MALDI-TOF | [148] |
| 11 | HSA | Fru-Lys | MALDI-MS | - | 6 mol/L guanidine-HCl, pH 8.5, 100 mmol/L ABC | DTT/IA | Lys-C | - | MALDI-TOF | [148] |
| 12 | HSA | AGEs | HPLC-MS, MALDI-MS | - | - | DTT | Lys-C | RP, C18 A: 0.1% aq. TFA B: 0.1% TFA in ACN | ESI-IT, MALDI | [156] |
| 13 | α-La, β-Lg | AGEs | MALDI-MS | - | - | DTT (after hydrolysis) | Asp-N | - | MALDI-TOF | [159] |
| 14 | plasma proteins | Fru-Lys | nano-HPLC-MS | BAC | 8 mol/L urea, 0.5 mmol/L EDTA, 100 mmol/L ABC | DTT/IA | Arg-C | RP, C18 A: 0.2% aq. HOAc, or 0.05% aq. TFA or 0.1% aq. TFA B: 90% ACN | ESI-LIT | [160] |
| 15 | ubiquitin | Fru-Lys | cap-HPLC/off-line FIA-MS | - | - | - | pepsin | RP, C18 A: 0.1% aq. TFA B: 0.1% TFA in ACN | ESI-FT–ICR | [151] |
| 16 | HSA | AGEs | HPLC-MS | - | - | - | proteina-se K | RP, C18 A: 0.1% aq. TFA B: 0.1% TFA in ACN | ESI-QIT | [152] |
| 17 | IgG, plasma proteins | Fru-Lys | MALDI-MS | ultra-filtration | - | - | papain | - | MALDI-TOF | [153] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soboleva, A.; Schmidt, R.; Vikhnina, M.; Grishina, T.; Frolov, A. Maillard Proteomics: Opening New Pages. Int. J. Mol. Sci. 2017, 18, 2677. https://doi.org/10.3390/ijms18122677
Soboleva A, Schmidt R, Vikhnina M, Grishina T, Frolov A. Maillard Proteomics: Opening New Pages. International Journal of Molecular Sciences. 2017; 18(12):2677. https://doi.org/10.3390/ijms18122677
Chicago/Turabian StyleSoboleva, Alena, Rico Schmidt, Maria Vikhnina, Tatiana Grishina, and Andrej Frolov. 2017. "Maillard Proteomics: Opening New Pages" International Journal of Molecular Sciences 18, no. 12: 2677. https://doi.org/10.3390/ijms18122677
APA StyleSoboleva, A., Schmidt, R., Vikhnina, M., Grishina, T., & Frolov, A. (2017). Maillard Proteomics: Opening New Pages. International Journal of Molecular Sciences, 18(12), 2677. https://doi.org/10.3390/ijms18122677