Computational Methods for Modeling Aptamers and Designing Riboswitches

Abstract
:
1. Introduction
2. Computational Method for Predicting Aptamer Structures
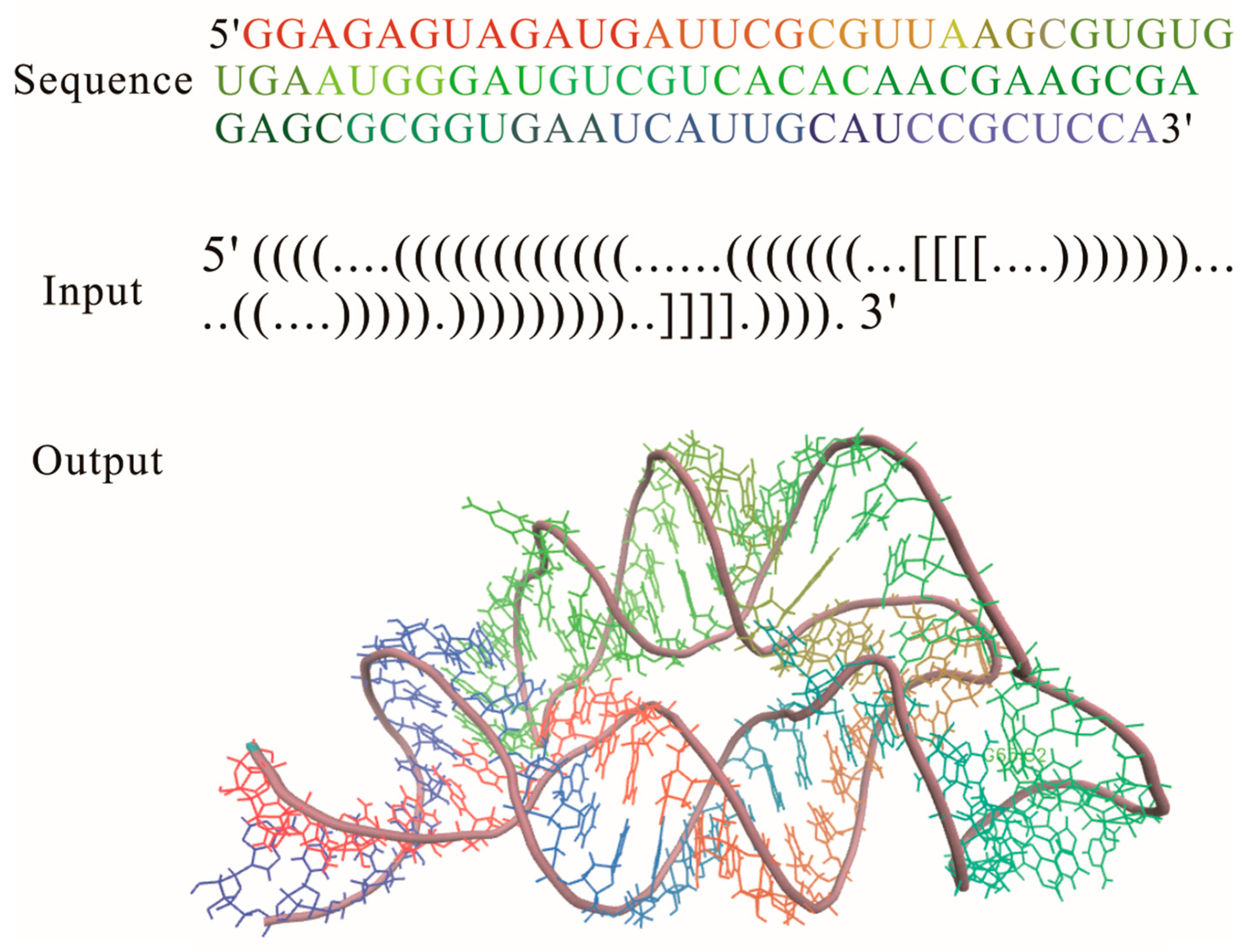
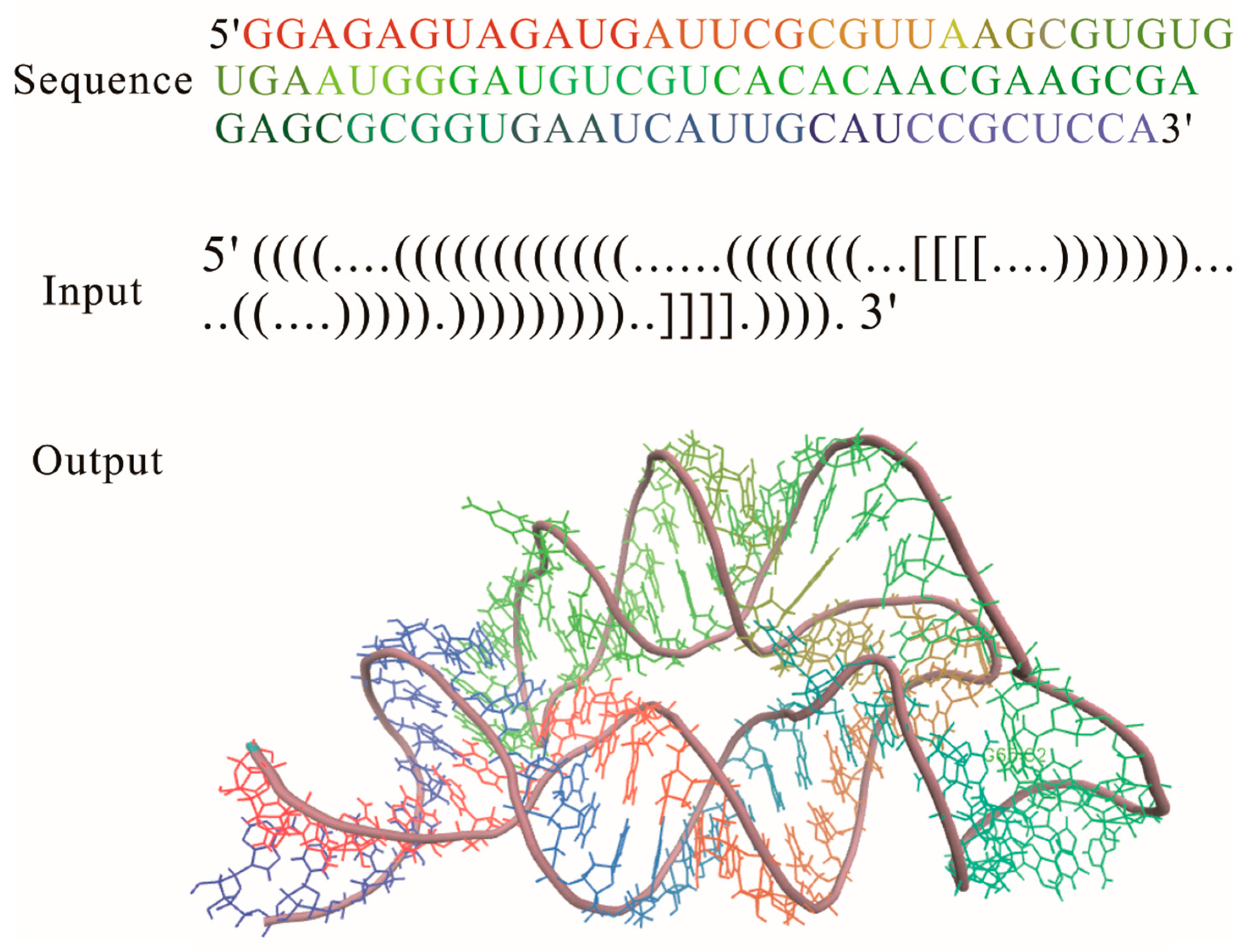
2.1. RNAComposer
2.2. Rosetta and Discrete Molecular Dynamics
3. Computation Model to Characterize Structural Changes in Aptamers
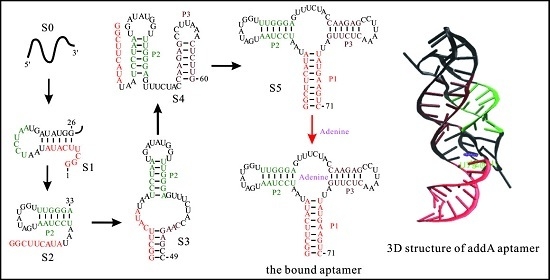
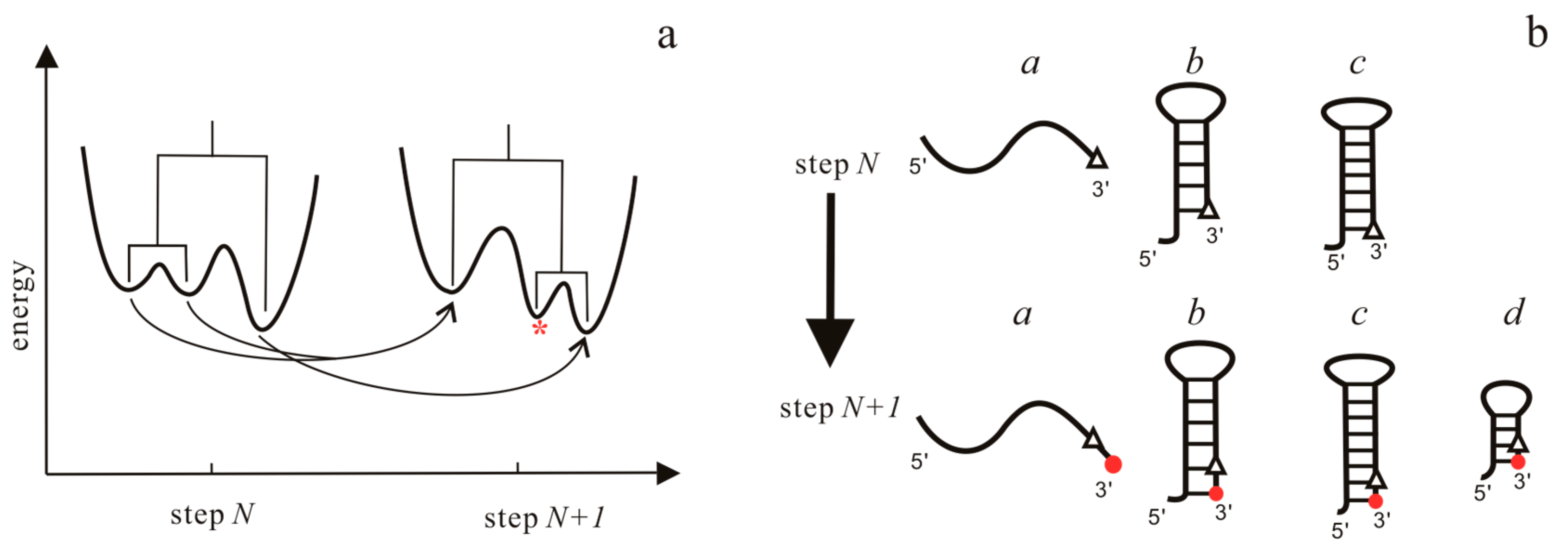
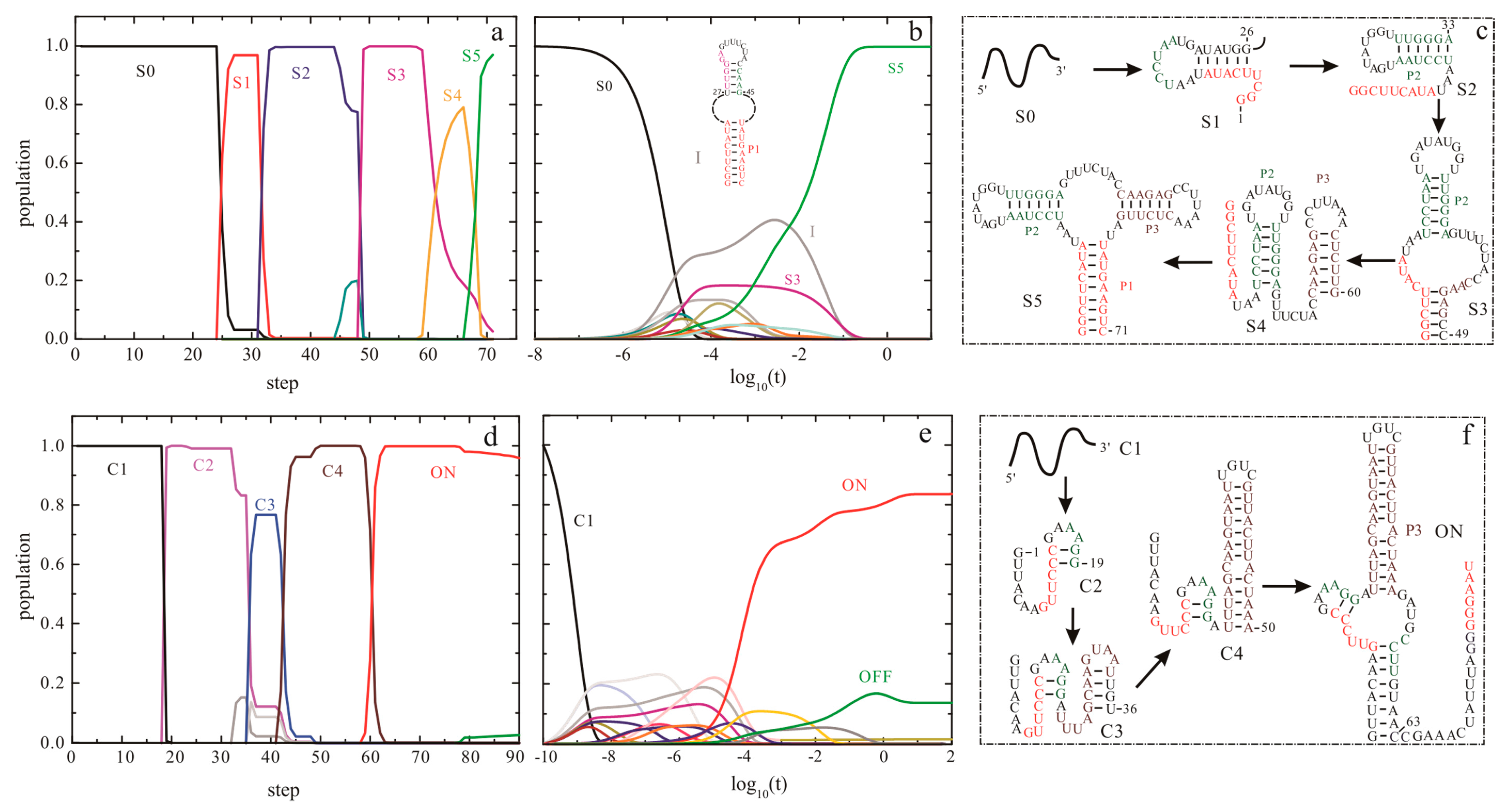
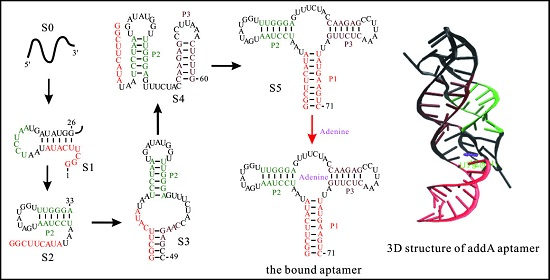
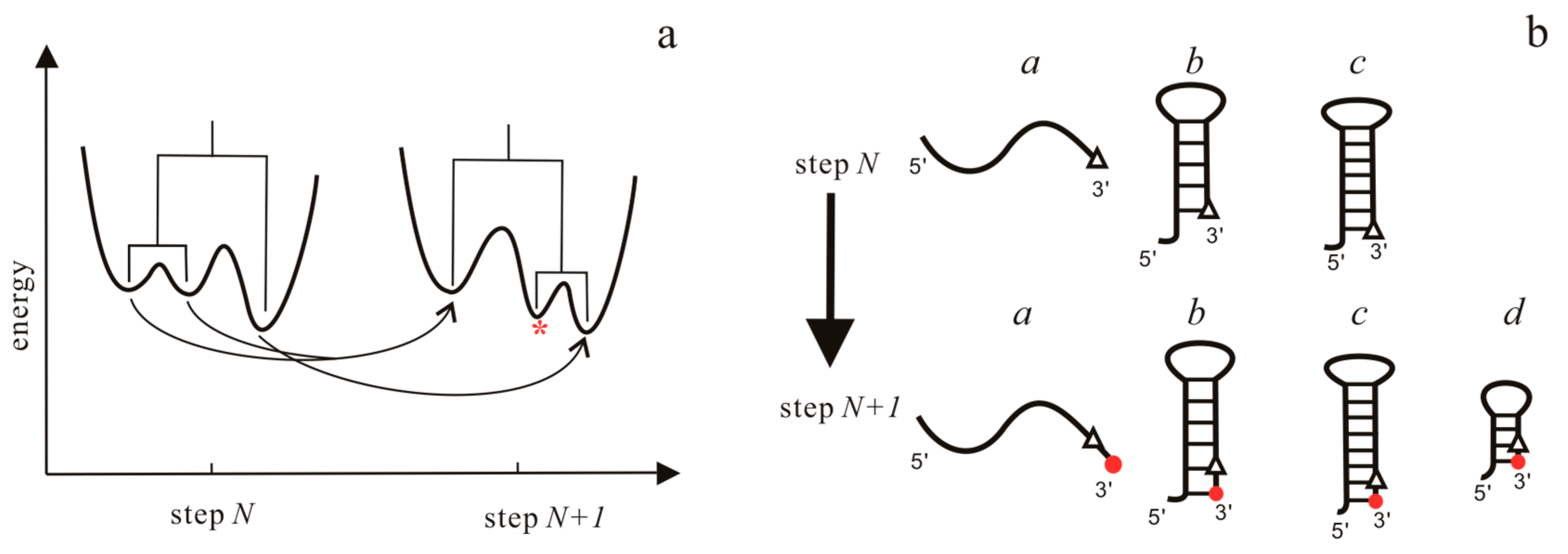
3.1. The Master Equation Approach
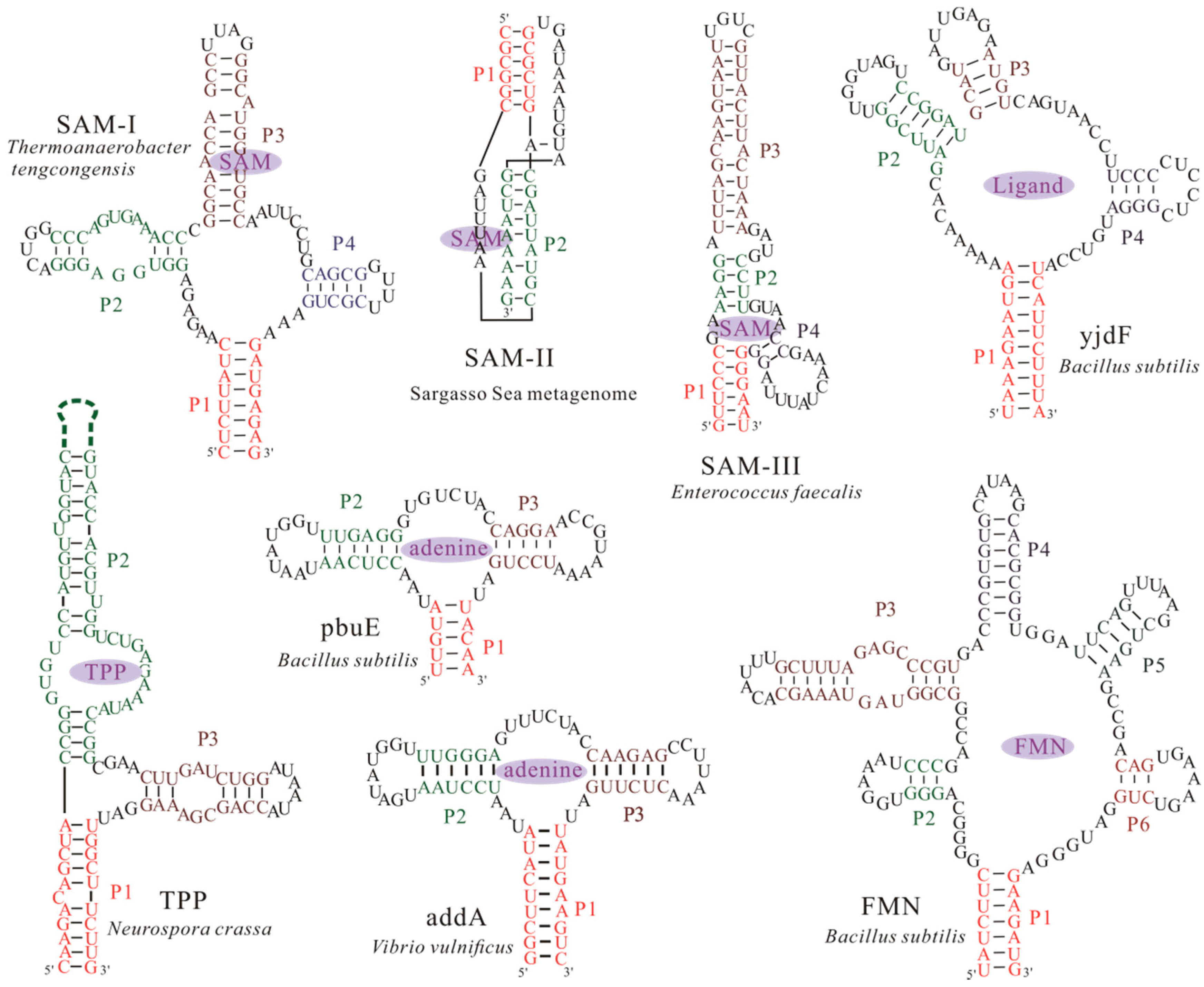
3.2. Coarse-Grained SOP Model
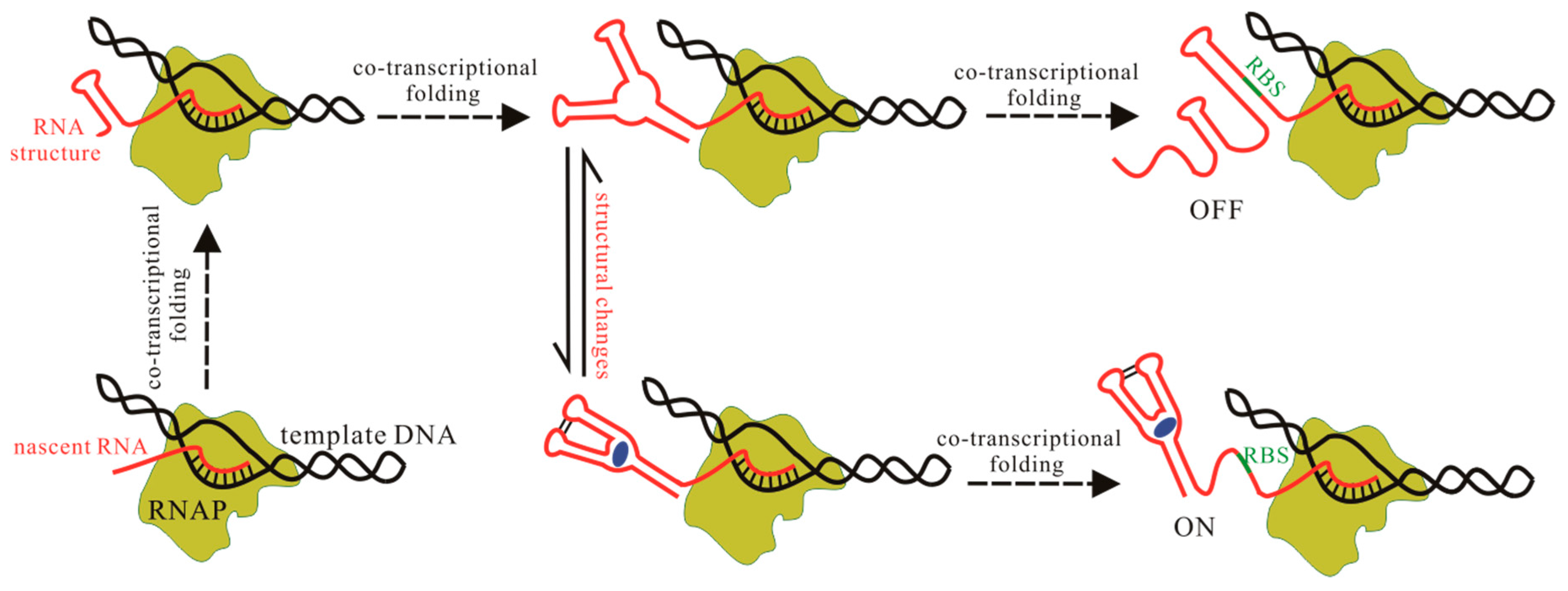
4. Methods to Predict the Structure Transitions during Transcription
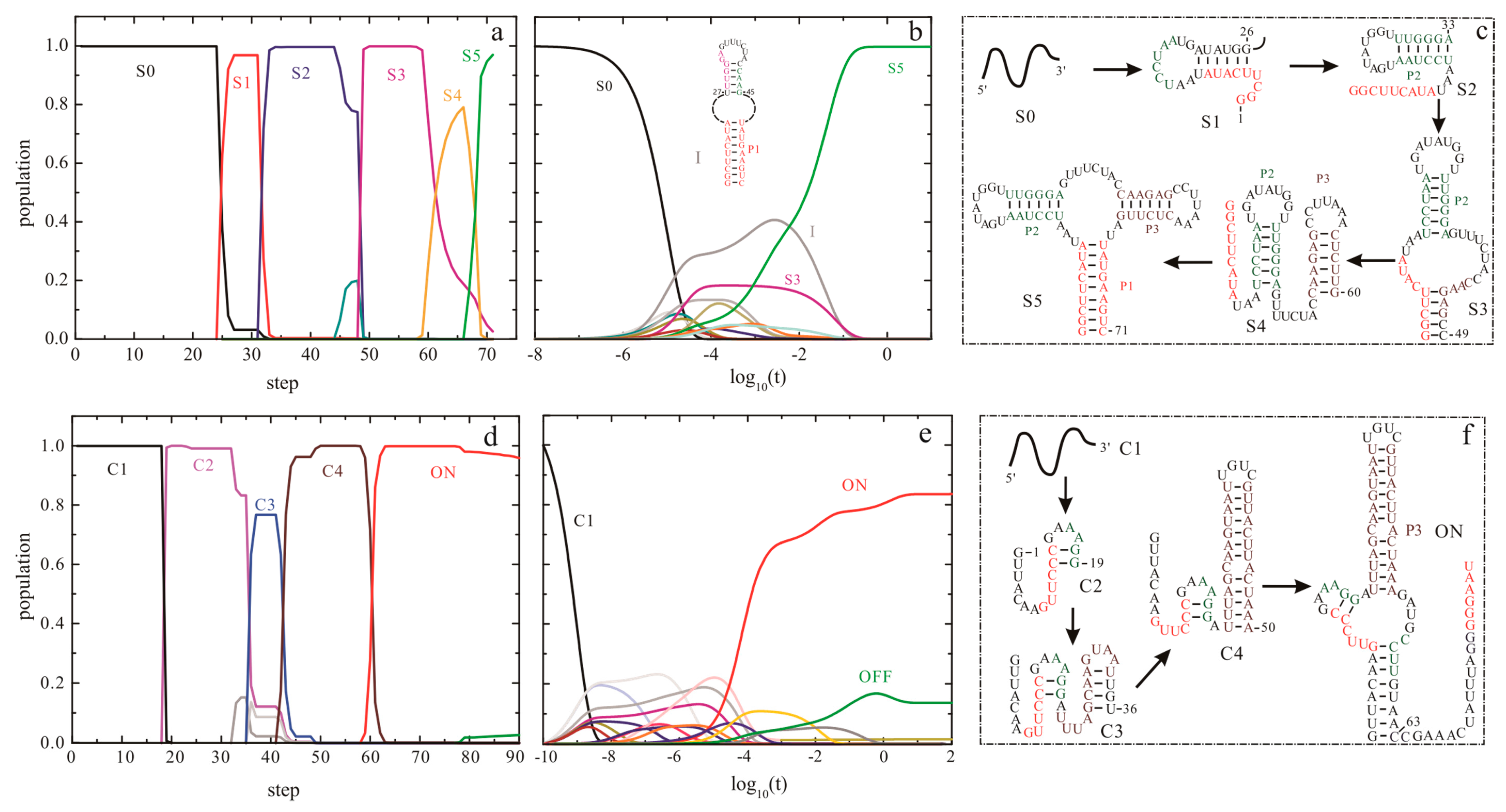
4.1. BarMap
4.2. The Helix-Based Computational Method
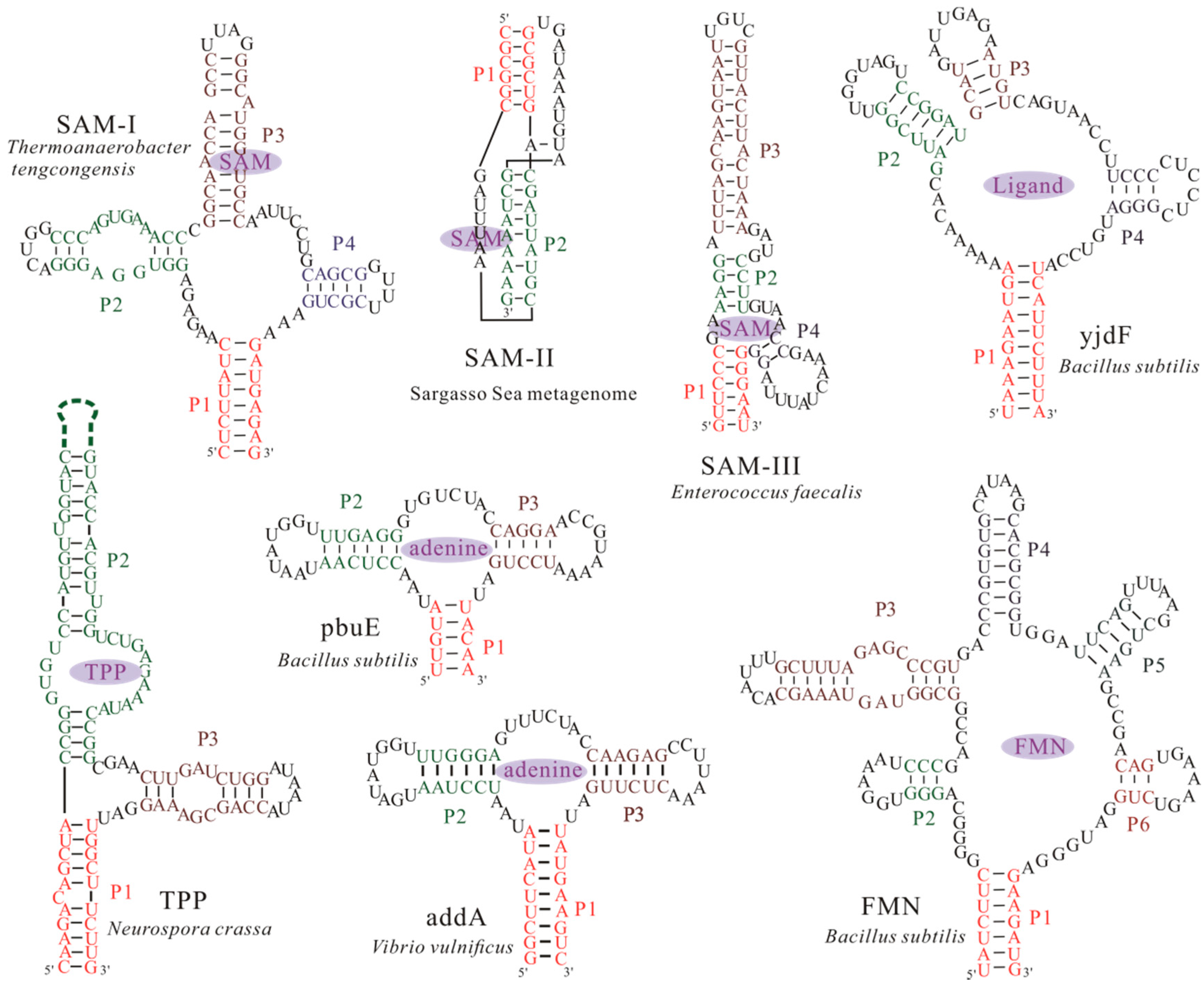
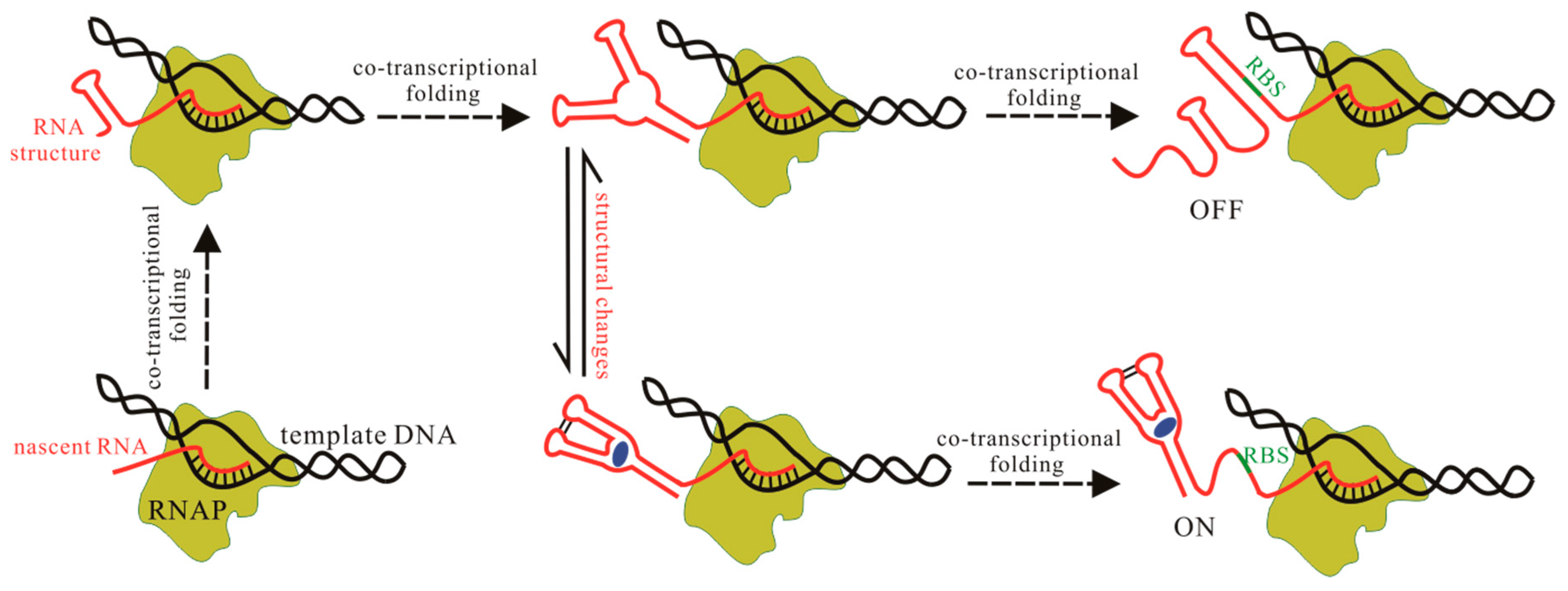
5. Computation Design for Synthetic Riboswitches
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ncRNA | noncoding RNA |
| SOP | Self-Organized Polymer |
| MC | Monte Carlo |
| FMN | Flavin Mononucleotide |
| SAM | S-adenosylmethionine |
| TPP | Thiamine Pyrophosphate |
| NMR | Nuclear Magnetic Resonance |
| MD | Molecular Dynamics |
| RNAP | RNA polymerase |
| RMSD | Root Mean Square Deviation |
| FARNA | Fragment Assembly of RNA |
| RAGTOP | RNA-As-Graph-Topologies |
References
- Breaker, R.R. Riboswitches and the RNA world. Cold Spring Harb. Perspect. Biol. 2012, 4, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Winkler, W.C. Riboswitches and the role of noncoding RNAs in bacterial metabolic control. Curr. Opin. Chem. Biol. 2005, 9, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Cech, T.R.; Steitz, J.A. The noncoding RNA revolution—Trashing old rules to forge new ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gottesman, S. Riboswitch regulates RNA. Science 2014, 345, 876–877. [Google Scholar] [CrossRef] [PubMed]
- Tucker, B.J.; Breaker, R.R. Riboswitches as versatile gene control elements. Curr. Opin. Struct. Biol. 2005, 15, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.L.; Sudarsan, N.; Weinberg, Z.; Roth, A.; Stockbridge, R.B.; Breaker, R.R. Widespread Genetic Switches and Toxicity Resistance Proteins for Fluoride. Science 2012, 335, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Dambach, M.; Sandoval, M.; Updegrove, T.B.; Anantharaman, V.; Aravind, L.; Waters, L.S.; Storz, G. The Ubiquitous yybP-ykoY Riboswitch Is a Manganese-Responsive Regulatory Element. Mol. Cell 2015, 57, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Dann, C.E.; Wakeman, C.A.; Sieling, C.L.; Baker, S.C.; Irnov, I.; Winkler, W.C. Structure and Mechanism of a Metal-Sensing Regulatory RNA. Cell 2007, 130, 878–892. [Google Scholar] [CrossRef] [PubMed]
- Price, I.R.; Gaballa, A.; Ding, F.; Helmann, J.D.; Ke, A. Mn2+-Sensing Mechanisms of yybP-ykoY Orphan Riboswitches. Mol. Cell 2015, 57, 1110–1123. [Google Scholar] [CrossRef] [PubMed]
- Cromie, M.J.; Shi, Y.; Latifi, T.; Groisman, E.A. An RNA Sensor for Intracellular Mg2+. Cell 2006, 125, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Breaker, R.R. Identification of 15 candidate structured noncoding RNA motifs in fungi by comparative genomics. BMC Genom. 2017, 18, 785. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, Z.; Lünse, C.E.; Corbino, K.A.; Ames, T.D.; Nelson, J.W.; Roth, A.; Perkins, K.R.; Sherlock, M.E.; Breaker, R.R. Detection of 224 candidate structured RNAs by comparative analysis of specific subsets of intergenic regions. Nucleic Acids Res. 2017, 45, 10811–10823. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, R.T.; Grundy, F.J.; Henkin, T.M. S-adenosylmethionine directly inhibits binding of 30S ribosomal subunits to the SMK box translational riboswitch RNA. Proc. Natl. Acad. Sci. USA 2007, 104, 4876–4880. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Smith, A.M.; Ding, F.; Chowdhury, A.; Henkin, T.M.; Ke, A. Variable sequences outside the SAM-binding core critically influence the conformational dynamics of the SAM-III/SMK box riboswitch. J. Mol. Biol. 2011, 409, 786–799. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.C.; Smith, A.M.; Fuchs, R.T.; Kleckner, I.R.; Henkin, T.M.; Foster, M.P. Tuning riboswitch regulation through conformational selection. J. Mol. Biol. 2011, 405, 926–938. [Google Scholar] [CrossRef] [PubMed]
- DebRoy, S.; Gebbie, M.; Ramesh, A.; Goodson, J.R.; Cruz, M.R.; van Hoof, A.; Winkler, W.C.; Garsin, D.A. A riboswitch-containing sRNA controls gene expression by sequestration of a response regulator. Science 2014, 345, 937–940. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, A.J.; Lund, P.E.; Blanco, M.R.; Walter, N.G. The Shine-Dalgarno sequence of riboswitch-regulated single mRNAs shows ligand-dependent accessibility bursts. Nat. Commun. 2016, 7, 8976. [Google Scholar] [CrossRef] [PubMed]
- Barrick, J.E.; Breaker, R.R. The distributions, mechanisms, and structures of metabolite-binding riboswitches. Genome Biol. 2007, 8, R239. [Google Scholar] [CrossRef] [PubMed]
- Breaker, R.R. Prospects for Riboswitch Discovery and Analysis. Mol. Cell 2011, 43, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hwang, X.Y.; Stav, S.; Breaker, R.R. The yjdF riboswitch candidate regulates gene expression by binding diverse azaaromatic compounds. RNA 2016, 22, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Serganov, A.; Nudler, E. A Decade of Riboswitches. Cell 2013, 152, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Deigan, K.E.; FerrÉ-D’AmarÉ, A.R. Riboswitches: Discovery of Drugs That Target Bacterial Gene-Regulatory RNAs. Acc. Chem. Res. 2011, 44, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Liberman, J.A.; Wedekind, J.E. Riboswitch structure in the ligand-free state. Wiley Interdiscip. Rev. RNA 2012, 3, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, S.D.; Rambo, R.P.; Van Tyne, D.; Batey, R.T. Structure of the SAM-II riboswitch bound to S-adenosylmethionine. Nat. Struct. Mol. Biol. 2008, 15, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Cheah, M.T.; Wachter, A.; Sudarsan, N.; Breaker, R.R. Control of alternative RNA splicing and gene expression by eukaryotic riboswitches. Nature 2007, 447, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M. A Glycine-Dependent Riboswitch That Uses Cooperative Binding to Control Gene Expression. Science 2004, 306, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Diegelman-Parente, A.; Bevilacqua, P.C. A mechanistic framework for Co-transcriptional folding of the HDV genomic ribozyme in the presence of downstream sequence. J. Mol. Biol. 2002, 324, 1–16. [Google Scholar] [CrossRef]
- Lemay, J.-F.; Desnoyers, G.; Blouin, S.; Heppell, B.; Bastet, L.; St-Pierre, P.; Massé, E.; Lafontaine, D.A. Comparative Study between Transcriptionally- and Translationally-Acting Adenine Riboswitches Reveals Key Differences in Riboswitch Regulatory Mechanisms. PLoS Genet. 2011, 7, e1001278. [Google Scholar] [CrossRef] [PubMed]
- Popenda, M.; Szachniuk, M.; Antczak, M.; Purzycka, K.J.; Lukasiak, P.; Bartol, N.; Blazewicz, J.; Adamiak, R.W. Automated 3D structure composition for large RNAs. Nucleic Acids Res. 2012, 40, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zuo, X.; Wang, Y.-X.; Dayie, T.K. Multiple conformations of SAM-II riboswitch detected with SAXS and NMR spectroscopy. Nucleic Acids Res. 2012, 40, 3117–3130. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhao, P.; Chen, S.J. Vfold: A web server for RNA structure and folding thermodynamics prediction. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010, 11, 129. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Baker, D. Automated de novo prediction of native-like RNA tertiary structures. Proc. Natl. Acad. Sci. USA 2007, 104, 14664–14669. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Karanicolas, J.; Baker, D. Atomic accuracy in predicting and designing noncanonical RNA structure. Nat. Methods 2010, 7, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Jonikas, M.A.; Radmer, R.J.; Altman, R.B. Knowledge-based instantiation of full atomic detail into coarse-grain RNA 3D structural models. Bioinformatics 2009, 25, 3259–3266. [Google Scholar] [CrossRef] [PubMed]
- Parisien, M.; Major, F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature 2008, 452, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.-Z.; Wang, F.-H.; Wu, Y.-Y.; Tan, Z.-J. A coarse-grained model with implicit salt for RNAs: Predicting 3D structure, stability and salt effect. J. Chem. Phys. 2014, 141, 105102. [Google Scholar] [CrossRef] [PubMed]
- Purzycka, K.J.; Popenda, M.; Szachniuk, M.; Antczak, M.; Lukasiak, P.; Blazewicz, J.; Adamiak, R.W. Automated 3D RNA Structure Prediction Using the RNAComposer Method for Riboswitches. In Methods in Enzymology; Elsevier Inc.: Waltham, MA, USA, 2015; Volume 553, pp. 3–34. ISBN 0076-6879. [Google Scholar]
- Ding, F.; Lavender, C.A.; Weeks, K.M.; Dokholyan, N.V. Three-dimensional RNA structure refinement by hydroxyl radical probing. Nat. Methods 2012, 9, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Higgs, P.G. RNA secondary structure: Physical and computational aspects. Q. Rev. Biophys. 2000, 33, 199–253. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Bulusu, G.; Mitra, A. MD simulations of ligand-bound and ligand-free aptamer: Molecular level insights into the binding and switching mechanism of the add A-riboswitch. RNA 2009, 15, 1673–1692. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-C.; Thirumalai, D. Relative stability of helices determines the folding landscape of adenine riboswitch aptamers. J. Am. Chem. Soc. 2008, 130, 14080–14081. [Google Scholar] [CrossRef] [PubMed]
- Hyeon, C.; Thirumalai, D. Mechanical Unfolding of RNA: From Hairpins to Structures with Internal Multiloops. Biophys. J. 2007, 92, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-C.; Hyeon, C.; Thirumalai, D. Sequence-dependent folding landscapes of adenine riboswitch aptamers. Phys. Chem. Chem. Phys. 2014, 16, 6376–6382. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-C.C.; Thirumalai, D. Kinetics of allosteric transitions in S-adenosylmethionine riboswitch are accurately predicted from the folding landscape. J. Am. Chem. Soc. 2013, 135, 16641–16650. [Google Scholar] [CrossRef] [PubMed]
- Proctor, J.R.; Meyer, I.M. CoFold: An RNA secondary structure prediction method that takes co-transcriptional folding into account. Nucleic Acids Res. 2013, 41, e102. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Zhang, W.-B.; Chen, S.-J. Predicting Secondary Structural Folding Kinetics for Nucleic Acids. Biophys. J. 2010, 98, 1617–1625. [Google Scholar] [CrossRef] [PubMed]
- Holtkamp, W.; Kokic, G.; Jäger, M.; Mittelstaet, J.; Komar, A.A.; Rodnina, M.V. Cotranslational protein folding on the ribosome monitored in real time. Science 2015, 350, 1104–1107. [Google Scholar] [CrossRef] [PubMed]
- Frieda, K.L.; Block, S.M. Direct Observation of Cotranscriptional Folding in an Adenine Riboswitch. Science 2012, 338, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Wickiser, J.K.; Winkler, W.C.; Breaker, R.R.; Crothers, D.M. The Speed of RNA Transcription and Metabolite Binding Kinetics Operate an FMN Riboswitch. Mol. Cell 2005, 18, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Hennelly, S.P.; Novikova, I.V.; Sanbonmatsu, K.Y. The expression platform and the aptamer: Cooperativity between Mg2+ and ligand in the SAM-I riboswitch. Nucleic Acids Res. 2013, 41, 1922–1935. [Google Scholar] [CrossRef] [PubMed]
- Lemay, J.-F.; Penedo, J.C.; Tremblay, R.; Lilley, D.M.J.; Lafontaine, D.A. Folding of the Adenine Riboswitch. Chem. Biol. 2006, 13, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Watters, K.E.; Strobel, E.J.; Yu, A.M.; Lis, J.T.; Lucks, J.B. Cotranscriptional folding of a riboswitch at nucleotide resolution. Nat. Struct. Mol. Biol. 2016, 23, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Lutz, B.; Faber, M.; Verma, A.; Klumpp, S.; Schug, A. Differences between cotranscriptional and free riboswitch folding. Nucleic Acids Res. 2014, 42, 2687–2696. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Wang, Y.; Wang, Z.; Wang, Y.; Zhang, W. Reversible-Switch Mechanism of the SAM-III Riboswitch. J. Phys. Chem. B 2016, 120, 12305–12311. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Wang, Y.; Zhang, W. Kinetic regulation mechanism of pbuE riboswitch. J. Chem. Phys. 2015, 142, 15103. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Wang, Y.; Zhang, W. The regulation mechanism of yitJ and metF riboswitches. J. Chem. Phys. 2015, 143, 45103. [Google Scholar] [CrossRef] [PubMed]
- Hofacker, I.L.; Flamm, C.; Heine, C.; Wolfinger, M.T.; Scheuermann, G.; Stadler, P.F. BarMap: RNA folding on dynamic energy landscapes. RNA 2010, 16, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Zhang, W.; Chen, S.-J. Cotranscriptional folding kinetics of ribonucleic acid secondary structures. J. Chem. Phys. 2011, 135, 245101. [Google Scholar] [CrossRef] [PubMed]
- Wachsmuth, M.; Findeiß, S.; Weissheimer, N.; Stadler, P.F.; Mörl, M. De novo design of a synthetic riboswitch that regulates transcription termination. Nucleic Acids Res. 2013, 41, 2541–2551. [Google Scholar] [CrossRef] [PubMed]
- Espah Borujeni, A.; Mishler, D.M.; Wang, J.; Huso, W.; Salis, H.M. Automated physics-based design of synthetic riboswitches from diverse RNA aptamers. Nucleic Acids Res. 2016, 44, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Huang, Y.; Gong, Z.; Wang, Y.; Man, J.; Xiao, Y. Automated and fast building of three-dimensional RNA structures. Sci. Rep. 2012, 2, 734. [Google Scholar] [CrossRef] [PubMed]
- Hofacker, I.L. RNA Secondary Structure Analysis Using the Vienna RNA Package. In Current Protocols in Bioinformatics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; pp. 1–16. ISBN 0471250953. [Google Scholar]
- Puton, T.; Kozlowski, L.P.; Rother, K.M.; Bujnicki, J.M. CompaRNA: A server for continuous benchmarking of automated methods for RNA secondary structure prediction. Nucleic Acids Res. 2013, 41, 4307–4323. [Google Scholar] [CrossRef] [PubMed]
- Bernhart, S.H.; Hofacker, I.L.; Will, S.; Gruber, A.R.; Stadler, P.F. RNAalifold: Improved consensus structure prediction for RNA alignments. BMC Bioinform. 2008, 9, 474. [Google Scholar] [CrossRef] [PubMed]
- Sükösd, Z.; Knudsen, B.; Vaerum, M.; Kjems, J.; Andersen, E.S. Multithreaded comparative RNA secondary structure prediction using stochastic context-free grammars. BMC Bioinform. 2011, 12, 103. [Google Scholar] [CrossRef] [PubMed]
- Popenda, M.; Szachniuk, M.; Blazewicz, M.; Wasik, S.; Burke, E.K.; Blazewicz, J.; Adamiak, R.W. RNA FRABASE 2.0: An advanced web-accessible database with the capacity to search the three-dimensional fragments within RNA structures. BMC Bioinform. 2010, 11, 231. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Rios, J.; Navarro, M.; Soberon, M. A conserved RNA structure (thi box) is involved in regulation of thiamin biosynthetic gene expression in bacteria. Proc. Natl. Acad. Sci. USA 2001, 98, 9736–9741. [Google Scholar] [CrossRef] [PubMed]
- Zirbel, C.L.; Roll, J.; Sweeney, B.A.; Petrov, A.I.; Pirrung, M.; Leontis, N.B. Identifying novel sequence variants of RNA 3D motifs. Nucleic Acids Res. 2015, 43, 7504–7520. [Google Scholar] [CrossRef] [PubMed]
- Rahrig, R.R.; Leontis, N.B.; Zirbel, C.L. R3D align: Global pairwise alignment of RNA 3D structures using local superpositions. Bioinformatics 2010, 26, 2689–2697. [Google Scholar] [CrossRef] [PubMed]
- Kladwang, W.; Chou, F.C.; Das, R. Automated RNA structure prediction uncovers a kink-turn linker in double glycine riboswitches. J. Am. Chem. Soc. 2012, 134, 1404–1407. [Google Scholar] [CrossRef] [PubMed]
- Cruz, J.A.; Westhof, E. Sequence-based identification of 3D structural modules in RNA with RMDetect. Nat. Methods 2011, 8, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Zahran, M.; Schlick, T. Computational Prediction of Riboswitch Tertiary Structures Including Pseudoknots by RAGTOP. In Methods in Enzymology; Elsevier Inc.: Waltham, MA, USA, 2015; Volume 553, pp. 115–135. ISBN 1557-7988 (Electronic) 0076-6879. [Google Scholar]
- Krokhotin, A.; Houlihan, K.; Dokholyan, N.V. iFoldRNA v2: Folding RNA with constraints. Bioinformatics 2015, 31, 2891–2893. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Laing, C.; Elmetwaly, S.; Jung, S.; Curuksu, J.; Schlick, T. Graph-based sampling for approximating global helical topologies of RNA. Proc. Natl. Acad. Sci. USA 2014, 111, 4079–4084. [Google Scholar] [CrossRef] [PubMed]
- Krokhotin, A.; Dokholyan, N. V Computational methods toward accurate RNA structure prediction using coarse-grained and all-atom models. Methods Enzymol. 2015, 553, 65–89. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gong, S.; Wang, Z.; Zhang, W. The thermodynamics and kinetics of a nucleotide base pair. J. Chem. Phys. 2016, 144, 115101. [Google Scholar] [CrossRef] [PubMed]
- Gusarov, I.; Nudler, E. Control of intrinsic transcription termination by N and NusA: The basic mechanisms. Cell 2001, 107, 437–449. [Google Scholar] [CrossRef]
- Chen, J.; Gong, S.; Wang, Y.; Zhang, W. Kinetic partitioning mechanism of HDV ribozyme folding. J. Chem. Phys. 2014, 140, 25102. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, W. Kinetic analysis of the effects of target structure on siRNA efficiency. J. Chem. Phys. 2012, 137, 225102. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; SantaLucia, J.; Burkard, M.E.; Kierzek, R.; Schroeder, S.J.; Jiao, X.; Cox, C.; Turner, D.H. Thermodynamic Parameters for an Expanded Nearest-Neighbor Model for Formation of RNA Duplexes with Watson−Crick Base Pairs. Biochemistry 1998, 37, 14719–14735. [Google Scholar] [CrossRef] [PubMed]
- Mathews, D.H.; Sabina, J.; Zuker, M.; Turner, D.H. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999, 288, 911–940. [Google Scholar] [CrossRef] [PubMed]
- Hyeon, C.; Morrison, G.; Thirumalai, D. Force-dependent hopping rates of RNA hairpins can be estimated from accurate measurement of the folding landscapes. Proc. Natl. Acad. Sci. USA 2008, 105, 9604–9609. [Google Scholar] [CrossRef] [PubMed]
- Hyeon, C.; Dima, R.I.; Thirumalai, D. Pathways and Kinetic Barriers in Mechanical Unfolding and Refolding of RNA and Proteins. Structure 2006, 14, 1633–1645. [Google Scholar] [CrossRef] [PubMed]
- Rieder, R.; Lang, K.; Graber, D.; Micura, R. Ligand-Induced Folding of the Adenosine Deaminase A-Riboswitch and Implications on Riboswitch Translational Control. ChemBioChem 2007, 8, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, R.; Grossberger, R.; Pichler, A.; Waldsich, C. RNA folding in vivo. Curr. Opin. Struct. Biol. 2002, 12, 296–300. [Google Scholar] [CrossRef]
- Badelt, S.; Hammer, S.; Flamm, C.; Hofacker, I.L. Thermodynamic and kinetic folding of riboswitches. Methods Enzymol. 2015, 553, 193–213. [Google Scholar] [CrossRef] [PubMed]
- Wolfinger, M.T.; Svrcek-Seiler, W.A.; Flamm, C.; Hofacker, I.L.; Stadler, P.F. Efficient computation of RNA folding dynamics. J. Phys. A. Math. Gen. 2004, 37, 4731–4741. [Google Scholar] [CrossRef]
- Flamm, C.; Hofacker, I.L.; Stadler, P.F.; Wolfinger, M.T. Barrier Trees of Degenerate Landscapes. Z. Phys. Chem. 2002, 216, 155. [Google Scholar] [CrossRef]
- Geis, M.; Flamm, C.; Wolfinger, M.T.; Tanzer, A.; Hofacker, I.L.; Middendorf, M.; Mandl, C.; Stadler, P.F.; Thurner, C. Folding kinetics of large RNAs. J. Mol. Biol. 2008, 379, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Kim, J.; Jha, S.; Aboul-ela, F. A mechanism for S-adenosyl methionine assisted formation of a riboswitch conformation: A small molecule with a strong arm. Nucleic Acids Res. 2009, 37, 6528–6539. [Google Scholar] [CrossRef] [PubMed]
- Chauvier, A.; Picard-Jean, F.; Berger-Dancause, J.-C.; Bastet, L.; Naghdi, M.R.; Dubé, A.; Turcotte, P.; Perreault, J.; Lafontaine, D.A. Transcriptional pausing at the translation start site operates as a critical checkpoint for riboswitch regulation. Nat. Commun. 2017, 8, 13892. [Google Scholar] [CrossRef] [PubMed]
- Perdrizet II, G.A.; Artsimovitch, I.; Furman, R.; Sosnick, T.R.; Pan, T. Transcriptional pausing coordinates folding of the aptamer domain and the expression platform of a riboswitch. Proc. Natl. Acad. Sci. USA 2012, 109, 3323–3328. [Google Scholar] [CrossRef] [PubMed]
- Hollands, K.; Sevostiyanova, A.; Groisman, E.A. Unusually long-lived pause required for regulation of a Rho-dependent transcription terminator. Proc. Natl. Acad. Sci. USA 2014, 111, E1999–E2007. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.N.; Pan, T. RNA Folding During Transcription: Protocols and Studies. In Methods in Enzymology; Elsevier Inc.: San Diego, CA, USA, 2009; Volume 468, pp. 167–193. ISBN 1557-7988 (Electronic)r0076-6879. [Google Scholar]
- Ketzer, P.; Kaufmann, J.K.; Engelhardt, S.; Bossow, S.; von Kalle, C.; Hartig, J.S.; Ungerechts, G.; Nettelbeck, D.M. Artificial riboswitches for gene expression and replication control of DNA and RNA viruses. Proc. Natl. Acad. Sci. USA 2014, 111, E554–E562. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Carothers, J.M.; Keasling, J.D. Design of a dynamic sensor-regulator system for production of chemicals and fuels derived from fatty acids. Nat. Biotechnol. 2012, 30, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Carothers, J.M.; Goler, J.A.; Juminaga, D.; Keasling, J.D. Model-Driven Engineering of RNA Devices to Quantitatively Program Gene Expression. Science 2011, 334, 1716–1719. [Google Scholar] [CrossRef] [PubMed]
- Townshend, B.; Kennedy, A.B.; Xiang, J.S.; Smolke, C.D. High-throughput cellular RNA device engineering. Nat. Methods 2015, 12, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Endoh, T.; Sugimoto, N. Rational Design and Tuning of Functional RNA Switch to Control an Allosteric Intermolecular Interaction. Anal. Chem. 2015, 87, 7628–7635. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.Y.; Smolke, C.D. Engineering dynamic cell cycle control with synthetic small molecule-responsive RNA devices. J. Biol. Eng. 2015, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Espah Borujeni, A.; Channarasappa, A.S.; Salis, H.M. Translation rate is controlled by coupled trade-offs between site accessibility, selective RNA unfolding and sliding at upstream standby sites. Nucleic Acids Res. 2014, 42, 2646–2659. [Google Scholar] [CrossRef] [PubMed]
- McKeague, M.; Wong, R.S.; Smolke, C.D. Opportunities in the design and application of RNA for gene expression control. Nucleic Acids Res. 2016, 44, 2987–2999. [Google Scholar] [CrossRef] [PubMed]
- Zemora, G.; Waldsich, C. RNA folding in living cells. RNA Biol. 2010, 7, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Herschlag, D. RNA Chaperones and the RNA Folding Problem. J. Biol. Chem. 1995, 270, 20871–20874. [Google Scholar] [CrossRef] [PubMed]
- Danilova, L.V.; Pervouchine, D.D.; Favorov, A.V.; Mironov, A.A. RNAKinetics: A web server that models secondary structure kinetics of an elongating RNA. J. Bioinform. Comput. Biol. 2006, 4, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Xayaphoummine, A.; Bucher, T.; Isambert, H. Kinefold web server for RNA/DNA folding path and structure prediction including pseudoknots and knots. Nucleic Acids Res. 2005, 33, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Kramer, F.R.; Mills, D.R. Secondary structure formation during RNA synthesis. Nucleic Acids Res. 1981, 9, 5109–5124. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Zhao, Y.; Chen, C.; Xiao, Y. Role of Ligand Binding in Structural Organization of Add A-riboswitch Aptamer: A Molecular Dynamics Simulation. J. Biomol. Struct. Dyn. 2011, 29, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Philips, A.; Milanowska, K.; Lach, G.; Boniecki, M.; Rother, K.; Bujnicki, J.M. MetalionRNA: Computational predictor of metal-binding sites in RNA structures. Bioinformatics 2012, 28, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Philips, A.; Milanowska, K.; Ach, G.L.; Bujnicki, J.M. LigandRNA : Computational predictor of RNA—Ligand interactions. RNA 2013, 1605–1616. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Description | Availability | Reference |
|---|---|---|---|
| RNAComposer | A motif library-based method that uses the dictionary tailored from RNA FRABASED database to build initial 3D structure. | Web server | [39] |
| Rosetta | A fragment-based method that uses FARFAR optimizes RNA conformations in the context of a physically realistic energy function. | Local installation | [72] |
| RMdetect | A bioinformatics tool for identifying known 3D structural modules on genomic sequences. | Local installation | [73] |
| JAR3D | Scoring sequences to motif groups based on sequences’ ability to form the same pattern of interactions in motif. | Web server | [70] |
| RAGTOP | Predicting RNA topologies by a coarse-grained sampling of 3D graphs guided by statistical knowledge-based potentials. | Not available online | [74] |
| iFoldRNA | Incorporating SHAPE into discrete molecular dynamics to predict RNA structure. | Web server | [75] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, S.; Wang, Y.; Wang, Z.; Zhang, W. Computational Methods for Modeling Aptamers and Designing Riboswitches. Int. J. Mol. Sci. 2017, 18, 2442. https://doi.org/10.3390/ijms18112442
Gong S, Wang Y, Wang Z, Zhang W. Computational Methods for Modeling Aptamers and Designing Riboswitches. International Journal of Molecular Sciences. 2017; 18(11):2442. https://doi.org/10.3390/ijms18112442
Chicago/Turabian StyleGong, Sha, Yanli Wang, Zhen Wang, and Wenbing Zhang. 2017. "Computational Methods for Modeling Aptamers and Designing Riboswitches" International Journal of Molecular Sciences 18, no. 11: 2442. https://doi.org/10.3390/ijms18112442
APA StyleGong, S., Wang, Y., Wang, Z., & Zhang, W. (2017). Computational Methods for Modeling Aptamers and Designing Riboswitches. International Journal of Molecular Sciences, 18(11), 2442. https://doi.org/10.3390/ijms18112442