
Bispecific Antibodies as a Development Platform for New Concepts and Treatment Strategies

Abstract
:1. Introduction
2. Diverse Formats of Bispecific Antibodies Meet the Optimal Biological Activity and Clinical Purpose
2.1. Single-Chain-Based Formats
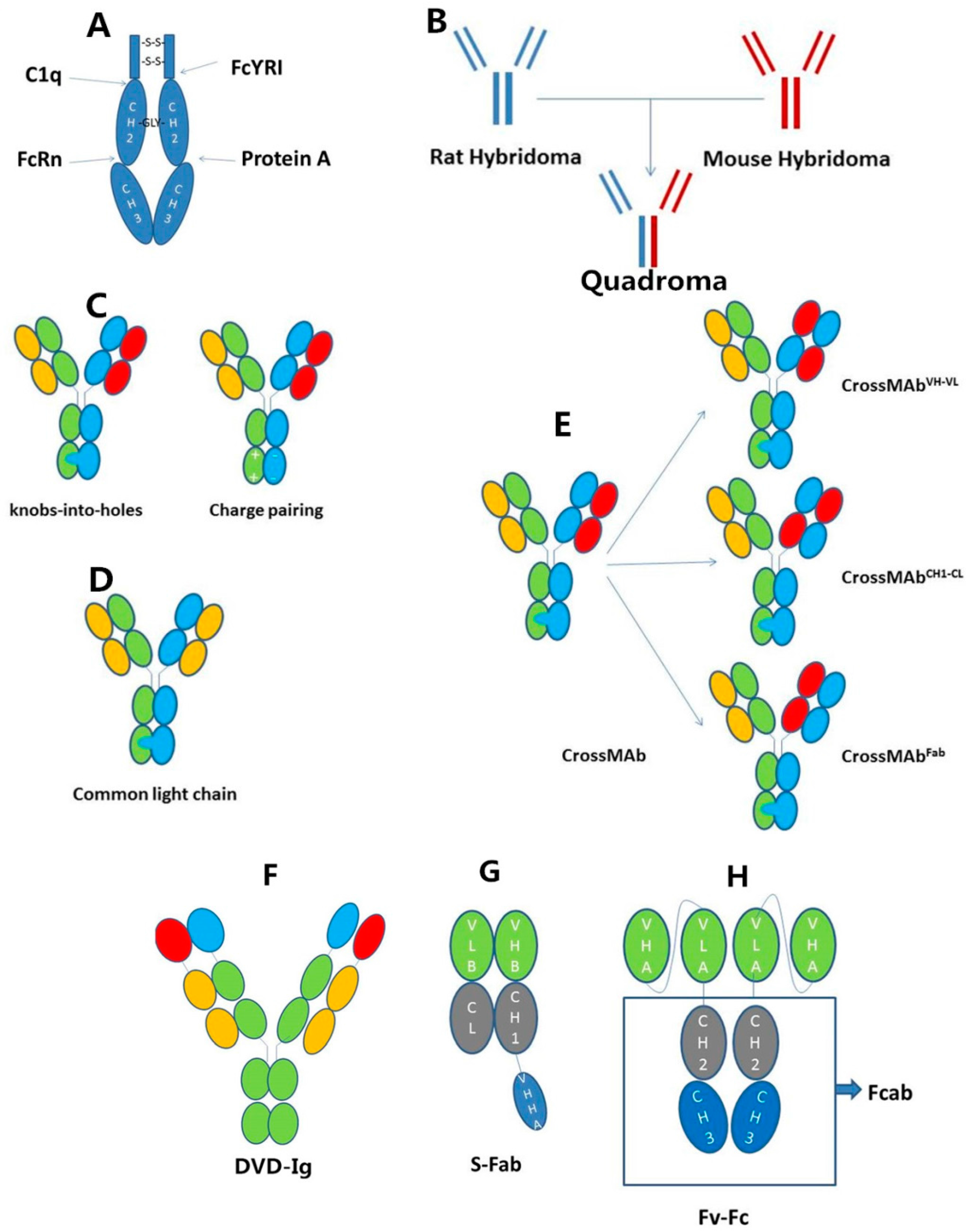
2.2. IgG-Based Formats
3. The Mechanism of Action for Bispecific Antibodies Is Like a Bridge Linking Two Cells or Blocking Two Antigens in the Same Cell
3.1. Retargeting Cellular Immunity towards the Malignant Cells
3.2. Delivering Cytotoxic Entities to the Malignant Cells
3.3. Modifying the Host Response against Drug Resistance
3.4. Anti-Tumor Angiogenesis
3.5. Directly Targeting the Malignant Cells
3.6. Bispecific Antibodies Used Besides for Tumors
4. Conclusions
Conflicts of Interest
References
- Elgundi, Z.; Reslan, M.; Cruz, E.; Sifniotis, V.; Kayser, V. The state-of-play and future of antibody therapeutics. Adv. Drug Deliv. Rev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Alexander, P.B.; Chen, R.; Gong, C.; Yuan, L.; Jasper, J.S.; Ding, Y.; Markowitz, G.J.; Yang, P.; Xu, X.; McDonnell, D.P.; et al. Distinct receptor tyrosine kinase subsets mediate anti-HER2 drug resistance in breast cancer. J. Biol. Chem. 2016. [Google Scholar] [CrossRef] [PubMed]
- Merchant, A.M.; Zhu, Z.; Yuan, J.Q.; Goddard, A.; Adams, C.W.; Presta, L.G.; Carter, P. An efficient route to human bispecific IgG. Nat. Biotechnol. 1998, 16, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Coloma, M.J.; Morrison, S.L. Design and production of novel tetravalent bispecific antibodies. Nat. Biotechnol. 1997, 15, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, J.B.; Presta, L.G.; Carter, P. “Knobs-into-holes” engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996, 9, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Lindhofer, H.; Mocikat, R.; Steipe, B.; Thierfelder, S. Preferential species-restricted heavy/light chain pairing in rat/mouse quadromas. Implications for a single-step purification of bispecific antibodies. J. Immunol. 1995, 155, 219–225. [Google Scholar] [PubMed]
- Orlandi, R.; Gussow, D.H.; Jones, P.T.; Winter, G. Cloning immunoglobulin variable domains for expression by the polymerase chain reaction. Proc. Natl. Acad. Sci. USA 1989, 86, 3833–3837. [Google Scholar] [CrossRef] [PubMed]
- Loffler, A.; Kufer, P.; Lutterbuse, R.; Zettl, F.; Daniel, P.T.; Schwenkenbecher, J.M.; Riethmuller, G.; Dorken, B.; Bargou, R.C. A recombinant bispecific single-chain antibody, CD19 × CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood 2000, 95, 2098–2103. [Google Scholar] [PubMed]
- Nagorsen, D.; Kufer, P.; Baeuerle, P.A.; Bargou, R. Blinatumomab: A historical perspective. Pharmacol. Ther. 2012, 136, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Brandl, C.; Haas, C.; D Argouges, S.; Fisch, T.; Kufer, P.; Brischwein, K.; Prang, N.; Bargou, R.; Suzich, J.; Baeuerle, P.A.; et al. The effect of dexamethasone on polyclonal T cell activation and redirected target cell lysis as induced by a CD19/CD3-bispecific single-chain antibody construct. Cancer Immunol. Immunother. 2007, 56, 1551–1563. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.A.; Zhang, W.; Rainey, G.J.; Burke, S.; Li, H.; Huang, L.; Gorlatov, S.; Veri, M.C.; Aggarwal, S.; Yang, Y.; et al. Application of dual affinity retargeting molecules to achieve optimal redirected T cell killing of B cell lymphoma. Blood 2011, 117, 4542–4551. [Google Scholar] [CrossRef] [PubMed]
- Reusch, U.; Burkhardt, C.; Fucek, I.; le Gall, F.; le Gall, M.; Hoffmann, K.; Knackmuss, S.H.; Kiprijanov, S.; Little, M.; Zhukovsky, E.A. A novel tetravalent bispecific TandAb (CD30/CD16A) efficiently recruits NK cells for the lysis of CD30+ tumor cells. mAbs 2014, 6, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Reusch, U.; Harrington, K.; Gudgeon, C.; Fucek, I.; Ellwanger, K.; Weichel, M.; Knackmuss, S.; Zhukovsky, E.; Fox, J.A.; Kunkel, L.A.; et al. Characterization of CD33/CD3 tetravalent bispecific tandem diabodies (TandAbs) for the treatment of acute myeloid leukemia. Clin. Cancer Res. 2016, 22, 5829–5838. [Google Scholar] [CrossRef] [PubMed]
- Reusch, U.; Duell, J.; Ellwanger, K.; Herbrecht, C.; Knackmuss, S.H.; Fucek, I.; Eser, M.; McAleese, F.; Molkenthin, V.; le Gall, F.; et al. A tetravalent bispecific TandAb (CD19/CD3), AFM11, efficiently recruits T cells for the potent lysis of CD19+ tumor cells. mAbs 2015, 7, 584–604. [Google Scholar] [CrossRef] [PubMed]
- Kontermann, R.E.; Brinkmann, U. Bispecific antibodies. Drug Discov. Today 2015, 20, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Offner, S.; Hofmeister, R.; Romaniuk, A.; Kufer, P.; Baeuerle, P.A. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol. Immunol. 2006, 43, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Bacac, M.; Fauti, T.; Sam, J.; Colombetti, S.; Weinzierl, T.; Ouaret, D.; Bodmer, W.; Lehmann, S.; Hofer, T.; Hosse, R.J.; et al. A novel carcinoembryonic antigen T cell bispecific antibody (CEA TCB) for the treatment of solid tumors. Clin. Cancer Res. 2016, 22, 3286–3297. [Google Scholar] [CrossRef] [PubMed]
- Stieglmaier, J.; Benjamin, J.; Nagorsen, D. Utilizing the bite (bispecific T cell engager) platform for immunotherapy of cancer. Expert Opin. Biol. Ther. 2015, 15, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.S.; Gokbuget, N.; Stein, A.S.; Zugmaier, G.; O’Brien, S.; Bargou, R.C.; Dombret, H.; Fielding, A.K.; Heffner, L.; Larson, R.A.; et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: A multicentre, single-arm, phase 2 study. Lancet Oncol. 2015, 16, 57–66. [Google Scholar] [CrossRef]
- McDonagh, C.F.; Huhalov, A.; Harms, B.D.; Adams, S.; Paragas, V.; Oyama, S.; Zhang, B.; Luus, L.; Overland, R.; Nguyen, S.; et al. Antitumor activity of a novel bispecific antibody that targets the ERBB2/ERBB3 oncogenic unit and inhibits heregulin-induced activation of ERBB3. Mol. Cancer Ther. 2012, 11, 582–593. [Google Scholar] [CrossRef] [PubMed]
- Vincent, K.J.; Zurini, M. Current strategies in antibody engineering: FC engineering and pH-dependent antigen binding, bispecific antibodies and antibody drug conjugates. Biotechnol. J. 2012, 7, 1444–1450. [Google Scholar] [CrossRef] [PubMed]
- Lobner, E.; Traxlmayr, M.W.; Obinger, C.; Hasenhindl, C. Engineered IgG1-FC—One fragment to bind them all. Immunol. Rev. 2016, 270, 113–131. [Google Scholar] [CrossRef] [PubMed]
- Subedi, G.P.; Hanson, Q.M.; Barb, A.W. Restricted motion of the conserved immunoglobulin G1 N-glycan is essential for efficient FcγrIIIa binding. Structure 2014, 22, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Yamane-Ohnuki, N.; Satoh, M. Production of therapeutic antibodies with controlled fucosylation. mAbs 2009, 1, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal FC receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Ying, T.; Ju, T.W.; Wang, Y.; Prabakaran, P.; Dimitrov, D.S. Interactions of IgG1 CH2 and CH3 domains with FcRn. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Tustian, A.D.; Endicott, C.; Adams, B.; Mattila, J.; Bak, H. Development of purification processes for fully human bispecific antibodies based upon modification of protein a binding avidity. mAbs 2016, 8, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Gunasekaran, K.; Pentony, M.; Shen, M.; Garrett, L.; Forte, C.; Woodward, A.; Ng, S.B.; Born, T.; Retter, M.; Manchulenko, K.; et al. Enhancing antibody fc heterodimer formation through electrostatic steering effects: Applications to bispecific molecules and monovalent IgG. J. Biol. Chem. 2010, 285, 19637–19646. [Google Scholar] [CrossRef] [PubMed]
- Spiess, C.; Merchant, M.; Huang, A.; Zheng, Z.; Yang, N.-Y.; Peng, J.; Ellerman, D.; Shatz, W.; Reilly, D.; Yansura, D.G.; et al. Bispecific antibodies with natural architecture produced by co-culture of bacteria expressing two distinct half-antibodies. Nat. Biotechnol. 2013, 31, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, W.; Regula, J.T.; Bahner, M.; Schanzer, J.; Croasdale, R.; Durr, H.; Gassner, C.; Georges, G.; Kettenberger, H.; Imhof-Jung, S.; et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc. Natl. Acad. Sci. USA 2011, 108, 11187–11192. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Ying, H.; Grinnell, C.; Bryant, S.; Miller, R.; Clabbers, A.; Bose, S.; McCarthy, D.; Zhu, R.-R.; Santora, L.; et al. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat. Biotechnol. 2007, 25, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Jakob, C.G.; Edalji, R.; Judge, R.A.; DiGiammarino, E.; Li, Y.; Gu, J.; Ghayur, T. Structure reveals function of the dual variable domain immunoglobulin (DVD-IG) molecule. mAbs 2013, 5, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Dmitriev, O.Y.; Lutsenko, S.; Muyldermans, S. Nanobodies as probes for protein dynamicsin vitro and in cells. J. Biol. Chem. 2016, 291, 3767–3775. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Xing, J.; Li, L.; Zhou, C.; Dong, B.; He, P.; Li, Q.; Wang, Z. A single-domain antibody-linked fab bispecific antibody HER2-S-FAB has potent cytotoxicity against HER2-expressing tumor cells. AMB Express 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; He, P.; Zhou, C.; Jing, L.; Dong, B.; Chen, S.; Zhang, N.; Liu, Y.; Miao, J.; Wang, Z.; et al. A novel bispecific antibody, S-fab, induces potent cancer cell killing. J. Immunother. 2015, 38, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; He, Y.; Zhang, G.; Ma, J.; Liu, C.; He, W.; Wang, W.; Han, H.; Boruah, B.M.; Gao, B. Retargeting T cells for HER2-positive tumor killing by a bispecific FV-FC antibody. PLoS ONE 2013, 8, e75589. [Google Scholar] [CrossRef] [PubMed]
- Wozniak-Knopp, G.; Bartl, S.; Bauer, A.; Mostageer, M.; Woisetschlager, M.; Antes, B.; Ettl, K.; Kainer, M.; Weberhofer, G.; Wiederkum, S.; et al. Introducing antigen-binding sites in structural loops of immunoglobulin constant domains: FC fragments with engineered HER2/NEU-binding sites and antibody properties. Protein Eng. Des. Sel. 2010, 23, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; Perera, R.; Grimm, H.P.; Sam, J.; Colombetti, S.; Fauti, T.; Fahrni, L.; Schaller, T.; Freimoser-Grundschober, A.; Zielonka, J.; et al. In vivo imaging of the activity of CEA TCB, a novel T cell bispecific antibody, reveals specific tumor targeting and fast induction of T cell mediated tumor killing. Clin. Cancer Res. 2016, 22, 4417–4427. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Perry, S.R.; Muniz-Medina, V.; Wang, X.; Wetzel, L.K.; Rebelatto, M.C.; Hinrichs, M.J.M.; Bezabeh, B.Z.; Fleming, R.L.; Dimasi, N.; et al. A biparatopic HER2-targeting antibody-drug conjugate induces tumor regression in primary models refractory to or ineligible for HER2-targeted therapy. Cancer Cell 2016, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Sengers, B.G.; McGinty, S.; Nouri, F.Z.; Argungu, M.; Hawkins, E.; Hadji, A.; Weber, A.; Taylor, A.; Sepp, A. Modeling bispecific monoclonal antibody interaction with two cell membrane targets indicates the importance of surface diffusion. mAbs 2016, 8, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Boutros, C.; Tarhini, A.; Routier, E.; Lambotte, O.; Ladurie, F.L.; Carbonnel, F.; Izzeddine, H.; Marabelle, A.; Champiat, S.; Berdelou, A.; et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat. Rev. Clin. Oncol. 2016, 13, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, P.; Hofmeister, R.; Brischwein, K.; Brandl, C.; Crommer, S.; Bargou, R.; Itin, C.; Prang, N.; Baeuerle, P.A. Serial killing of tumor cells by cytotoxic T cells redirected with a CD19-/CD3-bispecific single-chain antibody construct. Int. J. Cancer 2005, 115, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.; Thayer, W.O.; Liu, L.; Silvestri, G.; Nordstrom, J.L.; Garcia, J.V. CD19 × CD3 dart protein mediates human B cell depletion in vivo in humanized blt mice. Mol. Ther. Oncol. 2016, 3, 15024. [Google Scholar] [CrossRef] [PubMed]
- Przepiorka, D.; Ko, C.W.; Deisseroth, A.; Yancey, C.L.; Candau-Chacon, R.; Chiu, H.J.; Gehrke, B.J.; Gomez-Broughton, C.; Kane, R.C.; Kirshner, S.; et al. FDA approval: Blinatumomab. Clin. Cancer Res. 2015, 21, 4035–4039. [Google Scholar] [CrossRef] [PubMed]
- Klinger, M.; Brandl, C.; Zugmaier, G.; Hijazi, Y.; Bargou, R.C.; Topp, M.S.; Gokbuget, N.; Neumann, S.; Goebeler, M.; Viardot, A.; et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific bite antibody blinatumomab. Blood 2012, 119, 6226–6233. [Google Scholar] [CrossRef] [PubMed]
- Klinger, M.; Benjamin, J.; Kischel, R.; Stienen, S.; Zugmaier, G. Harnessing T cells to fight cancer with bite(r) antibody constructs—Past developments and future directions. Immunol. Rev. 2016, 270, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Brischwein, K.; Schlereth, B.; Guller, B.; Steiger, C.; Wolf, A.; Lutterbuese, R.; Offner, S.; Locher, M.; Urbig, T.; Raum, T.; et al. Mt110: A novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol. Immunol. 2006, 43, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Osada, T.; Hsu, D.; Hammond, S.; Hobeika, A.; Devi, G.; Clay, T.M.; Lyerly, H.K.; Morse, M.A. Metastatic colorectal cancer cells from patients previously treated with chemotherapy are sensitive to T cell killing mediated by CEA/CD3-bispecific T cell-engaging bite antibody. Br. J. Cancer 2010, 102, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Oberst, M.D.; Fuhrmann, S.; Mulgrew, K.; Amann, M.; Cheng, L.; Lutterbuese, P.; Richman, L.; Coats, S.; Baeuerle, P.A.; Hammond, S.A. CEA/CD3 bispecific antibody Medi-565/Amg 211 activation of T cells and subsequent killing of human tumors is independent of mutations commonly found in colorectal adenocarcinomas. mAbs 2014, 6, 1571–1584. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Raum, T.; Lutterbuese, R.; Voelkel, M.; Deegen, P.; Rau, D.; Kischel, R.; Hoffmann, P.; Brandl, C.; Schuhmacher, J.; et al. Regression of human prostate cancer xenografts in mice by Amg 212/Bay2010112, a novel PSMA/CD3-bispecific bite antibody cross-reactive with non-human primate antigens. Mol. Cancer Ther. 2012, 11, 2664–2673. [Google Scholar] [CrossRef] [PubMed]
- Al-Hussaini, M.; Rettig, M.P.; Ritchey, J.K.; Karpova, D.; Uy, G.L.; Eissenberg, L.G.; Gao, F.; Eades, W.C.; Bonvini, E.; Chichili, G.R.; et al. Targeting CD123 in acute myeloid leukemia using a T cell-directed dual-affinity retargeting platform. Blood 2016, 127, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Rothe, A.; Sasse, S.; Topp, M.S.; Eichenauer, D.A.; Hummel, H.; Reiners, K.S.; Dietlein, M.; Kuhnert, G.; Kessler, J.; Buerkle, C.; et al. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory hodgkin lymphoma. Blood 2015, 125, 4024–4031. [Google Scholar] [CrossRef] [PubMed]
- Seimetz, D.; Lindhofer, H.; Bokemeyer, C. Development and approval of the trifunctional antibody catumaxomab (anti-EPCAM × anti-CD3) as a targeted cancer immunotherapy. Cancer Treat. Rev. 2010, 36, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Kurbacher, C.M.; Horn, O.; Kurbacher, J.A.; Herz, S.; Kurbacher, A.T.; Hildenbrand, R.; Bollmann, R. Outpatient intraperitoneal catumaxomab therapy for malignant ascites related to advanced gynecologic neoplasms. Oncologist 2015, 20, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Diermeier-Daucher, S.; Ortmann, O.; Buchholz, S.; Brockhoff, G. Trifunctional antibody ertumaxomab: Non-immunological effects on HER2 receptor activity and downstream signaling. mAbs 2014, 4, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Schuster, F.R.; Stanglmaier, M.; Woessmann, W.; Winkler, B.; Siepermann, M.; Meisel, R.; Schlegel, P.G.; Hess, J.; Lindhofer, H.; Borkhardt, A.; et al. Immunotherapy with the trifunctional anti-CD20 × anti-CD3 antibody FBTA05 (lymphomun) in paediatric high-risk patients with recurrent CD20-positive b cell malignancies. Br. J. Haematol. 2015, 169, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Vallera, D.A.; Chen, H.; Sicheneder, A.R.; Panoskaltsis-Mortari, A.; Taras, E.P. Genetic alteration of a bispecific ligand-directed toxin targeting human CD19 and CD22 receptors resulting in improved efficacy against systemic b cell malignancy. Leuk. Res. 2009, 33, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Bachanova, V.; Frankel, A.E.; Cao, Q.; Lewis, D.; Grzywacz, B.; Verneris, M.R.; Ustun, C.; Lazaryan, A.; McClune, B.; Warlick, E.D.; et al. Phase I study of a bispecific ligand-directed toxin targeting CD22 and CD19 (DT2219) for refractory B cell malignancies. Clin. Cancer Res. 2015, 21, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, M.; Dorado, J.; Baeuerle, P.A.; Heeschen, C. Epcam/CD3-bispecific T cell engaging antibody MT110 eliminates primary human pancreatic cancer stem cells. Clin. Cancer Res. 2012, 18, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Burke, S.; Huang, L.; Gorlatov, S.; Li, H.; Wang, W.; Zhang, W.; Tuaillon, N.; Rainey, J.; Barat, B.; et al. Effector cell recruitment with novel FV-based dual-affinity re-targeting protein leads to potent tumor cytolysis and in vivo B cell depletion. J. Mol. Biol. 2010, 399, 436–449. [Google Scholar] [CrossRef] [PubMed]
- Stadler, C.R.; Bähr-Mahmud, H.; Plum, L.M.; Schmoldt, K.; Kölsch, A.C.; Türeci, Ö.; Sahin, U. Characterization of the first-in-class T cell-engaging bispecific single-chain antibody for targeted immunotherapy of solid tumors expressing the oncofetal protein claudin 6. OncoImmunology 2015, 5, e1091555. [Google Scholar] [CrossRef] [PubMed]
- Schmohl, J.U.; Felices, M.; Taras, L.; Miller, J.S.; Vallera, D.A. Enhanced ADCC and NK cell activation of an anti-carcinoma bispecific antibody by genetic insertion of a modified IL-15 cross-linker. Mol. Ther. 2016, 24, 1312–1322. [Google Scholar] [CrossRef] [PubMed]
- Von Strandmann, E.P.; Hansen, H.P.; Reiners, K.S.; Schnell, R.; Borchmann, P.; Merkert, S.; Simhadri, V.R.; Draube, A.; Reiser, M.; Purr, I.; et al. A novel bispecific protein (ULBP2-BB4) targeting the NKG2D receptor on natural killer (NK) cells and CD138 activates nk cells and has potent antitumor activity against human multiple myeloma in vitro and in vivo. Blood 2006, 107, 1955–1962. [Google Scholar] [CrossRef] [PubMed]
- Vyas, M.; Schneider, A.C.; Shatnyeva, O.; Reiners, K.S.; Tawadros, S.; Kloess, S.; Kohl, U.; Hallek, M.; Hansen, H.P.; Pogge von Strandmann, E. Mono- and dual-targeting triplebodies activate natural killer cells and have anti-tumor activity in vitro and in vivo against chronic lymphocytic leukemia. Oncoimmunology 2016, 5, e1211220. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Sun, F.; Xie, W.; Tang, M.; He, H.; Jia, X.; Tian, X.; Wang, M.; Zhang, J. A bispecific protein RG7S-mica recruits natural killer cells and enhances NKG2D-mediated immunosurveillance against hepatocellular carcinoma. Cancer Lett. 2016, 372, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Borlak, J.; Langer, F.; Spanel, R.; Schondorfer, G.; Dittrich, C. Immune-mediated liver injury of the cancer therapeutic antibody catumaxomab targeting EpCAM, CD3 and Fcγ receptors. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, J.; Thommen, D.S.; Herzig, P.; Bacac, M.; Klein, C.; Roller, A.; Belousov, A.; Levitsky, V.; Savic, S.; Moersig, W.; et al. Expression of inhibitory receptors on intratumoral T cells modulates the activity of a T cell-bispecific antibody targeting folate receptor. Oncoimmunology 2016, 5, e1062969. [Google Scholar] [CrossRef] [PubMed]
- Köhnke, T.; Krupka, C.; Tischer, J.; Knösel, T.; Subklewe, M. Increase of PD-L1 expressing B-precursor all cells in a patient resistant to the CD19/CD3-bispecific T cell engager antibody blinatumomab. J. Hematol. Oncol. 2015, 8. [Google Scholar] [CrossRef] [PubMed]
- Krupka, C.; Kufer, P.; Kischel, R.; Zugmaier, G.; Lichtenegger, F.S.; Kohnke, T.; Vick, B.; Jeremias, I.; Metzeler, K.H.; Altmann, T.; et al. Blockade of the PD-1/PD-l1 axis augments lysis of aml cells by the CD33/CD3 bite antibody construct Amg 330: Reversing a T cell-induced immune escape mechanism. Leukemia 2016, 30, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Kalathil, S.; Lugade, A.A.; Miller, A.; Iyer, R.; Thanavala, Y. Higher frequencies of GARP+CTLA-4+FOXP3+ T regulatory cells and myeloid-derived suppressor cells in hepatocellular carcinoma patients are associated with impaired T cell functionality. Cancer Res. 2013, 73, 2435–2444. [Google Scholar] [CrossRef] [PubMed]
- Yano, H.; Thakur, A.; Tomaszewski, E.N.; Choi, M.; Deol, A.; Lum, L.G. Ipilimumab augments antitumor activity of bispecific antibody-armed T cells. J. Transl. Med. 2014, 12, 191. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, D.; Lum, L.G. Activated T cells armed with bispecific antibodies kill tumor targets. Curr. Opin. Hematol. 2015, 22, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Zitron, I.M.; Thakur, A.; Norkina, O.; Barger, G.R.; Lum, L.G.; Mittal, S. Targeting and killing of glioblastoma with activated T cells armed with bispecific antibodies. BMC Cancer 2013, 13, 83. [Google Scholar] [CrossRef] [PubMed]
- Lum, L.G.; Thakur, A.; Al-Kadhimi, Z.; Colvin, G.A.; Cummings, F.J.; Legare, R.D.; Dizon, D.S.; Kouttab, N.; Maizel, A.; Colaiace, W.; et al. Targeted T cell therapy in stage IV breast cancer: A phase I clinical trial. Clin. Cancer Res. 2015, 21, 2305–2314. [Google Scholar] [CrossRef] [PubMed]
- Urbanska, K.; Lynn, R.C.; Stashwick, C.; Thakur, A.; Lum, L.G.; Powell, D.J. Targeted cancer immunotherapy via combination of designer bispecific antibody and novel gene-engineered T cells. J. Transl. Med. 2014, 12, 347. [Google Scholar] [CrossRef] [PubMed]
- Roybal, K.T.; Williams, J.Z.; Morsut, L.; Rupp, L.J.; Kolinko, I.; Choe, J.H.; Walker, W.J.; McNally, K.A.; Lim, W.A. Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell 2016, 167, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Dengl, S.; Sustmann, C.; Brinkmann, U. Engineered hapten-binding antibody derivatives for modulation of pharmacokinetic properties of small molecules and targeted payload delivery. Immunol. Rev. 2016, 270, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Metz, S.; Haas, A.K.; Daub, K.; Croasdale, R.; Stracke, J.; Lau, W.; Georges, G.; Josel, H.P.; Dziadek, S.; Hopfner, K.P.; et al. Bispecific digoxigenin-binding antibodies for targeted payload delivery. Proc. Natl. Acad. Sci. USA 2011, 108, 8194–8199. [Google Scholar] [CrossRef] [PubMed]
- Thorey, I.S.; Grote, M.; Mayer, K.; Brinkmann, U. Hapten-binding bispecific antibodies for the targeted delivery of siRNA and siRNA-containing nanoparticles. Methods Mol. Biol. 2016, 1364, 219–234. [Google Scholar] [PubMed]
- Schneider, B.; Grote, M.; John, M.; Haas, A.; Bramlage, B.; Lckenstein, L.M.; Jahn-Hofmann, K.; Bauss, F.; Cheng, W.; Croasdale, R.; et al. Targeted siRNA delivery and mRNA knockdown mediated by bispecific digoxigenin-binding antibodies. Mol. Ther. Nucleic Acids 2012, 1, e45. [Google Scholar] [CrossRef] [PubMed]
- Cheal, S.M.; Xu, H.; Guo, H.F.; Zanzonico, P.B.; Larson, S.M.; Cheung, N.K. Preclinical evaluation of multistep targeting of diasialoganglioside GD2 using an IgG-SCFV bispecific antibody with high affinity for GD2 and dota metal complex. Mol. Cancer Ther. 2014, 13, 1803–1812. [Google Scholar] [CrossRef] [PubMed]
- Cheal, S.M.; Xu, H.; Guo, H.-F.; Lee, S.-G.; Punzalan, B.; Chalasani, S.; Fung, E.K.; Jungbluth, A.; Zanzonico, P.B.; Carrasquillo, J.A.; et al. Theranostic pretargeted radioimmunotherapy of colorectal cancer xenografts in mice using picomolar affinity 86Y- or 177LU-Dota-BN binding SCFV C825/GPA33 IgG bispecific immunoconjugates. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 925–937. [Google Scholar] [CrossRef] [PubMed]
- MacDiarmid, J.A.; Mugridge, N.B.; Weiss, J.C.; Phillips, L.; Burn, A.L.; Paulin, R.P.; Haasdyk, J.E.; Dickson, K.A.; Brahmbhatt, V.N.; Pattison, S.T.; et al. Bacterially derived 400 nm particles for encapsulation and cancer cell targeting of chemotherapeutics. Cancer Cell 2007, 11, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.J.; Desai, J.; Rosenthal, M.; McArthur, G.A.; Pattison, S.T.; Pattison, S.L.; MacDiarmid, J.; Brahmbhatt, H.; Scott, A.M. A first-time-in-human phase I clinical trial of bispecific antibody-targeted, paclitaxel-packaged bacterial minicells. PLoS ONE 2015, 10, e0144559. [Google Scholar] [CrossRef] [PubMed]
- Poovassery, J.S.; Kang, J.C.; Kim, D.; Ober, R.J.; Ward, E.S. Antibody targeting of HER2/HER3 signaling overcomes heregulin-induced resistance to PI3K inhibition in prostate cancer. Int. J. Cancer 2015, 137, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Picarda, E.; Ohaegbulam, K.C.; Zang, X. Molecular pathways: Targeting B7-H3 (CD276) for human cancer immunotherapy. Clin. Cancer Res. 2016, 22, 3425–3431. [Google Scholar] [CrossRef] [PubMed]
- Loo, D.; Alderson, R.F.; Chen, F.Z.; Huang, L.; Zhang, W.; Gorlatov, S.; Burke, S.; Ciccarone, V.; Li, H.; Yang, Y.; et al. Development of an Fc-enhanced anti-B7-H3 monoclonal antibody with potent antitumor activity. Clin. Cancer Res. 2012, 18, 3834–3845. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Ma, P.; Zhao, C.; Xue, X.; Han, H.; Liu, C.; Tao, H.; Xiu, W.; Cai, J.; Zhang, M. B7-H3 as a promising target for cytotoxicity T cell in human cancer therapy. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Bivona, T.G. Mechanisms of resistance to epidermal growth factor receptor inhibitors and novel therapeutic strategies to overcome resistance in nsclc patients. Chemother. Res. Pract. 2012, 2012, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Lee, S.H.; Hwang, J.W.; Oh, S.J.; Kim, B.; Jung, S.; Shim, S.H.; Lin, P.W.; Lee, S.B.; Cho, M.Y.; et al. Novel strategy for a bispecific antibody: Induction of dual target internalization and degradation. Oncogene 2016, 35, 4437–4446. [Google Scholar] [CrossRef] [PubMed]
- Ratner, M. Genentech discloses safety concerns over avastin. Nat. Biotechnol. 2004, 22, 1198. [Google Scholar] [CrossRef] [PubMed]
- Chae, S.S.; Kamoun, W.S.; Farrar, C.T.; Kirkpatrick, N.D.; Niemeyer, E.; de Graaf, A.M.A.; Sorensen, A.G.; Munn, L.L.; Jain, R.K.; Fukumura, D. Angiopoietin-2 interferes with anti-VEGFR2-induced vessel normalization and survival benefit in mice bearing gliomas. Clin. Cancer Res. 2010, 16, 3618–3627. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, S.E.; Perez-Soler, R. Mechanisms of resistance to vascular endothelial growth factor blockade. Cancer 2012, 118, 3455–3467. [Google Scholar] [CrossRef] [PubMed]
- Kloepper, J.; Riedemann, L.; Amoozgar, Z.; Seano, G.; Susek, K.; Yu, V.; Dalvie, N.; Amelung, R.L.; Datta, M.; Song, J.W.; et al. Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc. Natl. Acad. Sci. USA 2016, 113, 4476–4481. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Kim, D.; Choi, Y.B.; Kang, K.; Sung, E.S.; Ahn, J.H.; Goo, J.; Yeom, D.H.; Jang, H.S.; Moon, K.D.; et al. Simultaneous blockade of VEGF and DLL4 by HD105, a bispecific antibody, inhibits tumor progression and angiogenesis. mAbs 2016, 8, 892–904. [Google Scholar] [CrossRef] [PubMed]
- Moores, S.L.; Chiu, M.; Bushey, B.S.; Chevalier, K.; Luistro, L.; Dorn, K.; Brezski, R.J.; Haytko, P.; Kelly, T.; Wu, S.J.; et al. A novel bispecific antibody targeting EGFR and cMet that is effective against EGFR inhibitor-resistant lung tumors. Cancer Res. 2016, 76, 3942–3953. [Google Scholar] [CrossRef] [PubMed]
- Shima, M.; Hanabusa, H.; Taki, M.; Matsushita, T.; Sato, T.; Fukutake, K.; Fukazawa, N.; Yoneyama, K.; Yoshida, H.; Nogami, K. Factor VIII-mimetic function of humanized bispecific antibody in hemophilia A. N. Engl. J. Med. 2016, 374, 2044–2053. [Google Scholar] [CrossRef] [PubMed]
- Nogami, K. Bispecific antibody mimicking factor VIII. Thromb. Res. 2016, 141, S34–S35. [Google Scholar] [CrossRef]
- Florio, M.; Gunasekaran, K.; Stolina, M.; Li, X.; Liu, L.; Tipton, B.; Salimi-Moosavi, H.; Asuncion, F.J.; Li, C.; Sun, B.; et al. A bispecific antibody targeting sclerostin and DKK-1 promotes bone mass accrual and fracture repair. Nat. Commun. 2016, 7, 11505. [Google Scholar] [CrossRef] [PubMed]
- Thaden, J.T.; Keller, A.E.; Shire, N.J.; Camara, M.M.; Otterson, L.; Huband, M.; Guenther, C.M.; Zhao, W.; Warrener, P.; Stover, C.K.; et al. Pseudomonas aeruginosa bacteremic patients exhibit nonprotective antibody titers against therapeutic antibody targets PCRV and PSL exopolysaccharide. J. Infect. Dis. 2016, 213, 640–648. [Google Scholar] [CrossRef] [PubMed]
- DiGiandomenico, A.; Keller, A.E.; Gao, C.; Rainey, G.J.; Warrener, P.; Camara, M.M.; Bonnell, J.; Fleming, R.; Bezabeh, B.; Dimasi, N.; et al. A multifunctional bispecific antibody protects against pseudomonas aeruginosa. Sci. Transl. Med. 2014, 6, 262ra155. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.J.; Atwal, J.K.; Zhang, Y.; Tong, R.K.; Wildsmith, K.R.; Tan, C.; Bien-Ly, N.; Hersom, M.; Maloney, J.A.; Meilandt, W.J.; et al. Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci. Transl. Med. 2014, 6, 261ra154. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yu, J.; Lanzi, A.; Yao, X.; Andrews, C.D.; Tsai, L.; Gajjar, M.R.; Sun, M.; Seaman, M.S.; Padte, N.N.; et al. Engineered bispecific antibodies with exquisite HIV-1-neutralizing activity. Cell 2016, 165, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Bournazos, S.; Gazumyan, A.; Seaman, M.S.; Nussenzweig, M.C.; Ravetch, J.V. Bispecific anti-HIV-1 antibodies with enhanced breadth and potency. Cell 2016, 165, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- Gramer, M.J.; van den Bremer, E.T.; van Kampen, M.D.; Kundu, A.; Kopfmann, P.; Etter, E.; Stinehelfer, D.; Long, J.; Lannom, T.; Noordergraaf, E.H.; et al. Production of stable bispecific IgG1 by controlled FAB-arm exchange. mAbs 2014, 5, 962–973. [Google Scholar] [CrossRef] [PubMed]
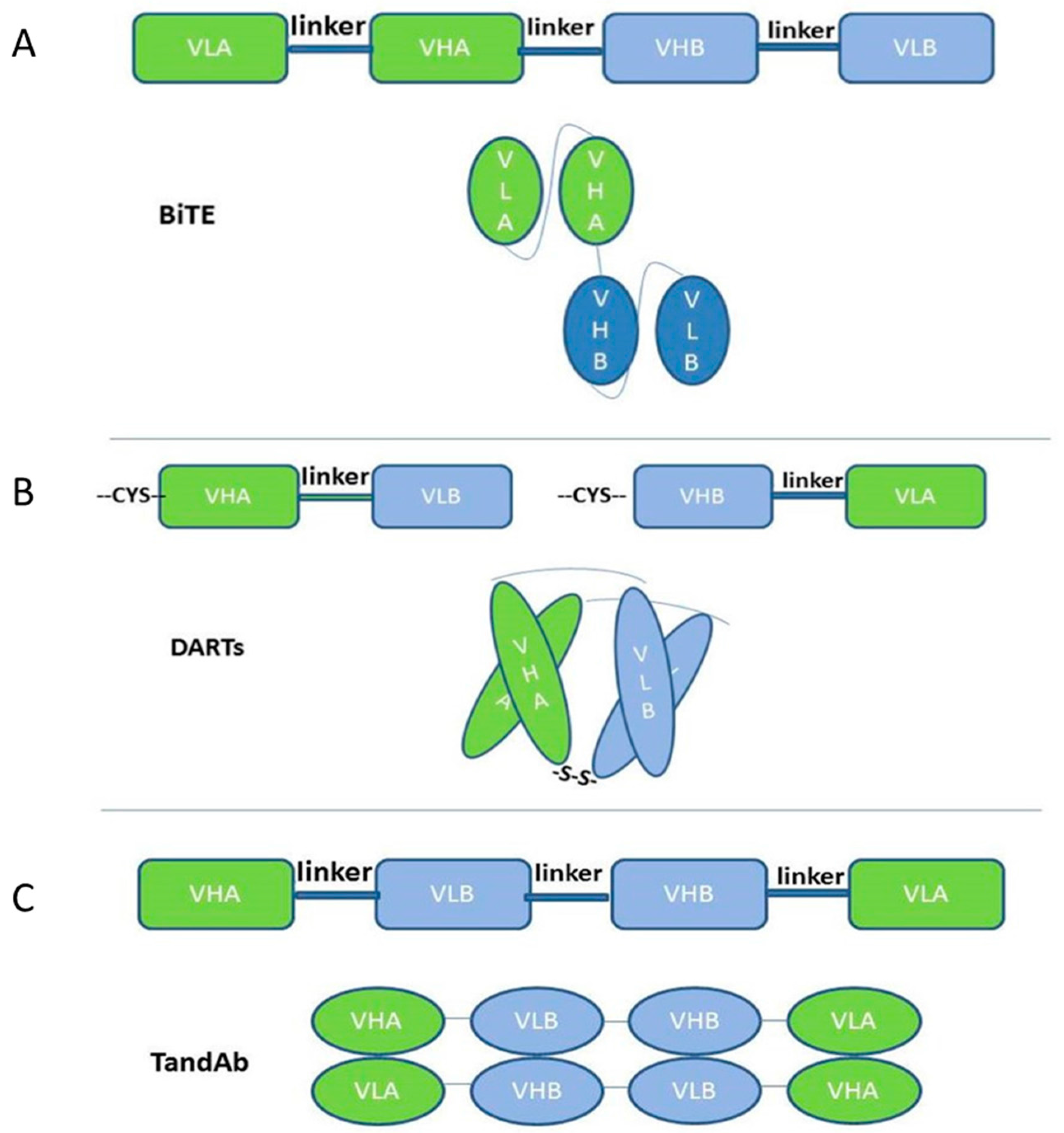

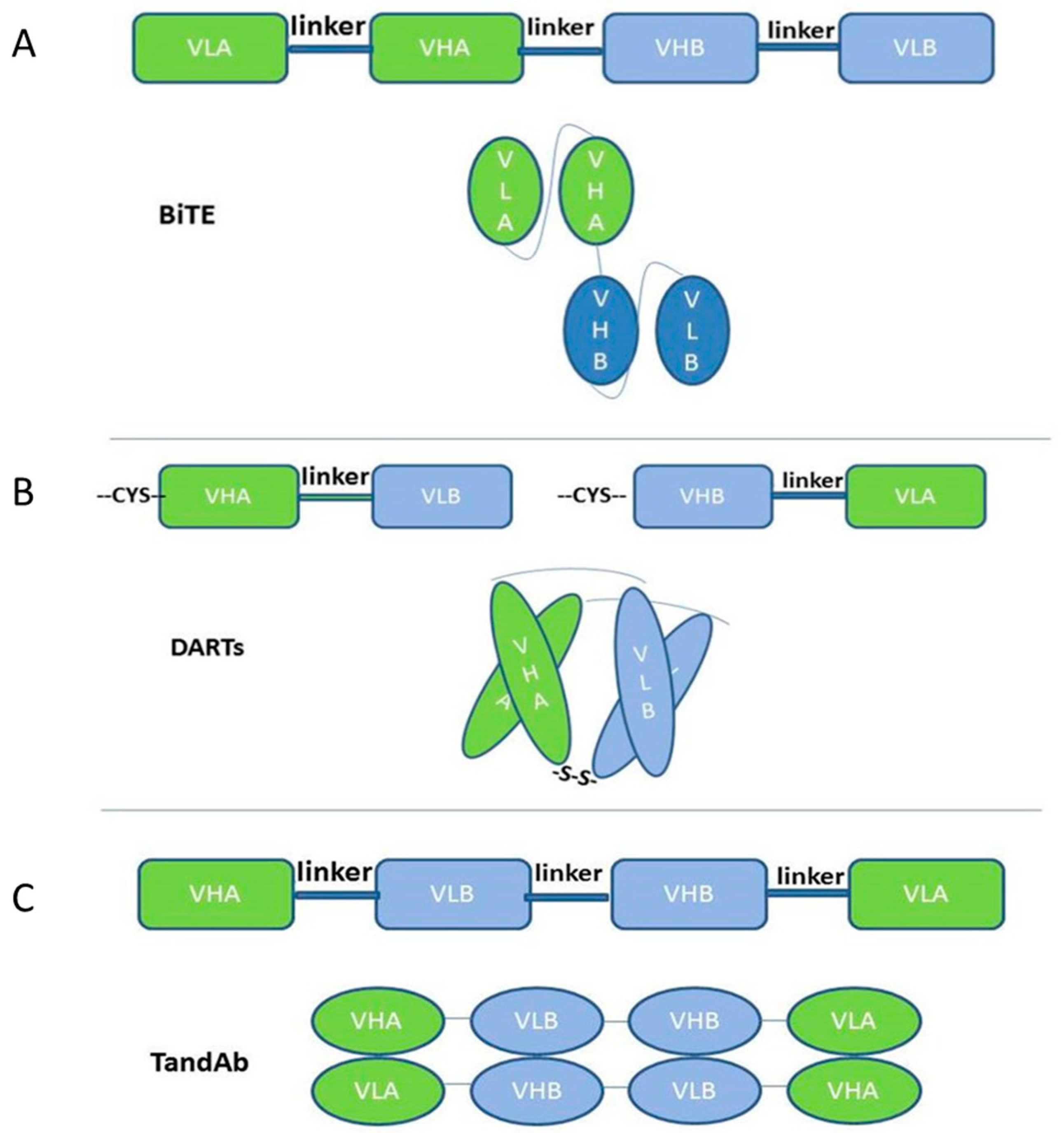{kind=link}
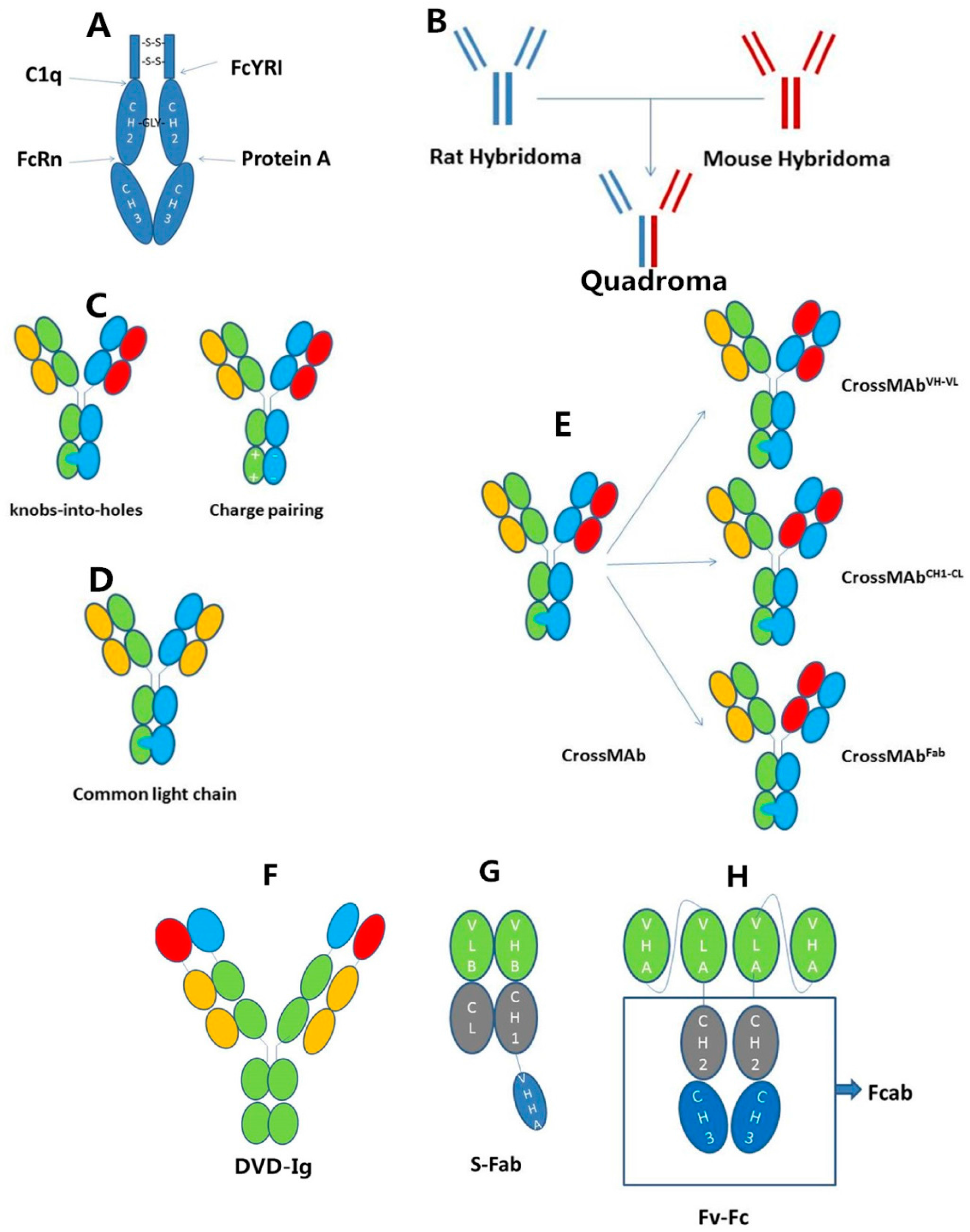{kind=link}
| Molecule | Targets | Format | Status of Clinical Trail | Disease | Citation |
|---|---|---|---|---|---|
| Blinatumomab | CD19 + CD3 | BiTE | approved | Acute lymphoblastic leukemia | [8,44,45,46] |
| MT-110 | EpCAM + CD3 | BiTE | Phase I (completed) | Lung, gastric, colorectal, breast, hormone-refractory prostate cancer, and ovarian cancer | [47] (ClinicalTrials.gov: NCT00635596) |
| MT-111 (MEDI-565/AMG 211) | CEA + CD3 | BiTE | Phase I (completed) | Gastrointestinal adenocarcinomas | [48,49] (ClinicalTrials.gov: NCT01284231) |
| AMG 112 (BAY2010112) | PSMA + CD3 | BiTE | Phase I (recruiting) | Prostate cancer | [50] (ClinicalTrials.gov: NCT01723475) |
| MGD006 | CD123 + CD3 | DARTs | Phase I (recruiting) | Acute myeloid leukemia | [51] (ClinicalTrials.gov identifier: NCT02152956) |
| AFM11 | CD19 + CD3 | TandAbs | Phase I (recruiting) | Relapsed and/or refractory CD19-positive B-cell NHL | [14] (ClinicalTrials.gov: NCT02106091) |
| AFM13 | CD30 + CD16A | TandAbs | Phase II (recruiting) | Hodgkin lymphoma | [52] (ClinicalTrials.gov: NCT02321592, NCT01221571, NCT02665650) |
| CEA TCB | CEA + CD3 | Knobs into Holes, CrossMAb | Phase I (recruiting) | Locally advanced and/or metastatic solid tumors | [17,38] (ClinicalTrials.gov: NCT02324257) |
| catumaxomab | EpCAM + CD3 | quadroma | Approved | Malignant ascites (MA) | [53,54] |
| ertumaxomab | HER2 + CD3 | quadroma | Phase II | Breast cancer | [55] (clinicaltrials.gov: NCT00351858) |
| FBTA05 | CD20 + CD3 | quadroma | Phase I/II | NHL | [56] (clinicaltrials.gov: NCT01138579) |
| MM-111 | HER2 + HER3 | (scFv)2-human serum albumin | Phase I | HER2-positive tumors | [20] (ClinicalTrials.gov: NCT00911898,NCT01304784) |
| DT2219 and DT2219ARL | CD22 + CD19 | (scFv)2 | Phase I | B-lineage leukemia or lymphoma | [57,58] (ClinicalTrials.gov: NCT02370160, NCT00889408) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, F.; Wen, W.; Qin, W. Bispecific Antibodies as a Development Platform for New Concepts and Treatment Strategies. Int. J. Mol. Sci. 2017, 18, 48. https://doi.org/10.3390/ijms18010048
Yang F, Wen W, Qin W. Bispecific Antibodies as a Development Platform for New Concepts and Treatment Strategies. International Journal of Molecular Sciences. 2017; 18(1):48. https://doi.org/10.3390/ijms18010048
Chicago/Turabian StyleYang, Fa, Weihong Wen, and Weijun Qin. 2017. "Bispecific Antibodies as a Development Platform for New Concepts and Treatment Strategies" International Journal of Molecular Sciences 18, no. 1: 48. https://doi.org/10.3390/ijms18010048
APA StyleYang, F., Wen, W., & Qin, W. (2017). Bispecific Antibodies as a Development Platform for New Concepts and Treatment Strategies. International Journal of Molecular Sciences, 18(1), 48. https://doi.org/10.3390/ijms18010048