Advanced Glycation End Products in the Pathogenesis of Psoriasis


{kind=link}
Abstract
:1. Introduction
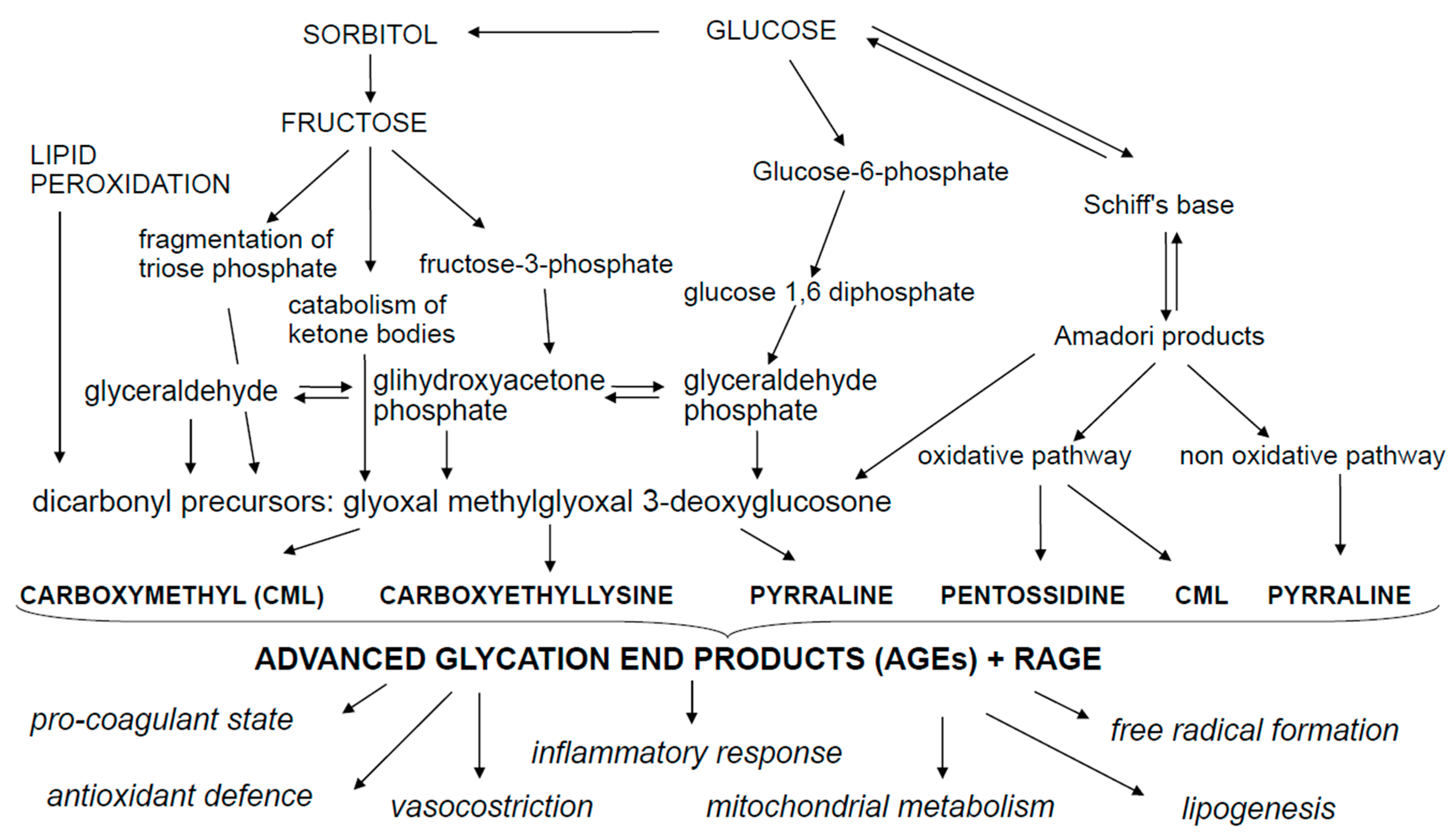
2. Advanced Glycation End Products (AGEs)
3. AGE, Diabetes Mellitus and Chronic Disorders
4. AGEs and Psoriasis
5. Conclusions
Supplementary Materials
Conflicts of Interest
Abbreviations
| AGEs | Advanced Glycation End Products |
| CML | Carboxymethyl |
| CEL | Carboxylethylisine |
| MG | Methylglyoxal |
| ALEs | Advanced Lipoxidation End-Products |
| RAGE | Membrane Receptors of AGEs |
| PKT | Tyrosine Kinase Protein |
References
- Papagrigoraki, A.; Del Giglio, M.; Cosma, C.; Maurelli, M.; Girolomoni, G.; Lapolla, A. Advanced Glycation End Products are Increased in the Skin and Blood of Patients with Severe Psoriasis. Acta Derm. Venereol. 2017, 97, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Monnier, V.M.; Cerami, A. Nonenzymatic browning in vivo-possible process for aging of long-lived proteins. Science 1981, 211, 491–493. [Google Scholar] [CrossRef] [PubMed]
- Da Moura Semedo, C.; Webb, M.; Waller, H.; Khunti, K.; Davies, M. Skin autofluorescence, a non-invasive marker of advanced glycation end products: Clinical relevance and limitations. Postgrad. Med. J. 2017, 93, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Damasiewicz-Bodzek, A.; Wielkoszyński, T. Advanced protein glycation in psoriasis. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced glycation endproducts: A review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Dyer, D.G.; Blackledge, J.A.; Thorpe, S.R.; Baynes, J.W. Formation of pentosidine during nonenzymatic browning of proteins by glucose. Identification of glucose and other carbohydrates as possible precursors of pentosidine in vivo. J. Biol. Chem. 1991, 266, 11654–11660. [Google Scholar] [PubMed]
- Miyata, T.; Inagi, R.; Asahi, K.; Yamada, Y.; Horie, K.; Sakai, H.; Uchida, K.; Kurokawa, K. Generation of protein carbonyls by glycoxidation and lipoxidation reactions with autoxidation products of ascorbic acid and polyunsaturated fatty acids. FEBS Lett. 1998, 437, 24–28. [Google Scholar] [CrossRef]
- Pizzimenti, S.; Ciamporcero, E.; Daga, M.; Pettazzoni, P.; Arcaro, A.; Cetrangolo, G.; Minelli, R.; Dianzani, C.; Lepore, A.; Gentile, F.; et al. Interaction of aldehydes derived from lipid peroxidation and membrane proteins. Front. Physiol. 2013, 4, 242. [Google Scholar] [CrossRef] [PubMed]
- Lapolla, A.; Reitano, R.; Baccarin, L.; Sartore, G.; Plebani, M.; Fedele, D. Pentosidine plasma levels and relation with metabolic control in diabetic patients. Horm. Metab. Res. 2005, 37, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Brings, S.; Fleming, T.; Freichel, M.; Muckenthaler, M.U.; Herzig, S.; Nawroth, P.P. Dicarbonyls and Advanced Glycation End-Products in the Development of Diabetic Complications and Targets for Intervention. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, S.R.; Baynes, J.W. Maillard reaction products in tissue proteins: New products and new perspectives. Amino Acids 2003, 25, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Vistoli, G.; De Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): An overview of their mechanisms of formation. Free Radic. Res. 2013, 47 (Suppl. 1), 3–27. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, S.; Roy, P.K.; Mitra, B.; Sen, S.; Sarkar, R.; Das, M.; Biswas, D.; Ghosh, P.; Biswas, A.; Chakraborty, S.; et al. Hyperlipidemia-mediated increased advanced Lipoxidation end products formation, an important factor associated with decreased erythrocyte glucose-6-phosphate dehydrogenase activity in mild Nonproliferative diabetic retinopathy. Can. J. Diabetes 2017, 41, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Byun, K.; Yoo, Y.; Son, M.; Lee, J.; Jeong, G.B.; Park, Y.M.; Salekdeh, G.H.; Lee, B. Advanced glycation end-products produced systemically and by macrophages: A common contributor to inflammation and degenerative diseases. Pharmacol. Ther. 2017, 177, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Koetsier, M.; Lutgers, H.L.; de Jonge, C.; Links, T.P.; Smit, A.J.; Graaff, R. Reference values of skin autofluorescence. Diabetes Technol. Ther. 2010, 12, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Sparvero, L.J.; Asafu-Adjei, D.; Kang, R.; Tang, D.; Amin, N.; Im, J.; Rutledge, R.; Lin, B.; Amoscato, A.A.; Zeh, H.J.; et al. RAGE (receptor for advanced glycation end products), RAGE ligands, and their role in cancer and inflammation. J. Transl. Med. 2009, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Mezentsev, A.V.; Bruskin, S.A.; Soboleva, A.G.; Sobolev, V.V.; Piruzian, E.S. Pharmacological control of receptor of advanced glycation end-products and its biological effects in psoriasis. Int. J. Biomed. Sci. 2013, 9, 112–122. [Google Scholar] [PubMed]
- Bergmann, C.; Strohbuecker, L.; Lotfi, R.; Sucker, A.; Joosten, I.; Koenen, H. High mobility group box 1 is increased in the sera of psoriatic patients with disease progression. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Soboleva, A.G.; Bruskin, S.A.; Nikolaev, A.A.; Sobolev, V.V.; Mezentsev, A.V. Role of receptor for advanced glycation endproducts in pathogenesis of psoriasis. Mol. Biol. 2013, 47, 743–753. [Google Scholar] [CrossRef]
- Shi, Y.; Sandoghchian, S.S.; Su, Z.; Liu, Y.; Tong, J.; Zheng, D.; Chen, J.; Liu, Y.; Xu, Y.; Jiao, Z.; et al. Enhanced HMGB1 expression may contribute to Th17 cells activation in rheumatoid arthritis. Clin. Dev. Immunol. 2012, 2012, 295081. [Google Scholar] [CrossRef] [PubMed]
- Botros, N.; Sluik, D.; van Waateringe, R.P.; de Vries, J.H.M.; Geelen, A.; Feskens, E.J.M. Advanced glycation end-products (AGEs) and associations with cardio-metabolic, lifestyle, and dietary factors in a general population: The NQplus study. Diabetes Metab. Res. Rev. 2017, 33. [Google Scholar] [CrossRef] [PubMed]
- Colhoun, H.M.; Betteridge, D.J.; Durrington, P.; Hitman, G.; Neil, A.; Livingstone, S. Total soluble and endogenous secretory receptor for advanced glycation end products as predictive biomarkers of coronary heart disease risk in patients with type 2 diabetes. Diabetes 2011, 60, 2379–2385. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Park, S.; Lee, M.J.; Song, Y.R.; Han, S.H.; Kim, S.G.; Kang, S.W.; Choi, K.H.; Kim, H.J.; Yoo, T.H. Plasma levels of soluble receptors for advanced glycation end products (sRAGE) and proinflammatory ligand for RAGE (ENRAGE) are associated with carotis atherosclerosis in patients with peritoneal dialysis. Atherosclerosis 2012, 220, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Jaisson, S.; Gillery, P. Evaluation of nonenzymatic posttranslational modification-derived products as biomarkers of molecular aging of proteins. Clin. Chem. 2010, 56, 1401–1412. [Google Scholar] [CrossRef] [PubMed]
- Sayej, W.N.; Knight, P.R.; Guo, W.A.; Mullan, B.; Ohtake, P.J.; Davidson, B.A.; Khan, A.; Baker, R.D.; Baker, S.S. Advanced glycation end products induce obesity and hepatosteatosis in CD-1 wild-type mice. BioMed Res. Int. 2016, 2016, 7867852. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.E.; Dordevic, A.L.; Tan, S.M.; Ryan, L.; Coughlan, M.T. Dietary advanced glycation end products and risk factors for chronic disease: A systematic review of randomised controlled trials. Nutrients 2016, 8, 125. [Google Scholar] [CrossRef] [PubMed]
- Kellow, N.J.; Savige, G.S. Dietary advanced glycation end-product restriction for the attenuation of insulin resistance, oxidative stress and endothelial dysfunction: A systematic review. Eur. J. Clin. Nutr. 2013, 67, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Peppa, M.; Vlassara, H. Advanced glycation end products and diabetic complications: A general overview. Hormones 2005, 4, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Piarulli, F.; Sartore, G.; Lapolla, A. Glyco-oxidation and cardiovascular complications in type 2 diabetes: A clinical update. Acta Diabetol. 2013, 50, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Nin, J.W.; Jorsal, A.; Ferreira, I.; Schalkwijk, C.G.; Prins, M.H.; Parving, H.H. Higher plasma levels of advanced glycation end products are associated with incident cardiovascular disease and all cause mortality in type 1 diabetes: A 12-year follow-up study. Diabetes Care 2011, 34, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Baynes, J.W.; Thorpe, S.R. Glycoxidation and lipoxidation in atherogenesis. Free Radic. Biol. Med. 2000, 28, 1708–1716. [Google Scholar] [CrossRef]
- Meerwaldt, R.; Links, T.; Zeebregts, C.; Tio, R.; Hillebrands, J.L.; Smit, A. The clinical relevance of assessing advanced glycation endproducts accumulation in diabetes. Cardiovasc. Diabetol. 2008, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H.; Striker, G.E. AGE restriction in diabetes mellitus: A paradigm shift. Nat. Rev. Endocrinol. 2011, 7, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Matsui, T. Advanced glycation end products, oxidative stress and diabetic nephropathy. Oxid. Med. Cell. Longev. 2010, 3, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Yagihashi, S.; Mizukami, H.; Sugimoto, K. Mechanism of diabetic neuropathy: Where are we now and where to go? J. Diabetes Investig. 2011, 2, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced glycation end products-sparking the development of diabetic vascular injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, V.; Beare, R.; Blizzard, L.; Phan, T.; Stapleton, J.; Chen, J.; Callisaya, M.; Martin, K.; Reutens, D. Cerebral white matter lesions, gait, and the risk of incident falls: A prospective population-based study. Stroke 2009, 40, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Weinstein, S.J.; Albanes, D.; Taylor, P.R.; Graubard, B.I.; Virtamo, J.; Stolzenberg-Solomon, R.Z. Evidence that serum levels of the soluble receptor for advanced glycation end products are inversely associated with pancreatic cancer risk: A prospective study. Cancer Res. 2011, 71, 3582–3589. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Zilmer, K.; Leping, V.; Zilmer, M. Serum methylglyoxal level and its association with oxidative stress and disease severity in patients with psoriasis. Arch. Dermatol. Res. 2013, 305, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Al Rifai, M.; Schneider, A.L.; Alonso, A.; Maruthur, N.; Parrinello, C.M.; Astor, B.C.; Hoogeveen, R.C.; Soliman, E.Z.; Chen, L.Y.; Ballantyne, C.M.; et al. sRAGE, inflammation, and risk of atrial fibrillation: Results from the Atherosclerosis Risk in Communities (ARIC) study. J. Diabetes Complic. 2015, 29, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Genuth, S.; Sun, W.; Cleary, P.; Sell, D.R.; Dahms, W.; Malone, J.; Sivitz, W.; Monnier, V.M.; DCCT Skin Collagen Ancillary Study Group. Glycation and carboxymethyllysine levels in skin collagen predict the risk of future 10-year progression of diabetic retinopathy and nephropathy in the diabetes control and complications trial and epidemiology of diabetes interventions and complications participants with type 1 diabetes. Diabetes 2005, 54, 3103–3111. [Google Scholar] [PubMed]
- Meerwaldt, R.; Hartog, J.W.; Graaff, R.; Huisman, R.J.; Links, T.P.; den Hollander, N.C.; Thorpe, S.R.; Baynes, J.W.; Navis, G.; Gans, R.O.; et al. Skin autofluorescence, a measure of cumulative metabolic stress and advanced glycation end products, predicts mortality in hemodialysis patients. J. Am. Soc. Nephrol. 2005, 16, 3687–3693. [Google Scholar] [CrossRef] [PubMed]
- Kiuchi, K.; Nejima, J.; Takano, T.; Ohta, M.; Hashimoto, H. Increased serum concentrations of advanced glycation end products: A marker of coronary artery disease activity in type 2 diabetic patients. Heart 2001, 85, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Schalkwijk, C.G.; Baidoshvili, A.; Stehouwer, C.D.; van Hinsbergh, V.W.; Niessen, H.W. Increased accumulation of the glycoxidation product Nepsilon-(carboxymethyl)lysine in hearts of diabetic patients: Generation and characterisation of a monoclonal anti-CML antibody. Biochim. Biophys. Acta 2004, 1636, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Cipollone, F.; Iezzi, A.; Fazia, M.; Zucchelli, M.; Pini, B.; Cuccurullo, C.; De Cesare, D.; De Blasis, G.; Muraro, R.; Bei, R.; et al. The receptor RAGE as a progression factor amplifying arachidonate-dependent inflammatory and proteolytic response in human atherosclerotic plaques: Role of glycemic control. Circulation 2003, 108, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Cuccurullo, C.; Iezzi, A.; Fazia, M.L.; De Cesare, D.; Di Francesco, A.; Muraro, R.; Bei, R.; Ucchino, S.; Spigonardo, F.; Chiarelli, F.; et al. Suppression of RAGE as a basis of simvastatin-dependent plaque stabilization in type 2 diabetes. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2716–2723. [Google Scholar] [CrossRef] [PubMed]
- Simm, A.; Wagner, J.; Gursinsky, T.; Nass, N.; Friedrich, I.; Schinzel, R.; Czeslik, E.; Silber, R.E.; Scheubel, R.J. Advanced glycation endproducts: A biomarker for age as an outcome predictor after cardiac surgery? Exp. Gerontol. 2007, 42, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.Y.; Kwon, H.M.; Ahn, C.W.; Lee, G.T.; Joung, B.; Hong, B.K.; Yoon, Y.W.; Kim, D.; Byun, K.H.; Kang, T.S.; et al. Serum levels of advanced glycation end products are associated with in-stent restenosis in diabetic patients. Yonsei Med. J. 2005, 46, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Hartog, J.W.; Voors, A.A.; Bakker, S.J.; Smit, A.J.; van Veldhuisen, D.J. Advanced glycation end-products (AGE) and heart failure: Pathophysiology and clinical implications. Eur. J. Heart Fail. 2007, 9, 1146–1155. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Basta, G.; Schmidt, A.M.; De Caterina, R. Advanced glycation end products and vascular inflammation: Implications for accelerated atherosclerosis in diabetes. Cardiovasc. Res. 2004, 63, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; He, J.C.; Zhu, L.; Peppa, M.; Lu, C.; Uribarri, J.; Vlassara, H. High levels of dietary advanced glycation end products transform low-density lipoprotein into a potent redox-sensitive mitogen-activated protein kinase stimulant in diabetic patients. Circulation 2004, 110, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Monnier, V.M.; Sell, D.R.; Genuth, S. Glycation products as markers and predictors of the progression of diabetic complications. Ann. N. Y. Acad. Sci. 2005, 1043, 567–581. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.I.; Bucciarelli, L.G.; Wendt, T.; Sakaguchi, T.; Lalla, E.; Qu, W.; Lu, Y.; Lee, L.; Stern, D.M.; Naka, Y.; et al. Blockade of receptor for advanced glycation endproducts: A new target for therapeutic intervention in diabetic complications and inflammatory disorders. Arch. Biochem. Biophys. 2003, 419, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Mulder, D.J.; Water, T.V.; Lutgers, H.L.; Graaff, R.; Gans, R.O.; Zijlstra, F.; Smit, A.J. Skin autofluorescence, a novel marker for glycemic and oxidative stress-derived advanced glycation endproducts: An overview of current clinical studies, evidence, and limitations. Diabetes Technol. Ther. 2006, 8, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Dimon-Gadal, S.; Gerbaud, P.; Thérond, P.; Guibourdenche, J.; Anderson, W.B.; Evain-Brion, D.; Raynaud, F. Increased oxidative damage to fibroblasts in skin with and without lesions in psoriasis. J. Investig. Dermatol. 2000, 114, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Gabr, S.A.; Al-Ghadir, A.H. Role of cellular oxidative stress and cytochrome c in the pathogenesis of psoriasis. Arch. Dermatol. Res. 2012, 304, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Lohwasser, C.; Neureiter, D.; Weigle, B.; Kirchner, T.; Schuppan, D. The receptor for advanced glycation end products is highly expressed in the skin and upregulated by advanced glycation end products and tumor necrosis factor-alpha. J. Investig. Dermatol. 2006, 126, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Batycka-Baran, A.; Hattinger, E.; Zwicker, S.; Summer, B.; Zack Howard, O.M.; Thomas, P.; Szepietowski, J.C.; Ruzicka, T.; Prinz, J.C.; Wolf, R. Leucocyte derived koebnerisin (S100A15) and psoriasin (S100A7) are systemic mediators of inflammation in psoriasis. J. Dermatol. Sci. 2015, 79, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Vasků, V.; Kanková, K.; Vasků, A.; Muzík, J.; Izakovicová Hollá, L.; Semrádová, V. Gene polymorphisms (G82S, 1704G/T, 2184A/G and 2245G/A) of the receptor of advanced glycation end products (RAGE) in plaque psoriasis. Arch. Dermatol. Res. 2002, 294, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Manigrasso, M.B.; Pan, J.; Rai, V.; Zhang, J.; Reverdatto, S.; Quadri, N.; DeVita, R.J.; Ramasamy, R.; Shekhtman, A.; Schmidt, A.M. Small molecule inhibition of ligand-stimulated RAGE-DIAPH1 signal transduction. Sci. Rep. 2016, 6, 22450. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papagrigoraki, A.; Maurelli, M.; Del Giglio, M.; Gisondi, P.; Girolomoni, G. Advanced Glycation End Products in the Pathogenesis of Psoriasis. Int. J. Mol. Sci. 2017, 18, 2471. https://doi.org/10.3390/ijms18112471
Papagrigoraki A, Maurelli M, Del Giglio M, Gisondi P, Girolomoni G. Advanced Glycation End Products in the Pathogenesis of Psoriasis. International Journal of Molecular Sciences. 2017; 18(11):2471. https://doi.org/10.3390/ijms18112471
Chicago/Turabian StylePapagrigoraki, Anastasia, Martina Maurelli, Micol Del Giglio, Paolo Gisondi, and Giampiero Girolomoni. 2017. "Advanced Glycation End Products in the Pathogenesis of Psoriasis" International Journal of Molecular Sciences 18, no. 11: 2471. https://doi.org/10.3390/ijms18112471
APA StylePapagrigoraki, A., Maurelli, M., Del Giglio, M., Gisondi, P., & Girolomoni, G. (2017). Advanced Glycation End Products in the Pathogenesis of Psoriasis. International Journal of Molecular Sciences, 18(11), 2471. https://doi.org/10.3390/ijms18112471