A Novel Mutation in the Fibrinogen Bβ Chain (c.490G>A; End of Exon 3) Causes a Splicing Abnormality and Ultimately Leads to Congenital Hypofibrinogenemia

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Patient with Kyoto IX
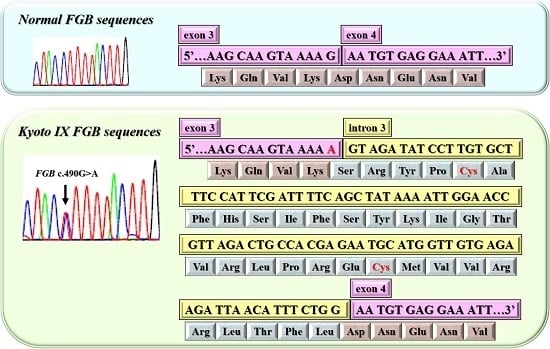
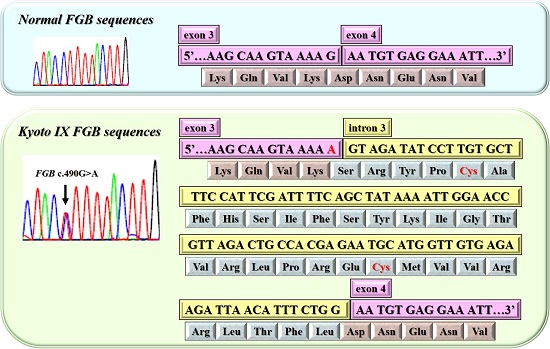
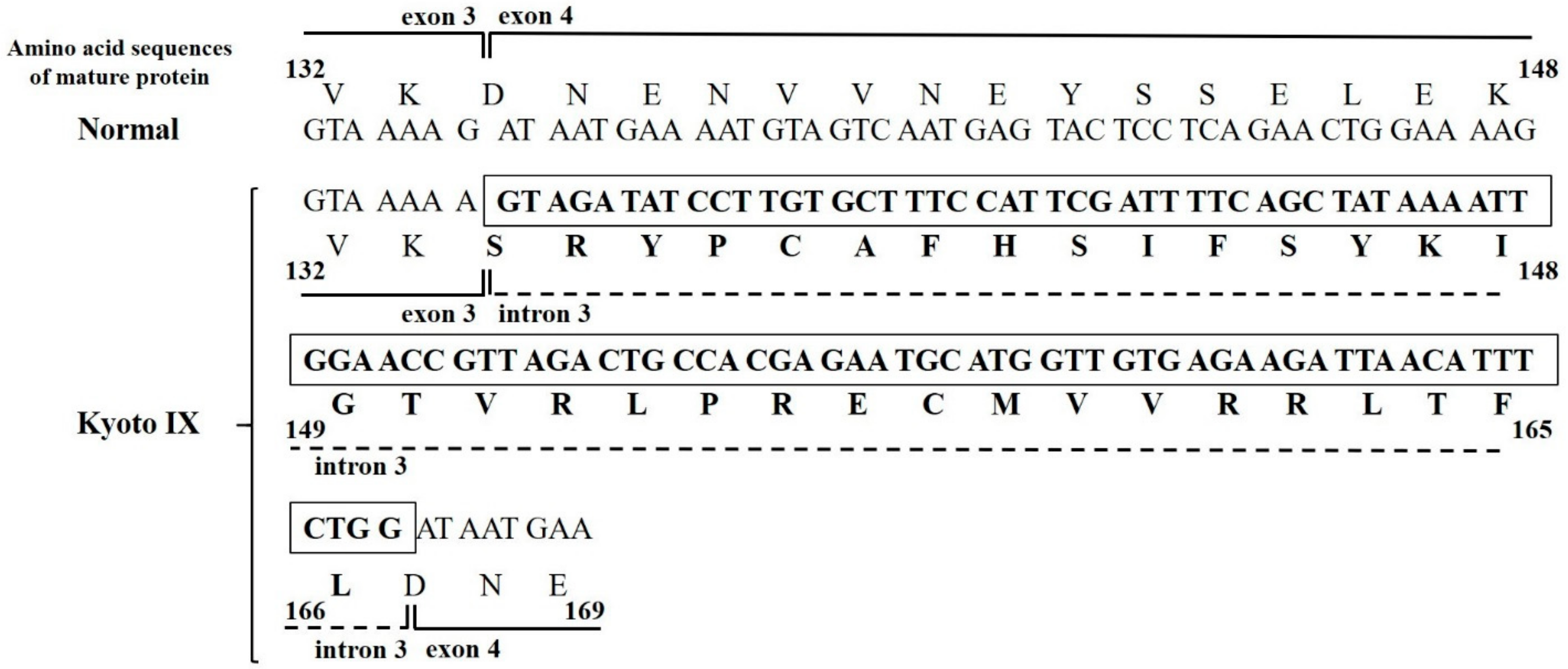
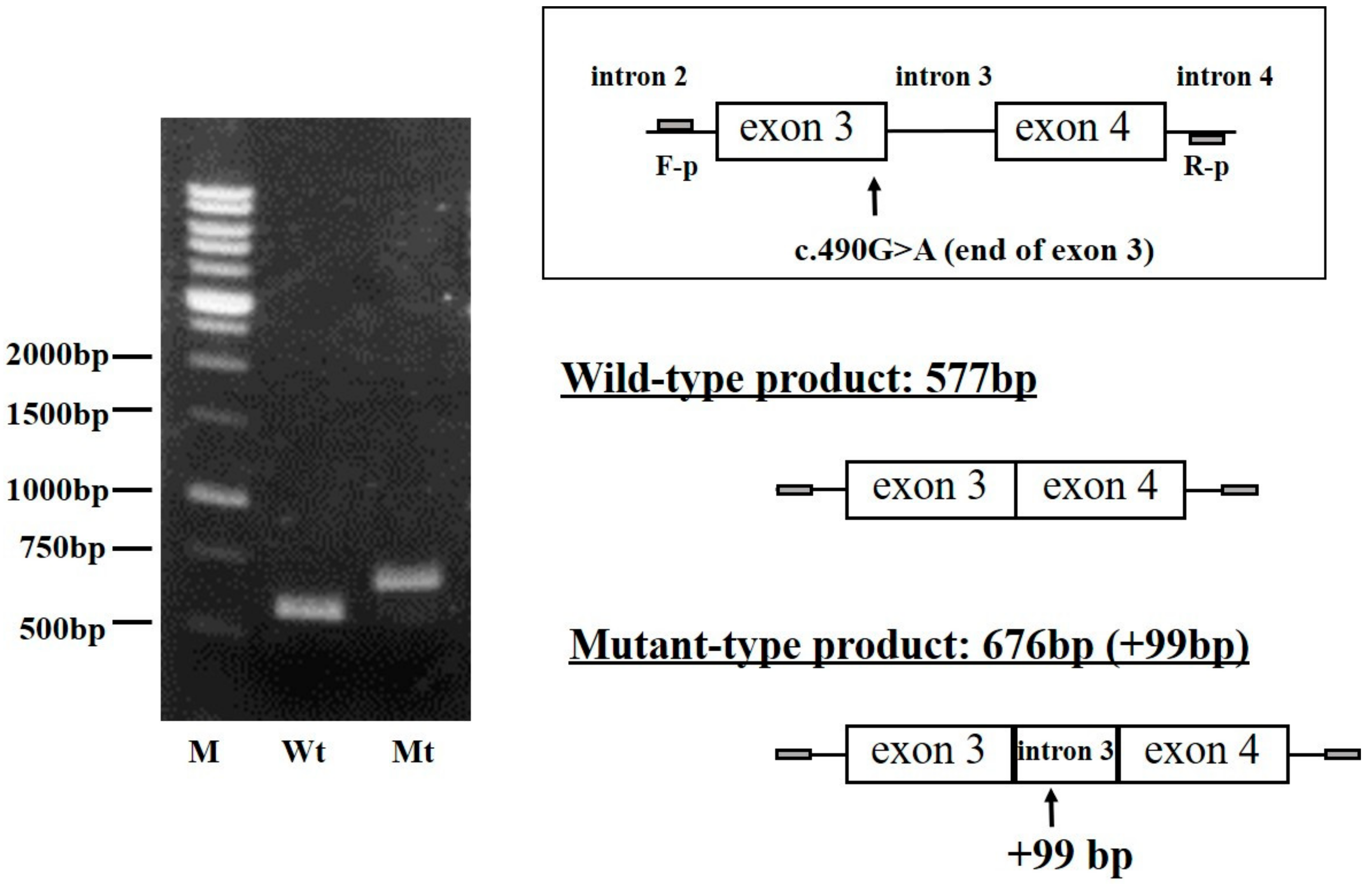
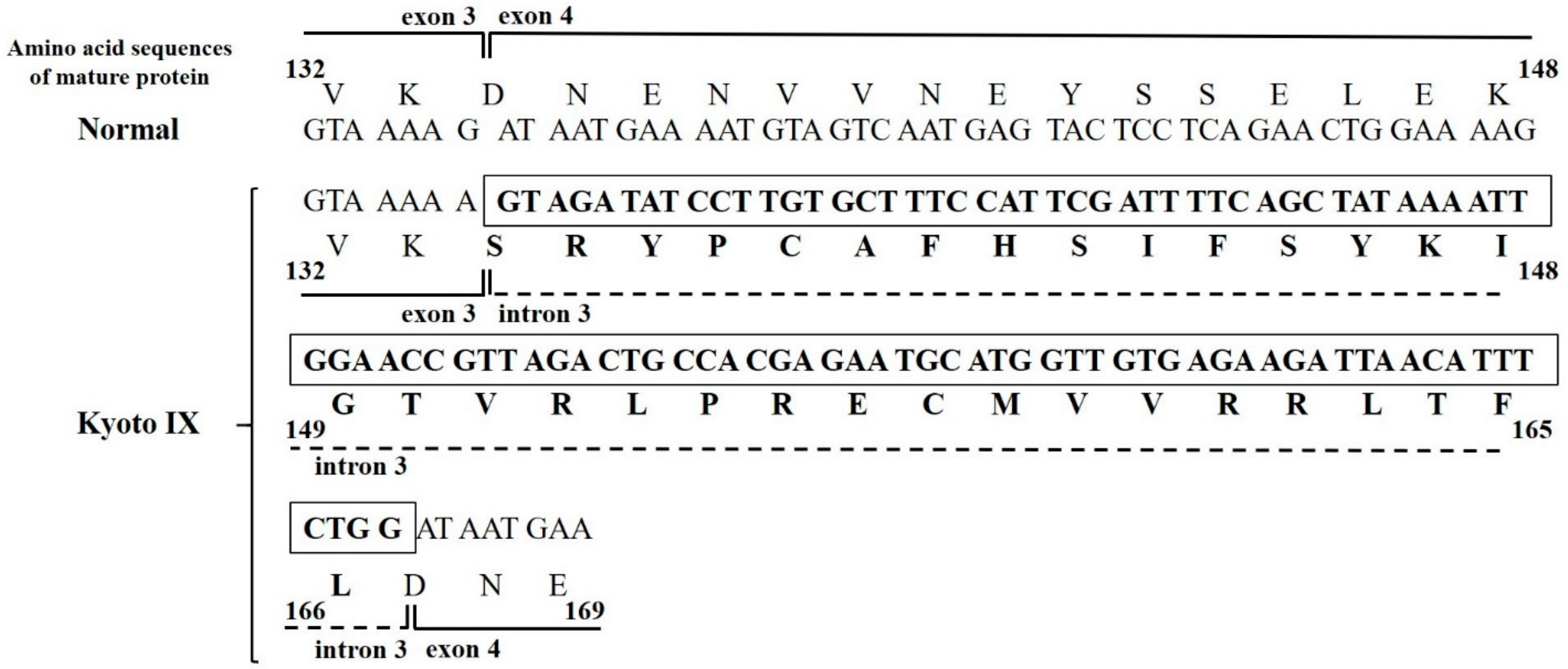
2.2. DNA Sequence Analysis of the Kyoto IX Patient
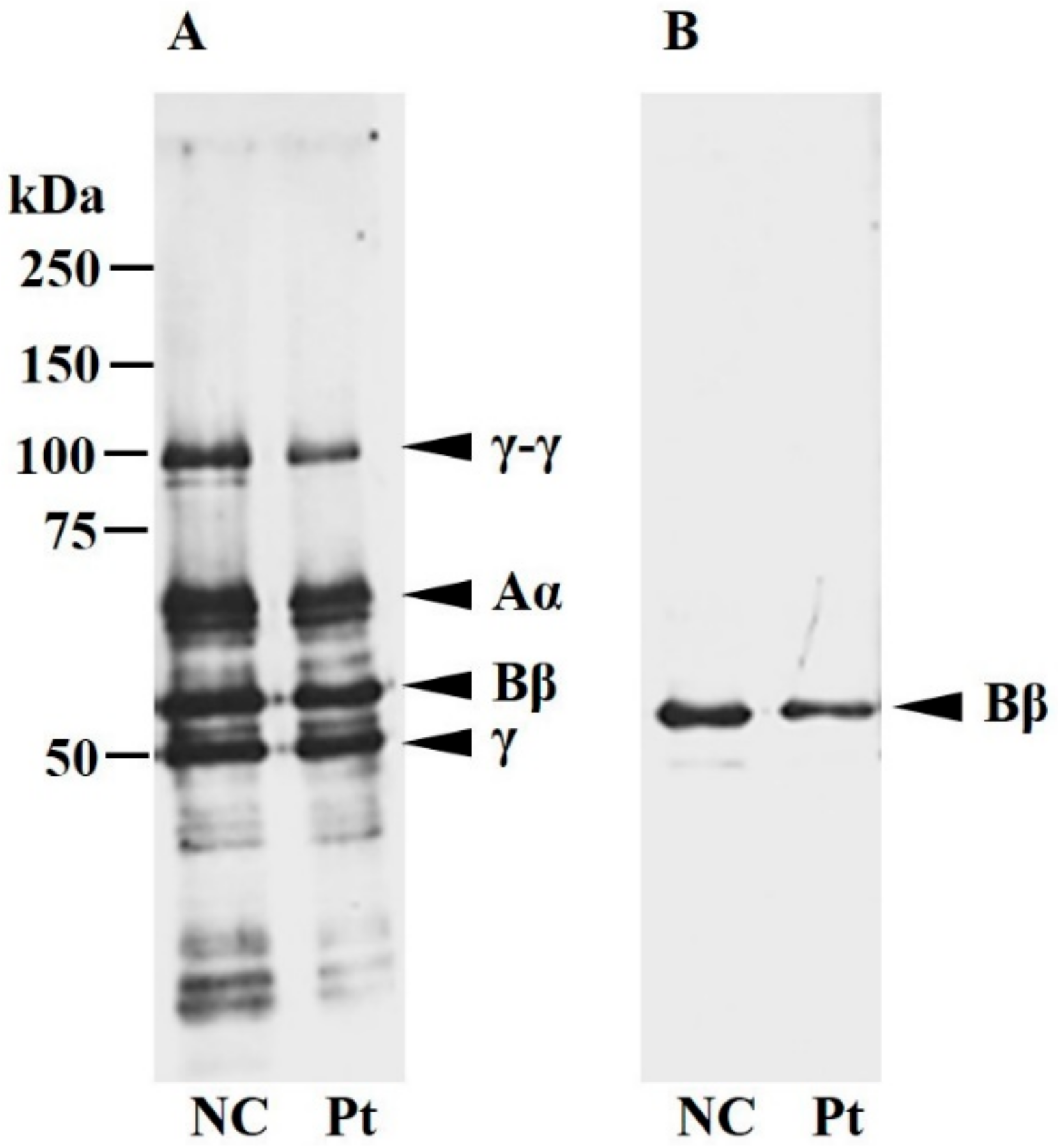
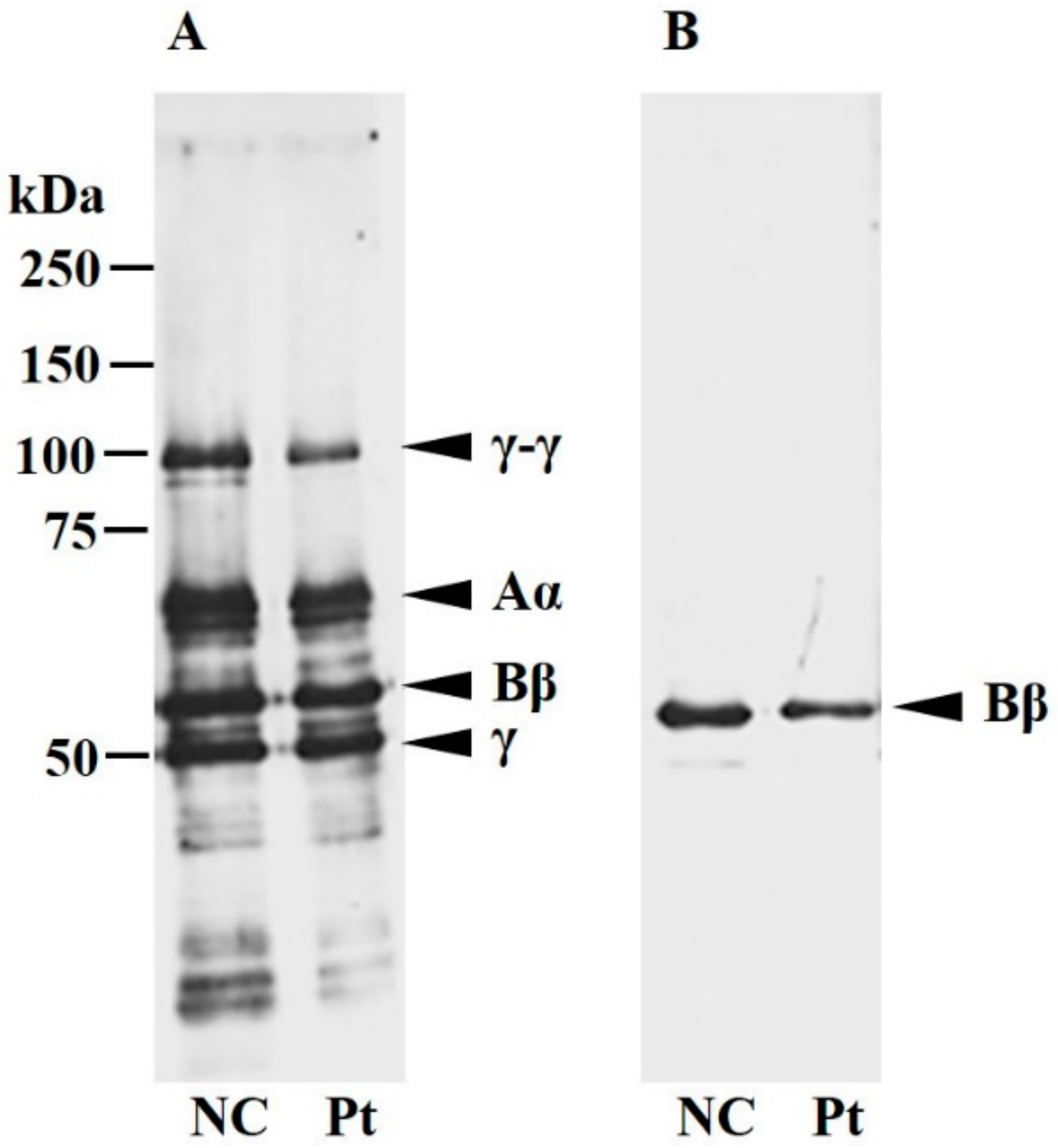
2.3. Characterization of Plasma Fibrinogen
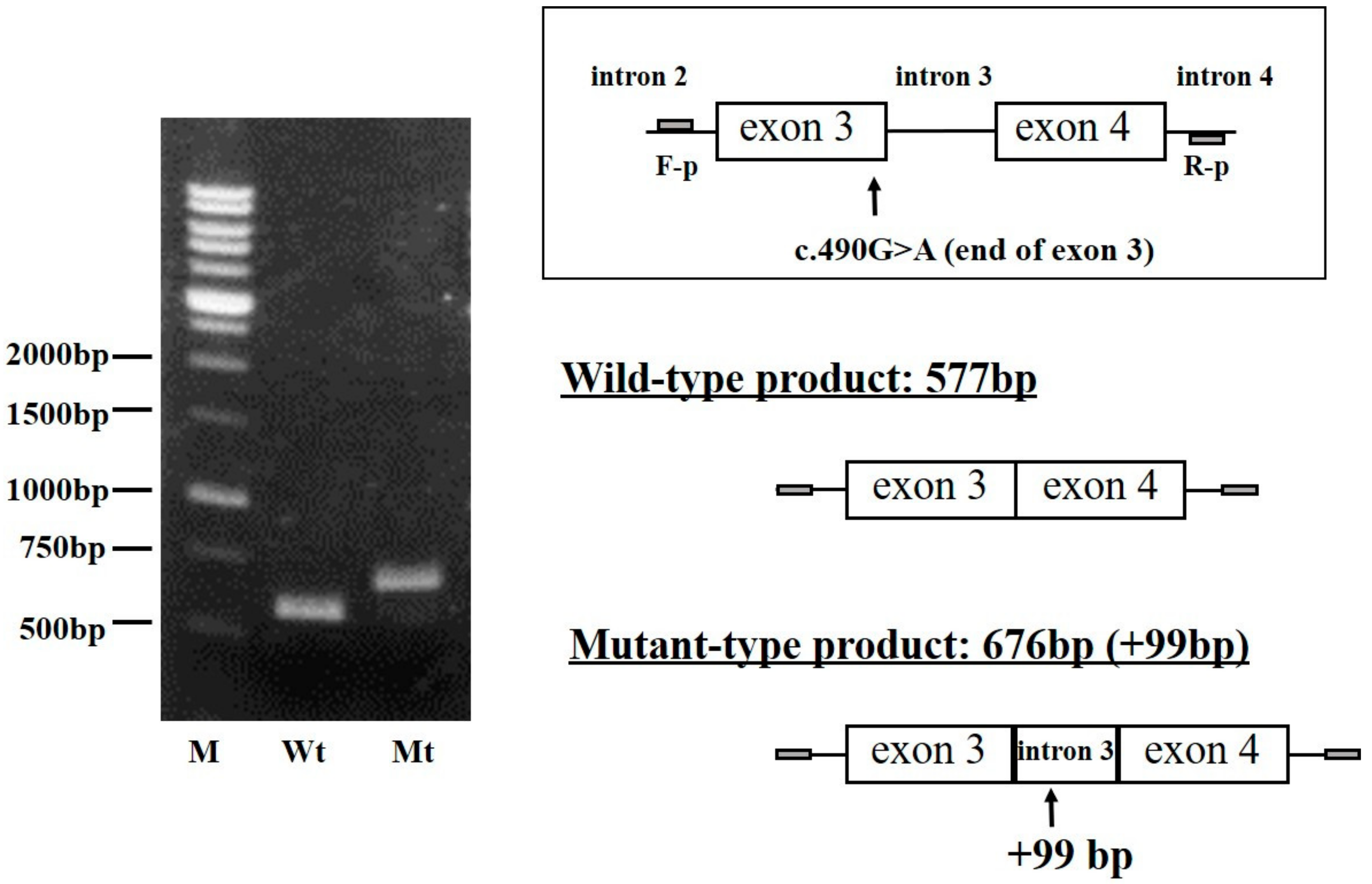
2.4. Analysis of FGB Gene Transcripts in the CHO Cell Line
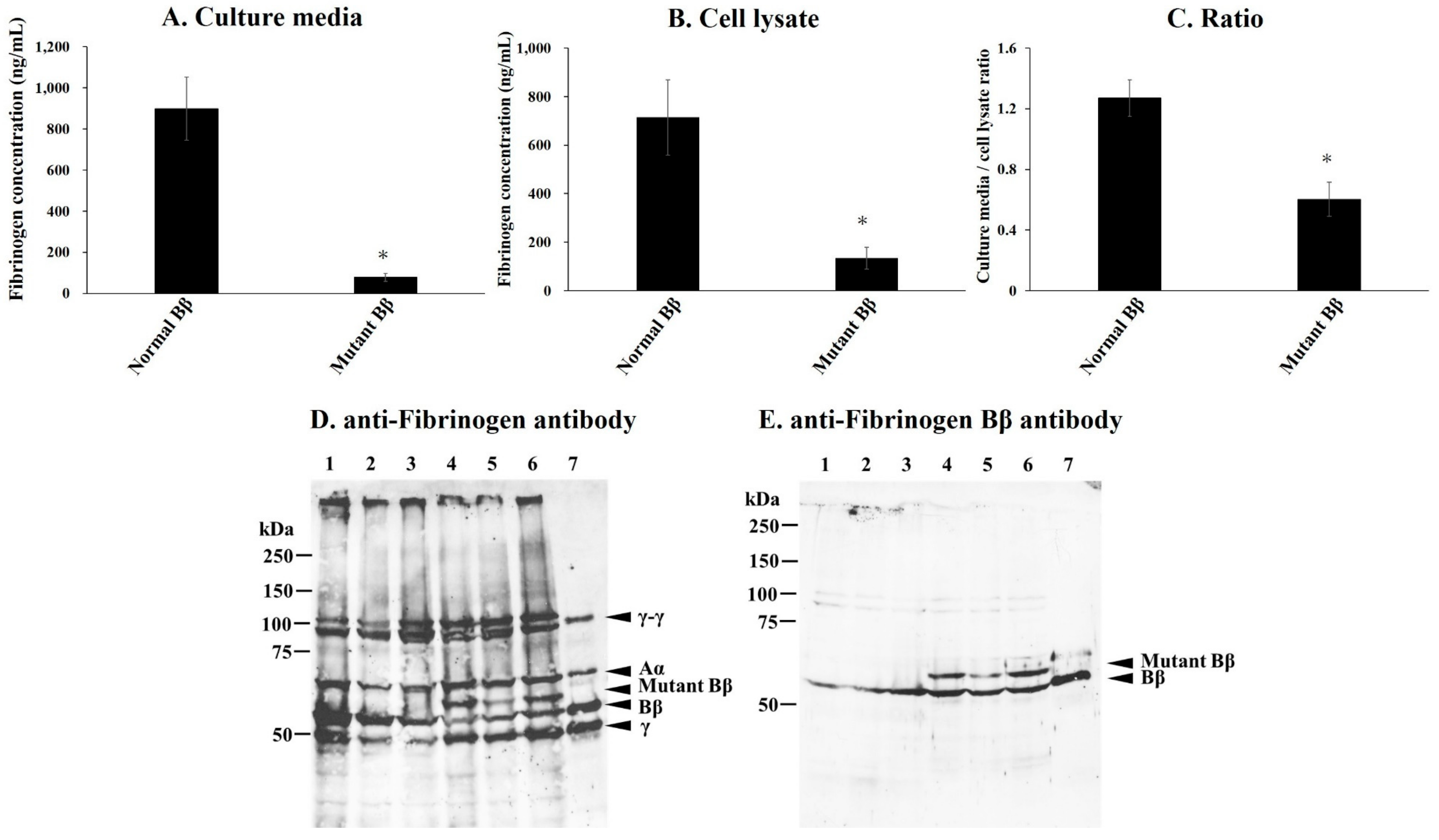
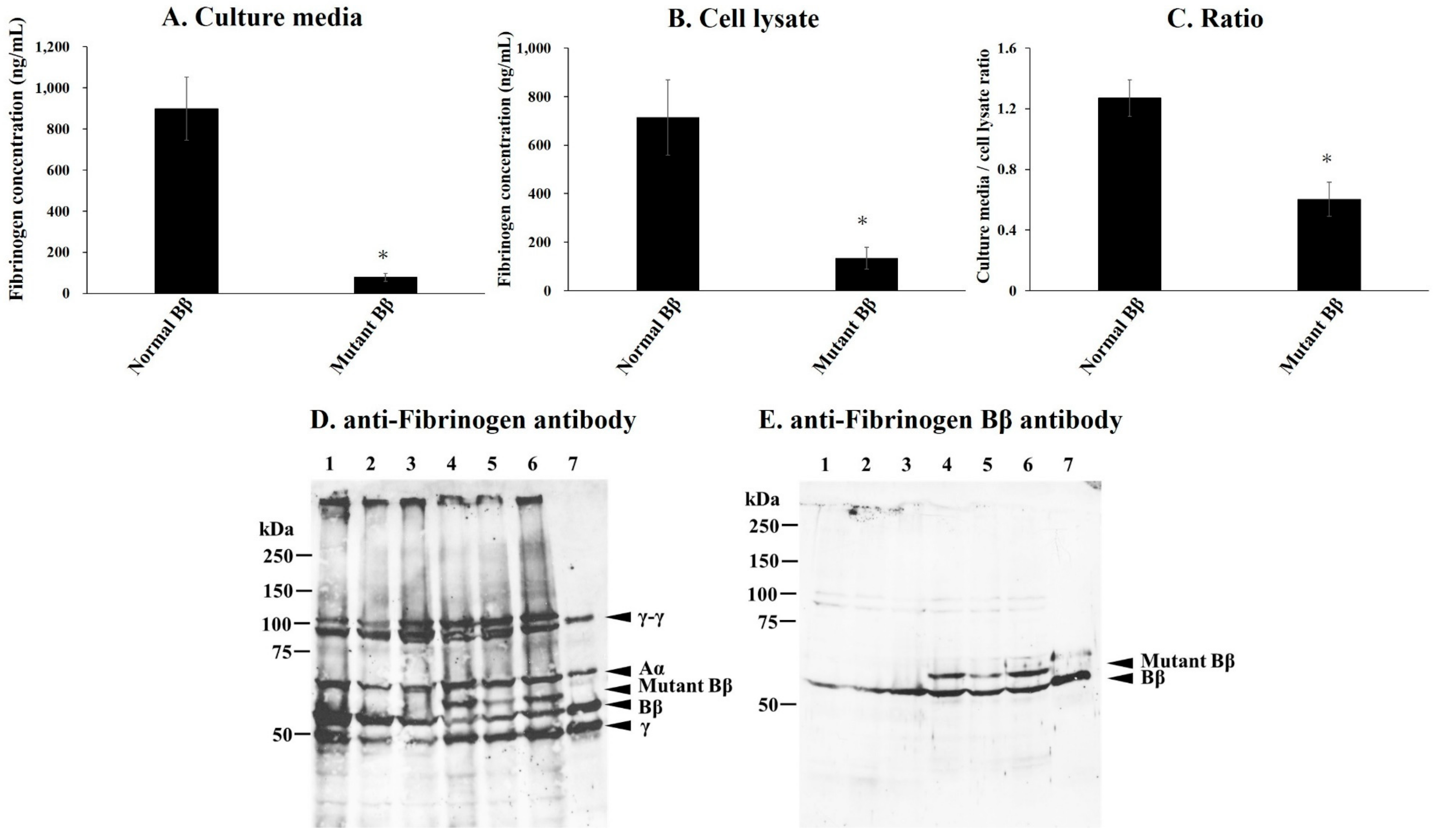
2.5. Synthesis and Secretion of Recombinant Variant Fibrinogen in CHO Cell Lines
3. Discussion
4. Materials and Methods
4.1. Characterization of Patient Plasma Fibrinogen
4.2. DNA Sequence Analysis
4.3. Construction of Mini-Gene and Expression Vectors
4.4. Production of Bβ-Chain mRNAs, RNA Extraction, and RT-PCR
4.5. Expression of Recombinant Mutant Fibrinogen
4.6. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CHO | Chinese hamster ovary |
| RT-PCR | Reverse transcriptase-polymerase chain reaction |
| SDS-PAGE | Sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| ELISA | Enzyme-linked immunosorbent assay |
References
- Weisel, J.W. Fibrinogen and fibrin. Adv. Protein. Chem. 2005, 70, 247–299. [Google Scholar] [PubMed]
- Weisel, J.W.; Litvinov, R.I. Mechanisms of fibrin polymerization and clinical implications. Blood 2013, 121, 1712–1719. [Google Scholar] [CrossRef] [PubMed]
- Kant, J.A.; Fornace, A.J., Jr.; Saxe, D.; Simon, M.I.; McBride, O.W.; Crabtree, G.R. Evolution and organization of the fibrinogen locus on chromosome 4: Gene duplication accompanied by transposition and inversion. Proc. Natl. Acad. Sci. USA 1985, 82, 2344–2348. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.S.; Dimichele, D.M. Rare inherited disorders of fibrinogen. Haemophilia 2008, 14, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
- Korte, W.; Poon, M.C.; Iorio, A.; Makris, M. Thrombosis in Inherited Fibrinogen Disorders. Transfus. Med. Hemother. 2017, 44, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Bornikova, L.; Peyvandi, F.; Allen, G.; Bernstein, J.; Manco-Johnson, M.J. Fibrinogen replacement therapy for congenital fibrinogen deficiency. J. Thromb. Haemost. 2011, 9, 1687–1704. [Google Scholar] [CrossRef] [PubMed]
- Groupe d’Etude sur 1’Hémostase et la Thrombose, Base de Données des Variants du Fibrinogène GFHT Web Site. Available online: http://site.geht.org/site/Pratiques-Professionnelles/Base-de-donnees-Fibrinogene/Database-English-Version/Fibrinogen-variants-Database-_79_.html (accessed on 3 April 2017).
- Asselta, R.; Duga, S.; Spena, S.; Peyvandi, F.; Castaman, G.; Malcovati, M.; Mannucci, P.M.; Tenchini, M.L. Missense or splicing mutation? The case of a fibrinogen Bbeta-chain mutation causing severe hypofibrinogenemia. Blood 2004, 103, 3051–3054. [Google Scholar] [CrossRef] [PubMed]
- Tompson, S.W.; Young, T.L. Assaying the Effects of Splice Site Variants by Exon Trapping in a Mammalian Cell Line. Bio-Protocol 2017, 7, e2281. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Lukowski, S.; Quintard, V.L.; Crutu, A.; Zak, M.; Regazzoni, S.; de Moerloose, P.; Neerman-Arbez, M. FGB mutations leading to congenital quantitative fibrinogen deficiencies: An update and report of four novel mutations. Thromb. Res. 2014, 133, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Brennan, S.O.; Davis, R.L.; Lowen, R.; Ruskova, A. Deletion of five residues from the coiled coil of fibrinogen (Bbeta Asn167_Glu171del) associated with bleeding and hypodysfibrinogenemia. Haematologica 2009, 94, 585–588. [Google Scholar] [CrossRef] [PubMed]
- Hanss, M.; Ffrench, P.; Vinciguerra, C.; Bertrand, M.A.; Mazancourt, P. Four cases of hypofibrinogenemia associated with four novel mutations. J. Thromb. Haemost. 2005, 3, 2347–2349. [Google Scholar] [CrossRef] [PubMed]
- Brennan, S.O.; Laurie, A.D.; Bell, J.A. Novel FGB mutation Bβ240Cys→Arg confirms importance of the Bβ211-240 disulphide for plasma expression of fibrinogen. Thromb. Res. 2016, 147, 94–96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Z.; Redman, C.M. Assembly and secretion of fibrinogen. Involvement of amino-terminal domains in dimer formation. J. Biol. Chem. 1996, 271, 12674–12680. [Google Scholar] [CrossRef] [PubMed]
- Hanss, M.; Pouymayou, C.; Blouch, M.T.; Lellouche, F.; Ffrench, P.; Rousson, R.; Abgrall, J.F.; Morange, P.E.; Quélin, F.; de Mazancourt, P. The natural occurrence of human fibrinogen variants disrupting inter-chain disulfide bonds (A{alpha}Cys36Gly, A{alpha}Cys36Arg and A{alpha}Cys45Tyr) confirms the role of N-terminal A{alpha} disulfide bonds in protein assembly and secretion. Haematologica 2011, 96, 1226–1230. [Google Scholar] [CrossRef] [PubMed]
- Platè, M.; Asselta, R.; Spena, S.; Spreafico, M.; Fagoonee, S.; Peyvandi, F.; Tenchini, M.L.; Duga, S. Congenital hypofibrinogenemia: Characterization of two missense mutations affecting fibrinogen assembly and secretion. Blood Cells Mol. Dis. 2008, 41, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Terasawa, F.; Fujita, K.; Okumura, N. Residue gamma153Cys is essential for the formation of the complexes Aalphagamma and Bbetagamma, assembly intermediates for the AalphaBbetagamma complex and intact fibrinogen. Clin. Chim. Acta 2005, 353, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Soya, K.; Takezawa, Y.; Okumura, N.; Terasawa, F. Nonsense-mediated mRNA decay was demonstrated in two hypofibrinogenemias caused by heterozygous nonsense mutations of FGG, Shizuoka III and Kanazawa II. Thromb. Res. 2013, 132, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Terasawa, F.; Kamijyo, Y.; Fujihara, N.; Yamauchi, K.; Kumagai, T.; Honda, T.; Shigematsu, S.; Okumura, N. In vitro transcription of compound heterozygous hypofibrinogenemia Matsumoto IX; first identification of FGB IVS6 deletion of 4 nucleotides and FGG IVS3-2A>G causing abnormal RNA splicing. Clin. Chim. Acta 2010, 411, 1325–1329. [Google Scholar] [CrossRef] [PubMed]
- Mukai, S.; Nagata, K.; Ikeda, M.; Arai, S.; Sugano, M.; Honda, T.; Okumura, N. Genetic analyses of novel compound heterozygous hypodysfibrinogenemia, Tsukuba I: FGG c.1129+62_65 del AATA and FGG c.1299+4 del A. Thromb. Res. 2016, 148, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Arai, S.; Mukai, S.; Takezawa, Y.; Terasawa, F.; Okumura, N. Novel heterozygous dysfibrinogenemia, Sumida (AαC472S), showed markedly impaired lateral aggregation of protofibrils and mildly lower functional fibrinogen levels. Thromb. Res. 2015, 135, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Mukai, S.; Ikeda, M.; Takezawa, Y.; Sugano, M.; Honda, T.; Okumura, N. Differences in the function and secretion of congenital aberrant fibrinogenemia between heterozygous γD320G (Okayama II) and γΔN319-ΔD320 (Otsu I). Thromb. Res. 2015, 136, 1318–1324. [Google Scholar] [CrossRef] [PubMed]
- Okumura, N.; Terasawa, F.; Tanaka, H.; Hirota, M.; Ota, H.; Kitano, K.; Kiyosawa, K.; Lord, S.T. Analysis of fibrinogen gamma-chain truncations shows the C-terminus, particularly gammaIle387, is essential for assembly and secretion of this multichain protein. Blood 2002, 99, 3654–3660. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taira, C.; Matsuda, K.; Arai, S.; Sugano, M.; Uehara, T.; Okumura, N. A Novel Mutation in the Fibrinogen Bβ Chain (c.490G>A; End of Exon 3) Causes a Splicing Abnormality and Ultimately Leads to Congenital Hypofibrinogenemia. Int. J. Mol. Sci. 2017, 18, 2470. https://doi.org/10.3390/ijms18112470
Taira C, Matsuda K, Arai S, Sugano M, Uehara T, Okumura N. A Novel Mutation in the Fibrinogen Bβ Chain (c.490G>A; End of Exon 3) Causes a Splicing Abnormality and Ultimately Leads to Congenital Hypofibrinogenemia. International Journal of Molecular Sciences. 2017; 18(11):2470. https://doi.org/10.3390/ijms18112470
Chicago/Turabian StyleTaira, Chiaki, Kazuyuki Matsuda, Shinpei Arai, Mitsutoshi Sugano, Takeshi Uehara, and Nobuo Okumura. 2017. "A Novel Mutation in the Fibrinogen Bβ Chain (c.490G>A; End of Exon 3) Causes a Splicing Abnormality and Ultimately Leads to Congenital Hypofibrinogenemia" International Journal of Molecular Sciences 18, no. 11: 2470. https://doi.org/10.3390/ijms18112470
APA StyleTaira, C., Matsuda, K., Arai, S., Sugano, M., Uehara, T., & Okumura, N. (2017). A Novel Mutation in the Fibrinogen Bβ Chain (c.490G>A; End of Exon 3) Causes a Splicing Abnormality and Ultimately Leads to Congenital Hypofibrinogenemia. International Journal of Molecular Sciences, 18(11), 2470. https://doi.org/10.3390/ijms18112470