Methylmalonyl-CoA Epimerase Deficiency Mimicking Propionic Aciduria


Abstract

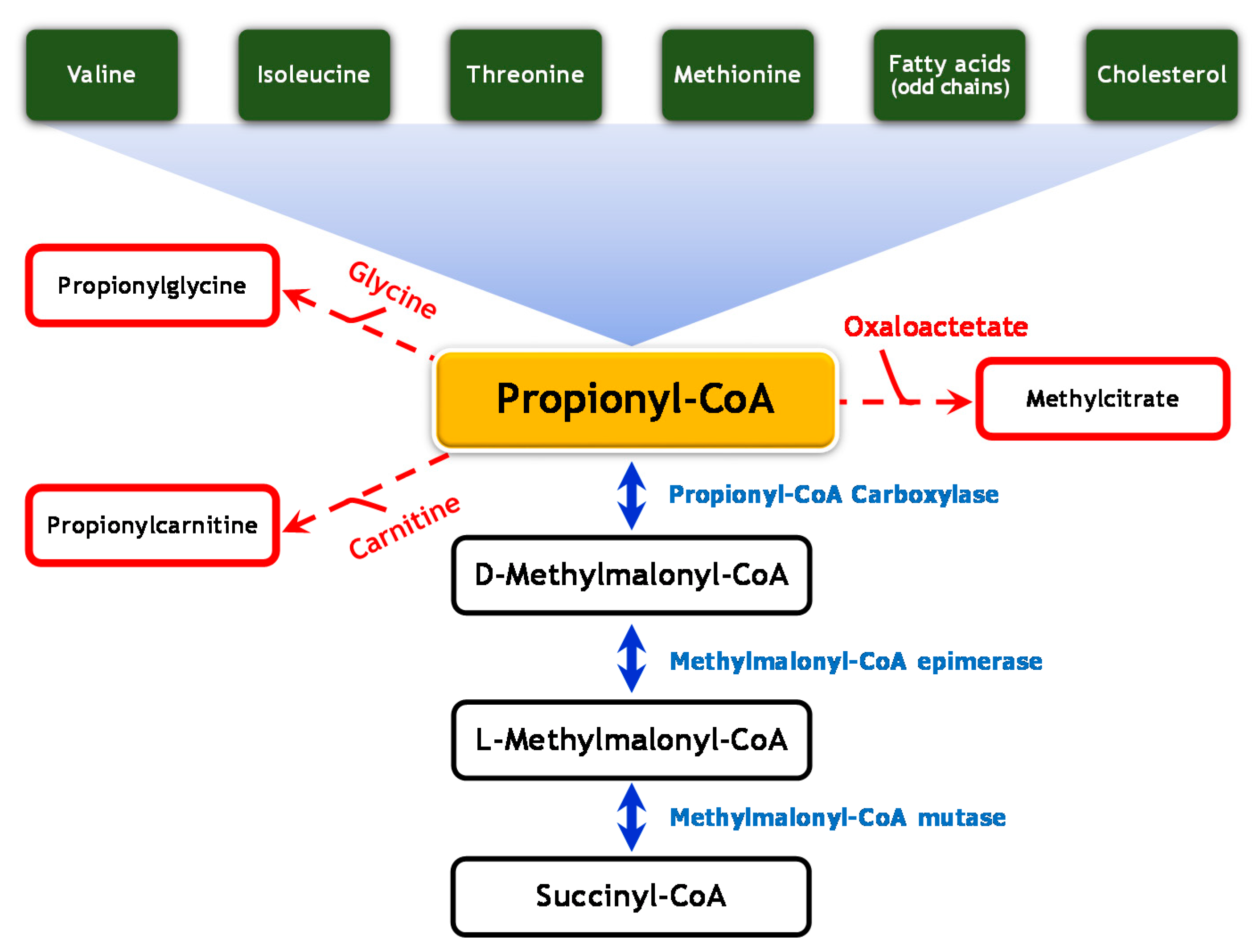
1. Introduction
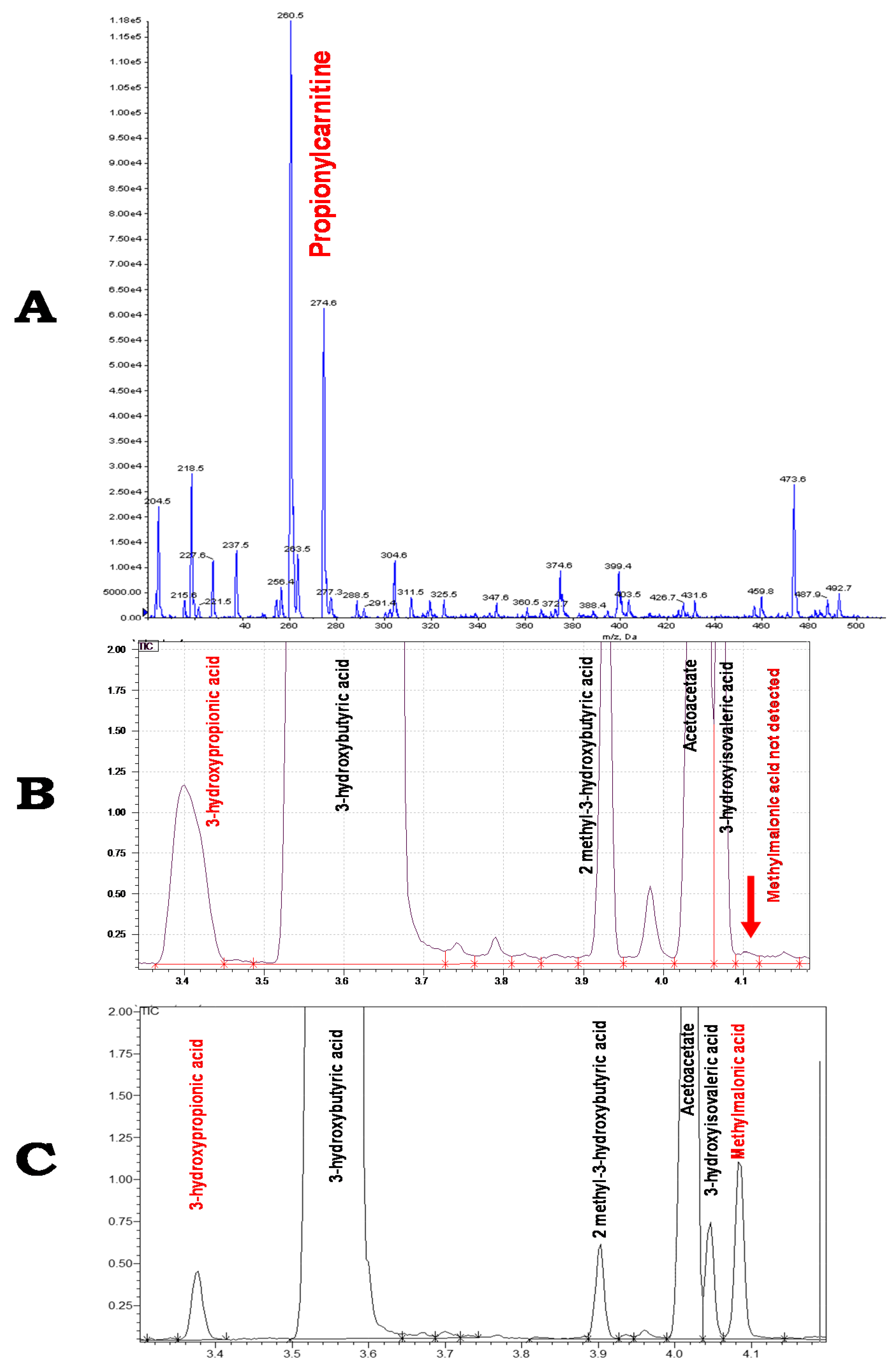
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Baumgartner, M.R.; Horster, F.; Dionisi-Vici, C.; Haliloglu, G.; Karall, D.; Chapman, K.A.; Huemer, M.; Hochuli, M.; Assoun, M.; Ballhausen, D.; et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J. Rare Dis. 2014, 9, 130. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, R.; Sasakawa, T.; Kaziro, Y.; Ochoa, S. A new enzyme in the conversion of propionyl coenzyme a to succinyl coenzyme a. J. Biol. Chem. 1961, 236, 53–55. [Google Scholar]
- Bikker, H.; Bakker, H.D.; Abeling, N.G.; Poll-The, B.T.; Kleijer, W.J.; Rosenblatt, D.S.; Waterham, H.R.; Wanders, R.J.; Duran, M. A homozygous nonsense mutation in the methylmalonyl-coa epimerase gene (mcee) results in mild methylmalonic aciduria. Hum. Mutat. 2006, 27, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M.; Gradinger, A.; Longo, N.; Wu, X.; Leclerc, D.; Lerner-Ellis, J.; Lemieux, M.; Belair, C.; Watkins, D.; Rosenblatt, D.S.; et al. Homozygous nonsense mutation in the mcee gene and sirna suppression of methylmalonyl-coa epimerase expression: A novel cause of mild methylmalonic aciduria. Mol. Genet. Metab. 2006, 88, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Gradinger, A.B.; Belair, C.; Worgan, L.C.; Li, C.D.; Lavallee, J.; Roquis, D.; Watkins, D.; Rosenblatt, D.S. Atypical methylmalonic aciduria: Frequency of mutations in the methylmalonyl coa epimerase gene (mcee). Hum. Mutat. 2007, 28, 1045. [Google Scholar] [CrossRef] [PubMed]
- Mazzuca, M.; Maubert, M.A.; Damaj, L.; Clot, F.; Cadoudal, M.; Dubourg, C.; Odent, S.; Benoit, J.F.; Bahi-Buisson, N.; Christa, L.; et al. Combined sepiapterin reductase and methylmalonyl-coa epimerase deficiency in a second patient: Cerebrospinal fluid polyunsaturated fatty acid level and follow-up under l-dopa, 5-htp and bh4 trials. JIMD Rep. 2015, 22, 47–55. [Google Scholar] [PubMed]
- Waters, P.J.; Thuriot, F.; Clarke, J.T.; Gravel, S.; Watkins, D.; Rosenblatt, D.S.; Levesque, S. Methylmalonyl-coa epimerase deficiency: A new case, with an acute metabolic presentation and an intronic splicing mutation in the mcee gene. Mol. Genet. Metab. Rep. 2016, 9, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Bobik, T.A.; Rasche, M.E. Identification of the human methylmalonyl-coa racemase gene based on the analysis of prokaryotic gene arrangements. Implications for decoding the human genome. J. Biol. Chem. 2001, 276, 37194–37198. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Patient | Isolated MCE | Age at Diagnosis | Acute Metabolic Acidosis | Psychomotor Development | Language Development | Neurological Impairment | Urinary MMA Concentration (µmol/mmol Creatinine) | MCEE Variants | Reference | |
|---|---|---|---|---|---|---|---|---|---|---|
| Allele 1 | Allele 2 | |||||||||
| P1 | Yes | 13.5 months | Yes | Normal | Normal | No | 180–1456 | c.139C > T-p.Arg47* | c.139C > T-p.Arg47* | [4] |
| P2 | Yes | 14 years (patient 1 sibling) Asymptomatic | No | Normal | Normal | No | 95–166 | c.139C > T-p.Arg47* | c.139C > T-p.Arg47* | [4] |
| P3 | Yes | Asymptomatic (patient 8 sibling) | No | Normal | Normal | No | 1400 | c.139C > T-p.Arg47* | c.139C > T-p.Arg47* | [5] |
| P4 | Yes (?) | 3 years | NC | Deteriorated motor function | Dysarthria | Mild spastic paraparesis Ataxia | 621 | c.178A > C-p.Lys60Gln | c.178A > C-p.Lys60Gln | [5] |
| P5 | Yes | 5 years | Yes | Normal | Normal | No | 47–151 | c.139C > T-p.Arg47* | c.379–644A > G-p.(?) | [7] |
| P6 | Yes | 5 years | Yes | Attentional difficulties | Moderate delay | No | No MMA excretion during the acute metabolic acidosis episode; afterwards 18-212 | c.139C > T-p.Arg47* | c.139C > T-p.Arg47* | This study |
| P7 | Combined with sepiapterin reductase deficiency | 2 years | No | Retardation | NC | Spasticity | 142 | c.139C > T-p.Arg47* | c.139C > T-p.Arg47* | [3] |
| P8 | Combined with sepiapterin reductase deficiency | 1 month | No | Retardation | Limited speech | Axial hypotonia Postural instability Oculogyric crisis Fatigability with sleep disorders | 60 | c.139C > T-p.Arg47* | c.139C > T-p.Arg47* | [6] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abily-Donval, L.; Torre, S.; Samson, A.; Sudrié-Arnaud, B.; Acquaviva, C.; Guerrot, A.-M.; Benoist, J.-F.; Marret, S.; Bekri, S.; Tebani, A. Methylmalonyl-CoA Epimerase Deficiency Mimicking Propionic Aciduria. Int. J. Mol. Sci. 2017, 18, 2294. https://doi.org/10.3390/ijms18112294
Abily-Donval L, Torre S, Samson A, Sudrié-Arnaud B, Acquaviva C, Guerrot A-M, Benoist J-F, Marret S, Bekri S, Tebani A. Methylmalonyl-CoA Epimerase Deficiency Mimicking Propionic Aciduria. International Journal of Molecular Sciences. 2017; 18(11):2294. https://doi.org/10.3390/ijms18112294
Chicago/Turabian StyleAbily-Donval, Lenaig, Stéphanie Torre, Aurélie Samson, Bénédicte Sudrié-Arnaud, Cécile Acquaviva, Anne-Marie Guerrot, Jean-François Benoist, Stéphane Marret, Soumeya Bekri, and Abdellah Tebani. 2017. "Methylmalonyl-CoA Epimerase Deficiency Mimicking Propionic Aciduria" International Journal of Molecular Sciences 18, no. 11: 2294. https://doi.org/10.3390/ijms18112294
APA StyleAbily-Donval, L., Torre, S., Samson, A., Sudrié-Arnaud, B., Acquaviva, C., Guerrot, A.-M., Benoist, J.-F., Marret, S., Bekri, S., & Tebani, A. (2017). Methylmalonyl-CoA Epimerase Deficiency Mimicking Propionic Aciduria. International Journal of Molecular Sciences, 18(11), 2294. https://doi.org/10.3390/ijms18112294