
PERM Hypothesis: The Fundamental Machinery Able to Elucidate the Role of Xenobiotics and Hormesis in Cell Survival and Homeostasis



Abstract
:1. Introduction
2. Organelles and Signalling Molecules of the Proterome
2.1. Role of Flavonoids as Stressing and Signalling Molecules
2.2. Role of Peroxisomes
2.3. Role of the Proteasome
2.4. Role of Mitochondria
2.5. Role of the Endoplasmic Reticulum (ER)
3. Proterome Function
3.1. The Role and Activity of the Proterome in the Autophagy/Apoptosis Balance
3.2. Activity of the Proterome: ROS as Signalling Molecules and the Role of Xenobiotics
3.3. Mitochondria in the Proterome Activity
3.4. Calcium Oscillation and Proterome Chaotic System
3.5. Chaotic Activity Elicited by ROS in the Proterome
4. Conclusions
Conflicts of Interest
References
- Marchi, S.; Patergnani, S.; Pinton, P. The endoplasmic reticulum-mitochondria connection: One touch, multiple functions. Biochim. Biophys. Acta 2014, 1837, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Howes, M.J.; Simmonds, M.S. The role of phytochemicals as micronutrients in health and disease. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Alasalvar, C.; Bolling, B.W. Review of nut phytochemicals, fat-soluble bioactives, antioxidant components and health effects. Br. J. Nutr. 2015, 113, S68–S78. [Google Scholar] [CrossRef]
- Shankar, E.; Kanwal, R.; Candamo, M.; Gupta, S. Dietary phytochemicals as epigenetic modifiers in cancer: Promise and challenges. Semin. Cancer Biol. 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Davinelli, S.; Maes, M.; Corbi, G.; Zarrelli, A.; Willcox, D.C.; Scapagnini, G. Dietary phytochemicals and neuro-inflammaging: From mechanistic insights to translational challenges. Immun. Ageing 2016, 13, 16. [Google Scholar] [CrossRef] [PubMed]
- Tyne, W.; Little, S.; Spurgeon, D.J.; Svendsen, C. Hormesis depends upon the life-stage and duration of exposure: Examples for a pesticide and a nanomaterial. Ecotoxicol. Environ. Saf. 2015, 120, 117–123. [Google Scholar] [CrossRef]
- Castillo-Quan, J.I.; Li, L.; Kinghorn, K.J.; Ivanov, D.K.; Tain, L.S.; Slack, C.; Kerr, F.; Nespital, T.; Thornton, J.; Hardy, J.; et al. Lithium Promotes Longevity through GSK3/NRF2-Dependent Hormesis. Cell Rep. 2016, 15, 638–650. [Google Scholar] [CrossRef] [PubMed]
- Møller, A.P.; Mousseau, T.A. Are Organisms Adapting to Ionizing Radiation at Chernobyl? Trends Ecol. Evol. 2016, 31, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Parsons, P.A. Hormesis: An adaptive expectation with emphasis on ionizing radiation. J. Appl. Toxicol. 2000, 20, 103–112. [Google Scholar] [CrossRef]
- Parsons, P.A. The hormetic zone: An ecological and evolutionary perspective based upon habitat characteristics and fitness selection. Q. Rev. Biol. 2001, 76, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Parsons, P.A. Metabolic efficiency in response to environmental agents predicts hormesis and invalidates the linear no-threshold premise: Ionizing radiation as a case study. Crit. Rev. Toxicol. 2003, 33, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Shalini, V.; Pushpan, C.K.; Sindhu, G.; Jayalekshmy, A.; Helen, A. Tricin, flavonoid from Njavara reduces inflammatory responses in hPBMCs by modulating the p38MAPK and PI3K/Akt pathways and prevents inflammation associated endothelial dysfunction in HUVECs. Immunobiology 2016, 221, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Huang, S. Inhibition of PI3K/Akt/mTOR signaling by natural products. Anticancer Agents Med. Chem. 2013, 13, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Hu, Y.Z. PI3K/Akt/mTOR pathway inhibitors in cancer: A perspective on clinical progress. Curr. Med. Chem. 2010, 17, 4326–4341. [Google Scholar] [CrossRef] [PubMed]
- Pandurangan, A.K. Potential targets for prevention of colorectal cancer: A focus on PI3K/Akt/mTOR and Wnt pathways. Asian Pac. J. Cancer Prev. 2013, 14, 2201–2205. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Pelling, J.C. Targeting the PI3K/Akt/mTOR axis by apigenin for cancer prevention. Anticancer Agents Med. Chem. 2013, 13, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.H.; Chiang, Y.C.; Tsai, M.H.; Liang, C.J.; Hsu, L.F.; Li, S.Y.; Wang, M.C.; Yen, F.L.; Lee, C.W. Eupafolin, a skin whitening flavonoid isolated from Phyla nodiflora, downregulated melanogenesis: Role of MAPK and Akt pathways. J. Ethnopharmacol. 2014, 151, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.L.; Tong, Q.; Wang, W.Q.; Shi, C.Y.; Xiong, W.; Chen, J.; Liu, X.; Fang, J.G. Suppression of Inflammatory Responses by Dihydromyricetin, a Flavonoid from Ampelopsis grossedentata, via Inhibiting the Activation of NF-κB and MAPK Signaling Pathways. J. Nat. Prod. 2015, 78, 1689–1696. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Nagappan, A.; Park, H.S.; Hong, G.E.; Yumnam, S.; Raha, S.; Saralamma, V.V.; Lee, W.S.; Kim, E.H.; Kim, G.S. Flavonoids isolated from Citrus platymamma induce mitochondrial-dependent apoptosis in AGS cells by modulation of the PI3K/AKT and MAPK pathways. Oncol. Rep. 2015, 34, 1517–1525. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Wang, C.; Peng, J.; Li, W.; Jin, Y.; Liu, Q.; Meng, Q.; Liu, K.; Sun, H. Naringin regulates cholesterol homeostasis and inhibits inflammation via modulating NF-κB and ERK signaling pathways in vitro. Pharmazie 2016, 71, 101–108. [Google Scholar]
- Xu, M.; Wang, S.; Song, Y.U.; Yao, J.; Huang, K.; Zhu, X. Apigenin suppresses colorectal cancer cell proliferation, migration and invasion via inhibition of the Wnt/β-catenin signaling pathway. Oncol. Lett. 2016, 11, 3075–3080. [Google Scholar] [CrossRef] [PubMed]
- Bremner, P.; Heinrich, M. Natural products as targeted modulators of the nuclear factor-βB pathway. J. Pharm. Pharmacol. 2002, 54, 453–472. [Google Scholar] [CrossRef] [PubMed]
- Nam, N.H. Naturally occurring NF-κB inhibitors. Mini Rev. Med. Chem. 2006, 6, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Kawser Hossain, M.; Abdal Dayem, A.; Han, J.; Yin, Y.; Kim, K.; Kumar Saha, S.; Yang, G.M.; Choi, H.Y.; Cho, S.G. Molecular Mechanisms of the Anti-Obesity and Anti-Diabetic Properties of Flavonoids. Int. J. Mol. Sci. 2016, 17, 569. [Google Scholar] [CrossRef] [PubMed]
- Vezza, T.; Rodríguez-Nogales, A.; Algieri, F.; Utrilla, M.P.; Rodriguez-Cabezas, M.E.; Galvez, J. Flavonoids in Inflammatory Bowel Disease: A Review. Nutrients 2016, 8, 211. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Gerevini, G.T.; Repossi, G.; Dain, A.; Tarres, M.C.; Das, U.N.; Eynard, A.R. Beneficial action of resveratrol: How and why? Nutrition 2016, 32, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Bell, L.; Lamport, D.J.; Butler, L.T.; Williams, C.M. A Review of the Cognitive Effects Observed in Humans Following Acute Supplementation with Flavonoids, and Their Associated Mechanisms of Action. Nutrients 2015, 7, 10290–10306. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Russo, G.L.; Daglia, M.; Kasi, P.D.; Ravi, S.; Nabavi, S.F.; Nabavi, S.M. Understanding genistein in cancer: The “good” and the “bad” effects: A review. Food Chem. 2016, 196, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Rendeiro, C.; Rhodes, J.S.; Spencer, J.P. The mechanisms of action of flavonoids in the brain: Direct versus indirect effects. Neurochem. Int. 2015, 89, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Al Mofleh, I.A. Spices, herbal xenobiotics and the stomach: Friends or foes? World J. Gastroenterol. 2010, 16, 2710–2719. [Google Scholar] [CrossRef] [PubMed]
- Fruehauf, J.P.; Meyskens, F.L., Jr. Reactive oxygen species: A breath of life or death? Clin. Cancer Res. 2007, 13, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, J.J.; Xu, D.P.; Zhou, T.; Zhou, Y.; Li, S.; Li, H.B. Bioactivities and Health Benefits of Wild Fruits. Int. J. Mol. Sci. 2016, 17, 1258. [Google Scholar] [CrossRef] [PubMed]
- Peluso, I.; Palmery, M. Flavonoids at the pharma-nutrition interface: Is a therapeutic index in demand? Biomed. Pharmacother. 2015, 71, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, G.B.; Smagghe, G.; Grootaert, C.; Zotti, M.; Raes, K.; van Camp, J. Flavonoid interactions during digestion, absorption, distribution and metabolism: A sequential structure-activity/property relationship-based approach in the study of bioavailability and bioactivity. Drug Metab. Rev. 2015, 47, 175–190. [Google Scholar] [CrossRef] [PubMed]
- Rietjens, I.M.; Al Huseiny, W.; Boersma, M.G. Flavonoids and alkenylbenzenes: New concepts in bioactivation studies. Chem. Biol. Interact. 2011, 192, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Rietjens, I.M.; Boersma, M.G.; van der Woude, H.; Jeurissen, S.M.; Schutte, M.E.; Alink, G.M. Flavonoids and alkenyl-benzenes: Mechanisms of mutagenic action and carcinogenic risk. Mutat. Res. 2005, 574, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Horobin, R.W. Predicting mitochondrial targeting by small molecule xenobiotics within living cells using QSAR models. Methods Mol. Biol. 2015, 1265, 13–23. [Google Scholar] [PubMed]
- Cribb, A.E.; Peyrou, M.; Muruganandan, S.; Schneider, L. The endoplasmic reticulum in xenobiotic toxicity. Drug Metab. Rev. 2005, 37, 405–442. [Google Scholar] [CrossRef] [PubMed]
- Frezza, M.; Schmitt, S.; Dou, Q.P. Targeting the ubiquitin-proteasome pathway: An emerging concept in cancer therapy. Curr. Top. Med. Chem. 2011, 11, 2888–2905. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Dai, X.; Zhang, Z.; Jiang, Y.; Ma, X.; Cai, X.; Li, Y. Proanthocyanidins protect against early diabetic peripheral neuropathy by modulating endoplasmic reticulum stress. J. Nutr. Biochem. 2014, 25, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Suliman, H.B.; Piantadosi, C.A. Mitochondrial Quality Control as a Therapeutic Target. Pharmacol. Rev. 2016, 68, 20–48. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; van Aken, O.; Schwarzländer, M.; Belt, K.; Millar, A.H. The Roles of Mitochondrial Reactive Oxygen Species in Cellular Signaling and Stress Response in Plants. Plant Physiol. 2016, 171, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Russell, E.G.; Cotter, T.G. Chapter Six-New Insights into the role of reactive oxygen species (ROS) in cellular-signal transduction processes. Int. Rev. Cell Mol. Biol. 2015, 319, 221–254. [Google Scholar] [PubMed]
- Siraki, A.G.; Pourahmad, J.; Chan, T.S.; Khan, S.; O’Brien, P.J. Endogenous and endobiotic induced reactive oxygen species formation by isolated hepatocytes. Free Radic. Biol. Med. 2002, 32, 2–10. [Google Scholar] [CrossRef]
- Bonekamp, N.A.; Völkl, A.; Fahimi, H.D.; Schrader, M. Reactive oxygen species and peroxisomes: Struggling for balance. Biofactors 2009, 35, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Schrader, M.; Fahimi, H.D. Peroxisomes and oxidative stress. Biochim. Biophys. Acta 2006, 1763, 1755–1766. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim. Biophys. Acta 2012, 1822, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Del Río, L.A.; Corpas, F.J.; Sandalio, L.M.; Palma, J.M.; Gómez, M.; Barroso, J.B. Reactive oxygen species, antioxidant systems and nitric oxide in peroxisomes. J. Exp. Bot. 2002, 53, 1255–1272. [Google Scholar] [CrossRef] [PubMed]
- Sandalio, L.M.; Romero-Puertas, M.C. Peroxisomes sense and respond to environmental cues by regulating ROS and RNS signalling networks. Ann. Bot. 2015, 116, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Sandalio, L.M.; Rodríguez-Serrano, M.; Romero-Puertas, M.C.; del Río, L.A. Role of peroxisomes as a source of reactive oxygen species (ROS) signaling molecules. Subcell. Biochem. 2013, 69, 231–255. [Google Scholar] [PubMed]
- Wang, B.; van Veldhoven, P.P.; Brees, C.; Rubio, N.; Nordgren, M.; Apanasets, O.; Kunze, M.; Baes, M.; Agostinis, P.; Fransen, M. Mitochondria are targets for peroxisome-derived oxidative stress in cultured mammalian cells. Free Radic. Biol. Med. 2013, 65, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Pomatto, L.C.; Raynes, R.; Davies, K.J. The peroxisomal Lon protease LonP2 in aging and disease: Functions and comparisons with mitochondrial Lon protease LonP1. Biol. Rev. 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Barrett, A.J.; Bateman, A. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2012, 40, D343–D350. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.; Gibellini, L.; Nasi, M.; de Biasi, S.; Bortolotti, C.A.; Iannone, A.; Cossarizza, A. Emerging role of Lon protease as a master regulator of mitochondrial functions. Biochim. Biophys. Acta 2016, 1857, 1300–1306. [Google Scholar] [CrossRef] [PubMed]
- Strauss, K.A.; Jinks, R.N.; Puffenberger, E.G.; Venkatesh, S.; Singh, K.; Cheng, I.; Mikita, N.; Thilagavathi, J.; Lee, J.; Sarafianos, S.; et al. CODAS syndrome is associated with mutations of LONP1, encoding mitochondrial AAA+ Lon protease. Am. J. Hum. Genet. 2015, 96, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Ngo, J.K.; Pomatto, L.C.; Davies, K.J. Upregulation of the mitochondrial Lon Protease allows adaptation to acute oxidative stress but dysregulation is associated with chronic stress, disease, and aging. Redox Biol. 2013, 1, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Ngo, J.K.; Davies, K.J. Importance of the lon protease in mitochondrial maintenance and the significance of declining lon in aging. Ann. N. Y. Acad. Sci. 2007, 1119, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.; Gibellini, L.; Guaraldi, G.; Orlando, G.; Gant, T.W.; Morselli, E.; Nasi, M.; Salomoni, P.; Mussini, C.; Cossarizza, A. Upregulation of nuclear-encoded mitochondrial LON protease in HAART-treated HIV-positive patients with lipodystrophy: Implications for the pathogenesis of the disease. AIDS 2010, 24, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.Y.; Chiu, Y.C.; Lee, A.Y.; Hwang, T.L. Mitochondrial Lon protease controls ROS-dependent apoptosis in cardiomyocyte under hypoxia. Mitochondrion 2015, 23, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.; Waterham, H.R.; Ferdinandusse, S. Metabolic Interplay between Peroxisomes and Other Subcellular Organelles Including Mitochondria and the Endoplasmic Reticulum. Front. Cell Dev. Biol. 2016, 3, 83. [Google Scholar] [CrossRef] [PubMed]
- Niforou, K.; Cheimonidou, C.; Trougakos, I.P. Molecular chaperones and proteostasis regulation during redox imbalance. Redox Biol. 2014, 2, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Aiken, C.T.; Kaake, R.M.; Wang, X.; Huang, L. Oxidative stress-mediated regulation of proteasome complexes. Mol. Cell. Proteom. 2011, 10, R110.006924. [Google Scholar] [CrossRef] [PubMed]
- Adams, J. The proteasome: Structure, function, and role in the cell. Cancer Treat. Rev. 2003, 29, 3–9. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [PubMed]
- Reyskens, K.M.; Essop, M.F. HIV protease inhibitors and onset of cardiovascular diseases: A central role for oxidative stress and dysregulation of the ubiquitin-proteasome system. Biochim. Biophys. Acta 2014, 1842, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Segref, A.; Kevei, É.; Pokrzywa, W.; Schmeisser, K.; Mansfeld, J.; Livnat-Levanon, N.; Ensenauer, R.; Glickman, M.H.; Ristow, M.; Hoppe, T. Pathogenesis of human mitochondrial diseases is modulated by reduced activity of the ubiquitin/proteasome system. Cell Metab. 2014, 19, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Warnatsch, A.; Bergann, T.; Krüger, E. Oxidation matters: The ubiquitin proteasome system connects innate immune mechanisms with MHC class I antigen presentation. Mol. Immunol. 2013, 55, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Voges, D.; Zwickl, P.; Baumeister, W. The 26S proteasome: A molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 1999, 68, 1015–1068. [Google Scholar] [CrossRef] [PubMed]
- Livnat-Levanon, N.; Kevei, É.; Kleifeld, O.; Krutauz, D.; Segref, A.; Rinaldi, T.; Erpapazoglou, Z.; Cohen, M.; Reis, N.; Hoppe, T.; et al. Reversible 26S proteasome disassembly upon mitochondrial stress. Cell Rep. 2014, 7, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yen, J.; Kaiser, P.; Huang, L. Regulation of the 26S proteasome complex during oxidative stress. Sci. Signal. 2010, 3, ra88. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R.; Chakrabarti, O. Regulation of Mitofusin1 by Mahogunin Ring Finger-1 and the proteasome modulates mitochondrial fusion. Biochim. Biophys. Acta 2016, 1863, 3065–3083. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R.; Chakrabarti, O. Ubiquitin-mediated regulation of the E3 ligase GP78 by MGRN1 in trans affects mitochondrial homeostasis. J. Cell Sci. 2016, 129, 757–773. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; St-Pierre, P.; Shankar, J.; Wang, P.T.; Joshi, B.; Nabi, I.R. Regulation of mitophagy by the Gp78 E3 ubiquitin ligase. Mol. Biol. Cell 2013, 24, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A role for ubiquitin in selective autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Chhangani, D.; Mishra, A. Mahogunin ring finger-1 (MGRN1) suppresses chaperone-associated misfolded protein aggregation and toxicity. Sci. Rep. 2013, 3, 1972. [Google Scholar] [CrossRef] [PubMed]
- Csordás, G.; Hajnóczky, G. SR/ER-mitochondrial local communication: Calcium and ROS. Biochim. Biophys. Acta 2009, 1787, 1352–1362. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.; Huang, Z.; Chen, L.; Wang, W.; Zhang, B.; Xu, Y.; Pan, S.; Mao, Z.; Hu, H.; Geng, Q. Associations between autophagy, the ubiquitin-proteasome system and endoplasmic reticulum stress in hypoxia-deoxygenation or ischemia-reperfusion. Eur. J. Pharmacol. 2016, 791, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.Y.; Zhou, X.D.; Yang, J.; Chen, L.X.; Ran, D.H. Inhibition of autophagy enhances heat-induced apoptosis in human non-small cell lung cancer cells through ER stress pathways. Arch. Biochem. Biophys. 2016, 607, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Chen, C.; Jiang, X.; Zhang, Z. ROS-mediated endoplasmic reticulum stress and mitochondrial dysfunction underlie apoptosis induced by resveratrol and arsenic trioxide in A549 cells. Chem. Biol. Interact. 2016, 245, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Yang, J.; Zhao, J.; Xiao, C.; Xu, C.; Xiang, Y. The switch from ER stress-induced apoptosis to autophagy via ROS-mediated JNK/p62 signals: A survival mechanism in methotrexate-resistant choriocarcinoma cells. Exp. Cell Res. 2015, 334, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Li, H.; Zhang, Y.; Ron, D.; Walter, P. Divergent effects of PERK and IRE1 signaling on cell viability. PLoS ONE 2009, 4, e4170. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.C.; Kihara, A.H.; Goulart, V.A.; Tonelli, F.M.; Gomes, K.N.; Ulrich, H.; Resende, R.R. Calcium signaling and cell proliferation. Cell. Signal. 2015, 27, 2139–2149. [Google Scholar] [CrossRef]
- Giorgi, C.; Bonora, M.; Sorrentino, G.; Missiroli, S.; Poletti, F.; Suski, J.M.; Galindo Ramirez, F.; Rizzuto, R.; Di Virgilio, F.; Zito, E.; et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc. Natl. Acad. Sci. USA 2015, 112, 1779–1784. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Liu, H.; Jiang, C.C.; Fang, L.; Chen, C.; Zhang, X.D.; Jiang, Z.W. Connecting endoplasmic reticulum stress to autophagy through IRE1/JNK/beclin-1 in breast cancer cells. Int. J. Mol. Med. 2014, 34, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Moretti, L.; Cha, Y.I.; Niermann, K.J.; Lu, B. Switch between apoptosis and autophagy radiation-induced endoplasmic reticulum stress? Cell Cycle 2007, 6, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Dupont, G.; Combettes, L. Fine tuning of cytosolic Ca2+ oscillations. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Pecze, L.; Blum, W.; Schwaller, B. Routes of Ca2+ shuttling during Ca2+ oscillations: Focus on the role of mitochondrial Ca2+ handling and cytosolic Ca2+ buffers. J. Biol. Chem. 2015, 290, 28214–28230. [Google Scholar] [CrossRef] [PubMed]
- Dupont, G.; Combettes, L.; Bird, G.S.; Putney, J.W. Calcium oscillations. Cold Spring Harb. Perspect. Biol. 2011, 3, a004226. [Google Scholar] [CrossRef] [PubMed]
- Dupont, G. Modeling the intracellular organization of calcium signaling. WileyInterdiscip. Rev. Syst. Biol. Med. 2014, 6, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, N.; Kurths, J.; Diaz-Guilera, A. synchronization of mobile chaotic oscillator networks. Chaos 2016, 26, 094824. [Google Scholar] [CrossRef] [PubMed]
- Cortassa, S.; Aon, M.A.; Winslow, R.L.; O’Rourke, B. A mitochondrial oscillator dependent on reactive oxygen species. Biophys. J. 2004, 87, 2060–2073. [Google Scholar] [CrossRef] [PubMed]
- Aon, M.A.; Cortassa, S.; O’Rourke, B. Mitochondrial oscillations in physiology and pathophysiology. Adv. Exp. Med. Biol. 2008, 641, 98–117. [Google Scholar] [PubMed]
- Gong, C.; Li, C.; Qi, X.; Song, Z.; Wu, J.; Hughes, M.E.; Li, X. The daily rhythms of mitochondrial gene expression and oxidative stress regulation are altered by aging in the mouse liver. Chronobiol. Int. 2015, 32, 1254–1263. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamada, T.; Tsukita, S.; Kaneko, K.; Shirai, Y.; Munakata, Y.; Ishigaki, Y.; Imai, J.; Uno, K.; Hasegawa, Y.; et al. Chronic mild stress alters circadian expressions of molecular clock genes in the liver. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E301–E309. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Aon, M.A.; Almas, T.; Cortassa, S.; Winslow, R.L.; O’Rourke, B. A reaction-diffusion model of ROS-induced ROS release in a mitochondrial network. PLoS Comput. Biol. 2010, 6, e1000657. [Google Scholar] [CrossRef] [PubMed]
- Mortenson, M.M.; Schlieman, M.G.; Virudachalam, S.; Lara, P.N.; Gandara, D.G.; Davies, A.M.; Bold, R.J. Reduction in BCL-2 levels by 26S proteasome inhibition with bortezomib is associated with induction of apoptosis in small cell lung cancer. Lung Cancer. 2005, 49, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Meissner, C.; Lorenz, H.; Hehn, B.; Lemberg, M.K. Intramembrane protease PARL defines a negative regulator of PINK1- and PARK2/Parkin-dependent mitophagy. Autophagy 2015, 11, 1484–1498. [Google Scholar] [CrossRef] [PubMed]
- Lenhausen, A.M.; Wilkinson, A.S.; Lewis, E.M.; Dailey, K.M.; Scott, A.J.; Khan, S.; Wilkinson, J.C. Apoptosis Inducing Factor Binding Protein PGAM5 Triggers Mitophagic Cell Death That Is Inhibited by the Ubiquitin Ligase Activity of X-Linked Inhibitor of Apoptosis. Biochemistry 2016, 55, 3285–3302. [Google Scholar] [CrossRef] [PubMed]
- Horbay, R.; Bilyy, R. Mitochondrial dynamics during cell cycling. Apoptosis 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Cuffe, L.; Szegezdi, E.; Logue, S.E.; Neary, C.; Healy, S.; Samali, A. Mechanisms of ER Stress-Mediated Mitochondrial Membrane Permeabilization. Int. J. Cell Biol. 2010, 2010, 170215. [Google Scholar] [CrossRef] [PubMed]
- Manoj, K.M.; Parashar, A.; Gade, S.K.; Venkatachalam, A. Functioning of Microsomal Cytochrome P450s: Murburn Concept Explains the Metabolism of Xenobiotics in Hepatocytes. Front. Pharmacol. 2016, 7, 161. [Google Scholar] [CrossRef] [PubMed]
- Lantow, M.; Lupke, M.; Frahm, J.; Mattsson, M.O.; Kuster, N.; Simko, M. ROS release and Hsp70 expression after exposure to 1,800 MHz radiofrequency electromagnetic fields in primary human monocytes and lymphocytes. Radiat. Environ. Biophys. 2006, 45, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Wang, H.; Huang, L.; Zhao, Y.; Wang, J. Crosstalk between autophagy and intracellular radiation response (Review). Int. J. Oncol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Chirumbolo, S. The role of quercetin, flavonols and flavones in modulating inflammatory cell function. Inflamm. Allergy Drug Targets 2010, 9, 263–285. [Google Scholar] [CrossRef] [PubMed]
- Van der Heiden, E.; Bechoux, N.; Muller, M.; Sergent, T.; Schneider, Y.J.; Larondelle, Y.; Maghuin-Rogister, G.; Scippo, M.L. Food flavonoid aryl hydrocarbon receptor-mediated agonistic/antagonistic/synergic activities in human and rat reporter gene assays. Anal. Chim. Acta 2009, 637, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Zhou, Y.; Huang, S.K. SHP-2 phosphatase controls aryl hydrocarbon receptor-mediated ER stress response in mast cells. Arch. Toxicol. 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.; Xie, J.; Zhang, D.; Han, Y.; Wang, C. Polypeptide from Chlamys farreri suppresses ultraviolet-B irradiation-induced apoptosis through restoring ER redox homeostasis, scavenging ROS generation, and suppressing the PERK-eIF2a-CHOP pathway in HaCaT cells. J. Photochem. Photobiol. B 2015, 151, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Bujarrabal, A.; Schumacher, B. Hormesis running hot and cold. Cell Cycle 2016, 29, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Sthijns, M.M.; Weseler, A.R.; Bast, A.; Haenen, G.R. Time in Redox Adaptation Processes: From Evolution to Hormesis. Int. J. Mol. Sci. 2016, 17, 1649. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J. Preconditioning is hormesis part I: Documentation, dose-response features and mechanistic foundations. Pharmacol. Res. 2016, 110, 242–264. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J. Preconditioning is hormesis part II: How the conditioning dose mediates protection: Dose optimization within temporal and mechanistic frameworks. Pharmacol. Res. 2016, 110, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Kim, Y.; Park, J.; Shim, S.; Lee, J.; Hong, S.H.; Ahn, H.H.; Lee, H.; Jung, Y.K. iRhom1 regulates proteasome activity via PAC1/2 under ER stress. Sci. Rep. 2015, 5, 11559. [Google Scholar] [CrossRef] [PubMed]
- Spinazzi, M.; de Strooper, B. PARL: The mitochondrial rhomboid protease. Semin. Cell Dev. Biol. 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.Y.; McQuibban, G.A. The mitochondrial rhomboid protease: Its rise from obscurity to the pinnacle of disease-relevant genes. Biochim. Biophys. Acta 2013, 1828, 2916–2925. [Google Scholar] [CrossRef] [PubMed]
- Durcan, T.M.; Fon, E.A. The three “P”s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015, 29, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Concannon, C.G.; Koehler, B.F.; Reimertz, C.; Murphy, B.M.; Bonner, C.; Thurow, N.; Ward, M.W.; Villunger, A.; Strasser, A.; Kögel, D.; et al. Apoptosis induced by proteasome inhibition in cancer cells: Predominant role of the p53/PUMA pathway. Oncogene 2007, 26, 1681–1692. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M. Unraveling the truth about antioxidants: Mitohormesis explains ROS-induced health benefits. Nat. Med. 2014, 20, 709–711. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R.; Vanderauwera, S.; Suzuki, N.; Miller, G.; Tognetti, V.B.; Vandepoele, K.; Gollery, M.; Shulaev, V.; van Breusegem, F. ROS signaling: The new wave? Trends Plant Sci. 2011, 16, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R. ROS Are Good. Trends Plant Sci. 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Aon, M.A.; Cortassa, S.; Marbán, E.; O’Rourke, B. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J. Biol. Chem. 2003, 278, 44735–44744. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.C.; Chan, D.C. Parkin uses the UPS to ship off dysfunctional mitochondria. Autophagy 2011, 7, 771–772. [Google Scholar] [CrossRef] [PubMed]
- Kemeny, S.; Dery, D.; Loboda, Y.; Rovner, M.; Lev, T.; Zuri, D.; Finberg, J.P.; Larisch, S. Parkin promotes degradation of the mitochondrial pro-apoptotic ARTS protein. PLoS ONE 2012, 7, e38837. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Zhang, Q.; Yan, Z.; Chen, R.; Zeh, H.J., III; Kang, R.; Lotze, M.T.; Tang, D. Strange attractors: DAMPs and autophagy link tumor cell death and immunity. Cell Death Dis. 2013, 4, e966. [Google Scholar] [CrossRef] [PubMed]
- Marhl, M.; Haberichter, T.; Brumen, M.; Heinrich, R. Complex calcium oscillations and the role of mitochondria and cytosolic proteins. Biosystems 2000, 57, 75–86. [Google Scholar] [CrossRef]
- Tsigelny, I.F.; Nigam, S.K. Complex dynamics of chaperone-protein interactions under cellular stress. Cell Biochem. Biophys. 2004, 40, 263–276. [Google Scholar] [CrossRef]
- Yordi, E.G.; Pérez, E.M.; Matos, M.J.; Villares, E.U. Structural alerts for predicting clastogenic activity of pro-oxidant flavonoid compounds: Quantitative structure-activity relationship study. J. Biomol. Screen. 2012, 17, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Eghbaliferiz, S.; Iranshahi, M. Prooxidant Activity of Polyphenols, Flavonoids, Anthocyanins and Carotenoids: Updated Review of Mechanisms and Catalyzing Metals. Phytother. Res. 2016, 30, 1379–1391. [Google Scholar] [CrossRef] [PubMed]
- Manoj, K.M.; Parashar, A.; Venkatachalam, A.; Goyal, S.; Singh, P.G.; Gade, S.K.; Periyasami, K.; Jacob, R.S.; Sardar, D.; Singh, S.; et al. Atypical profiles and modulations of heme-enzymes catalyzed outcomes by low amounts of diverse additives suggest diffusible radicals’ obligatory involvement in such redox reactions. Biochimie 2016, 125, 91–111. [Google Scholar] [CrossRef] [PubMed]
- Olsen, L.F.; Degn, H. Chaos in an enzyme reaction. Nature 1977, 267, 177–178. [Google Scholar] [CrossRef] [PubMed]
- Epstein, I.R. The consequences of imperfect mixing in autocatalytic chemical and biological systems. Nature 1995, 374, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Klemm, K.; Bornholdt, S. Topology of biological networks and reliability of information processing. Proc. Natl. Acad. Sci. USA 2005, 102, 18414–18419. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Kumar, R.; Malhotra, N.; Singh, N.; Dada, R. Mild oxidative stress is beneficial for sperm telomere length maintenance. World J. Methodol. 2016, 6, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Kamata, H.; Hirata, H. Redox regulation of cellular signalling. Cell. Signal. 1999, 11, 1–14. [Google Scholar] [CrossRef]
- Kazemi, E.; Mortazavi, S.M.; Ali-Ghanbari, A.; Sharifzadeh, S.; Ranjbaran, R.; Mostafavi-Pour, Z.; Zal, F.; Haghani, M. Effect of 900 MHz Electromagnetic Radiation on the Induction of ROS in Human Peripheral Blood Mononuclear Cells. J. Biomed. Phys. Eng. 2015, 5, 105–114. [Google Scholar] [PubMed]
- Chen, Z.; Liu, X.; Ma, S. The Roles of Mitochondria in Autophagic Cell Death. Cancer Biother. Radiopharm. 2016, 31, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Lee, C.C.; Lin, H.H.; Chen, M.C.; Lin, C.C.; Chang, J.Y. Autophagy-Regulated ROS from Xanthine Oxidase Acts as an Early Effector for Triggering Late Mitochondria-Dependent Apoptosis in Cathepsin S-Targeted Tumor Cells. PLoS ONE 2015, 10, e0128045. [Google Scholar] [CrossRef] [PubMed]
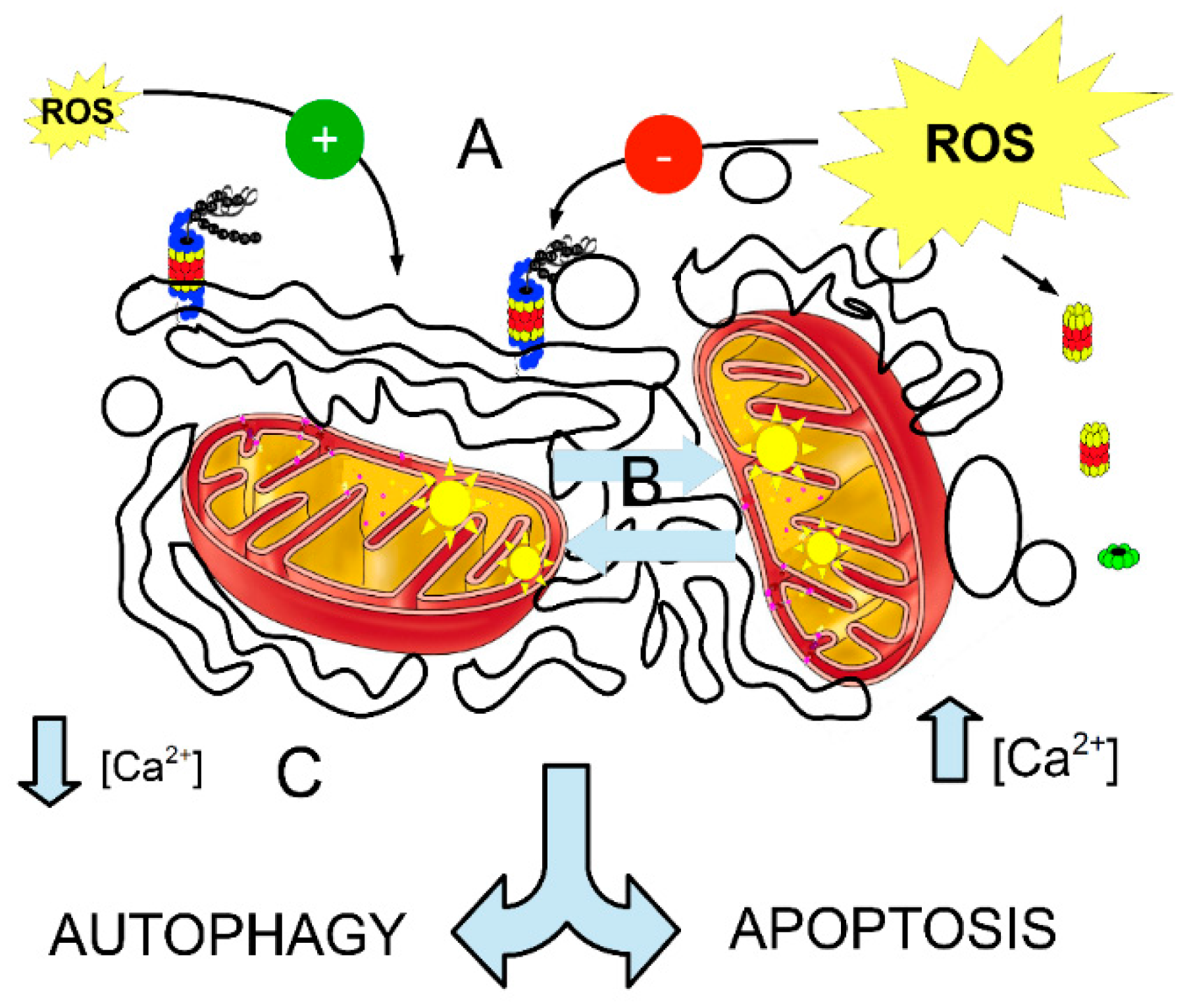


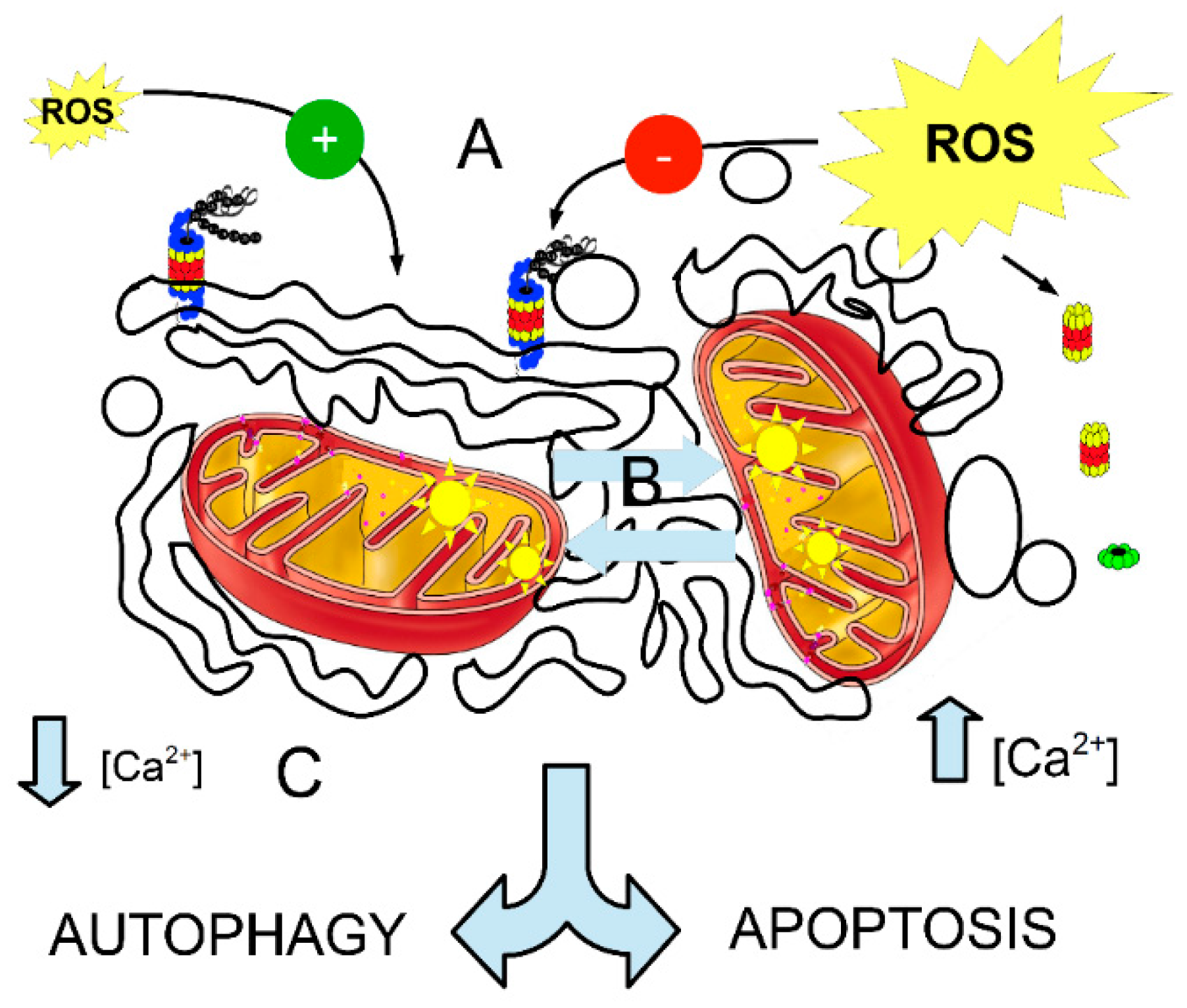{kind=link}
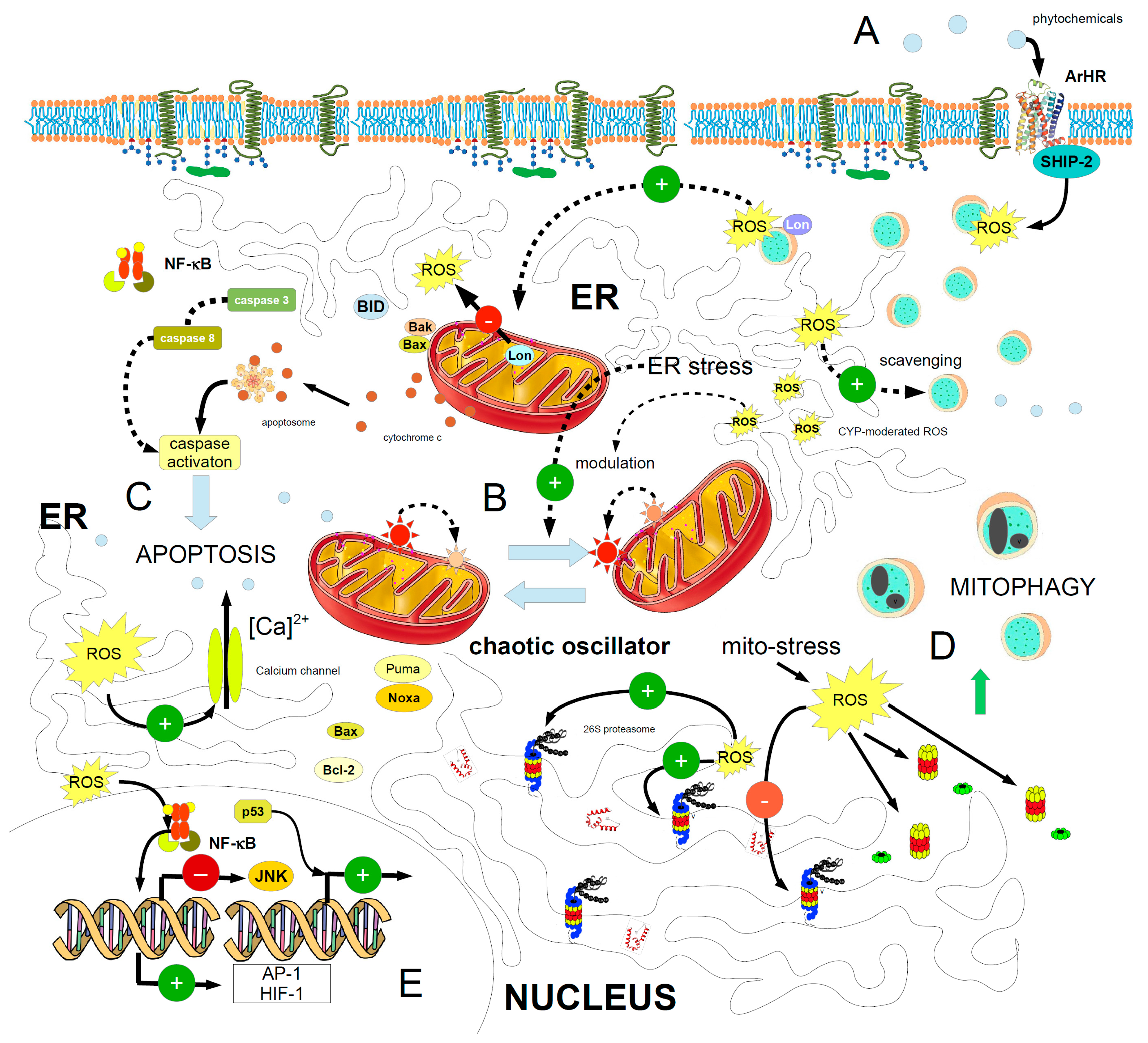{kind=link}
| System | Description | Working Structure | References |
|---|---|---|---|
| CYPs-ROS | Murburn hypothesis | Small amounts of ROS are able to switch on the chaotic network of cytochrome P450 groups | [61,62] |
| ROS-mitochondria | Chaotic synchronization of oscillation networks | The macroscopic property of the mitochondrial network is reproduced in a reaction-diffusion model of ROS-induced ROS-release | [97] |
| ROS-calcium | Chaotic interplay | Sub toxic levels of ROS interplay with calcium signaling network | [92] |
| Calcium oscillations | On the basis of the permeability of the ER channels and on the kinetic properties of calcium binding to the cytosolic proteins, different patterns of complex calcium oscillations occur | [47] | |
| Proterome | Chaotic synchronization | Synchronization of mobile chaotic oscillators in the bi-dimensional landscape | [98] |
| ROS signalling | Participation in the synchronization process | [99,100,101] | |
| Mitochondria | Dynamics in the network | [102,103,104] | |
| Proteasome and chaperones | Chaotic-type oscillatory system depending of ATP levels | [105] |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chirumbolo, S.; Bjørklund, G. PERM Hypothesis: The Fundamental Machinery Able to Elucidate the Role of Xenobiotics and Hormesis in Cell Survival and Homeostasis. Int. J. Mol. Sci. 2017, 18, 165. https://doi.org/10.3390/ijms18010165
Chirumbolo S, Bjørklund G. PERM Hypothesis: The Fundamental Machinery Able to Elucidate the Role of Xenobiotics and Hormesis in Cell Survival and Homeostasis. International Journal of Molecular Sciences. 2017; 18(1):165. https://doi.org/10.3390/ijms18010165
Chicago/Turabian StyleChirumbolo, Salvatore, and Geir Bjørklund. 2017. "PERM Hypothesis: The Fundamental Machinery Able to Elucidate the Role of Xenobiotics and Hormesis in Cell Survival and Homeostasis" International Journal of Molecular Sciences 18, no. 1: 165. https://doi.org/10.3390/ijms18010165
APA StyleChirumbolo, S., & Bjørklund, G. (2017). PERM Hypothesis: The Fundamental Machinery Able to Elucidate the Role of Xenobiotics and Hormesis in Cell Survival and Homeostasis. International Journal of Molecular Sciences, 18(1), 165. https://doi.org/10.3390/ijms18010165