Abstract
A series of mono-pyrrolo[2,3-d]pyrimidines 4a–4k, unsymmetrical bis-purine isosteres 5a–5e and symmetrical bis-pyrrolo[2,3-d]pyrimidines 6a and 6b connected via di(1,2,3-triazolyl)phenyl linker were synthesized by click chemistry. Whereas mono- 4g and bis-pseudopurine 5e showed selective inhibitory activities on cervical carcinoma (HeLa) cells, bis-pyrrolo[2,3-d]pyrimidine 6b exhibited potent and selective anti-proliferative effect in the nanomolar range on pancreatic carcinoma (CFPAC-1) cells. Among these, compound 6b induced a significant reduction in the expression level of CDK9 (cyclin-dependent kinase 9)/cyclin T1 in CFPAC-1 cells concomitant with attenuation of proliferative signaling mediated by c-Raf (rapidly accelerated fibrosarcoma) and p38 MAP (mitogen-activated protein) kinases. Our findings encourage further development of novel structurally related analog of 6b to obtain more selective anticancer agent for treating pancreatic cancer.
1. Introduction
Kinases have emerged as one of the most intensively pursued classes of drug targets with approximately 30 various kinase targets being developed to the stage ready for clinical trials. Among them, cyclin-dependent kinases (CDKs) belong to the serine/threonine kinases with fundamental role in the control of the cell cycle and/or proliferation and transcription [1]. The kinase activity of CDKs is tightly regulated by the binding to cyclins, the levels of which depend on the balance between protein synthesis and proteasomal degradation, which plays an important role in regulating cellular processes [2]. Overexpression of CDK activators such as cyclin D1 is a major cause for the excessive activation of CDKs [3]. Therefore, deregulation of CDK-cyclin activity in cancer cells has provided a rationale for the investigation of CDK inhibitors for therapeutic intervention in various types of cancer. Over the past two decades, several CDK inhibitors have been developed as potential cancer therapeutics and tested in numerous clinical trials for several tumor types [4,5,6]. The first CDK inhibitor, palbociclib, was pyrido[2,3-d]pyrimidinone derivative that was selective CDK4 and CDK6 inhibitor approved for the treatment of breast cancer [7,8]. Further investigations on purine and purine-like classes resulted in discovery of the 2,6,9-trisubstituted purine analog, roscovitine [CY-202, (R)-roscovitine, seliciclib] that was found to inhibit a subset of CDKs through a direct competition at the ATP-binding site [9,10,11,12]. This purine-based CDK inhibitor was among the first agents evaluated in the clinic [13]. Despite many successful preclinical studies, roscovitine did not meet the initial expectations for a CDK inhibitor in clinical trials [14]. Therefore, purine bioisosteres, as small-molecule CDKs inhibitors, have been widely explored [15,16,17]. Some of them that constitute pyrazolo[1,5-a]pyrimidine [18,19,20,21], benzimidazole [22,23] and pyrrolo[2,3-d]pyrimidine [24,25,26] scaffolds have been currently under clinical evaluation for the treatment of various cancers (Figure 1).
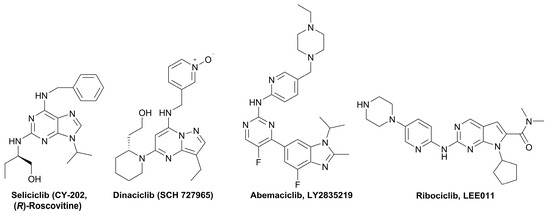
Figure 1.
(R)-Roscovitine and purine isosteres as small-molecule inhibitors of CDK (cyclin-dependent kinases) under clinical evaluations for the treatment of cancer.
Based on the aforementioned and our results on purinomimetics [27] and pyrimidine [28] with diverse substituents at N-1 of 1,2,3-triazole, we performed structural modification by designing pyrrolo[2,3-d]pyrimidines containing alkyl, varied substituted phenyl and phenyl sulfonamide pharmacophores at C-4, instead of N-1, of the 1,2,3-triazole moiety (Figure 2) in order to evaluate their contribution to the anti-proliferative activity. Encouraged by the findings that some heterocyclic dimers with acyclic and cyclic spacer were endowed with pronounced cytostatic activity [29,30,31,32,33,34,35,36] and the importance of halogenated compounds that have been widely exploited in drug discovery [37,38,39] the structural diversity was further extended to the synthesis of halogen-substituted bis-pseudopurines connected through 1,2,3-triazole linker. 1,2,3-Triazole heterocycle has been recognized as good amide bioisosteres and therefore has been widely applied in molecular hybridization approach as an important linker between selected pharmacophoric moieties to produce new hybrid molecules with improved biological properties [40,41,42,43]. Besides unsymmetrical bis-pseudopurine, symmetrical bis-pyrrolo[2,3-d]pyrimidines connected via di(ethylene-1,2,3-triazolyl)phenyl spacer were synthesized (Figure 2).
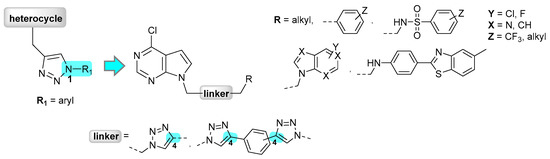
Figure 2.
Design and synthesis of mono- and bis-pseudopurines with C-4 substituted 1,2,3-triazole scaffold.
Therefore, a series of mono- (4a–4k) and bis-pseudopurines (5a–5e, 6a, 6b) were provided with the aim to evaluate their cytostatic activities and further investigate the effects of selected candidates on molecular targets CDK9/cyclin T1, p38 MAPK and c-Raf-1 kinases that regulate cell proliferation.
2. Results and Discussion
2.1. Chemistry
At the top of the working area, a focused library of 18 hybrids of mono- and bis-purine isosteres linked via 1,2,3-triazole moiety was prepared as shown in Scheme 1. N-Alkylation of 6-chloro-7-deazapurine (4-chloro-pyrrolo[2,3-d]pyrimidine) with 1,2-dibromoethane in the presence of K2CO3 afforded the 2-bromoethyl 7-deazapurine derivative 2, which was then converted to the key intermediate 7-deazapurine ethyl azide 3 using NaN3. Target regioselective 1,4-disubstituted 1,2,3-triazoles (4a–4k, 5a–5e, 6a and 6b) were prepared by Huisgen 1,3-dipolar cycloaddition under microwave irradiation using copper(I) catalyst, that was obtained from copper(II) sulfate and metallic copper. 4-Alkyl- (4a, 4b) and 4-aryl- (4c–4h) and 4-arylsulfonamide-substituted (4i–4k) 1,2,3-triazolyl pyrrolo[2,3-d]pyrimidine hybrids were obtained by Cu(I)-catalyzed click reaction of corresponding terminal alkynes and 7-deazapurine azide derivative 3, while bis-purine isosteres (5a–5e) were prepared using 7-deazapurine azide 3 and N-alkynyl heterocyclic base. N-Propargylated derivatives of aryl sulfonamides and heterocyclic bases, including 6-chloro-7-deazapurine, 6-chloropurine, 7-bromo-6-chloro-7-deazapurine, 5-fluoroindole and 5-methylbenzothiazole [44,45,46] were prepared according to known procedures given in literature. Symmetrical bis(6-chloro-7-deazapurine-1,2,3-triazol-1-ylethane) derivatives 6a and 6b connected through phenyl were prepared were synthesized by Cu(I)-catalyzed 1,3-cycloaddition of 1,4- and 1,3-diethynylbenzene with azide 3.
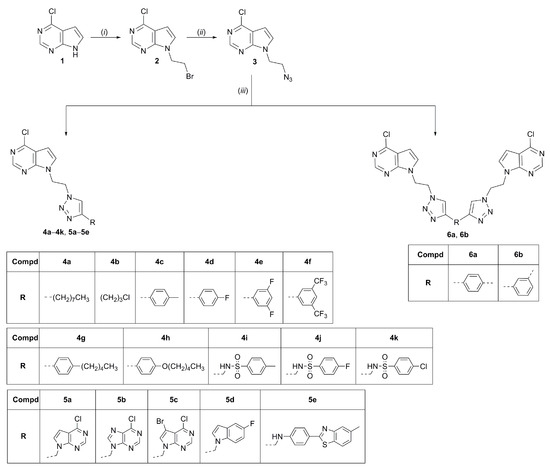
Scheme 1.
Reagents and conditions: (i) 1,2-dibromoethane, DMF (dimethylformamide), Ar atmosphere, rt (room temperature), 24 h; (ii) NaN3, acetonitrile, reflux, overnight; (iii) corresponding terminal alkyne, Cu(0), 1 M CuSO4 solution, tert-butanol:H2O = 1:1, DMF, MW (microwave), 300 W, 80 °C, 45 min. Compd: compound.
2.2. X-ray Crystal Structure Analysis
The structures of 4g, 5a and 5b are confirmed by single crystal X-ray diffraction method. In 4g, 5a and 5b triazole ring is bonded to the nitrogen N7 atom of the 7-deazapurine ring via ethylene spacer (Figure 3).
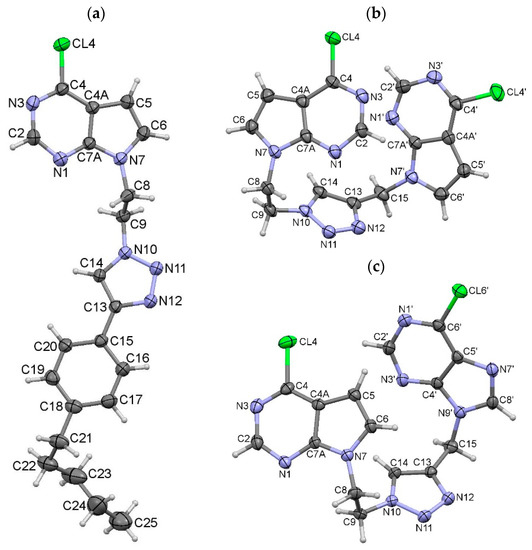
Figure 3.
Molecular structures of 4g (a), 5a (b), and 5b (c), with the atom-numbering schemes. Displacement ellipsoids for non-hydrogen atoms are drawn at the 30% probability level. Color code: blue, N; green, Cl; dark grey, C; light grey, H.
The C13 atom of the triazole ring has three different substituents. Pentylphenyl moiety is directly bonded to the triazole ring in 4g, while 7-deazapurine ring in 5a and purine ring in 5b are bonded to the triazole ring via methylene spacer. Equivalent bond lengths in these structures are very similar, but there are some conformational differences between them. Phenyl ring in 4g is coplanar with the triazole ring; the dihedral angle between the mean planes of the rings is 3.93(13)°. The dihedral angle between the 7-deazapurine and triazole rings in 4g is slightly bigger (Table S1). On the contrary, 7-deazapurine rings in 5a, as well as deazapurine and purine rings in 5b are twisted with respect to the triazole ring for 40° to 70°. It should be mentioned also that 7-deazapurine ring in 5a is rotated around N7–C8 bond for 187° compared to the ring in 5b, as defined by the C6–N7–C8–C9 torsion angle (Figure 3b,c; Table S1).
Weak C–H···N and C–H···Cl hydrogen bonds self-assemble the molecules of 4g, 5a and 5b (Table S2). Thus, six weak C–H···N hydrogen bonds and one C–H···Cl hydrogen bond participate in the supramolecular assembling of 4g (Figure 4a). Each molecule is linked to four neighboring molecules. The C6 atom is proton donor for two hydrogen bonds, as well as the N12 atom is proton acceptor for two hydrogen bonds. All these interactions form two-dimensional network (Figure S1a). Two π···π interactions established between triazole and phenyl rings of neighboring molecules are also included in the formation of such supramolecular structure (Table S3 and Figure S1b). The molecules of 5a are linked by three C–H···N hydrogen bonds and one C–H···Cl hydrogen bond (Table S2). Each molecule is linked by these four interactions to other three molecules (Figure 4b), so forming parallel arrangement of hydrogen-bonded chains (Figure S2). Crystal packing diagram reveals that chains are disposed in zig-zag manner and form two-dimensional network (Figure S2). As in 5a, each molecule of 5b is connected to three neighboring molecules by three C–H···N hydrogen bonds and one C–H···Cl hydrogen bond (Figure 4c and Table S2). Hydrogen-bonded molecules are mutually parallel, and form two-dimensional network very similar to that in 5a (Figure S3a). However, there is one distinct difference between these structures. Six-membered N1/C2/N3/C4/C4A/C7A rings of neighboring molecules are mutually parallel, so forming two π···π interactions (Table S3 and Figure S3b). These two interactions extend two-dimensional network into three-dimensional.
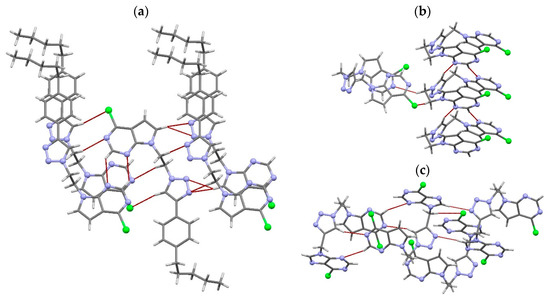
Figure 4.
Capped stick representation of 4g (a), 5a (b) and 5b (c), showing C–H···N and C–H···Cl hydrogen bonds. Nitrogen and chlorine atoms are presented in ball and stick style. Color code: blue, N; green, Cl; grey, C; light grey, H.
2.3. Biological Evaluations
2.3.1. Anti-Proliferative Evaluations
Results of anti-proliferative evaluations of compounds 4a–4k, 5a–5e, 6a and 6b on human tumor lung adenocarcinoma (A549), ductal pancreatic adenocarcinoma (CFPAC-1), cervical carcinoma (HeLa) and colorectal adenocarcinoma, metastatic (SW620) are presented in Table 1. Roscovitine was used as reference compound. The compound that found to exhibit the highest cytostatic activity and roscovitine were also evaluated on normal human foreskin fibroblasts (HFF).

Table 1.
In vitro growth inhibitory effects of synthesized compounds on selected tumor cell lines.
From the mono-pyrrolo[2,3-d]pyrimidines (4a–4k), compounds containing 4-alkyl (4a, 4b) and 4-aryl-substituted (4c–4f) 1,2,3-triazole showed only marginal cytostatic effects. It can be observed that type and position of substituents at phenyl ring in 4c–4f had no impact on the growth of tested tumor cell lines. Importantly, p-alkyl- and p-alkoxyphenyl substituents at C-4 of 1,2,3-triazole in 4g and 4h, respectively, considerably improved anti-proliferative effects on CFPAC-1, HeLa and SW620 cells. Thus, 7-deazapurine 4g with 4-(p-pentylphenyl)-1,2,3-triazole exhibited the highest potency (IC50 = 9.5 µM) on HeLa cells, whereas its analog 4h with p-pentoxyphenyl substituent at C-4 showed strong activity on CFPAC-1 (IC50 = 8.1 µM), HeLa (IC50 = 7.8 µM) and SW620 (IC50 = 6.9 µM) cells. Interestingly, electron-rich p-alkoxy unit in 4g led to significant improved inhibitory activity on CFPAC-1 cells relative to compound 4h (IC50 > 100 µM). The influence of substituent at phenylsulfonamide was also observed indicating that the electron-withdrawing halogen atoms enhanced the cytostatic effects compared with electron-donating methyl group. Namely, 4i with p-methylphenylsulfonamide was deprived of any cytostatic activity. Conversely, p-fluorophenylsulfonamide in 4j caused slight enhancement of potency on CFPAC-1, HeLa and SW620 cells and p-chlorophenylsulfonamide in 4k increased the activity on all tested cell lines with respect to 4i. From mono-pyrrolo[2,3-d]pyrimidine series (4a–4k), except for 4f, compounds 4g and 4h with best cytostatic effects showed also the highest ClogP (coefficients logP) values.
Among the series of bis-pseudopurines 5a–5e, 6a and 6b, the impact of bromine at C-7 of 7-deazapurine (7-BrDPu) in 5c was detected displaying higher activities of 5c against all tested cell lines than corresponding analog (7-DPu) 5a without bromine. These differences in activities were particularly expressed on HeLa and SW620 cells. Bis-purine isosteres bearing 2-aminophenylbenzothiazole (BTh) was endowed with strong and selective anti-proliferative effect (IC50 = 5.3 µM) on HeLa cells. On the contrary, 6-chloropurine (6-ClPu) and 5-fluoroindole (5-FIn) moieties in compounds 5b and 5d, respectively, decreased their anti-proliferative effects. Symmetrical bis(6-chloro-7-deazapurine-ethyl-1,2,3-triazole) connected via 1,4- and 1,3-phenyl spacer in 6a and 6b, respectively, exhibited the most pronounced inhibitory effect, particularly on CFPAC-1 cells (6a: IC50 = 3.6 µM, 6b: IC50 = 0.95 µM). Abovementioned structural requirements that influenced the cytostatic activities are summarized in Figure 5.
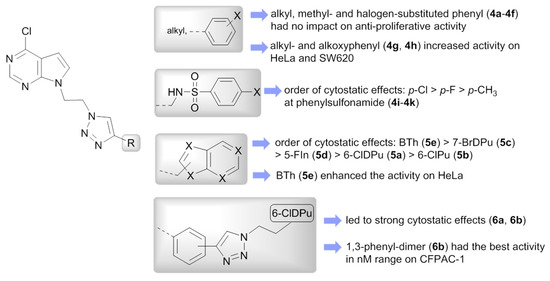
Figure 5.
Structure-activity relationship (SAR) of a series of mono- (4a–4k) and bis-pseudopurines (5a–5e, 6a and 6b).
In accord with findings for mono-pyrrolo[2,3-d]pyrimidine series (4a–4k), relationship between lipophilicity and anti-proliferative effect was also observed for bis-pseudopurine series (5a–5e, 6a and 6b) suggesting that compounds 5e, 6a and 6b with best cytostatic effect were rather lipophilic with ClogP values from 4.9 to 5.4.
Mono- 4g and bis-pseudopurine 5e with selective antitumor activities on HeLa cells, and symmetrical bis-pyrrolo[2,3-d]pyrimidines 6b exhibiting potent and selective cytostatic effect on CFPAC-1 cells were identified as candidates for further biological evaluations in order to investigate their mechanism of action on HeLa and CFPAC-1 cells. In comparison with cytostatic activities of compounds 4g, 5e and 6b, roscovitine exerted markedly weaker anti-proliferative effects on all tested cancer cell lines. However, compound 6b showing the cytostatic activity in the nanomolar range on CFPAC-1 cells was also cytotoxic to normal fibroblasts. Therefore, additional efforts are required to determine which structural modifications are required to retain anticancer activity and eliminate toxicity of 6b.
2.3.2. Western Blot Analysis of Predicted Protein Targets
The prediction of activity spectra for substances analysis (PASS) [47] was performed to reveal the probable protein targets of selected candidates 4g, 5e and 6b. Thus, CDK9/cyclin T1 was indicated with the highest probability as a potential target for 4g, 5e and 6b (Tables S4–S6). Western blot analyses (Figure 6) showed that compounds 5e and 6b significantly reduced the expression level of CDK9/cyclin T1 in human pancreatic cancer CFPAC-1 cells and human cervical cancer HeLa cells, respectively. In addition, compound 4g also induced down-regulation of CDK9/cyclin T1, although to a lesser extent. Thus, we can conclude that compounds 4g, 5e and 6b exert their antitumor effects by targeting CDK9/cyclin T1. Similarly, among the most advanced CDK4 and CDK6 inhibitors, purinomimetics abemaciclib (LY2835219) [22,23] and ribociclib (LEE011) (Figure 1) [23,48] showed to be potent CDK9 inhibitors. In addition, the therapeutic potential of selective CDK inhibition on pancreatic cancer cells was revealed by other structurally related small molecule CDK inhibitor, dinaciclib (SCH727965) (Figure 1) [21,49]. Furthermore, PASS analysis also revealed that some of selected compounds could potentially inhibit p38 MAP kinase with higher (compound 6b) or lower probability (compound 4g) (Tables S4–S6). For compound 5e, PASS also revealed the possibility to inhibit MAPK with high probability, and for compounds 4g and 6b with lower probabilities. Since c-Raf is a member of MAPK family [50], this protein was tested as a potential target for all three compounds. Previously demonstrated roles of p38 MAPK and c-Raf in promoting cancer cell proliferation [51] provide additional rationale for examining their involvement in mediating anti-proliferative effects of tested compounds revealed by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay.
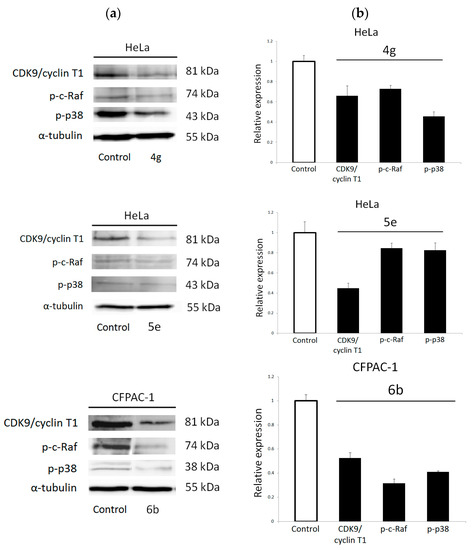
Figure 6.
Western blot analysis of predicted protein targets of compounds 4g, 5e and 6b. (a) Representative Western blots are shown detecting the cellular levels of selected proteins before and after treatment of the pancreatic adenocarcinoma (CFPAC-1) and HeLa cells with indicated compounds at their 2 × IC50 values for 48 h. Approximate molecular weights (kDa) are indicated. (b) Relative protein expressions, as determined by densitometric analysis of protein bands and normalized to the α-tubulin loading control. Two independent experiments were performed with similar results. Data are presented as mean values ± SD.
Our results revealed that symmetrical bis-pyrrolo[2,3-d]pyrimidine 6b elicited significant decrease in the levels of phospho-c-Raf and phospho-p38 kinases, which points to abrogation of their activities (Figure 6). Modest inhibition of c-Raf activity was also observed with mono-pyrrolo[2,3-d]pyrimidine 4g and unsymmetrical bis-purine isostere 5e. Importantly, 4g led to a marked reduction in p38 MAPK activity in HeLa cells. These differences in the inhibitory effects on c-Raf and p38 kinases activities between evaluated compounds could be ascribed to both purine isostere and substituent at C-4 of 1,2,3-triazole moiety. It is interesting to note that symmetrical bis(pyrrolo[2,3-d]pyrimidine) hybrid 6b with the most pronounced expression of c-Raf and p38 kinases also showed the most potent cytostatic effects among these compounds as evident from their IC50 values (Table 1). Altogether, Western blot study has demonstrated that anti-proliferative effect of compound 6b in CFPAC-1 cells could be attributed to inhibition of CDK9/cyclin T1 and attenuation of oncogenic signaling propagated by p38 MAPK and c-Raf, which encourages further development of compound 6b as a novel anticancer agent for treating pancreatic cancer.
2.3.3. Apoptosis Detection
Annexin V assay was carried out to determine if the growth-inhibitory activity of compound 6b exhibiting the most pronounced potency could be attributed to induction of apoptosis. Obtained results (Table 2, Figure 7) showed that compound 6b reduced viable cell population by 19.48% concurrent with a marked increase in late apoptotic/primary necrotic and secondary necrotic cells by 12.24% and 10.28%, respectively. Therefore, obtained results showed that antitumor effects of compound 6b in CFPAC-1 cells could be ascribed to its ability to induce late apoptosis and necrosis.
Table 2.
Annexin V assay for apoptosis detection in CFPAC-1 cells a.
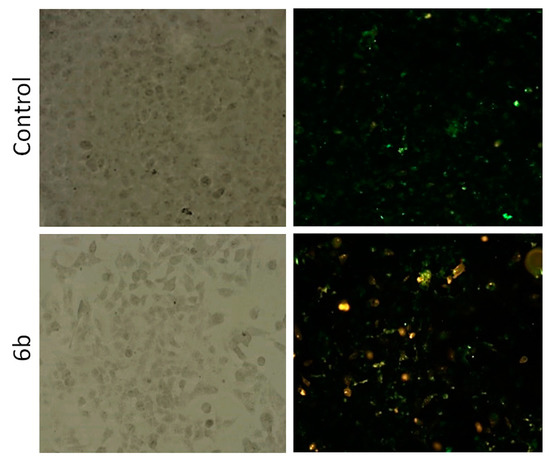
Figure 7.
Detection of apoptosis induced by compound 6b in human pancreatic adenocarcinoma CFPAC-1 cell line using Annexin V-FITC assay. Cells were visualized by fluorescence microscope at 40× magnification before and after treatment with indicated compounds at the concentration of 2 × IC50 for 48 h. PI staining was used as a nuclear marker. Shown here are bright-field images (left) and late apoptotic/primary necrotic cells (right).
3. Materials and Methods
3.1. General
All solvents were purified using appropriate drying agents and stored over 3 Å molecular sieves. Thin layer chromatography (TLC) was carried out using Merck silica gel 60F-254 plates (Merck, Darmstadt, Germany), while Fluka 0.063–0.2 mm silica gel (Fluka, Neu-Ulm, Germany) was applied for purification by column chromatography. Melting points (m.p.) were determined on Kofler micro hot-stage (Reichert, Wien, Austria). 1H and 13C APT (Attached Proton Test) NMR (nuclear magnetic resonance) spectra were recorded in DMSO-d6 on a Varian Gemini 300 (at 300 and 75 MHz) or Varian Gemini 600 (at 600 and 150 MHz) (Figures S4–S23). Chemical shifts (б) were referenced to the residual solvent signal of DMSO at б 2.50 ppm for 1H and б 39.50 ppm for 13C. Individual resonances were assigned on the basis of their chemical shifts, signal intensities, multiplicity of resonances and H-H coupling constants. High performance liquid chromatography was performed on an Agilent 1100 series system with UV detection (photodiode array detector). Zorbax C18 reverse-phase analytical column (2.1 × 30 mm, 3.5 µm) was used. All novel compounds showed 95% purity in this HPLC (high performance liquid chromatography) system. Microwave-assisted syntheses were performed in a Milestone start S microwave oven using quartz cuvettes.
3.2. Experimental Procedures for the Synthesis of Compounds
7-(2-Bromoethyl)-4-chloro-7H-pyrrolo[2,3-d]pyrimidine (2). 6-Chloro-7-deazapurine (500 mg; 3.26 mmol) and K2CO3 (540 mg; 3.91 mmol) were dissolved in 10 mL DMF and stirred under argon atmosphere for 1 h. 1,2-Dibromoethane (0.34 mL; 3.91 mmol) was added to the mixture and stirred for 24 h at room temperature. The solvent was evaporated to dryness and the residue was purified by column chromatography (hexane:ethyl acetate = 8:1) to give compound 1 as white powder (347.38 mg, 41%, m.p. = 91–93 °C). 1H NMR (300 MHz, DMSO) δ 8.66 (1H, s, H2), 7.85 (1H, d, J = 3.6 Hz, H6), 6.69 (1H, d, J = 3.6 Hz, H5), 4.71 (2H, t, J = 6.2 Hz, CH2), 3.96 (2H, t, J = 6.2 Hz, CH).13C NMR (151 MHz, DMSO) δ 150.7, 150.6, 150.3, 131.4, 116.8, 98.6, 45.9, 31.6.
7-(2-Azidoethyl)-4-chloro-7H-pyrrolo[2,3-d]pyrimidine (3). Compound 2 (247.8 mg, 0.95 mmol) and NaN3 (167.4 mg, 1.90 mmol) were stirred in acetonitrile (10 mL) under reflux overnight. The solvent was evaporated to dryness and the residue was purified by column chromatography (hexane: ethyl acetate = 1:1) to obtain 3 as crude oil (189.2, 95%). 1H NMR (300 MHz, DMSO) δ 8.67 (1H, s, H2), 7.83 (1H, d, J = 3.6 Hz, H6), 6.71 (1H, d, J = 3.6 Hz, H5), 4.49 (2H, t, J = 6.2 Hz, CH2), 3.81 (2H, t, J = 6.2 Hz, CH2). 13C NMR (151 MHz, DMSO) δ 151.0, 150.8, 150.5, 131.5, 116.9, 98.9, 50.1, 43.9.
3.2.1. General Procedure for the Synthesis of Compounds (4a–4k and 5a–5e)
The azido derivative 3 was dissolved in 0.5 mL DMF and t-BuOH:H2O = 1:1 (2–3 mL), Cu(0) (0.3 eq.), 1 M CuSO4 (0.8 eq.). The corresponding terminal alkyne (1.2 eq.) was added and stirred under microwave irradiation for 45 min at 80 °C and 300 W. The solvent was evaporated and the residue was purified by column chromatography using CH2Cl2:CH3OH = 100:1, as an initial eluent, and CH2Cl2:CH3OH = 10:1, as final eluent.
4-Chloro-7-(2-(4-octyl-1H-1,2,3-triazol-1-yl)ethyl)-7H-pyrrolo[2,3-d]pyrimidine (4a). Compound 4a was prepared using the above-mentioned procedure using compound 3 (30 mg, 0.14 mmol) and decyne (0.03 mL, 0.17 mmol) to obtain 4a as brown solid (13.6 mg, 29%, m.p. = 115–118 °C). 1H NMR (600 MHz, DMSO) δ 8.55 (1H, s, H2), 7.55 (1H, s, H5′), 7.52 (1H, d, J = 3.6 Hz, H6), 6.59 (1H, d, J = 3.6 Hz, H5), 4.84–4.76 (2H, m, CH2), 4.75–4.68 (2H, m, CH2), 2.46 (2H, t, J = 7.4 Hz, CH2′′), 1.46–1.38 (2H, m, CH2′′), 1.31–1.22 (8H, m, CH2′′), 1.17–1.12 (2H, m, CH2′′), 0.86 (3H, t, J = 7.1 Hz, CH3′′). 13C NMR (75 MHz, DMSO) δ 153.2, 150.7, 150.3, 147.1, 131.2, 122.2, 116.8, 99.0, 49.0, 44.8, 31.6, 29.0, 28.8, 28.7, 28.4, 24.9, 22.3, 14.1. Anal. calcd. (analytically calculated) for C18H25ClN6: C, 59.91; H, 6.98; N, 23.29. Found: C, 60.13; H, 7.07; N, 23.22.
4-Chloro-7-(2-(4-(3-chloropropyl)-1H-1,2,3-triazol-1-yl)ethyl)-7H-pyrrolo[2,3-d]pyrimidine (4b). Compound 4b was prepared using the above-mentioned procedure using compound 3 (30 mg, 0.14 mmol) and 5-chloropent-1-yne (0.02 mL, 0.17 mmol) to obtain 4b as white crystals (34.9 mg, 77%, m.p. = 114–116 °C). 1H NMR (300 MHz, DMSO) δ 8.55 (1H, s, H2), 7.66 (1H, s, H5′), 7.53 (1H, d, J = 3.6 Hz, H6), 6.60 (1H, d, J = 3.6 Hz, H5), 4.80 (2H, m, CH2), 4.77–4.70 (2H, m, CH2), 3.54 (2H, t, J = 6.5 Hz, CH2′′), 2.64 (2H, t, J = 7.3 Hz, CH2′′), 2.03–1.77 (2H, m, CH2′′). 13C NMR (75 MHz, DMSO) δ 150.8, 150.7, 150.3, 145.5, 131.2, 122.5, 116.8, 98.9, 49.0, 44.8, 44.5, 31.8, 22.1. Anal. calcd. for C13H14Cl2N6: C, 48.01; H, 4.34; N, 25.84. Found: C, 47.86; H, 4.39; N, 25.71.
4-Chloro-7-(2-(4-(p-tolyl)-1H-1,2,3-triazol-1-yl)ethyl)-7H-pyrrolo[2,3-d]pyrimidine (4c). Compound 4c was prepared using the above-mentioned procedure using compound 3 (30 mg, 0.14 mmol) and 1-ethynyl-4-methylbenzene (0.02 mL, 0.17 mmol) to obtain 4c as white powder (15 mg, 31%, m.p. = 198–201 °C). 1H NMR (300 MHz, DMSO) δ 8.56 (1H, s, H2), 8.32 (1H, s, H5′), 7.60 (2H, d, J = 8.1 Hz, Ph′′), 7.56 (1H, d, J = 3.6 Hz, H6), 7.22 (2H, d, J = 7.9 Hz, Ph′′), 6.61 (1H, d, J = 3.6 Hz, H5), 4.93–4.85 (2H, m, CH2), 4.85–4.76 (2H, m, CH2), 2.31 (3H, s, CH3′′). 13C NMR (75 MHz, DMSO) δ 150.8, 150.7, 150.3, 146.5, 137.3, 131.3, 129.5, 127.8, 125.1, 121.3, 116.9, 98.9, 49.2, 44.8, 20.9. Anal. calcd. for C17H15ClN6: C, 60.27; H, 4.46; N, 24.80. Found: C, 60.35; H, 4.43; N, 24.98.
4-Chloro-7-(2-(4-(4-fluorophenyl)-1H-1,2,3-triazol-1-yl)ethyl)-7H-pyrrolo[2,3-d]pyrimidine (4d). Compound 4d was prepared using the above-mentioned procedure using compound 3 (30 mg, 0.14 mmol) and 1-ethynyl-4-fluorobenzene (0.02 mL, 0.17 mmol) to obtain 4d as white crystals (25.8 mg, 54%, m.p. = 200–202 °C). 1H NMR (300 MHz, DMSO) δ 8.56 (1H, s, H2), 8.38 (1H, s, H5′), 7.79–7.73 (2H, m, Ph′′), 7.56 (1H, d, J = 3.6 Hz, H6), 7.26 (2H, t, J = 8.9 Hz, Ph′′), 6.61 (1H, d, J = 3.6 Hz, H5), 4.93–4.87 (2H, m, CH2), 4.84–4.78 (2H, m, CH2). 13C NMR (151 MHz, DMSO) δ 162.7; 161.1 (d, JCF = 244.6 Hz), 150.8, 150.7, 150.3, 145.6, 131.3, 127.3; 127.2 (d, JCF = 8.2 Hz), 127.2, 121.7, 116.9, 116.0; 115.8 (d, JCF = 21.7 Hz), 99.0, 49.3, 44.8. Anal. calcd. for C16H12ClFN6: C, 56.07; H, 3.53; N, 24.52. Found: C, 55.83; H, 3.80; N, 24.26.
4-Chloro-7-(2-(4-(3,5-difluorophenyl)-1H-1,2,3-triazol-1-yl)ethyl)-7H-pyrrolo[2,3-d]pyrimidine (4e). Compound 4e was prepared using the above-mentioned procedure using compound 3 (30 mg, 0.14 mmol) and 1-ethynyl-3,5-difluorobenzene (0.02 mL, 0.17 mmol) to obtain 4e as white crystals (37.3 mg, 74%, m.p. = 187–190 °C). 1H NMR (300 MHz, DMSO) δ 8.56 (1H, s, H2), 8.52 (1H, s, H5′), 7.57 (1H, d, J = 3.6 Hz, H6), 7.47–7.42 (2H, m, Ph′′), 7.27–7.13 (1H, m, Ph′′), 6.61 (1H, d, J = 3.6 Hz, H5), 4.96–4.89 (2H, m, CH2), 4.84–4.78 (2H, m, CH2). 13C NMR (75 MHz, DMSO) δ 164.7; 161.4 (d, JCF = 245.7 Hz) 164.5; 161.3 (d, JCF = 245.7 Hz), 150.9, 150.7, 150.3, 144.5, 134.1, 131.4, 123.2, 116.9, 108.3; 108.2; 108.1, 108.0 (dd, JCF = 26.3, 8.6 Hz), 103.6; 103.3; 102.9 (t, JCF = 25.9 Hz), 99.0, 49.5, 44.8. Anal. calcd. for C16H11ClF2N6: C, 53.27; H, 3.07; N, 29.30. Found: C, 53.42; H, 3.01; N, 29.20.
7-(2-(4-(3,5-Bis(trifluoromethyl)phenyl)-1H-1,2,3-triazol-1-yl)ethyl)-4-chloro-7H-pyrrolo[2,3-d]pyrimidine (4f). Compound 4f was prepared using the above-mentioned procedure using compound 3 (30 mg, 0.14 mmol) and 1-ethynyl-3,5-bis(trifluoromethyl)benzene to obtain 4f as white power (60.9 mg, 94%, m.p. = 215–218 °C). 1H NMR (300 MHz, DMSO) δ 8.80 (1H, s, H5′), 8.55 (1H, s, H2), 8.39 (2H, s, Ph′′), 8.05 (1H, s, Ph′′), 7.58 (1H, d, J = 3.6 Hz, H6), 6.61 (1H, d, J = 3.6 Hz, H5), 4.96–4.94 (2H, m, CH2), 4.84–4.80 (2H, m, CH2). 13C NMR (151 MHz, DMSO) δ 150.9, 150.9, 150.4, 143.9, 133.2, 131.6; 131.4; 131.2; 131.0 (q, JCF = 33.3 Hz), 131.4, 126.1; 124.3; 122.5; 120.7 (q, JCF = 273.0 Hz), 125.5; 125.4 (d, JCF = 3.6 Hz), 123.9, 121.4, 121.4; 121.4 (m), 117.0, 99.2, 49.7, 44.9. Anal. calcd. for C18H11ClF6N6: C, 46.92; H, 2.41; N, 18.24. Found: C, 47.21; H, 2.16; N, 18.37.
4-Chloro-7-(2-(4-(4-pentylphenyl)-1H-1,2,3-triazol-1-yl)ethyl)-7H-pyrrolo[2,3-d]pyrimidine (4g). Compound 4g was prepared using the above-mentioned procedure using compound 3 (30 mg, 0.14 mmol) and 1-ethynyl-4-pentylbenzene (0.03 mL, 0.17 mmol) to obtain 4g as colorless crystals (40.2 mg, 72%, m.p. = 170–173 °C). 1H NMR (300 MHz, DMSO) δ 8.57 (1H, s, H2), 8.33 (1H, s, H5′), 7.62 (2H, d, J = 8.1 Hz, Ph′′), 7.54 (1H, d, J = 3.7 Hz, H6), 7.23 (2H, d, J = 8.1 Hz, Ph′′), 6.60 (1H, d, J = 3.6 Hz, H5), 4.92–4.86 (2H, m, CH2), 4.84–4.78 (2H, m,CH2), 2.61–2.54 (2H, m, CH2′′), 1.58 (2H, dt, J = 14.5, 7.1 Hz, CH2′′), 1.35–1.24 (4H, m, CH2′′), 0.86 (3H, t, J = 6.8 Hz, CH3′′). 13C NMR (151 MHz, DMSO) δ 150.9, 150.7, 150.4, 146.6, 142.3, 131.4, 128.9, 128.1, 125.2, 121.4, 116.9, 99.0, 49.2, 44.8, 35.0, 31.0, 30.7, 22.1, 14.1. Anal. calcd. for C21H23ClN6: C, 63.87; H, 5.87; N, 21.28. Found: C, 63.82; H, 6.00; N, 20.96.
4-Chloro-7-(2-(4-(4-(pentyloxy)phenyl)-1H-1,2,3-triazol-1-yl)ethyl)-7H-pyrrolo[2,3-d]pyrimidine (4h). Compound 4h was prepared using the above-mentioned procedure using compound 3 (30 mg, 0.14 mmol) and 1-ethynyl-4-(pentyloxy)benzene to obtain 4h as white powder (49.7 mg, 86%, m.p. = 166–168 °C). 1H NMR (300 MHz, DMSO) δ 8.57 (1H, s, H2), 8.26 (1H, s, H5′), 7.62 (2H, d, J = 8.7 Hz, Ph′′), 7.55 (1H, d, J = 3.6 Hz, H6), 6.96 (2H, d, J = 8.8 Hz, Ph′′), 6.60 (1H, d, J = 3.6 Hz, H5), 4.91–4.85 (2H, m, CH2), 4.83–4.77 (2H, m, CH2), 3.98 (2H, t, J = 6.5 Hz, CH2′′), 1.78–1.65 (2H, m, CH2′′), 1.46–1.28 (4H, m, CH2′′), 0.90 (3H, t, J = 7.0 Hz, CH2′′). 13C NMR (75 MHz, DMSO) δ 158.5, 150.8, 150.6, 150.3, 146.3, 131.3, 126.5, 123.0, 120.7, 116.8, 114.8, 98.8, 67.5, 49.1, 44.7, 28.4, 27.7, 21.9, 14.0. Anal. calcd. for C21H23ClN6O: C, 61.38; H, 5.64; N, 20.45. Found: C, 61.42; H, 5.55; N, 20.64.
N-((1-(2-(4-Chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)ethyl)-1H-1,2,3-triazol-4-yl)methyl)-4-methylbenzenesulfonamide (4i). Compound 4i was prepared using the above-mentioned procedure using compound 3 (20 mg, 0.1 mmol) and 4-methyl-N-(prop-2-ynyl)benzenesulfonamide (24.07 mg, 0.12 mmol) to obtain 4i as white powder (30.3 mg, 73%, m.p. = 190–192 °C). 1H NMR (600 MHz, DMSO) δ 8.59 (1H, s, H2), 7.98 (1H, t, J = 5.6 Hz, NH′′), 7.78 (1H, s, H5′′), 7.65 (2H, d, J = 8.2 Hz, Ph′′), 7.46 (1H, d, J = 3.6 Hz, H6), 7.37 (2H, d, J = 8.1 Hz, Ph′′), 6.59 (1H, d, J = 3.6 Hz, H5), 4.79 (2H, t, J = 5.8 Hz, CH2), 4.71 (2H, t, J = 5.7 Hz, CH2), 3.92 (2H, d, J = 5.5 Hz, CH2′′), 2.38 (3H, s, CH3′′). 13C NMR (151 MHz, DMSO) δ 150.7, 150.5, 150.3, 143.6, 142.6, 137.4, 131.1, 129.5, 126.6, 123.5, 116.7, 98.7, 48.7, 44.5, 38.0, 20.9. Anal. calcd. for C18H18ClN7O2S: C, 50.06; H, 4.20; N, 22.70. Found: C, 49.91; H, 4.52; N, 22.33.
N-((1-(2-(4-Chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)ethyl)-1H-1,2,3-triazol-4-yl)methyl)-4-fluorobenzenesulfonamide (4j). Compound 4j was prepared using the above-mentioned procedure using compound 3 (20 mg, 0.1 mmol) and 4-fluoro-N-(prop-2-ynyl)benzenesulfonamide (26.60 mg, 0.12 mmol) to obtain 4j as white powder (10.1 mg, 24%, m.p. = 178–179 °C). 1H NMR (600 MHz, DMSO) δ 8.58 (1H, s, H2), 8.13 (1H, s, NH′′), 7.85–7.79 (3H, m, J = 9.5, 6.2 Hz, H5′, Ph′′), 7.47 (1H, d, J = 3.5 Hz, H6), 7.40 (2H, t, J = 8.8 Hz, Ph′′), 6.59 (1H, d, J = 3.5 Hz, H5), 4.79 (2H, t, J = 5.7 Hz, CH2), 4.72 (2H, t, J = 5.8 Hz, CH2), 3.96 (2H, s, CH2′′). 13C NMR (151 MHz, DMSO) δ 164.9; 163.2 (JCF = 251.0 Hz), 150.7, 150.6, 150.2, 143.4, 136.8; 136.7 (JCF = 3.4 Hz), 131.1, 129.6, 129.5 (JCF = 9.5 Hz), 123.6, 116.7, 116.3; 116.1 (JCF = 22.7 Hz), 98.7, 48.7, 44.5, 37.9. Anal. calcd. for C17H15ClFN7O2S: C, 46.85; H, 3.47; N, 22.49. Found: C, 46.87; H, 3.19; N, 22.62.
N-((1-(2-(4-Chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)ethyl)-1H-1,2,3-triazol-4-yl)methyl)-4-chlorobenzenesulfonamide (4k). Compound 4k was prepared using the above-mentioned procedure using compound 3 (25 mg, 0.11 mmol), and 4-chloro-N-(prop-2-ynyl)benzenesulfonamide (28.83 mg, 0.13 mmol) to obtain 4k as white powder (35.3 mg, 71%, m.p. = 195–197 °C). 1H NMR (300 MHz, DMSO) δ 8.59 (1H, s, H2), 8.20 (1H, bs, NH′′), 7.80 (1H, s, H5′), 7.76 (2H, d, J = 8.7 Hz, Ph′′), 7.64 (2H, d, J = 8.7 Hz, Ph′′), 7.47 (1H, d, J = 3.6 Hz, H6), 6.60 (1H, d, J = 3.6 Hz, H5), 4.86–4.74 (2H, m, CH2), 4.74–4.63 (2H, m, CH2), 3.97 (2H, s, CH2′′). 13C NMR (75 MHz, DMSO) δ 150.8, 150.7, 150.3, 143.4, 139.3, 137.3, 131.2, 129.3, 128.6, 123.7, 116.8, 98.8, 48.9, 44.6, 37.9. Anal. calcd. for C17H15Cl2N7O2S: C, 45.14; H, 3.34; N, 21.68. Found: C, 44.89; H, 3.46; N, 21.60.
4-Chloro-7-((1-(2-(4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)ethyl)-1H-1,2,3-triazol-4-yl)methyl)-7H-pyrrolo[2,3-d]pyrimidine (5a). Compound 5a was prepared using the above-mentioned procedure using compound 3 (40 mg, 0.18 mmol) and 4-chloro-7-(prop-2-yn-1-yl)-7H-pyrrolo[2,3-d]pyrimidine (38.6 mg, 0.22 mmol) to obtain 5a as white powder (57.2 mg, 77%, m.p. = 202–204 °C). 1H NMR (600 MHz, DMSO) δ 8.64 (1H, s, H2′′), 8.41 (1H, s, H2), 7.82 (1H, s, H5′), 7.61 (1H, d, J = 3.6 Hz, H6′′), 7.47 (1H, d, J = 3.6 Hz, H6), 6.65 (1H, d, J = 3.6 Hz, H5′′), 6.51 (1H, d, J = 3.6 Hz, H5), 5.46 (2H, s, CH2′′), 4.81 (2H, t, J = 5.7 Hz, CH2), 4.70 (2H, t, J = 5.7 Hz, CH2). 13C NMR (75 MHz, DMSO) δ 150.7, 150.7, 150.5, 150.4, 150.3, 150.1, 142.6, 131.0, 123.9, 116.8, 116.6, 98.8, 98.8, 49.1, 44.6, 39.4. Anal. calcd. for C17H13Cl2N9: C, 49.29; H, 3.16; N, 30.43. Found: C, 49.59; H, 3.02; N, 30.27.
6-Chloro-9-((1-(2-(4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)ethyl)-1H-1,2,3-triazol-4-yl)methyl)-9H-purine (5b). Compound 5b was prepared using the above-mentioned procedure using compound 3 (16 mg, 0.09 mmol) and 6-chloro-9-(prop-2-yn-1-yl)-9H-purine (24.0 mg, 0.11 mmol) to obtain 5b as white powder (30.9 mg, 83%, m.p. = 214–216 °C). 1H NMR (300 MHz, DMSO) δ 8.77 (1H, s, H2′′), 8.68 (1H, s, H2), 8.40 (1H, s, H8′′), 7.94 (1H, s, H5′), 7.46 (1H, d, J = 3.6 Hz, H6), 6.51 (1H, d, J = 3.6 Hz, H5), 5.51 (2H, s, CH2′′), 4.85–4.80 (2H, m, J = 6.6, 4.4 Hz, CH2), 4.73–4.67 (2H, m, J = 6.6, 4.4 Hz, CH2). 13C NMR (75 MHz, DMSO) δ 151.6, 151.5, 150.6, 150.5, 150.0, 149.1, 147.2, 141.7, 131.0, 130.7, 124.1, 116.6, 98.7, 49.1, 44.6, 38.7. Anal. calcd. for C16H12Cl2N10: C, 46.28; H, 2.91; N, 33.73. Found: C, 46.27; H, 3.17; N, 33.77.
5-Bromo-4-chloro-7-((1-(2-(4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)ethyl)-1H-1,2,3-triazol-4-yl)methyl)-7H-pyrrolo[2,3-d]pyrimidine (5c). Compound 5c was prepared using the above-mentioned procedure using compound 3 (15 mg, 0.07 mmol) and 5-bromo-4-chloro-7-(prop-2-yn-1-yl)-7H-pyrrolo[2,3-d]pyrimidine (22.7 mg, 0.08 mmol) to obtain 5c as white crystals (26.2 mg, 76%, m.p. = 168–171 °C). 1H NMR (600 MHz, DMSO) δ 8.68 (1H, s, H2), 8.41 (1H, s, H2′′), 7.91 (1H, s, H5′), 7.86 (1H, s, H8′′), 7.46 (1H, d, J = 3.6 Hz, H6), 6.51 (1H, d, J = 3.6 Hz, H5), 5.45 (2H, s, CH2′′), 4.82–4.80 (2H, m, J = 6.6, 4.7 Hz, CH2), 4.72–4.68 (2H, m, J = 6.6, 4.8 Hz, CH2). 13C NMR (151 MHz, DMSO) δ 151.0, 150.6, 150.5, 150.0, 149.6, 142.1, 131.0, 130.8, 124.0, 116.6, 113.9, 98.6, 85.9, 49.0, 44.5, 39.5. Anal. calcd. for C17H12BrCl2N9: C, 41.40; H, 2.45; N, 25.56. Found: C, 41.69; H, 2.34; N, 25.72.
4-Chloro-7-(2-(4-((5-fluoro-1H-indol-1-yl)methyl)-1H-1,2,3-triazol-1-yl)ethyl)-7H-pyrrolo[2,3-d]pyrimidine (5d). Compound 5d was prepared using the above-mentioned procedure using compound 3 (40 mg, 0.18 mmol) and 5-fluoro-1-(prop-2-yn-1-yl)-1H-indole (37.4 mg, 0.22 mmol) to obtain 5d as white powder (70.2 mg, 98%, m.p. = 169–171 °C). 1H NMR (600 MHz, DMSO) δ 8.44 (1H, s, H2), 7.78 (1H, s, H5′), 7.46–7.41 (2H, m, H6, H7′′), 7.33 (1H, d, J = 3.1 Hz, H2′′), 7.29 (1H, dd, J = 9.9, 2.4 Hz, H4′′), 6.95 (1H, td, J = 9.3, 2.5 Hz, H6′′), 6.51 (1H, d, J = 3.6 Hz, H5), 6.41 (1H, dd, J = 3.2, 0.6 Hz, H3′′), 5.35 (2H, s, CH2′′), 4.82–4.79 (2H, m, CH2), 4.72–4.68 (2H, m, CH2). 13C NMR (75 MHz, DMSO) δ 158.7; 155.6 (d, JCF = 231.6 Hz), 150.7, 150.6, 150.2, 143.4, 132.3, 131.0, 130.3, 128.5; 128.4 (d, J = 10.4 Hz), 123.6, 116.6, 111.1; 111.0 (d, JCF = 9.9 Hz) 109.4; 109.0 (d, JCF = 26.1 Hz), 105.1; 104.8 (d, JCF = 23.1 Hz), 101.0, 101.0 (d, JCF = 4.7 Hz), 98.8, 49.0, 44.6, 40.9. Anal. calcd. for C19H15ClFN7: C, 57.65; H, 3.82; N, 24.77. Found: C, 57.36; H, 4.01; N, 25.02.
N-((1-(2-(4-Chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)ethyl)-1H-1,2,3-triazol-4-yl)methyl)-4-(5-methylbenzo[d]thiazol-2-yl)benzenamine (5e). Compound 5e was prepared using the above-mentioned procedure using compound 3 (30 mg, 0.14 mmol) and 4-(5-methylbenzo[d]thiazol-2-yl)-N-(prop-2-ynyl)benzenamine (48.74 mg, 0.17 mmol) to obtain 5e as yellow powder (27.3 mg, 38%, m.p. = 228–230 °C). 1H NMR (300 MHz, DMSO) δ 8.54 (1H, s, H2), 7.83–7.74 (5H, m, H5′; H4′′; H7′′; Ph′′), 7.40 (1H, d, J = 3.6 Hz, H6), 7.27 (1H, dd, J = 8.3, 1.2 Hz, H6′′), 6.82 (1H, t, J = 5.9 Hz, NH′′), 6.68 (2H, d, J = 8.8 Hz, Ph′′), 6.52 (1H, d, J = 3.6 Hz, H5), 4.87–4.76 (2H, m, CH2), 4.77–4.67 (2H, m, CH2), 4.31 (2H, d, J = 5.9 Hz, CH2′′), 2.43 (3H, s, CH3′′). 13C NMR (151 MHz, DMSO) δ 167.1, 152.0, 151.4, 150.9, 150.7, 150.3, 145.4, 134.1, 133.9, 131.2, 128.7, 127.7, 123.1, 121.6, 121.4, 120.8, 116.8, 114.1, 99.1, 49.7, 44.6, 38.0, 21.0. Anal. calcd. for C25H21ClN8S: C, 59.93; H, 4.22; N, 22.36. Found: C, 59.84; H, 4.15; N, 22.48.
3.2.2. General Procedure for the Synthesis of Compounds (6a and 6b)
The corresponding diethynylbenzene and azido derivative 3 (1.2 eq.) were dissolved in 0.5 mL DMF and t-BuOH:H2O = 1:1 (2 mL), Cu(0) (0.5 eq.), 1M CuSO4 (1 eq.) and stirred under microwave irradiation for 1.5 h at 80 °C and 300 W. The solvent was evaporated and the residue was purified by column chromatography using CH2Cl2:CH3OH = 100:1 as an initial eluent and CH2Cl2:CH3OH = 10:1, as final eluent.
1,4-Bis(1-(2-(4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)ethyl)-1H-1,2,3-triazol-4-yl)benzene (6a). Compound 6a was prepared in accord with the general procedure using compound 3 (44.1 mg, 0.20 mmol) and 1,4-diethynylbenzene (11.3 mg, 0.09 mmol) to obtain 6a as white powder ( 20.4 mg, 39%, m.p. > 250 °C). 1H NMR (600 MHz, DMSO) δ 8.57 (2H, s, H2, H2′′), 8.44 (2H, s, H5′), 7.78 (4H, s, Ph), 7.58 (2H, d, J = 3.6 Hz, H6, H6′′), 6.61 (2H, d, J = 3.6 Hz, H5, H5′′), 5.06–4.84 (4H, m, CH2, CH2′′), 4.87–4.73 (4H, m, CH2, CH2′′). 13C NMR (151 MHz, DMSO) δ 151.0, 150.7, 150.6, 145.9, 131.2, 130.0, 125.5, 122.1, 116.8, 98.8, 49.1, 44.6. Anal. calcd. for C26H20Cl2N12: C, 54.65; H, 3.53; N, 29.41. Found: C, 54.65; H, 3.31; N, 29.78.
1,3-Bis(1-(2-(4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)ethyl)-1H-1,2,3-triazol-4-yl)benzene (6b). Compound 6b was prepared in accord with the general procedure using compound 3 (53.9 mg, 0.24 mmol) and 1,3-diethynylbenzene (13.9 mg, 0.11 mmol) to obtain 6b as white powder (50.2 mg, 81%, m.p. = 193–196 °C). 1H NMR (300 MHz, DMSO) δ 8.59–8.54 (2H, m, H2, H2′′), 8.51–8.47 (2H, m, H5′), 7.80–7.75 (1H, m, Ph), 7.66 (1H, dd, J = 7.7, 1.5 Hz, Ph), 7.58–7.53 (2H, m, J = 3.6, 1.3 Hz, H6, H6′′), 7.51–7.42 (2H, m, Ph), 6.69–6.56 (2H, m, H5, H5′′), 4.97–4.75 (8H, m, CH2). 13C NMR (75 MHz, DMSO) δ 150.7, 150.6, 150.2, 146.0, 145.3, 131.3, 131.1, 131.0, 129.4, 128.1, 125.5, 124.6, 122.3, 122.2, 122.0, 116.8, 98.8, 49.2, 44.7, 44.7. Anal. calcd. for C26H20Cl2N12: C, 54.65; H, 3.53; N, 29.41. Found: C, 54.58; H, 3.76; N, 29.46.
3.3. X-ray Crystal Structure Analysis
Single crystals of 4g, 5a, and 5b suitable for single crystal X-ray structure analysis were obtained at room temperature by partial evaporation from ethanol, methanol and acetone, respectively. Data were collected at room temperature (295 K) on Oxford Diffraction Xcalibur Nova R diffractometer with mirror-monochromatized CuKα radiation (λ = 1.54184 Å). Data collection and processing was performed by using the CrysAlisPro program [52]. The intensities were corrected for absorption using two different absorption correction methods, multi-scan (for 4g and 5b) and analytical (5a) [52]. All structures were solved using direct methods with SIR-2004 [53] and refined by full-matrix least-squares calculations based on F2 using SHELXL-2016 [54] integrated in the WinGX program package [55]. All hydrogen atoms were included in calculated positions, with SHELXL-2016 [54] defaults. Geometric restraints and restraints on anisotropic thermal parameters were used in the refinement of slightly disordered butyl chain atoms in 4g. For structure analysis and molecular and crystal structure drawing preparation were used the PLATON [56] and Mercury [57] programs. The CCDC 1551719–1551721 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Crystal data for 4g: 0.315 × 0.163 × 0.072 mm3; C21H23ClN6, Mr = 394.90, triclinic, space group P −1 (No. 2); a = 4.6875(2) Å, b = 12.7403(6) Å, c = 17.7949(10) Å, α = 109.141(4)°, β = 92.486(4)°, γ = 91.871(4)°, V = 1001.78(9) Å3; Z = 2; ρ = 1.309 g cm−3, μ(CuKα) = 1.833 mm−1; θmax = 70.000°, 7842 reflections measured, 3785 unique reflections and 3298 with I ≥ 2σ(I), Rint = 0.0212; Final R indices [(I > 2σ(I)]: R = 0.0621, wR = 0.1785, [all data]: R = 0.0681, wR = 0.1876, S = 1.072 for 254 parameters and 11 restraints, largest diff. peak and hole 0.809/−0.528 e Å−3.
Crystal data for 5a: 0.622 × 0.083 × 0.030 mm3; C17H13Cl2N9, Mr = 414.26, monoclinic, space group P 21/n (No. 14); a = 5.0071(3) Å, b = 19.6228(17) Å, c = 18.2988(10) Å, β = 94.275(4)°, V = 1792.9(2) Å3; Z = 4; ρ = 1.535 g cm−3, μ(CuKα) = 3.475 mm−1; θmax = 69.985°, 6929 reflections measured, 3114 unique reflections and 2116 with I ≥ 2σ(I), Rint = 0.0637; Final R indices [(I > 2σ(I)]: R = 0.0610, wR = 0.0896, [all data]: R = 0.1564, wR = 0.1837, S = 1.003 for 253 parameters and 0 restraints, largest diff. peak and hole 0.266/−0.269 e Å−3.
Crystal data for 5b: 0.386 × 0.301 × 0.196 mm3; C16H12Cl2N10, Mr = 415.26, triclinic, space group P −1 (No. 2); a = 7.2549(3) Å, b = 8.6808(3) Å, c = 14.7203(6) Å, α = 89.171(3)°, β = 78.931(4)°, γ = 73.387(3)°, V = 871.03(6) Å3; Z = 2; ρ = 1.583 g cm−3, μ(CuKα) = 3.595 mm−1; θmax = 69.988°, 6701 reflections measured, 3171 unique reflections and 3022 with I ≥ 2σ(I), Rint = 0.0266; Final R indices [(I > 2σ(I)]: R = 0.0373, wR = 0.1021, [all data]: R = 0.0388, wR = 0.1040, S = 1.028 for 253 parameters and 0 restraints, largest diff. peak and hole 0.202/−0.270 e Å−3.
3.4. In Silico
Values of logP, n-octanol/water partition coefficients, for novel compounds were calculated by ChemAxon algorithm available within MarvinView Ver. 6.2.2. For predictions of plausible biological targets and pharmacological activities web-service PASS (Available online: http://www.pharmaexpert.ru/passonline/index.php; based on the identification of substructure features typical for active compounds was used [47].
3.5. Cell Culturing
Human cell lines A549 (lung carcinoma), HeLa (cervical carcinoma), SW620 (colorectal adenocarcinoma, metastatic) and CFPAC-1 (pancreatic cancer, derived from metastatic: liver) as well as on normal human foreskin fibroblasts (HFF) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured in humidified atmosphere at 37 °C with 5% CO2. As growth medium, Dulbecco’s modified Eagle medium (DMEM, Lonza Group, Basel, Switzerland) was used with the addition of fetal bovine serum (Lonza Group, Basel, Switzerland) (10%), l-glutamine (Lonza Group, Basel, Switzerland) (2 mM) and antibiotics: streptomycin (Lonza Group, Basel, Switzerland) (100 µg/mL) and penicillin (Lonza Group, Basel, Switzerland) (100 U/mL).
3.6. Proliferation Assay
Cells were seeded onto 96 well microtiter plates at a seeding density of 3000 cells/well for carcinoma cell lines, and 5000 cells/well for normal human fibroblasts. The next day, cells were treated with test agents in five different concentrations (0.01 to 100 µM) and further incubated for 72 h. Dimethyl sulfoxide (DMSO) (solvent), was tested for potential cytotoxic effect but it did not exceed 0.1%. Following 72 h incubation, the MTT assay was performed and measured absorbances were transformed into percentage of cell growth as described previously [58]. Results were obtained from three independent experiments. IC50 values were calculated using linear regression analysis and results were statistically analyzed by ANOVA, Tukey post-hoc test (p < 0.05).
3.7. Western Blot Analysis
Cells were seeded in 6-well plates in the concentration depending on tested cell line varying from 1 × 105 to 2 × 105 cells/well. Cells were treated for 48 h with 2 × IC50 concentrations of selected compounds. Following treatment, cells were lysed with RIPA (radioimmunoprecipitation assay) buffer supplemented with protease and phosphatase inhibitors (Roche, Basel, Switzerland). Total proteins (50 µg) were resolved on 10% or 12% polyacrilamide gels, deppending on protein size, and transferred onto PVDF (polyvinylidene fluoride) membranes (Bio-Rad, Hercules, CA, USA) that were blocked for 1h with either 4% BSA (bovine serum albumin, Sigma Aldrich, St. Louis, MO, USA) or 5% non-fat milk (Bio-Rad, Hercules, CA, USA) prepared in TBST (Tris-buffered saline, 0.1% Tween 20). Membranes were probed with primary antibodies against CDK9/cyclin T1 (Cell Signaling Technology, Danvers, MA, USA), p-c-Raf (Abcam, Cambridge, UK) and p-p38 MAPK (p-38 MAPK (Thr180/Tyr182) (Cell Signaling Technology) at 4 °C overnight. The next day, membranes were washed in TBST and probed with horseradish peroxidase-conjugated secondary antibodies goat anti-mouse (Santa Cruz Biotechnology, Dallas, TX, USA) or goat anti-rabbit (Santa Cruz Biotechnology, Dallas, TX, USA). Protein bands were visualised using chemiluminesncence supstrate and ImageQuant LAS 500 (GE Healthcare, Little Chalfont, UK). Following visualisation, protein band density was analyzed by Quantity One 1-D Analysis Software (Bio-Rad, Hercules, CA, USA).
3.8. Apoptosis Detection
Cells were seeded into 8 well chambers (Lab-tek II Chmaber Slides, Thermo Fisher Scientific, Waltham, MA, USA) in concentration of 2 × 104 cells per well and treated with 2 × IC50 concentrations of selected compounds for 48 h. Staining of the cells was performed by Annexin-V-FITC Staining kit (Santa Cruz Biotechnology, Dallas, TX, USA) according to the manufacturer’s instructions. Cells were visualized by fluorescent microscope (Olympus, Shinjuku, Tokyo, Japan) at magnification of 40×.
4. Conclusions
A series of mono-pyrrolo[2,3-d]pyrimidines comprising alkyl (4a, 4b), varied substituted aryl (4c–4h) and halogenphenylsulfonamide (4i–4k) attached to 1,2,3-triazole at C-4, unsymmetrical (5a–5e) and symmetrical bis-purine isosteres (6a, 6b) were designed and synthesized by 1,3-dipolar Huisgen cycloaddition reaction under microwave irradiation using copper(II) sulfate and metallic copper, as a catalyst. The stereostructures of mono- 4g, and bis-pseudopurines 5a and 5b were unambiguously confirmed by crystallographic analyses.
Mono-pyrrolo[2,3-d]pyrimidine 4g and bis-purine isosteres 5e exhibited selective anti-proliferative effects on cervical carcinoma cells (HeLa), while 6b showed strong and selective cytostatic activity (IC50 = 0.95 µM) on pancreatic cancer cells (CFPAC-1). Growth-inhibitory effects of symmetrical bis-purine isostere 6b against CFPAC-1 could be ascribed to induction of late apoptosis and necrosis. At the molecular level, this compound proved to be a potent inhibitor of CDK9/cyclin T1 and to suppress proliferative signaling transduced by p38 MAPK and c-Raf. Overall, new chemical entity based on symmetrical bis-pyrrolo[2,3-d]pyrimidines connected through di(ethylene-1,2,3-triazolyl)phenyl spacer was identified as a novel CDK9/cyclin T1 inhibitor for potential treatment of pancreatic cancer.
Supplementary Materials
Supplementary materials can be found at www.mdpi.com/1422-0067/18/11/2292/s1.
Acknowledgments
We greatly appreciate the financial support of the Croatian Science Foundation (grant number IP-2013-11-5596), University of Rijeka research grants 13.11.1.1.11. and 13.11.2.1.12., and the access to equipment owned by the University of Rijeka within the project RISK “Development of University of Rijeka campus laboratory research infrastructure”, financed by the European Regional Development Fund (ERDF).
Author Contributions
Andrea Bistrović participated in the synthesis and structural characterization of presented compounds; Anja Harej and Petra Grbčić performed biological evaluations, Sandra Kraljević Pavelić designed the biological assays; Mario Cetina was involved in the X-ray crystal structure analysis; Mirela Sedić analyzed and described the results of biological evaluations; Silvana Raić-Malić conceived this study and wrote the paper. All authors reviewed and approved the final manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Choi, Y.J.; Anders, L. Signaling through cyclin D-dependent kinases. Oncogene 2014, 33, 1890–1903. [Google Scholar] [CrossRef]
- Diaz-Moralli, S.; Tarrado-Castellarnau, M.; Miranda, A.; Cascante, M. Targeting cell cycle regulation in cancer therapy. Pharmacol. Ther. 2013, 138, 255–271. [Google Scholar] [CrossRef] [PubMed]
- Welburn, J.P.I.; Endicott, J.A. Inhibition of the cell cycle with chemical inhibitors: A targeted approach. Semin. Cell Dev. Biol. 2005, 16, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Sonawane, Y.A.; Taylor, M.A.; Napoleon, J.V.; Rana, S.; Contreras, J.I.; Natarajan, A. Cyclin dependent kinase 9 inhibitors for cancer therapy. J. Med. Chem. 2016, 59, 8667–8684. [Google Scholar] [CrossRef] [PubMed]
- Mariaule, G.; Belmont, P. Cyclin-dependent kinase inhibitors as marketed anticancer drugs: Where are we Now? A short survey. Molecules 2014, 19, 14366–14382. [Google Scholar] [CrossRef] [PubMed]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Palanisamy, R.P. Palbociclib: A new hope in the treatment of breast cancer. J. Cancer Res. Ther. 2016, 12, 1220–1223. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [PubMed]
- Gray, N.S.; Wodicka, L.; Thunnissen, A.M.W.; Norman, T.C.; Kwon, S.; Espinoza, F.H.; Morgan, D.O.; Barnes, G.; LeClerc, S.; Meijer, L.; et al. Exploiting chemical libraries, structure, and genomics in the search for kinase inhibitors. Science 1998, 281, 533–538. [Google Scholar] [CrossRef] [PubMed]
- McClue, S.J.; Blake, D.; Clarke, R.; Cowan, A.; Cummings, L.; Fischer, P.M.; MacKenzie, M.; Melville, J.; Stewart, K.; Wang, S.; et al. In vitro and in vivo antitumor properties of the cyclin dependent kinase inhibitor CYC202 (R-roscovitine). Int. J. Cancer 2002, 102, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Zatloukal, M.; Jorda, R.; Gucky, T.; Reznickova, E.; Voller, J.; Pospisil, T.; Malinkova, V.; Adamcova, H.; Krystof, V.; Strnad, M. Synthesis and in vitro biological evaluation of 2,6,9-trisubstituted purines targeting multiple cyclin-dependent kinases. Eur. J. Med. Chem. 2013, 61, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Singh, J.; Ojha, R.; Singh, H.; Kaur, M.; Bedi, P.; Nepali, K. Design strategies, structure activity relationship and mechanistic insights for purines as kinase inhibitors. Eur. J. Med. Chem. 2016, 112, 298–396. [Google Scholar] [CrossRef] [PubMed]
- Le Tourneau, C.; Faivre, S.; Laurence, V.; Delbaldo, C.; Vera, K.; Girre, V.; Chiao, J.; Armour, S.; Frame, S.; Green, S.R.; et al. Phase I evaluation of seliciclib (R-roscovitine), a novel oral cyclin-dependent kinase inhibitor, in patients with advanced malignancies. Eur. J. Cancer 2010, 46, 3243–3250. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J.; Kalyan, K.; Sorokinas, A.; Stankunas, E.; Levy, J.; Meskinyte, I.; Stankevicius, V.; Kaupinis, A.; Valius, M. Roscovitine in cancer and other diseases. Ann. Transl. Med. 2015, 3, 135. [Google Scholar] [CrossRef] [PubMed]
- Kryštof, V.; Moravcová, D.; Paprskářová, M.; Barbier, P.; Peyrot, V.; Hlobilková, A.; Havlíček, L.; Strnad, M. Synthesis and biological activity of 8-azapurine and pyrazolo[4,3-d]pyrimidine analogues of myoseverin. Eur. J. Med. Chem. 2006, 41, 1405–1411. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Dessalew, N.; Bharatam, P.V. 3D-QSAR CoMFA study on indenopyrazole derivatives as cyclin dependent kinase 4 (CDK4) and cyclin dependent kinase 2 (CDK2) inhibitors. Eur. J. Med. Chem. 2006, 41, 1310–1319. [Google Scholar] [CrossRef] [PubMed]
- Jorda, R.; Paruch, K.; Krystof, V. Cyclin-dependent kinase inhibitors inspired by roscovitine: Purine bioisosteres. Curr. Pharm. Des. 2012, 18, 2974–2980. [Google Scholar] [CrossRef] [PubMed]
- Paruch, K.; Dwyer, M.P.; Alvarez, C.; Brown, C.; Chan, T.Y.; Doll, R.J.; Keertikar, K.; Knutson, C.; McKittrick, B.; Rivera, J.; et al. Discovery of dinaciclib (SCH 727965): A potent and selective inhibitor of cyclin-dependent kinases. ACS Med. Chem. Lett. 2010, 1, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Parry, D.; Guzi, T.; Shanahan, F.; Davis, N.; Prabhavalkar, D.; Wiswell, D.; Seghezzi, W.; Paruch, K.; Dwyer, M.P.; Doll, R.; et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol. Cancer Ther. 2010, 9, 2344–2353. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.; Jones, J.; Johnson, A.J.; Andritsos, L.; Maddocks, K.; Jaglowski, S.; Hessler, J.; Grever, M.R.; Im, E.; Zhou, H.; et al. Dinaciclib is a novel cyclin-dependent kinase inhibitor with significant clinical activity in relapsed and refractory chronic lymphocytic leukemia. Leukemia 2015, 29, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, G.; Mishra, A.; Bisht, S.; Karikari, C.; Garrido-Laguna, I.; Rasheed, Z.; Ottenhof, N.A.; Dadon, T.; Alvarez, H.; Fendrich, V.; et al. Cyclin-dependent kinase inhibitor dinaciclib (SCH727965) inhibits pancreatic cancer growth and progression in murine xenograft models. Cancer Biol. Ther. 2011, 12, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Gelbert, L.M.; Cai, S.; Lin, X.; Sanchez-Martinez, C.; Del Prado, M.; Lallena, M.J.; Torres, R.; Ajamie, R.T.; Wishart, G.N.; Flack, R.S.; et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: In vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Investig. New Drugs 2014, 32, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Barroso-Sousa, R.; Shapiro, G.I.; Tolaney, S.M. Clinical development of the CDK4/6 inhibitors ribociclib and abemaciclib in breast cancer. Breast Care 2016, 11, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Ribociclib: First global approval. Drugs 2017, 77, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.X.; Sicinska, E.; Czaplinski, J.T.; Remillard, S.P.; Moss, S.; Wang, Y.; Brain, C.; Loo, A.; Snyder, E.L.; Demetri, G.D.; et al. Antiproliferative effects of CDK4/6 inhibition in CDK4-amplified human liposarcoma in vitro and in vivo. Mol. Cancer Ther. 2014, 13, 2184–2193. [Google Scholar] [CrossRef] [PubMed]
- Casimiro, M.C.; Velasco-Velázquez, M.; Aguirre-Alvarado, C.; Pestell, R.G. Overview of cyclins D1 function in cancer and the CDK inhibitor landscape: past and present. Expert Opin. Investig. Drugs 2014, 23, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Maračić, S.; Kraljević, T.G.; Paljetak, H.Č.; Perić, M.; Matijašić, M.; Verbanac, D.; Cetina, M.; Raić-Malić, S. 1,2,3-Triazole pharmacophore-based benzofused nitrogen/sulfur heterocycles with potential anti-Moraxella catarrhalis activity. Bioorg. Med. Chem. 2015, 23, 7448–7463. [Google Scholar] [CrossRef] [PubMed]
- Gregorić, T.; Sedić, M.; Grbčić, P.; Paravić, A.T.; Pavelić, S.K.; Cetina, M.; Vianello, R.; Raić-Malić, S. Novel pyrimidine-2,4-dione-1,2,3-triazole and furo[2,3-d]pyrimidine-2-one-1,2,3-triazole hybrids as potential anti-cancer agents: Synthesis, computational and X-ray analysis and biological evaluation. Eur. J. Med. Chem. 2017, 125, 1247–1267. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Luxami, V.; Paul, K. Purine-benzimidazole hybrids: Synthesis, single crystal determination and in vitro evaluation of antitumor activities. Eur. J. Med. Chem. 2015, 93, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Abbot, V.; Sharma, P.; Dhiman, S.; Noolvi, M.N.; Patelc, H.M.; Varun Bhardwaj, V. Small hybrid heteroaromatics: Resourceful biological tools in cancer research. RSC Adv. 2017, 7, 28313–28349. [Google Scholar] [CrossRef]
- Carbone, A.; Parrino, B.; Di Vita, G.; Attanzio, A.; Spanò, V.; Montalbano, A.; Barraja, P.; Tesoriere, L.; Livrea, M.A.; Diana, P.; et al. Synthesis and antiproliferative activity of thiazolyl-bis-pyrrolo[2,3-b]pyridines and indolyl-thiazolyl-pyrrolo[2,3-c]pyridines, nortopsentin analogues. Mar. Drugs 2015, 13, 460–492. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Tandon, V. Synthesis and biological activity of novel inhibitors of topoisomerase I: 2-Aryl-substituted 2-bis-1H-benzimidazoles. Eur. J. Med. Chem. 2011, 46, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Kode, N.; Chen, L.; Murthy, D.; Adewumi, D.; Phadtare, S. New bis-N9-(methylphenylmethyl)purine derivatives: Synthesis and antitumor activity. Eur. J. Med. Chem. 2007, 42, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Alpan, A.S.; Zencir, S.; Zupkó, I.; Coban, G.; Réthy, B.; Gunes, H.S.; Topcu, Z. Biological activity of bis-benzimidazole derivatives on DNA topoisomerase I and HeLa, MCF7 and A431 cells. J. Enzyme Inhib. Med. Chem. 2009, 24, 844–849. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.H.; Cheng, M.S.; Wang, Q.H.; Nie, H.; Liao, N.; Wang, J.; Chen, H. Design, synthesis, and anti-tumor evaluation of novel symmetrical bis-benzimidazoles. Eur. J. Med. Chem. 2009, 44, 1808–1812. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.T.; Hsei, I.J.; Chen, C. Synthesis and anticancer evaluation of bis(benzimidazoles), bis(benzoxazoles), and benzothiazoles. Bioorg. Med. Chem. 2006, 14, 6106–6119. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, P.; Hays, F.A.; Westhof, E.; Ho, P.S. Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 16789–16794. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Shi, T.; Wang, Y.; Yang, H.; Yan, X.; Luo, X.; Jiang, H.; Zhu, W. Halogen bondings—A novel interaction for rational drug design? J. Med. Chem. 2009, 52, 2854–2862. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef] [PubMed]
- Raić-Malić, S.; Meščić, A. Recent trends in 1,2,3-triazolo-nucleosides as promising anti-infective and anticancer agents. Curr. Med. Chem. 2015, 22, 1462–1499. [Google Scholar] [CrossRef] [PubMed]
- Kerru, N.; Singh, P.; Koorbanally, N.; Raj, R.; Kumar, V. Recent advances (2015–2016) in anticancer hybrids. Eur. J. Med. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, J.; Khan, A.A.; Ali, Z.; Haider, R.; Shahar Yar, M. Structureactivity relationship (SAR) study and design strategies of nitrogen-containing heterocyclic moieties for their anticancer activities. Eur. J. Med. Chem. 2017, 125, 143–189. [Google Scholar] [CrossRef] [PubMed]
- Dheer, D.; Singh, V.; Shankar, R. Medicinal attributes of 1,2,3-triazoles: Current developments. Bioorg. Chem. 2017, 71, 30–54. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Sander, K.; Galante, E.; Rajkumar, V.; Badar, A.; Robson, M.; El-Emir, E.; Lythgoe, M.F.; Pedley, R.B.; Årstad, E. A one-pot three-component radiochemical reaction for rapid assembly of 125I-labeled molecular probes. J. Am. Chem. Soc. 2013, 135, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Chittepu, P.; Sirivolu, V.R.; Seela, F. Nucleosides and oligonucleotides containing 1,2,3-triazole residues with nucleobase tethers: Synthesis via the azide-alkyne ‘click’ reaction. Bioorg. Med. Chem. 2008, 16, 8427–8439. [Google Scholar] [CrossRef] [PubMed]
- Kraljević, T.G.; Harej, A.; Sedić, M.; Pavelić, S.K.; Stepanić, V.; Drenjančević, D.; Talapko, J.; Raić-Malić, S. Synthesis, in vitro anticancer and antibacterial activities and in silico studies of new 4-substituted 1,2,3-triazole-coumarin hybrids. Eur. J. Med. Chem. 2016, 124, 794–808. [Google Scholar] [CrossRef] [PubMed]
- Filimonov, D.A.; Poroikov, V.V. Chemoinformatics Approaches to Virtual Screening; Varnek, A., Tropsha, A., Eds.; RSC Publishing: Cambridge, UK, 2008; pp. 182–216. [Google Scholar]
- Sumi, N.J.; Kuenzi, B.M.; Knezevic, C.E.; Rix, L.L.R.; Rix, U. Chemoproteomics reveals novel protein and lipid kinase targets of clinical CDK4/6 inhibitors in lung cancer. ACS Chem. Biol. 2015, 10, 2680–2686. [Google Scholar] [CrossRef] [PubMed]
- Kretz, A.L.; Schaum, M.; Richter, J.; Kitzig, E.F.; Engler, C.C.; Leithäuser, F.; Henne-Bruns, D.; Knippschild, U.; Lemke, J. CDK9 is a prognostic marker and therapeutic target in pancreatic cancer. Tumor Biol. 2017, 39, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, M.G.; Millar, J.B. Control of the eukaryotic cell cycle by MAP kinase signaling pathways. FASEB J. 2000, 14, 2147–2157. [Google Scholar] [CrossRef] [PubMed]
- Oxford Diffraction. Xcalibur CCD System, CrysAlisPro; Agilent Technologies: Abingdon, UK, 2015. [Google Scholar]
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; De Caro, L.; Giacovazzo, C.; Polidori, G.; Spagna, R. SIR2004: An improved tool for crystal structure determination and refinement. J. Appl. Crystallogr. 2005, 38, 381–388. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Gazivoda, T.; Raić-Malić, S.; Krištafor, V.; Makuc, D.; Plavec, J.; Kraljević-Pavelić, S.; Pavelić, K.; Naesens, L.; Andrei, G.; Snoeck, R.; et al. Synthesis, cytostatic and anti-HIV evaluations of the new unsaturated acyclic C-5 pyrimidine nucleoside analogues. Bioorg. Med. Chem. 2008, 16, 5624–5634. [Google Scholar] [CrossRef] [PubMed]
















© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).