The Impact of Synaptic Zn2+ Dynamics on Cognition and Its Decline


Abstract
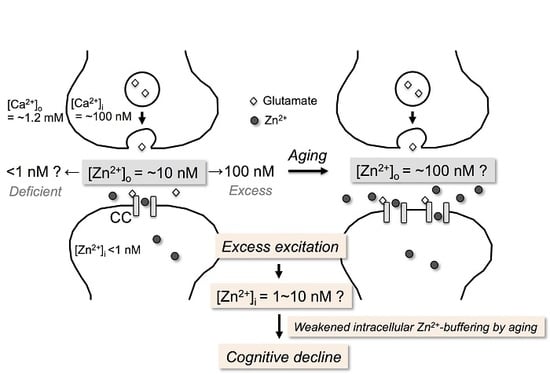
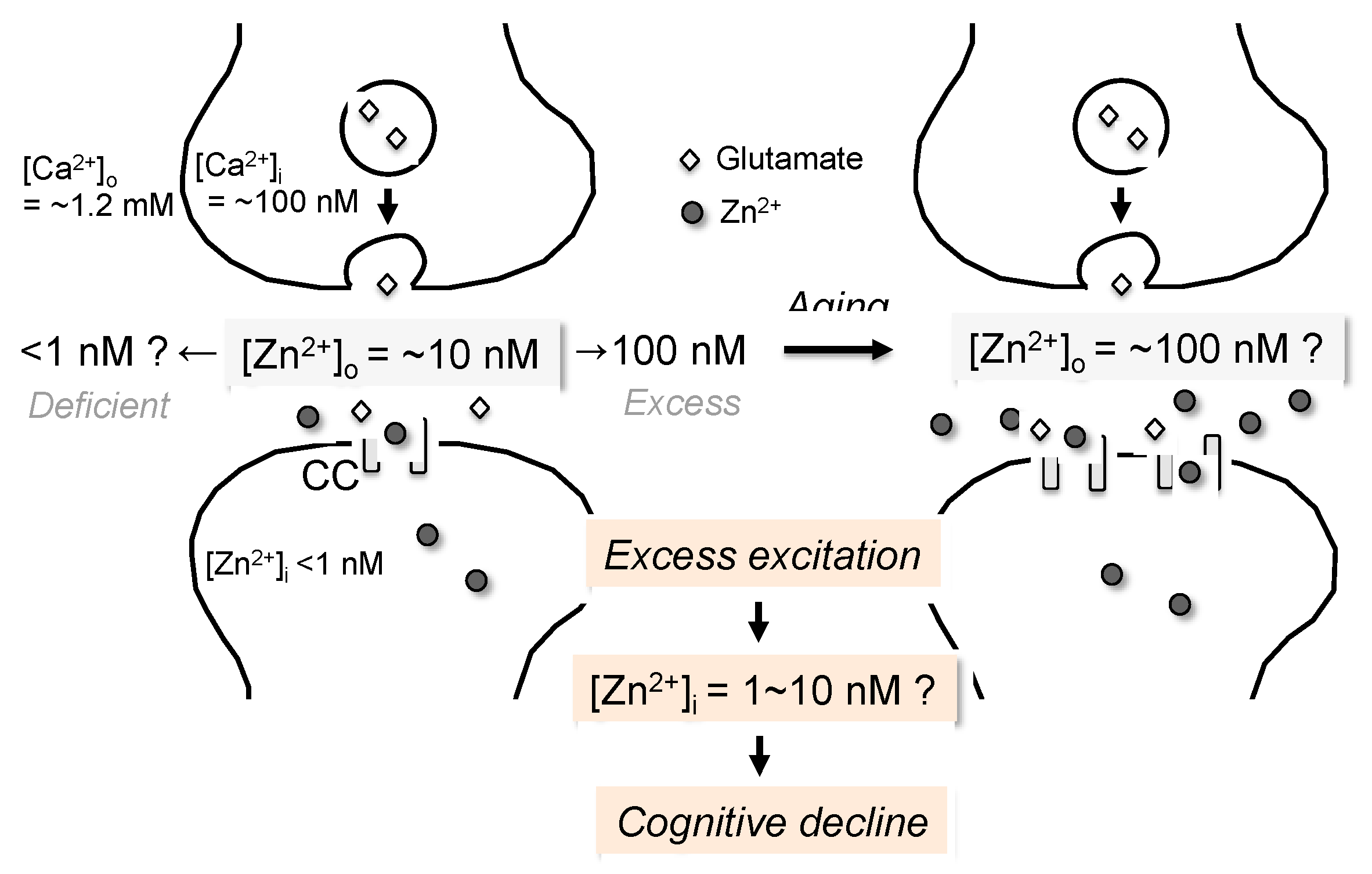
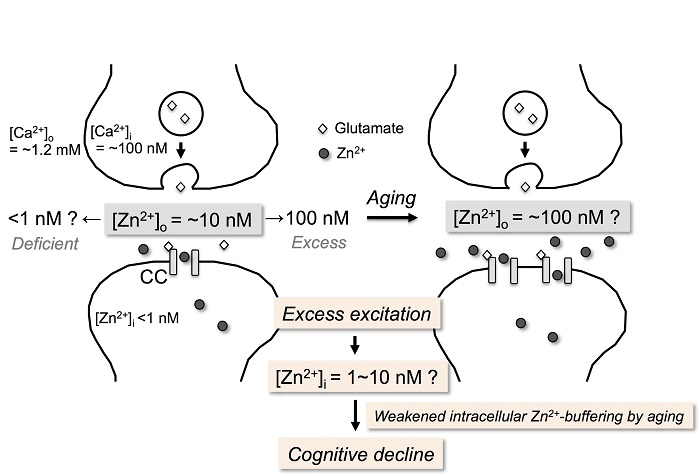{kind=link}
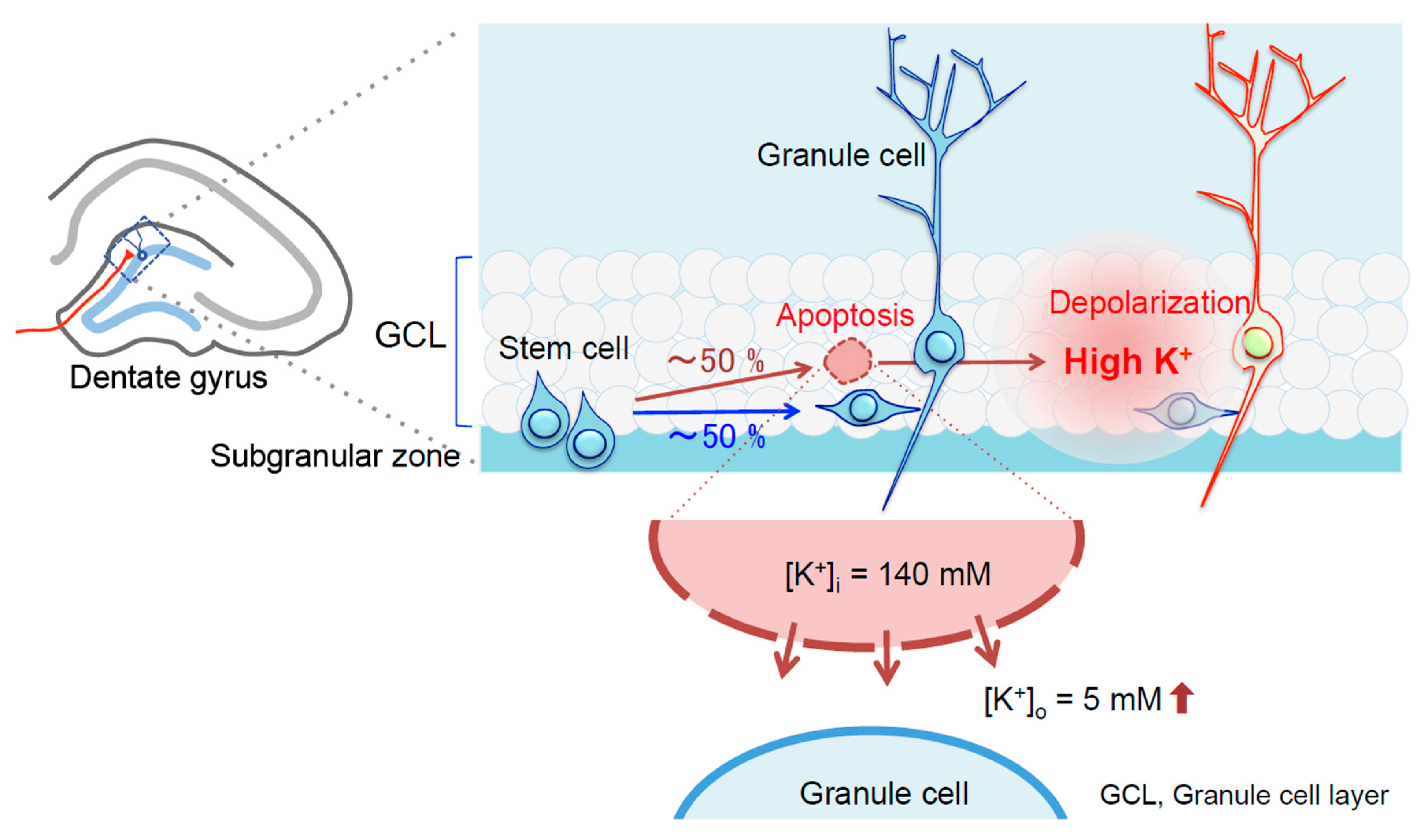
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Physiology of Brain Zn2+
3. Impact of Synaptic Zn2+ Dynamics on Cognition
4. Impact of Synaptic Zn2+ Dynamics on Cognitive Decline
5. Perspectives
Author Contributions
Conflicts of Interest
References
- Knierim, J.J. The hippocampus. Curr. Biol. 2015, 25, R1116–R1121. [Google Scholar] [CrossRef] [PubMed]
- Small, S.A.; Schobel, S.A.; Buxton, R.B.; Witter, M.P.; Barnes, C.A. A pathophysiological framework of hippocampal dysfunction in ageing and disease. Nat. Rev. Neurosci. 2011, 12, 585–601. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Leutgeb, S.; Leutgeb, J.K. Spatial and memory circuits in the medial entorhinal cortex. Curr. Opin. Neurobiol. 2015, 32, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Malenka, R.C.; Nicoll, R.A. Long-term potentiation—A decade of progress? Science 1999, 285, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Blitzer, R.D.; Iyengar, R.; Landau, E.M. Postsynaptic signaling networks: Cellular cogwheels underlying long-term plasticity. Biol. Psychiatry 2005, 57, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, R.A.; Malenka, R.C. Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature 1995, 377, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Maher, P. Chronic glutamate toxicity in neurodegenerative diseases—What is the evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef] [PubMed]
- Lo, E.H.; Dalkara, T.; Moskowitz, M.A. Mechanisms, challenges and opportunities in stroke. Nat. Rev. Neurosci. 2003, 4, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Malairaman, U.; Dandapani, K.; Katyal, A. Effect of Ca2EDTA on zinc mediated inflammation and neuronal apoptosis in hippocampus of an in vivo mouse model of hypobaric hypoxia. PLoS ONE 2014, 9, e110253. [Google Scholar] [CrossRef] [PubMed]
- Medvedeva, Y.V.; Weiss, J.H. Intramitochondrial Zn2+ accumulation via the Ca2+ uniporter contributes to acute ischemic neurodegeneration. Neurobiol. Dis. 2014, 68, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Tamano, H.; Koike, Y.; Nakada, H.; Shakushi, Y.; Takeda, A. Significance of synaptic Zn2+ signaling in zincergic and non-zincergic synapses in the hippocampus in cognition. J. Trace Elem. Med. Biol. 2016, 38, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Tamano, H. Significance of the degree of synaptic Zn2+ signaling in cognition. BioMetals 2016, 29, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Tamano, H. Insight into zinc signaling from dietary zinc deficiency. Brain Res. Rev. 2009, 62, 33–34. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, N.; Jeyaraju, D.V.; Peralta, M.R., 3rd; Seress, L.; Pellegrini, L.; Tóth, K. Vesicular zinc regulates the Ca2+ sensitivity of a subpopulation of presynaptic vesicles at hippocampal mossy fiber terminals. J. Neurosci. 2011, 31, 18251–18265. [Google Scholar] [CrossRef] [PubMed]
- Colbourne, F.; Grooms, S.Y.; Zukin, R.S.; Buchan, A.M.; Bennett, M.V. Hypothermia rescues hippocampal CA1 neurons and attenuates down-regulation of the AMPA receptor GluR2 subunit after forebrain ischemia. Proc. Natl. Acad. Sci. USA 2003, 100, 2906–2910. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Koh, J.Y.; Bush, A.I. The neurobiology of zinc in health and disease. Nat. Rev. Neurosci. 2005, 6, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Noh, K.M.; Yokota, H.; Mashiko, T.; Castillo, P.E.; Zukin, R.S.; Bennett, M.V. Blockade of calcium-permeable AMPA receptors protects hippocampal neurons against global ischemia-induced death. Proc. Natl. Acad. Sci. USA 2005, 102, 12230–12235. [Google Scholar] [CrossRef] [PubMed]
- Stork, C.J.; Li, Y.V. Rising zinc: A significant cause of ischemic neuronal death in the CA1 region of rat hippocampus. J. Cereb. Blood Flow Metab. 2009, 29, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Sensi, S.L.; Paoletti, P.; Bush, A.I.; Sekler, I. Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 2009, 10, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Krężel, A.; Maret, W. The biological inorganic chemistry of zinc ions. Arch. Biochem. Biophys. 2016, 611, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.K.; Robinson, H.P. Effects of divalent cations on slow unblock of native NMDA receptors in mouse neocortical pyramidal neurons. Eur. J. Neurosci. 2011, 34, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.C.; Keep, R.F. Brain fluid calcium concentration and response to acute hypercalcaemia during development in the rat. J. Physiol. 1988, 402, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Neves, G.; Cooke, S.F.; Bliss, T.V.P. Synaptic plasticity, memory and the hippocampus: A neural network approach to causality. Nat. Rev. Neurosci. 2008, 9, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Lisman, J.; Yasuda, R.; Raghavachari, S. Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 2012, 13, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Alberdi, E.; Sánchez-Gómez, M.V.; Cavaliere, F.; Pérez-Samartín, A.; Zugaza, J.L.; Trullas, R.; Domercq, M.; Matute, C. Amyloid β oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium 2010, 47, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Bickler, P.E.; Warren, D.E.; Clark, J.P.; Gabatto, P.; Gregersen, M.; Brosnan, H. Anesthetic protection of neurons injured by hypothermia and rewarming: Roles of intracellular Ca2+ and excitotoxicity. Anesthesiology 2012, 117, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Abushik, P.A.; Sibarov, D.A.; Eaton, M.J.; Skatchkov, S.N.; Antonov, S.M. Kainate-induced calcium overload of cortical neurons in vitro: Dependence on expression of AMPAR GluA2-subunit and down-regulation by subnanomolar ouabain. Cell Calcium 2013, 54, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Hershey, C.O.; Hershey, L.A.; Varnes, A.; Vibhakar, S.D.; Lavin, P.; Strain, W.H. Cerebrospinal fluid trace element content in dementia: Clinical, radiologic, and pathologic correlations. Neurology 1983, 33, 1350–1353. [Google Scholar] [CrossRef] [PubMed]
- Gellein, K.; Skogholt, J.H.; Aaseth, J.; Thoresen, G.B.; Lierhagen, S.; Steinnes, E.; Syversen, T.; Flaten, T.P. Trace elements in cerebrospinal fluid and blood from patients with a rare progressive central and peripheral demyelinating disease. J. Neurol. Sci. 2008, 266, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Michalke, B.; Nischwitz, V. Review on metal speciation analysis in cerebrospinal fluid-current methods and results: A review. Anal. Chim. Acta 2010, 682, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Giblin, L.J.; Krezel, A.; McAdoo, D.J.; Muelle, R.N.; Zeng, Y.; Balaji, R.V.; Masalha, R.; Thompson, R.B.; Fierke, C.A.; et al. Concentrations of extracellular free zinc (pZn)e in the central nervous system during simple anesthetization, ischemia and reperfusion. Exp. Neurol. 2006, 198, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Tamano, H. Significance of low nanomolar concentration of Zn2+ in artificial cerebrospinal fluid. Mol. Neurobiol. 2017, 54, 2477–2482. [Google Scholar] [CrossRef] [PubMed]
- Tamano, H.; Nishio, R.; Shakushi, Y.; Sasaki, M.; Koike, Y.; Osawa, M.; Takeda, A. In vitro and in vivo physiology of low nanomolar concentrations of Zn2+ in artificial cerebrospinal fluid. Sci. Rep. 2017, 7, 42897. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Danscher, G.; Jensen, M.S.; Thompson, R.; Motamedi, M.; Frederickson, C.J. Release of synaptic zinc is substantially depressed by conventional brain slice preparations. Brain Res. 2000, 879, 7–12. [Google Scholar] [CrossRef]
- Takeda, A.; Koike, Y.; Osaw, M.; Tamano, H. Characteristic of extracellular Zn2+ influx in the middle-aged dentate gyrus and its involvement in attenuation of LTP. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sensi, S.L.; Canzoniero, L.M.T.; Yu, S.P.; Ying, H.S.; Koh, J.Y.; Kerchner, G.A.; Choi, D.W. Measurement of intracellular free zinc in living cortical neurons: Routes of entry. J. Neurosci. 1997, 15, 9554–9564. [Google Scholar]
- Colvin, R.A.; Bush, A.I.; Volitakis, I.; Fontaine, C.P.; Thomas, D.; Kikuchi, K.; Holmes, W.R. Insights into Zn2+ homeostasis in neurons from experimental and modeling studies. Am. J. Physiol. Cell Physiol. 2008, 294, C726–C742. [Google Scholar] [CrossRef] [PubMed]
- Grabrucker, A.M.; Knight, M.J.; Proepper, C.; Bockmann, J.; Joubert, M.; Rowan, M.; Nienhaus, G.U.; Garner, C.C.; Bowie, J.U.; Kreutz, M.R.; et al. Concerted action of zinc and ProSAP/Shank in synaptogenesis and synapse maturation. EMBO J. 2011, 30, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Perrin, L.; Roudeau, S.; Carmona, A.; Domart, F.; Petersen, J.D.; Bohic, S.; Yang, Y.; Cloetens, P.; Ortega, R. Zinc and Copper Effects on Stability of Tubulin and Actin Networks in Dendrites and Spines of Hippocampal Neurons. ACS Chem. Neurosci. 2017, 8, 1490–1499. [Google Scholar] [CrossRef] [PubMed]
- Castillo, P.E.; Weisskopf, M.G.; Nicoll, R.A. The role of Ca2+ channels in hippocampal mossy fiber synaptic transmission and long-term potentiation. Neuron 1994, 12, 261–269. [Google Scholar] [CrossRef]
- Tong, G.; Malenka, R.C.; Nicoll, R.A. Long-term potentiation in cultures of single hippocampal granule cells: A presynaptic form of plasticity. Neuron 1996, 16, 1147–1157. [Google Scholar]
- Breustedt, J.; Vogt, K.E.; Miller, R.J.; Nicoll, R.A.; Schmitz, D. α1E-containing Ca2+ channels are involved in synaptic plasticity. Proc. Natl. Acad. Sci. USA 2003, 100, 12450–12455. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Storm, D.R. Calmodulin-regulated adenylyl cyclases: Cross-talk and plasticity in the central nervous system. Mol. Pharmacol. 2003, 63, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Z.; Pan, E.; Xiong, Z.Q.; McNamara, J.O. Zinc-mediated transactivation of TrkB potentiates the hippocampal mossy fiber-CA3 pyramid synapse. Neuron 2008, 57, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Pan, E.; Zhang, X.A.; Huang, Z.; Krezel, A.; Zhao, M.; Tinberg, C.E.; Lippard, S.J.; McNamara, J.O. Vesicular zinc promotes presynaptic and inhibits postsynaptic long-term potentiation of mossy fiber-CA3 synapse. Neuron 2011, 71, 1116–1126. [Google Scholar] [CrossRef] [PubMed]
- Malenka, R.C.; Bear, M.F. LTP and LTD: An embarrassment of riches. Neuron 2004, 44, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Cull-Candy, S.G.; Leszkiewicz, D.N. Role of distinct NMDA receptor subtypes at central synapses. Sci. STKE 2004, 255, re16. [Google Scholar] [CrossRef] [PubMed]
- Westbrook, G.L.; Mayer, M.L. Micromolar concentrations of Zn2+ antagonize NMDA and GABA responses of hippocampal neurons. Nature 1987, 328, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Moshaver, A.; Raymond, L.A. Differential sensitivity of recombinant N-methyl-d-aspartate receptor subtypes to zinc inhibition. Mol. Pharmacol. 1997, 51, 1015–1023. [Google Scholar] [PubMed]
- Paoletti, P.; Ascher, P.; Neyton, J. High-affinity zinc inhibition of NMDA NR1-NR2A receptors. J. Neurosci. 1998, 17, 5711–5725. [Google Scholar]
- Choi, Y.B.; Lipton, S.A. Identification and mechanism of action of two histidine residues underlying high-affinity Zn2+ inhibition of the NMDA receptor. Neuron 1999, 23, 171–180. [Google Scholar] [CrossRef]
- Hirano, T.; Kikuchi, K.; Urano, Y.; Nagano, T. Improvement and biological applications of fluorescent probes for zinc, ZnAFs. J. Am. Chem. Soc. 2002, 124, 6555–6562. [Google Scholar] [CrossRef] [PubMed]
- Ueno, S.; Tsukamoto, M.; Hirano, T.; Kikuchi, K.; Yamada, M.K.; Nishiyama, N.; Nagano, T.; Matsuki, N.; Ikegaya, Y. Mossy fiber Zn2+ spillover modulates heterosynaptic N-methyl-d-aspartate receptor activity in hippocampal CA3 circuits. J. Cell Biol. 2002, 158, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Suzuki, M.; Tempaku, M.; Ohashi, K.; Tamano, H. Influx of extracellular Zn2+ into the hippocampal CA1 neurons is required for cognitive performance via long-term potentiation. Neuroscience 2015, 304, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Remondes, M.; Schuman, E.M. Role for a cortical input to hippocampal area CA1 in the consolidation of a long-term memory. Nature 2004, 431, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.; Rivest, A.J.; Nakashiba, T.; Tominaga, T.; Tonegawa, S. Entorhinal cortex layer III input to the hippocampus is crucial for temporal association memory. Science 2011, 334, 1415–1420. [Google Scholar] [CrossRef] [PubMed]
- Vago, D.R.; Kesner, R.P. Disruption of the direct perforant path input to the CA1 subregion of the dorsal hippocampus interferes with spatial working memory and novelty detection. Behav. Brain Res. 2008, 189, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Sindreu, C.B.; Varoqui, H.; Erickson, J.D.; Pérez-Clausell, J. Boutons containing vesicular zinc define a subpopulation of synapses with low AMPAR content in rat hippocampus. Cereb. Cortex 2003, 13, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Tamano, H.; Nishio, R.; Takeda, A. Involvement of intracellular Zn2+ signaling in LTP at perforant pathway-CA1 pyramidal cell synapse. Hippocampus 2017, 27, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, J.; Morales, I.S.; Villarreal, D.M.; Derrick, B.E. Low-frequency stimulation induces long-term depression and slow onset long-term potentiation at perforant path-dentate gyrus synapses in vivo. J. Neurophysiol. 2014, 111, 1259–1273. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Ren, T.; Yu, X. Disruption of calmodulin-dependent protein kinase II α/brain-derived neurotrophic factor (α-CaMKII/BDNF) signalling is associated with zinc deficiency-induced impairments in cognitive and synaptic plasticity. Br. J. Nutr. 2013, 110, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Tamano, H.; Ogawa, T.; Takada, S.; Nakamura, M.; Fujii, H.; Ando, M. Intracellular Zn2+ signaling in the dentate gyrus is required for object recognition memory. Hippocampus 2014, 24, 1404–1412. [Google Scholar] [CrossRef] [PubMed]
- Tamano, H.; Minamino, T.; Fujii, H.; Takada, S.; Nakamura, M.; Ando, M.; Takeda, A. Blockade of intracellular Zn2+ signaling in the dentate gyrus erases recognition memory via impairment of maintained LTP. Hippocampus 2015, 25, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Hieber, C.; Jonas, P.; Bischofberger, J. Enhanced synaptic plasticity in newly generated granule cells of the adult hippocampus. Nature 2004, 429, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Kheirbek, M.A.; Tannenholz, L.; Hen, R. NR2B-dependent plasticity of adult-born granule cells is necessary for context discrimination. J. Neurosci. 2012, 32, 8696–8702. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Sawashita, J.; Okada, S. Biological half-lives of zinc and manganese in rat brain. Brain Res. 1995, 695, 53–58. [Google Scholar] [CrossRef]
- Suh, S.W.; Won, S.J.; Hamby, A.M.; Yoo, B.H.; Fan, Y.; Sheline, C.T.; Tamano, H.; Takeda, A.; Liu, J. Decreased brain zinc availability reduces hippocampal neurogenesis in mice and rats. J. Cereb. Blood Flow Metab. 2009, 29, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.; Corniola, R.S.; Gower-Winter, S.D.; Morgan, T.J., Jr.; Bishop, B.; Levenson, C.W. Zinc deficiency induces apoptosis via mitochondrial p53- and caspase-dependent pathways in human neuronal precursor cells. J. Trace Elem. Med. Biol. 2015, 30, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Cope, E.C.; Morris, D.R.; Gower-Winter, S.D.; Brownstein, N.C.; Levenson, C.W. Effect of zinc supplementation on neuronal precursor proliferation in the rat hippocampus after traumatic brain injury. Exp. Neurol. 2016, 279, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Mocchegiani, E.; Bertoni-Freddari, C.; Marcellini, F.; Malavolta, M. Brain, aging and neurodegeneration: Role of zinc ion availability. Prog. Neurobiol. 2005, 75, 367–390. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Parncutt, J.M.; Finkelstein, D.I.; Bush, A.I. Cognitive loss in zinc transporter-3 knock-out mice: A phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J. Neurosci. 2010, 30, 1631–1636. [Google Scholar] [CrossRef] [PubMed]
- Sindreu, C.; Palmiter, R.D.; Storm, D.R. Zinc transporter ZnT-3 regulates presynaptic Erk1/2 signaling and hippocampus-dependent memory. Proc. Natl. Acad. Sci. USA 2012, 108, 3366–3370. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Sedjahtera, A.; Gunawan, L.; Bray, L.; Hare, D.; Lear, J.; Doble, P.; Bush, A.I.; Finkelstein, D.I.; Cherny, R.D. A novel approach to rapidly prevent age-related cognitive decline. Aging Cell 2014, 13, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Parncutt, J.; Lal, V.; James, S.; Hare, D.; Doble, P.; Finkelstein, D.I.; Bush, A.I. Metal chaperones prevent zinc-mediated cognitive decline. Neurobiol. Dis. 2015, 81, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Stork, C.J.; Li, Y.V. Intracellular zinc elevation measured with a “calcium-specific” indicator during ischemia and reperfusion in rat hippocampus: A question on calcium overload. J. Neurosci. 2006, 26, 10430–10437. [Google Scholar] [CrossRef] [PubMed]
- Brickman, A.M.; Khan, U.A.; Provenzano, F.A.; Yeung, L.K.; Suzuki, W.; Schroeter, H.; Wall, M.; Sloan, R.P.; Small, S.A. Enhancing dentate gyrus function with dietary flavanols improves cognition in older adults. Nat. Neurosci. 2014, 17, 1798–1803. [Google Scholar] [CrossRef] [PubMed]
- Jinno, S. Aging affects new cell production in the adult hippocampus: A quantitative anatomic review. J. Chem. Neuroanat. 2016, 76, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Fujise, Y.; Tsuchiya, Y.; Tamano, H.; Takeda, A. Excess influx of Zn2+ into dentate granule cells affects object recognition memory via attenuated LTP. Neurochem. Int. 2015, 87, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Tamano, H.; Hisatsune, M.; Murakami, T.; Nakada, H.; Fujii, H. Maintained LTP and memory are lost by Zn2+ influx into dentate granule cells, but not Ca2+ influx. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Tamano, H.; Murakami, T.; Nakada, H.; Minamino, T.; Koike, Y. Weakened intracellular Zn2+-buffering in the aged dentate gyrus and its involvement in erasure of maintained LTP. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Takada, S.; Nakamura, M.; Suzuki, M.; Tamano, H.; Ando, M.; Oku, N. Transient increase in Zn2+ in hippocampal CA1 pyramidal neurons causes reversible memory deficit. PLoS ONE 2011, 6, e28615. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Lau, L.; Wei, J.; Zhu, D.; Zou, S.; Sun, H.S.; Fu, Y.; Liu, F.; Lu, Y. Expression of Ca(2+)-permeable AMPA receptor channels primes cell death in transient forebrain ischemia. Neuron 2004, 43, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.H. Ca permeable AMPA channels in diseases of the nervous system. Front. Mol. Neurosci. 2011, 4, 42. [Google Scholar] [CrossRef] [PubMed]
- Pagliusi, S.R.; Gerrard, P.; Abdallah, M.; Talabot, D.; Catsicas, S. Age-related changes in expression of AMPA-selective glutamate receptor subunits: Is calcium-permeability altered in hippocampal neurons? Neuroscience 1994, 61, 429–433. [Google Scholar] [CrossRef]
- Foster, T.C. Calcium homeostasis and modulation of synaptic plasticity in the aged brain. Aging Cell 2007, 6, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Bodhinathan, K.; Foster, T.C. Susceptibility to calcium dysregulation during brain aging. Front. Aging Neurosci. 2009, 1, 2. [Google Scholar] [CrossRef] [PubMed]
- Toescu, E.C.; Vreugdenhil, M. Calcium and normal brain ageing. Cell Calcium 2010, 47, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Thibault, O.; Landfield, P.W. Increase in single L-type calcium channels in hippocampal neurons during aging. Science 1996, 272, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Dysregulation of neural calcium signaling in Alzheimer disease, bipolar disorder and schizophrenia. Prion 2013, 7, 2–13. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takeda, A.; Tamano, H. The Impact of Synaptic Zn2+ Dynamics on Cognition and Its Decline. Int. J. Mol. Sci. 2017, 18, 2411. https://doi.org/10.3390/ijms18112411
Takeda A, Tamano H. The Impact of Synaptic Zn2+ Dynamics on Cognition and Its Decline. International Journal of Molecular Sciences. 2017; 18(11):2411. https://doi.org/10.3390/ijms18112411
Chicago/Turabian StyleTakeda, Atsushi, and Hanuna Tamano. 2017. "The Impact of Synaptic Zn2+ Dynamics on Cognition and Its Decline" International Journal of Molecular Sciences 18, no. 11: 2411. https://doi.org/10.3390/ijms18112411
APA StyleTakeda, A., & Tamano, H. (2017). The Impact of Synaptic Zn2+ Dynamics on Cognition and Its Decline. International Journal of Molecular Sciences, 18(11), 2411. https://doi.org/10.3390/ijms18112411