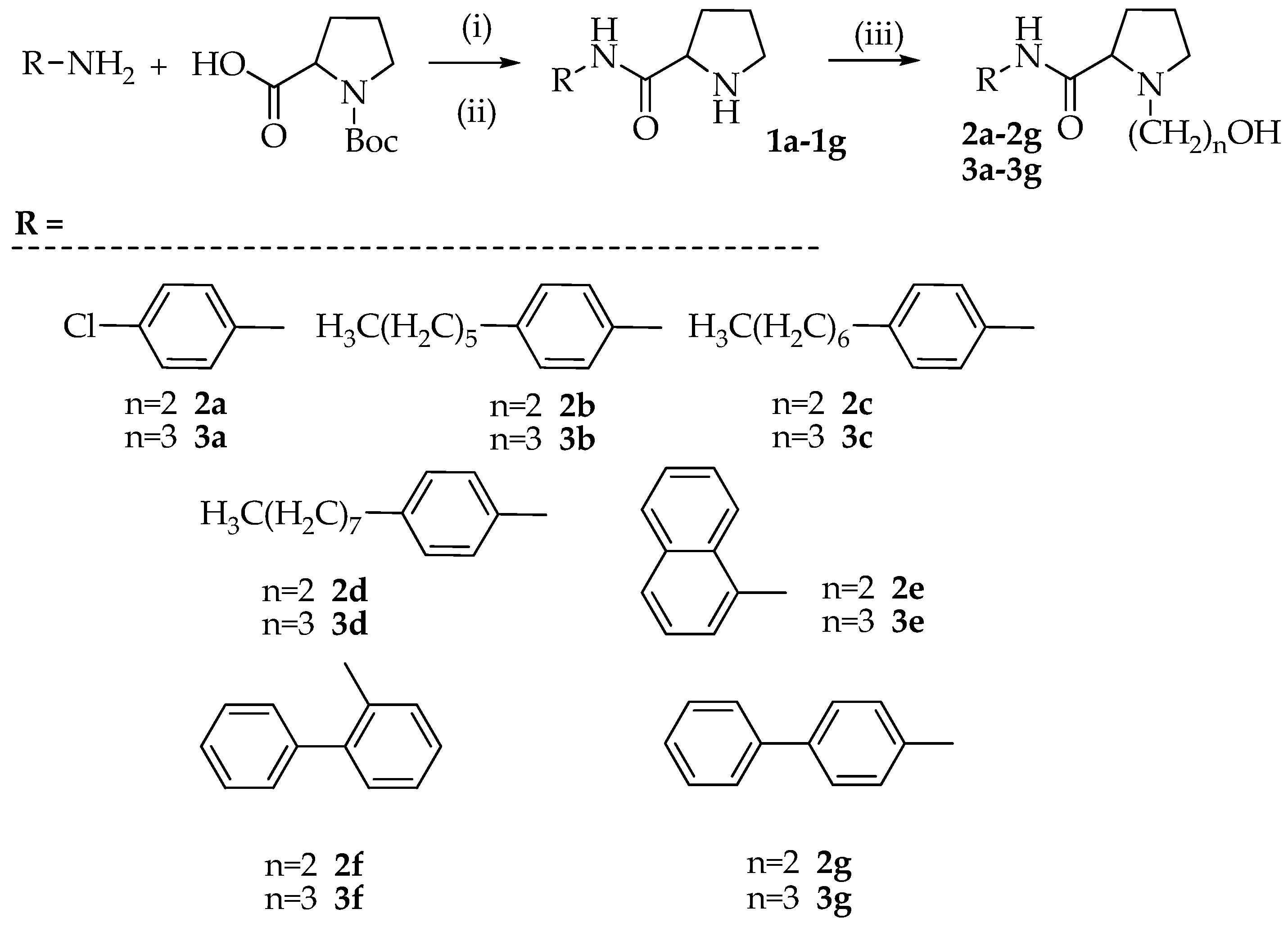
3.2. Synthesis of N-(aryl)-1-(hydroxyalkyl)pyrrolidine-2-carboxamide Derivatives (Compounds 2a–2g and 3a–3g)
3.2.1. Synthesis of N-(4-chlorophenyl)pyrrolidine-2-carboxamide (1a)
To a solution of Boc-Pro-OH (1 mmol) and p-chloro-aniline (1 mmol) dissolved in anhydrous DMF (15 mL) were added TBTU (1.1 mmol), HOBt (1.1 mmol), and N, N-diisopropylethylamine (DIPEA) (1.1 mmol). The reaction mixture was stirred for 12 h at room temperature. After solvent removal, the residue was dissolved in ethyl acetate and extracted with 10% citric acid, 5% NaHCO3, and brine. The organic phase was dried with Na2SO4, filtered, and evaporated to dryness. The residue was purified on a silica gel column using DCM/MeOH (9.5/0.5 v/v) as an eluent. The crystallization with diethyl ether provided the Boc protected product that was dissolved in 40% TFA in DCM (10 mL) and the mixture was stirred at room temperature for 2 h. After evaporation of the solvent, the desired product 1a was obtained as a white solid by crystallization with diethyl ether (yield 47%, calculated on two steps).
Using the above described procedure intermediates 1b–1g were prepared starting from 4-hexylaniline (1b), 4-heptylaniline (1c), 4-octylaniline (1d), α-naphtylamine (1e), 2-aminobiphenyl (1f), and 4-aminobiphenyl (1g).
3.2.2. N-(4-chlorophenyl)-1-(2-hydroxyethyl)pyrrolidine-2-carboxamide (2a)
A solution of 2-bromoethanol (1 mmol) and NaI (1.5 mmol) dissolved in 15 mL of DMF was stirred under reflux for 30 min. Then, a solution of 1a (1 mmol) and K2CO3 (1.5 mmol) in 10 mL of DMF was added drop wise. After 12 h, the reaction mixture was cooled at room temperature, the solid was filtered off, and the solvent was evaporated under vacuum. The obtained residue was dissolved in ethyl acetate and the solution was extracted with brine. The organic phase was then dried with Na2SO4, filtered, and evaporated under vacuum, furnishing an oily residue. This residue was purified by chromatography on a silica gel column using a mixture of DCM/MeOH (9.5/0.5; v/v) as an eluent, thus obtaining the final product 2a as an oil. Yield: 67%. 1H-NMR δ = 9.77 (s, 1H, -NH), 7.54 (d, J = 8.1 Hz, 2H), 7.19 (d, J = 8.1 Hz, 2H), 3.76–3.71 (m, 1H), 3.66–3.63 (m, 1H), 3.27–3.26 (m, 1H), 3.17–3.15 (m, 1H), 2.85–2.80 (m, 1H), 2.70–2.66 (m, 1H), 2.40–2.36 (m, 1H), 2.22–2.17 (m, 1H), 1.98–1.96 (m, 1H), 1.79–1.73 (m, 2H); 13C-NMR δ = 173.45, 136.99, 131.47, 129.11, 120.84, 68.18, 61.07, 57.89, 54.32, 30.87, 24.70. ESI-MS calculated: 268.74; Found: 269.3 [M + H]+. Anal. (C13H17ClN2O2), C, H, N.
3.2.3. N-(4-hexylphenyl)-1-(2-hydroxyethyl)pyrrolidine-2-carboxamide (2b)
Compound 2b was obtained following the procedure described for 2a, starting from 1b and 2-bromoethanol. Yield: 60%; oil. 1H-NMR δ = 9.65 (s, 1H, -NH), 7.53 (d, J = 8.1 Hz, 2H), 7.11 (d, J = 8.1 Hz, 2H), 3.78–3.75 (m, 1H), 3.72–3.70 (m, 1H), 3.33–3.31 (m, 1H), 3.24–3.20 (m, 1H), 2.88 (m, 1H), 2.74–2.71 (m, 1H), 2.56 (t, J = 7.0 Hz, J = 7.7 Hz, 2H), 2.46–2.43 (m, 1H), 2.20–2.25 (m, 1H), 2.01–2.05 (m, 1H), 1.82–1.80 (m, 2H), 1.28 (bs, 8H), 0.87 (t, J = 7.0 Hz, 3H); 13C-NMR δ = 173.24, 138.85, 135.88, 129.02, 119.54, 68.21, 61.09, 57.93, 54.36, 35.62, 31.97, 31.83, 30.88, 29.14, 24.65, 22.87, 14.38. ESI-MS calculated: 318.45; Found: 319.3 [M + H]+. Anal. (C19H30N2O2), C, H, N.
3.2.4. N-(4-heptylphenyl)-1-(2-hydroxyethyl)pyrrolidine-2-carboxamide (2c)
Compound 2c was obtained following the procedure described for 2a, starting from 1c and 2-bromoethanol. Yield: 54%; oil. 1H-NMR δ = 9.62 (s, 1H, -NH), 7.56 (d, J = 7.8 Hz, 2H), 7.11 (d, J = 7.8 Hz, 2H), 3.86–3.76 (m, 3H), 3.34–3.28 (m, 1H), 3.21–3.13 (m, 1H), 2.58 (t, J = 7.0 Hz, 2H), 2.45–2.39 (m, 1H), 2.09–2.01 (m, 1H), 2.03–1.97 (m, 1H), 1.78–1.73 (m, 2H), 1.69–1.65 (m, 1H), 1.52–1.45 (m, 2H), 1.30 (bs, 8H), 0.87 (t, J = 7.0 Hz, 3H); 13C-NMR δ = 172.65, 138.85, 135.41, 129.07, 119.50, 68.24, 61.10, 58.01, 54.37, 35.36, 31.99, 31.77, 30.87, 29.68, 29.14, 24.64, 22.64, 14.07. ESI-MS calculated: 332.48; Found: 333.4 [M + H]+. Anal. (C20H32N2O2), C, H, N.
3.2.5. N-(4-octylphenyl)-1-(2-hydroxyethyl)pyrrolidine-2-carboxamide (2d)
Compound 2d was obtained following the procedure described for 2a, starting from 1d and 2-bromoethanol. Yield: 73%; oil. 1H-NMR δ = 9.62 (s, 1H, -NH), 7.53 (d, J = 8.1 Hz, 2H), 7.10 (d, J = 8.1 Hz, 2H), 3.80–3.75 (m, 1H), 3.71–3.70 (m, 1H), 3.32–3.30 (m, 1H), 3.23–3.20 (m, 1H), 2.88 (m, 1H), 2.74–2.71 (m, 1H), 2.55 (t, J = 7.3 Hz, 2H), 2.47–2.41 (m, 1H), 2.24–2.22 (m, 1H), 2.03–2.00 (m, 1H), 1.82–1.80 (m, 2H), 1.49–1.52 (m, 2H), 1.26 (bs, 10H), 0.86 (t, J = 7.0 Hz, 3H); 13C-NMR δ = 173.04, 138.81, 135.90, 128.99, 119.52, 68.29, 61.13, 57.95, 54.38, 35.60, 32.10, 31.81, 30.87, 29.69, 29.49, 29.45, 24.63, 22.88, 14.32. ESI-MS calculated: 332.48; Found: 333.4 [M + H]+. Anal. (C21H34N2O2), C, H, N.
3.2.6. 1-(2-hydroxyethyl)-N-(naphthalen-1-yl)pyrrolidine-2-carboxamide (2e)
Compound 2e was obtained following the procedure described for 2a, starting from 1e and 2-bromoethanol. Yield: 66%; oil. 1H-NMR δ = 10.23 (s, 1H, –NH), 8.23 (d, J = 7.3 Hz, 1H), 8.01 (d, J = 7.7 Hz, 1H), 7.86 (d, 7.7 Hz, 1H), 7.65 (d, J = 8.1 Hz, 1H), 7.53–7.45 (m, 3H), 3.87–3.77 (m, 2H), 3.45–3.44 (m, 1H), 3.39 (dd, J = 13.9, 4.0 Hz, 1H), 3.05–3.00 (m, 1H), 2.82–2.77 (m, 1H), 2.56–2.50 (m, 1H), 2.36–2.26 (m, 1H), 2.13–2.10 (m, 1H), 1.93–1.90 (m, 2H); 13C-NMR δ = 173.65, 134.30, 132.90, 128.90, 126.54, 126.29, 126.16, 126.04,124.95, 120.91, 118.56, 68.90, 61.29, 58.26, 54.62, 31.09, 24.87. ESI-MS calculated: 284.15; Found: 285.3 [M + H]+. Anal. (C17H20N2O2), C, H, N.
3.2.7. N-(biphenyl-2-yl)-1-(2-hydroxyethyl)pyrrolidine-2-carboxamide (2f)
Compound 2f was obtained following the procedure described for 2a, starting from 1f and 2-bromoethanol. Yield: 72%; oil. 1H-NMR δ = 9.67 (s, 1H,–NH), 8.45 (d, J = 8.1 Hz, 1H), 7.50 (d, J = 7.3 Hz, 2H), 7.45–7.24 (m, 5H), 7.18 (t, J = 6.9 Hz, 1H), 3.28–3.25 (m, 3H), 3.19 (d, J = 9.9 Hz, 1H), 2.97–2.93 (m, 2H), 2.68–2.65 (m, 1H), 2.48–2.45 (m, 1H), 2.31–2.29 (m, 1H), 2.16–2.14 (m, 1H), 2.09–2.00 (m, 1H), 1.65–1.63 (m, 1H); 13C-NMR δ = 173.14, 139.08, 135.17, 132.25, 130.17, 129.80, 128.98, 128.80, 127.95, 124.23, 120.60, 68.88, 61.14, 58.16, 54.27, 30.85, 24.95. ESI-MS calculated: 310.39; Found: 311.4 [M + H]+. Anal. (C19H22N2O2), C, H, N.
3.2.8. N-(biphenyl-4-yl)-1-(2-hydroxyethyl)pyrrolidine-2-carboxamide (2g)
Compound 2g was obtained following the procedure described for 2a, starting from 1g and 2-bromoethanol. Yield: 69%; oil. 1H-NMR δ = 9.83 (s, 1H, –NH), 7.73 (d, J = 8.4 Hz, 2H), 7.57–7.53 (m, 4H), 7.43 (t, J = 7.3, 7.7 Hz, 2H), 7.33 (t, J = 7.3 Hz, 1H), 3.81–3.78 (m, 1H), 3.75–3.73 (m, 1H), 3.38–3.36 (m, 1H), 3.29–3.28 (m, 1H), 2.92–2.90 (m, 1H), 2.80–2.78 (m, 1H), 2.49–2.47 (m, 1H), 2.30–2.26 (m, 1H), 2.07–2.05 (m, 1H), 1.86–1.82 (m, 2H); 13C-NMR δ = 173.36, 140.92, 137.63, 136.95, 128.97, 128.80, 127.76, 127.04, 119.88, 68.28, 61.09, 57.93, 54.38, 30.89, 24.67. ESI-MS calculated: 310.39; Found: 311.3 [M + H]+. Anal. (C19H22N2O2), C, H, N.
3.2.9. N-(4-chlorophenyl)-1-(3-hydroxypropyl)pyrrolidine-2-carboxamide (3a)
Compound 3a was obtained following the procedure described for 2a, starting from 1a and 3-bromopropanol. Yield: 82%; oil. 1H-NMR δ = 9.47 (s, 1H, –NH), 7.60 (d, J = 8.1 Hz, 2H), 7.27 (d, J = 7.7 Hz, 2H), 3.83–3.76 (m, 2H), 3.30–3.27 (m, 1H), 3.17–3.14 (m, 1H), 2.93–2.86 (m, 1H), 2.61–2.58 (m, 1H), 2.40–2.33 (m, 1H), 2.30–2.20 (m, 1H), 1.98–1.96 (m, 1H), 1.83–1.78 (m, 4H); 13C-NMR δ = 173.20, 136.83, 131.47, 129.13, 120.86, 68.92, 61.39, 53.95, 53.14, 31.26, 30.71, 24.36. ESI-MS calculated: 282.77; Found: 283.3 [M + H]+. Anal. (C14H19ClN2O2), C, H, N.
3.2.10. N-(4-hexylphenyl)-1-(3-hydroxypropyl)pyrrolidine-2-carboxamide (3b)
Compound 3b was obtained following the procedure described for 2a, starting from 1b and 3-bromopropanol. Yield: 56%; oil. 1H-NMR δ = 9.65 (s, 1H, –NH), 7.53 (d, J = 8.1 Hz, 2H), 7.11 (d, J = 8.1 Hz, 2H), 3.78–3.75 (m, 2H), 3.72–3.70 (m, 1H), 3.33–3.31 (m, 1H), 3.24–3.20 (m, 1H), 2.88 (t, 2H), 2.74–2.71 (m, 1H), 2.56 (t, 2H), 2.46–2.43 (m, 1H), 2.20–2.25 (m, 1H), 2.01–2.05 (m, 1H), 1.82–1.80 (m, 1H), 1.28 (bs, 8H), 0.87 (t, 3H); 13C-NMR δ = 173.24, 138.98, 135.41, 128.89, 119.37, 68.21, 61.09, 53.89, 53.03, 35.35, 31.71, 31.51, 30.35, 29.68, 28.86, 23.93, 22.59, 14.08. ESI-MS calculated: 332.48; Found: 333.3 [M + H]+. Anal. (C20H32N2O2), C, H, N.
3.2.11. N-(4-heptylphenyl)-1-(3-hydroxypropyl)pyrrolidine-2-carboxamide (3c)
Compound 3c was obtained following the procedure described for 2a, starting from 1c and 3-bromopropanol. Yield: 84%; oil. 1H-NMR δ = 9.62 (s, 1H, –NH), 7.58 (d, J = 7.8 Hz, 2H), 7.12 (d, J = 7.8 Hz, 2H), 3.80–3.76 (m, 3H), 3.49–3.46 (m, 1H), 3.11–3.07 (m, 1H), 2.55 (t, J = 7.0 Hz, 2H), 2.47–2.40 (m, 1H), 2.14–2.09 (m, 1H), 2.03–1.97 (m, 1H), 1.82–1.80 (m, 2H), 1.70–1.66 (m, 1H), 1.58–1.55 (m, 2H), 1.30 (bs, 10H), 0.87 (t, J = 7.0 Hz, 3H); 13C-NMR δ = 172.80, 138.48, 135.41, 129.07, 119.50, 68.24, 61.08, 53.80, 53.13, 35.36, 31.50, 30.52, 29.67, 29.45, 29.24, 29.19, 24.14, 22.68, 14.07. ESI-MS calculated: 346.51; Found: 347.4 [M + H]+. Anal. (C21H34N2O2), C, H, N.
3.2.12. N-(4-octylphenyl)-1-(3-hydroxypropyl)pyrrolidine-2-carboxamide (3d)
Compound 3d was obtained following the procedure described for 2a, starting from 1d and 3-bromopropanol. Yield: 61%; oil. 1H-NMR δ = 9.62 (s, 1H, –NH), 7.50 (d, J = 7.8 Hz, 2H), 7.13 (d, J = 7.8 Hz, 2H), 3.90–3.85 (m, 2H), 3.76–3.72 (m, 1H), 3.64–3.62 (m, 1H), 3.50–3.49 (m, 1H), 3.26–3.24 (m, 1H), 3.12–3.10 (m, 1H), 3.02–2.99 (m, 1H), 2.53 (t, J = 6.6 Hz, 2H), 2.03–2.00 (m, 1H), 1.92–1.94 (m, 2H), 1.76–1.78 (m, 2H), 1.49–1.52 (m, 2H), 1.26 (bs, 10H), 0.86 (t, J = 7.0 Hz, 3H); 13C-NMR δ = 172.93, 138.99, 135.48, 128.87, 119.45, 68.54, 60.82, 53.80, 52.88, 35.35, 31.86, 31.55, 30.52, 29.67, 29.45, 29.24, 29.19, 24.15, 22.64, 14.08. ESI-MS calculated: 360.53; Found: 361.6 [M + H]+. Anal. (C22H36N2O2), C, H, N.
3.2.13. 1-(3-hydroxypropyl)-N-(naphthalen-1-yl)pyrrolidine-2-carboxamide (3e)
Compound 3e was obtained following the procedure described for 2a, starting from 1e and 3-bromopropanol. Yield: 52%; oil. 1H-NMR δ = 10.11 (s, 1H, –NH), 8.20 (d, J = 7.3 Hz, 1H), 7.87–7.84 (m, 2H), 7.66 (d, J = 8.1 Hz, 1H), 7.55–7.46 (m, 3H), 3.77–3.74 (m, 2H), 3.42–3.40 (m, 1H), 3.35 (dd, J = 13.9, 4.0 Hz, 1H), 3.02–2.96 (m, 1H), 2.76–2.70 (m, 1H), 2.52–2.46 (m, 1H), 2.34–2.29 (m, 1H), 2.11–2.08 (m, 1H), 1.91–1.88 (m, 4H); 13C-NMR δ = 173.52, 134.32, 132.72, 129.08, 126.54, 126.38, 126.23, 126.07, 125.01, 120.42, 118.76, 69.09, 61.24, 54.44, 53.40, 32.00, 31.02, 24.79. ESI-MS calculated: 298.29; Found: 299.1 [M + H]+. Anal. (C18H22N2O2), C, H, N.
3.2.14. N-(biphenyl-2-yl)-1-(3-hydroxypropyl)pyrrolidine-2-carboxamide (3f)
Compound 3f was obtained following the procedure described for 2a, starting from 1f and 3-bromopropanol. Yield: 62%; oil. 1H-NMR δ = 9.71 (s, 1H, –NH), 8.37 (d, J = 8.1 Hz, 1H), 7.46 (d, J = 6.6 Hz, 2H), 7.39–7.23 (m, 5H), 7.18 (t, J = 6.9 Hz, 1H), 3.32–3.16 (m, 3H), 2.96–2.91 (m, 2H), 2.58–2.55 (m, 1H), 2.43–2.40 (m, 1H), 2.28–2.24 (m, 1H), 2.16–2.14 (m, 1H), 2.02–1.94 (m, 1H), 1.78–1.75 (m, 2H), 1.63–1.60 (m, 1H); 13C-NMR δ = 173.14, 139.10, 135.17, 132.25, 130.16, 129.63, 128.98, 128.80, 127.95, 124.33, 120.61, 68.90, 61.15, 54.24, 53.38, 31.25, 30.85, 24.95. ESI-MS calculated: 324.42; Found: 325.4 [M + H]+. Anal. (C20H24N2O2), C, H, N.
3.2.15. N-(biphenyl-4-yl)-1-(3-hydroxypropyl)pyrrolidine-2-carboxamide (3g)
Compound 3g was obtained following the procedure described for 2a, starting from 1g and 3-bromopropanol. Yield: 54%; oil. 1H-NMR δ = 9.76 (s, 1H, –NH), 7.74 (d, J = 8.2 Hz, 2H), 7.55–7.53 (m, 4H), 7.42 (t, J = 7.4, 7.0 Hz, 2H), 7.33 (t, J = 7.4 Hz, 1H), 3.82–3.78 (m, 2H), 3.65–3.63 (m, 1H), 3.48–3.42 (m, 1H), 3.06–3.00 (m, 1H), 2.87–2.84 (m, 2H), 2.62–2.60 (m, 1H), 2.40–2.37 (m, 1H), 2.05–2.03 (m, 1H), 1.90–1.83 (m, 3H); 13C-NMR δ = 173.36, 140.48, 137.13, 137.05, 128.77, 127.55, 127.09, 126.80, 119.99, 68.54, 61.02, 54.04, 53.37, 30.45, 29.68, 23.91. ESI-MS calculated: 324.42; Found: 325.5 [M + H]+. Anal. (C20H24N2O2), C, H, N.
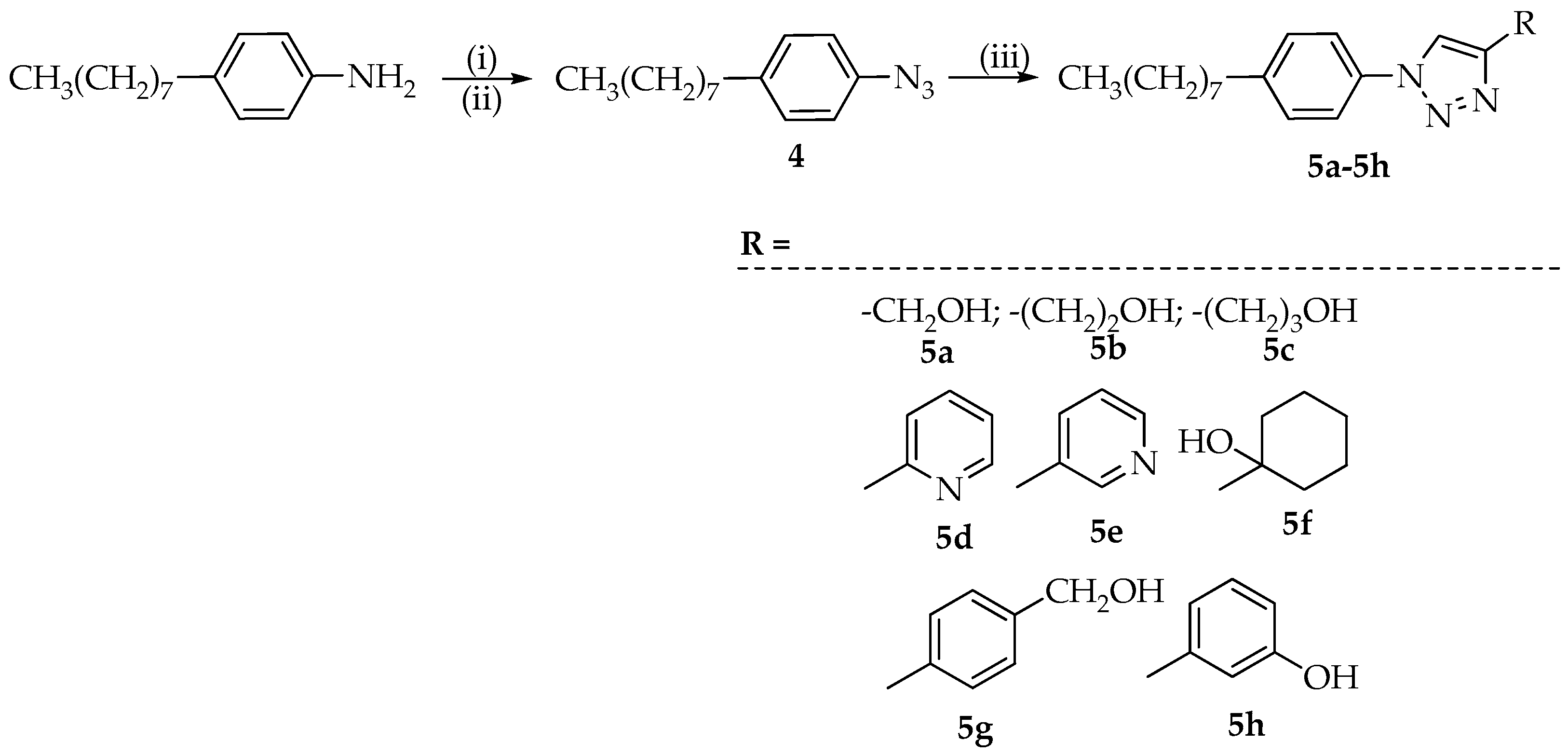
3.3. Synthesis of 1,4-Disubstituted 1,2,3-Triazoles 5a–5h
3.3.1. Synthesis of 1-Azido-4-octylbenzene (4)
One mmol of 4-octylaniline was suspended in 1 mL of H2O and 0.370 mL of concentrated HCl. To the reaction mixture, stirred and cooled to 0 °C, a solution of NaNO2 (1.04 mmol) in 1 mL of H2O was then added dropwise. After 30 min, the reaction mixture containing the formed diazonium salt, was poured into a solution of sodium azide (1.12 mmol) in 0.2 mL of H2O and ice. The formation of an intense foam, due to the release of N2, was observed. The mixture was stirred for 12 h, then the solution was extracted with DCM (three times) and the collected organic phases were dried with Na2SO4. Evaporation of the solvent left the desired intermediate 4, with a yield of 68%.
3.3.2. Synthesis of (1-(4-Octylphenyl)-1H-1,2,3-triazol-4-yl)methanol (5a)
2-Propin-1-ol (1 mmol) and 4 (1 mmol) were suspended in a 1:1 mixture of water and tert-butyl alcohol (12 mL). Sodium ascorbate (0.05 mmol) and copper sulfate pentahydrate (0.01 mmol) dissolved in 6 mL of H2O:tBuOH (1/1, v/v) were added. The heterogeneous mixture was stirred vigorously for 48 h, at which point it cleared and TLC analysis indicated complete consumption of the reactants. The reaction mixture was diluted with water (15 mL), cooled in ice, and the white precipitate was collected by filtration. After washing the precipitate with cold water (2 × 5 mL), it was dried under vacuum to afford the pure product as an off-white powder. Yield: 85%; mp 69–70 °C. 1H-NMR δ = 8.59 (s, 1H), 7.76 (d, J = 7.0 Hz, 2H), 7.37 (d, J = 6.6 Hz, 2H), 5.27 (s, 2H), 4.58 (s, 1H, –OH), 2.61 (m, J = 7.8 Hz, J = 6.3 Hz, 2H), 1.57 (m, 2H), 1.26 (bs, 10 H), 0.82 (t, J = 6.3 Hz, J = 7.0 Hz, 3H). 13C-NMR δ = 149.66, 143.59, 135.36, 130.24, 121.55, 120.56, 55.65, 35.24, 31.94, 31.48, 29.49, 29.33, 29.29, 22.75, 14.63. ESI-MS calculated: 287.40; Found: 288.0 [M + H]+. Anal. (C17H25N3O), C, H, N.
3.3.3. 2-(1-(4-Octylphenyl)-1H-1,2,3-triazol-4-yl)ethanol (5b)
Compound 5b was obtained following the procedure described for 5a, starting from 4 and 3-butyn-1-ol. Yield: 90%; mp 90–91 °C. 1H-NMR δ = 8.48 (s, 1H), 7.74 (d, J = 7.7 Hz, 2H), 7.37 (d, J = 7.7 Hz, 2H), 4.72 (s, 1H, –OH), 3.68 (m, 2H), 2.84 (t, J = 6.2 Hz, J = 6.6 Hz, 2H), 2.61 (t, J = 6.9 Hz, 2H), 1.58 (m, 2H), 1.27 (bs, 10 H), 0.83 (t, J = 6.3 Hz, J = 7.0 Hz, 3H). 13C-NMR δ = 146.17, 143.44, 135.38, 130.23, 121.30, 120.44, 60.93, 35.24, 31.95, 31.47, 29.88, 29.49, 29.33, 29.28, 22.76, 14.63. ESI-MS calculated: 301.43; Found: 302.1 [M + H]+. Anal. (C18H27N3O) C, H, N.
3.3.4. 3-(1-(4-Octylphenyl)-1H-1,2,3-triazol-4-yl)propan-1-ol (5c)
Compound 5c was obtained following the procedure described for 5a, starting from 4 and 4-pentyn-1-ol. Yield: 77%; mp 59–60 °C. 1H-NMR δ = 8.48 (s, 1H), 7.74 (d, J = 8.2 Hz, 2H), 7.37 (d, J = 7.7 Hz, 2H), 4.50 (t, 1H, –OH, J = 5.1 Hz), 3.48 (m, 2H), 2.73 (t, 2H, J = 7.4 Hz, J = 7.8 Hz), 2.63 (t, 2H, J = 7.4 Hz, J = 7.8 Hz), 1.82 (m, 2H), 1.57 (m, 2H), 1.27 (bs, 10 H), 0.85 (t, 3H, J = 6.3 Hz, J = 6.6 Hz). 13C-NMR δ = 148.35, 143.17, 135.20, 129.97, 120.43, 120.18, 60.46, 34.99, 32.58, 31.70, 31.25, 29.25, 29.09, 29.04, 22.52, 22.14, 14.40. ESI-MS calculated: 315.45; Found: 316.1 [M + H]+; 338.3 [M + Na]+; 354.3 [M + K]+. Anal. (C19H29N3O), C, H, N.
3.3.5. 2-(1-(4-Octylphenyl)-1H-1,2,3-triazol-4-yl)pyridine (5d)
Compound 5d was obtained following the procedure described for 5a, starting from 4 and 2-ethynylpyridine. Yield: 83%; mp 85–86 °C. 1H-NMR δ = 9.25 (s, 1H), 8.64 (d, J = 3.9 Hz, 1H), 8.10 (d, J = 7.8 Hz, 1H), 7.92 (m, 3H), 7.41 (m, 3H), 2.65 (t, J = 7.4 Hz, J = 7.8 Hz, 2H), 1.58 (m, 2H), 1.28 (bs, 10 H), 0.83 (t, J = 6.3 Hz, J = 6.6 Hz, 3H). 13C-NMR δ = 150.10, 150.03, 148.54, 143.78, 137.77, 134.93, 130.05, 123.74, 121.58, 120.57, 120.21, 35.03, 31.71, 31.24, 29.26, 29.10, 29.06, 22.52, 14.39. ESI-MS calculated: 334.46; Found: 335.2 [M + H]+. Anal. (C21H26N4), C, H, N.
3.3.6. 3-(1-(4-Octylphenyl)-1H-1,2,3-triazol-4-yl)pyridine (5e)
Compound 5e was obtained following the procedure described for 5a, starting from 4 and 3-ethynylpyridine. Yield: 85%; mp 125–126 °C. 1H-NMR δ = 9.35 (s, 1H), 9.12 (s, 1H), 8.57 (m, 2H), 8.29 (d, J = 7.7 Hz, 1H), 7.81 (d, J = 8.1 Hz, 2H), 7.44 (d, J = 8.1 Hz, 2H), 2.64 (t, J = 7.4 Hz, J = 7.8 Hz, 2H), 1.59 (m, 2H), 1.27 (bs, 10 H), 0.83 (t, J = 6.3 Hz, J = 6.6 Hz, 3H). 13C-NMR δ = 149.7, 146.97, 144.87, 143.88, 134.90, 132.97, 130.13, 124.54, 120.78, 120.50, 79.62, 35.05, 31.71, 31.25, 29.26, 29.11, 22.53, 14.40. ESI-MS calculated: 334.46; Found: 335.2 [M + H]+. Anal. (C21H26N4), C, H, N.
3.3.7. 1-(1-(4-Octylphenyl)-1H-1,2,3-triazol-4-yl)cyclohexanol (5f)
Compound 5f was obtained following the procedure described for 5a, starting from 4 and 1-ethynylcyclohexanol. Yield: 78%; mp 119–120 °C. 1H-NMR δ = 8.51 (s, 1H), 7.77 (d, J = 8.1 Hz, 2H), 7.36 (d, J = 8.1 Hz, 2H), 4.94 (s, 1H), 2.62 (t, J = 7.0 Hz, J = 7.7 Hz, 2H), 1.94–1.63 (mm, 6H), 1.57 (m, 2H), 1.43 (m, 4H), 1.26 (bs, 10 H), 0.82 (t, J = 6.3 Hz, J = 6.6 Hz, 3H). 13C-NMR δ = 157.35, 143.41, 135.46, 130.21, 120.44, 119.85, 68.64, 38.37, 35.24, 31.95, 31.47, 29.49, 29.30, 25.93, 22.76, 22.38, 14.63. ESI-MS calculated: 355.52; Found: 356.4 [M + H]+. Anal. (C22H33N3O), C, H, N.
3.3.8. (4-(1-(4-Octylphenyl)-1H-1,2,3-triazol-4-yl)phenyl)methanol (5g)
Compound 5g was obtained following the procedure described for 5a, starting from 4 and (4-ethynylphenyl)methanol. Yield: 87%; mp 160–161 °C. 1H-NMR δ = 9.20 (s, 1H), 7.89 (d, J = 7.0 Hz, 2H), 7.83 (d, J = 7.4 Hz, 2H), 7.43 (d, J = 7.4 Hz, 4H) 5.23 (s, 1H, -OH), 4.53 (d, J = 4.7 Hz, 2H), 2.66 (t, J = 7.0 Hz, J = 6.6 Hz, 2H), 1.60 (m, 2H), 1.28 (bs, 10 H), 0.84 (t, J = 6.3 Hz, J = 6.6 Hz, 3H). 13C-NMR δ = 147.67, 143.60, 143.07, 135.03, 130.06, 129.18, 127.41, 125.52, 120.34, 119.70, 63.10, 35.03, 31.70, 31.24, 29.25, 29.10, 29.06, 22.51, 14.39. ESI-MS calculated: 363.50; Found: 364.1 [M + H]+. Anal. (C23H29N3O), C, H, N.
3.3.9. 3-(1-(4-Octylphenyl)-1H-1,2,3-triazol-4-yl)phenol (5h)
Compound 5h was obtained following the procedure described for 5a, starting from 4 and 2-ethynylphenol. Yield: 78%; mp 126–127 °C. 1H-NMR δ = 9.57 (s, 1H, –OH), 9.15 (s, 1H), 7.82 (d, J = 7.3 Hz, 2H), 7.41 (d, J = 7.4 Hz, 2H), 7.35 (s, 1H), 7.32 (d, J = 6.6 Hz, 1H), 7.27 (t, J = 7.9 Hz, J = 6.3 Hz, J = 7.0 Hz, 1H), 6.75 (d, J = 7.4 Hz, 1H), 2.63 (t, J = 7.0 Hz, J = 6.6 Hz, 2H), 1.58 (m, 2H), 1.27 (bs, 10 H), 0.83 (t, J = 6.3 Hz, J = 6.6 Hz, 3H). 13C-NMR δ = 158.27, 147.75, 143.59, 135.03, 131.96, 130.45, 130.05, 120.37, 119.86, 116.67, 115.67, 112.53, 35.03, 31.71, 31.25, 29.26, 29.10, 29.05, 22.52, 14.39. ESI-MS calculated: 349.42; Found: 350.2 [M + H]+. Anal. (C22H27N3O), C, H, N.
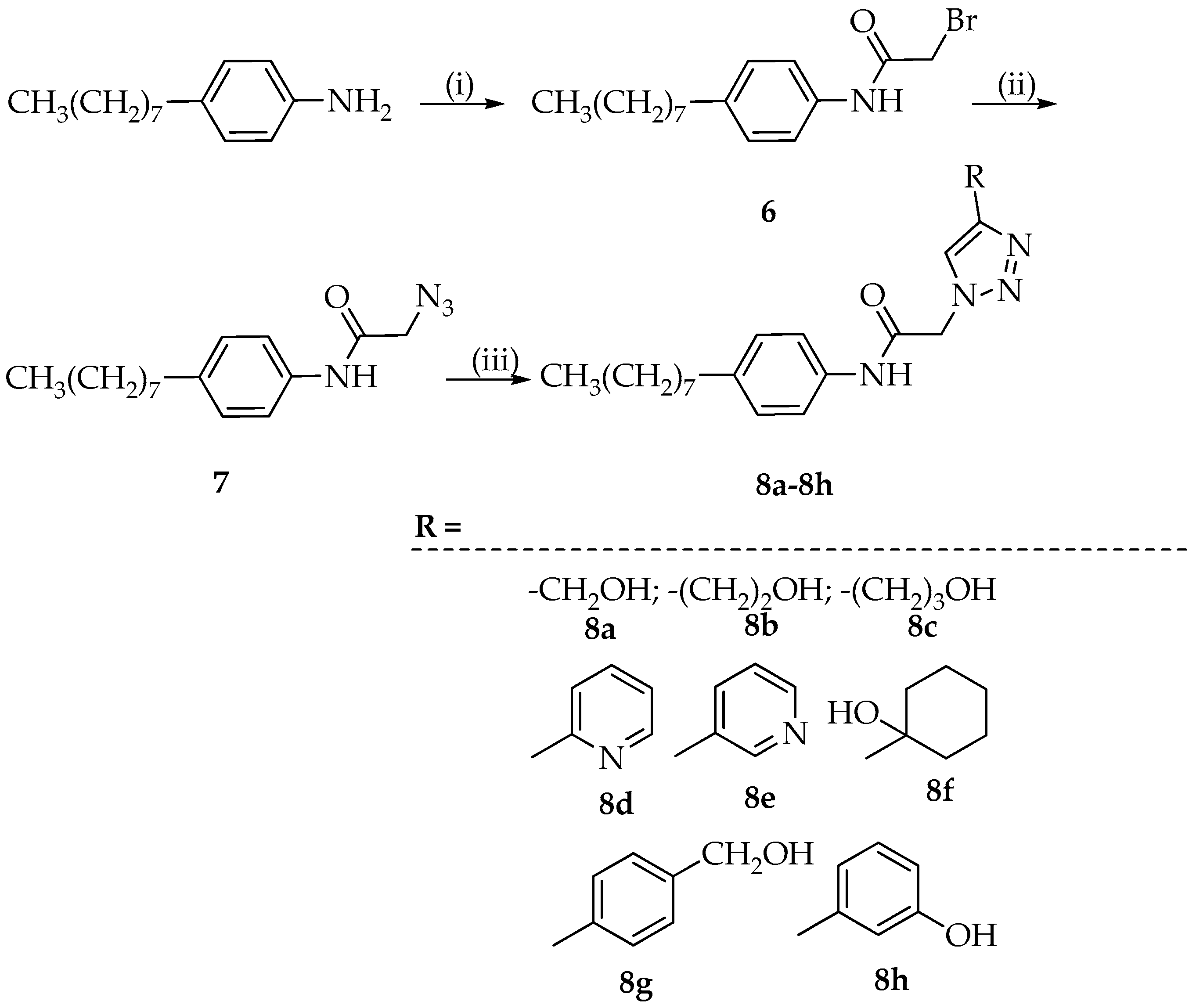
3.4. Synthesis of 1,4-Disubstituted 1,2,3-Triazoles 8a–8h
3.4.1. Synthesis of 2-Bromo-N-(4-octylphenyl)acetamide (6)
The 2-bromoacetyl bromide (1.3 mmol), dissolved in toluene (5 mL), was added dropwise to a solution of 4-octyl-aniline (1 mmol) in toluene (5 mL). The reaction mixture was heated to 110 °C, and after 4 h, ethyl acetate was added and the resulting solution was extracted with brine (three times). The organic phase, dried with Na2SO4, was filtered and evaporated to dryness. The crystallization with ethyl ether provided the desired product 6 (yield: 83%).
3.4.2. Synthesis of 2-Azido-N-(4-octylphenyl)acetamide (7)
The intermediate 6 (1 mmol) was dissolved in ethanol (10 mL), sodium azide (1.25 mmol) was added, and the reaction was refluxed for 2 h and 30 min. Then, it was cooled and diethyl ether (10 mL) was added. The precipitated NaBr was filtered off and the filtrate was evaporated to dryness. The residue was crystallized with hexane providing the desired product with 7 (yield: 86%).
3.4.3. 2-(4-(Hydroxymethyl)-1H-1,2,3-triazol-1-yl)-N-(4-octylphenyl)acetamide (8a)
Compound 8a was obtained following the procedure described for 5a, starting from 7 and 2-propin-1-ol. Yield: 75%; mp 171–172 °C. 1H-NMR δ = 10.34 (s, 1H, –NH), 7.95 (s, 1H), 7.46 (d, J = 6.2 Hz, 2H), 7.12 (d, J = 6.3 Hz, 2H), 5.26 (s, 2H), 5.18 (s, 1H, –OH), 4.52 (s, 2H), 2.48 (t, 2H, J = 7.8 Hz, J = 6.3 Hz), 1.51 (m, 2H), 1.24 (bs, 10 H), 0.83 (t, 3H, J = 6.3 Hz, J = 7.0 Hz). 13C-NMR δ = 169.46, 153.23, 143.19, 141.54, 134.03, 129.76, 124.66, 60.47, 57.52, 39.97, 36.69, 36.40, 34.25, 34.09, 34.03, 27.51, 19.38. ESI-MS calculated: 344.45; Found: 345.5 [M + H]+. Anal. (C19H28N4O2), C, H, N.
3.4.4. 2-(4-(2-Hydroxyethyl)-1H-1,2,3-triazol-1-yl)-N-(4-octylphenyl)acetamide (8b)
Compound 8b was obtained following the procedure described for 5a, starting from 7 and 3-butyn-1-ol. Yield: 80%; mp 170–171 °C. 1H-NMR δ = 10.32 (s, 1H, –NH), 7.85 (s, 1H), 7.45 (d, J = 7.0 Hz, 2H), 7.12 (d, J = 7.0 Hz, 2H), 5.21 (s, 2H), 4.67 (s, 1H, –OH), 3.63 (m, 2H), 2.77 (t, 2H, J = 6.6 Hz, J = 6.2 Hz), 2.49 (t, 2H, J = 7.8 Hz, J = 6.3 Hz), 1.51 (m, 2H), 1.23 (bs, 10 H), 0.82 (t, 3H, J = 6.3 Hz, J = 7.0 Hz). 13C-NMR δ = 164.50, 144.64, 138.19, 136.54, 129.04, 124.46, 119.67, 60.84, 52.51, 34.98, 31.70, 31.40, 29.59, 29.26, 29.10, 29.04, 22.51, 14.39. ESI-MS calculated: 358.48; Found: 359.3 [M + H]+. Anal. (C20H30N4O2), C, H, N.
3.4.5. 2-(4-(3-Hydroxypropyl)-1H-1,2,3-triazol-1-yl)-N-(4-octylphenyl)acetamide (8c)
Compound 8c was obtained following the procedure described for 5a, starting from 7 and 4-pentyn-1-ol. Yield: 82%; mp 171–172 °C. 1H-NMR δ = 10.30 (s, 1H, –NH), 7.81 (s, 1H), 7.43 (d, J = 7.0 Hz, 2H), 7.10 (d, J = 7.0 Hz, 2H), 5.19 (s, 2H), 4.44 (s, 1H, –OH), 3.40 (m, 2H), 2.62 (t, 2H, J = 7.4 Hz, J = 7.8 Hz), 1.71 (t, 2H, J = 7.4 Hz, J = 7.8 Hz), 1.49 (m, 2H), 1.21 (bs, 10 H), 0.81 (t, 3H, J = 6.3 Hz, J = 7.0 Hz). 13C-NMR δ = 164.53, 146.99, 138.19, 136.54, 129.03, 123.88, 119.67, 60.49, 52.50, 34.98, 32.74, 31.70, 31.40, 29.26, 29.10, 29.04, 22.51, 22.07, 14.39. ESI-MS calculated: 372.50; Found: 373.3 [M + H]+. Anal. (C21H32N4O2), C, H, N.
3.4.6. N-(4-octylphenyl)-2-(4-(pyridin-2-yl)-1H-1,2,3-triazol-1-yl)acetamide (8d)
Compound 8d was obtained following the procedure described for 5a, starting from 7 and 2-ethynylpyridine. Yield: 78%; mp 199–200 °C. 1H-NMR δ = 10.40 (s, 1H, –NH), 8.59 (s, 1H), 8.04 (d, J = 7.7 Hz, 1H), 7.91 (m, 2H), 7.47 (d, J = 8.1 Hz, 2H), 7.35 (t, 1H, J = 5.5 Hz, J = 6.2 Hz), 7.13 (d, J = 8.1 Hz, 2H), 5.38 (s, 2H), 2.51 (t, 2H, J = 7.8 Hz, J = 6.3 Hz), 1.51 (m, 2H), 1.24 (bs, 10 H), 0.84 (t, 3H, J = 6.3 Hz, J = 7.0 Hz). 13C-NMR δ = 164.53, 150.68, 150.35, 147.74, 138.49, 137.89, 136.76, 129.30, 125.65, 123.63, 120.08, 119.96, 52.99, 35.23, 31.94, 31.64, 29.50, 29.34, 29.28, 22.75, 14.63. ESI-MS calculated: 391.51; Found: 392.2 [M + H]+. Anal. (C23H29N5O), C, H, N.
3.4.7. N-(4-octylphenyl)-2-(4-(pyridin-3-yl)-1H-1,2,3-triazol-1-yl)acetamide (8e)
Compound 8e was obtained following the procedure described for 5a, starting from 7 and 3-ethynylpyridine. Yield: 85%; mp 196–197 °C. 1H-NMR δ = 10.42 (s, 1H, –NH), 9.06 (s, 1H), 8.71 (s, 1H), 8.53 (m, 2H), 8.23 (d, J = 7.7 Hz, 1H), 7.47 (d, J = 8.1 Hz, 2H), 7.13 (d, J = 8.1 Hz, 2H), 5.37 (s, 2H), 2.51 (t, 2H, J = 7.8 Hz, J = 6.3 Hz), 1.51 (m, 2H), 1.23 (bs, 10 H), 0.84 (t, 3H, J = 6.3 Hz, J = 7.0 Hz). 13C-NMR δ = 164.48, 149.57, 147.04, 144.10, 138.55, 136.72, 133.09, 129.31, 124.72, 124.42, 119.97, 94.57, 53.11, 35.21, 31.93, 31.63, 29.49, 29.33, 29.27, 22.75, 14.63. ESI-MS calculated: 391.51; Found: 392.1 [M + H]+. Anal. (C23H29N5O), C, H, N.
3.4.8. 2-(4-(1-Hydroxycyclohexyl)-1H-1,2,3-triazol-1-yl)-N-(4-octylphenyl)acetamide (8f)
Compound 8f was obtained following the procedure described for 5a, starting from 7 and 1-ethynylcyclohexanol. Yield: 83%; mp 154–155 °C. 1H-NMR δ = 10.33 (s, 1H, –NH), 7.86 (s, 1H), 7.46 (d, J = 8.2 Hz, 2H), 7.12 (d, J = 8.2 Hz, 2H), 5.23 (s, 2H), 4.86 (s, 1H, –OH), 2.51 (t, 2H, J = 7.8 Hz, J = 6.3 Hz), 1.85–1.64 (mm, 6H), 1.51 (m, 2H), 1.41 (m, 4H), 1.24 (bs, 10 H), 0.83 (t, 3H, J = 6.3 Hz, J = 7.0 Hz). 13C-NMR δ = 164.51, 156.08, 138.19, 136.57, 129.04, 122.97, 119.65, 68.44, 52.55, 38.29, 34.98, 31.70, 31.41, 29.26, 29.10, 29.04, 25.70, 22.52, 22.09, 14.39. ESI-MS calculated: 412.57; Found: 413.3 [M + H]+. Anal. (C24H36N4O2), C, H, N.
3.4.9. 2-(4-(4-(Hydroxymethyl)phenyl)-1H-1,2,3-triazol-1-yl)-N-(4-octylphenyl)acetamide (8g)
Compound 8g was obtained following the procedure described for 5a, starting from 7 and (4-ethynylphenyl)methanol. Yield: 78%; mp 238–239 °C. 1H-NMR δ = 10.39 (s, 1H, –NH), 8.52 (s, 1H), 7.81 (d, J = 7.8 Hz, 2H), 7.47 (d, J = 8.6 Hz, 2H), 7.38 (d, J = 7.8 Hz, 2H), 7.12 (d, J = 8.6 Hz, 2H), 5.32 (s, 2H), 5.20 (t, 1H, –OH, J = 5.9 Hz, J = 5.9 Hz), 4.51 (d, J = 5.5 Hz, 2H) 2.51 (t, 2H, J = 7.8 Hz, J = 6.3 Hz), 1.50 (m, 2H), 1.23 (bs, 10 H), 0.82 (t, 3H, J = 6.3 Hz, J = 7.0 Hz). 13C-NMR δ = 164.36, 146.65, 142.68, 138.26, 136.51, 129.61, 129.06, 127.40, 125.34, 123.22, 119.71, 63.11, 52.778, 34.98, 31.69, 31.39, 29.25, 29.10, 29.03, 22.51, 14.38. ESI-MS calculated: 420.55; Found: 421.2 [M + H]+. Anal. (C25H32N4O2), C, H, N.
3.4.10. 2-(4-(3-Hydroxyphenyl)-1H-1,2,3-triazol-1-yl)-N-(4-octylphenyl)acetamide (8h)
Compound 8h was obtained following the procedure described for 5a, starting from 7 and 2-ethynylphenol. Yield: 83%; mp 200–201 °C. 1H-NMR δ = 10.39 (s, 1H, –NH), 9.51 (s, 1H, –OH), 8.48 (s, 1H), 7.47 (d, J = 7.8 Hz, 2H), 7.27-7.21 (m, 3H), 7.13 (d, J = 8.2 Hz, 2H), 6.72 (s, 1H), 5.31 (s, 2H), 2.51 (t, 2H, J = 7.8 Hz, J = 6.3 Hz), 1.51 (m, 2H), 1.23 (bs, 10 H), 0.82 (t, 3H, J = 6.3 Hz, J = 7.0 Hz). 13C-NMR δ = 164.36, 158.22, 146.71, 138.26, 136.52, 132.38, 130.41, 129.06, 123.41, 119.72, 116.48, 115.34, 112.30, 52.75, 34.98, 31.71, 31.40, 29.26, 29.10, 29.04, 22.51, 14.39. ESI-MS calculated: 406.52; Found: 407.3 [M + H]+. Anal. (C24H30N4O2), C, H, N.

 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





