
Melanoma Brain Metastasis: Mechanisms, Models, and Medicine


Abstract
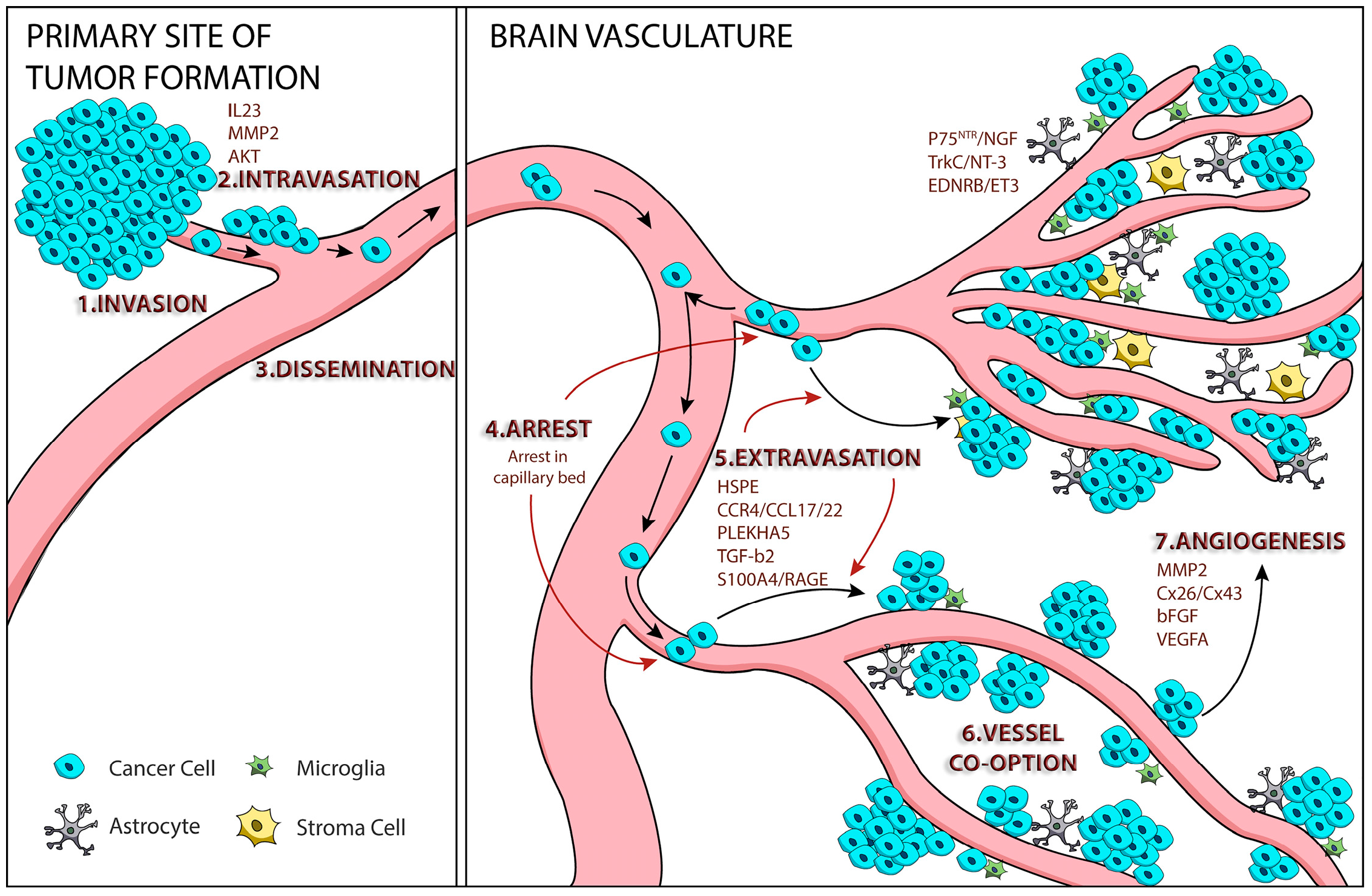
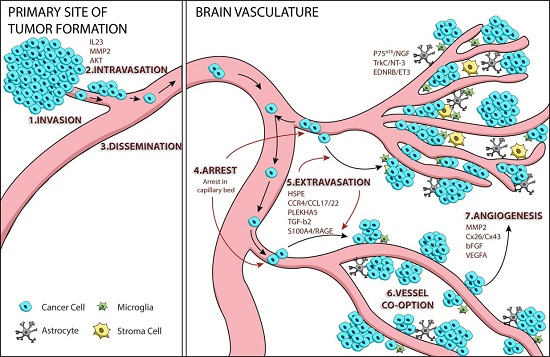
: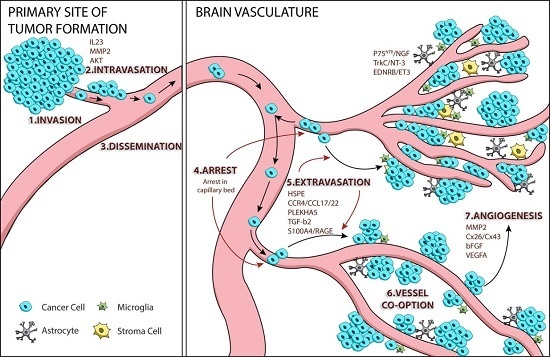
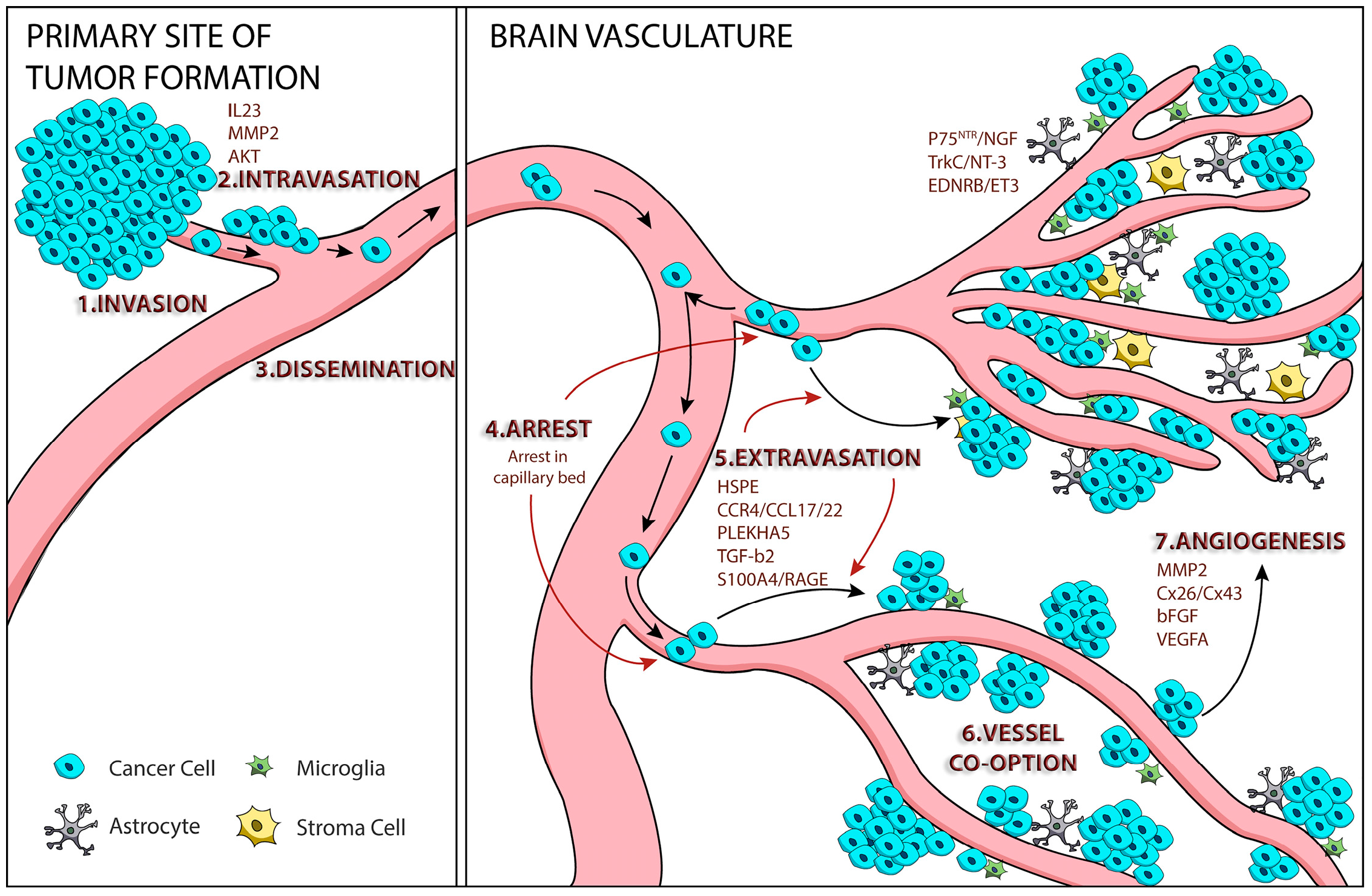
1. Tumor Cell Metastasis
2. Mechanisms of Melanoma Brain Metastasis
2.1. Melanoma Cell Arrest within the Brain Microvasculature
2.2. Extravasation
2.3. Perivascular Positioning and Growth by Vessel Co-Option
3. AKT Signaling and Melanoma Brain Metastasis
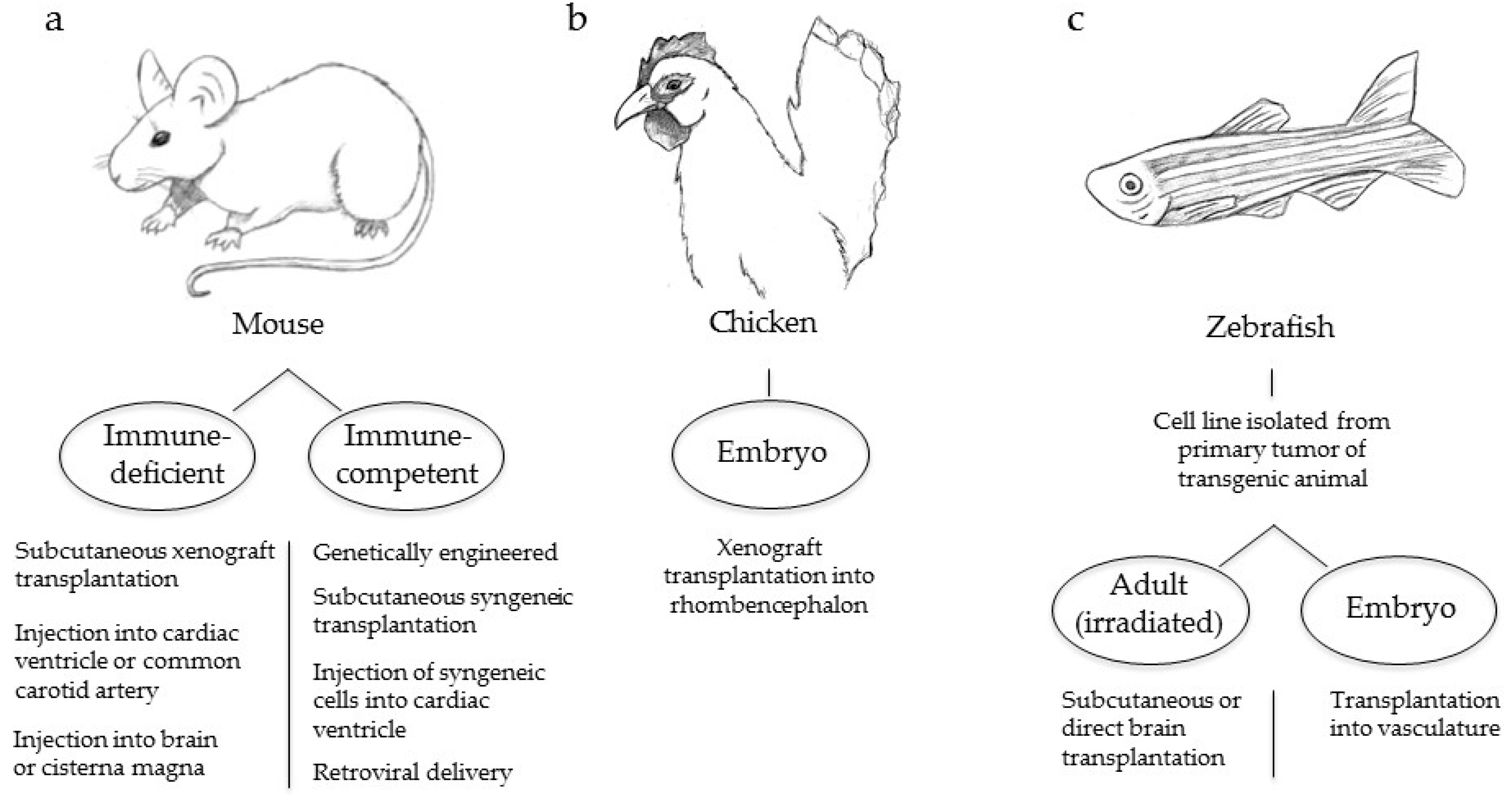
4. Animal Models of Melanoma Brain Metastasis
5. Melanoma Brain Metastasis Therapies
5.1. Surgery
5.2. Stereotactic Radiosurgery
5.3. Whole-Brain Radiation Therapy
5.4. Chemotherapy
5.5. Targeted Therapies
5.6. MAPK Pathway Inhibitors
5.7. Immune-Based Therapies
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nguyen, D.X.; Bos, P.D.; Massague, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Maman, S.; Edry-Botzer, L.; Sagi-Assif, O.; Meshel, T.; Yuan, W.; Lu, W.; Witz, I.P. The metastatic microenvironment: Lung-derived factors control the viability of neuroblastoma lung metastasis. Int. J. Cancer 2013, 133, 2296–2306. [Google Scholar] [CrossRef] [PubMed]
- Klein-Goldberg, A.; Maman, S.; Witz, I.P. The role played by the microenvironment in site-specific metastasis. Cancer Lett. 2014, 352, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yu, D. Microenvironment determinants of brain metastasis. Cell Biosci. 2011, 1, 8. [Google Scholar] [CrossRef]
- Colombino, M.; Capone, M.; Lissia, A.; Cossu, A.; Rubino, C.; de Giorgi, V.; Massi, D.; Fonsatti, E.; Staibano, S.; Nappi, O.; et al. BRAF/NRAS mutation frequencies among primary tumors and metastases in patients with melanoma. J. Clin. Oncol. 2012, 30, 2522–2529. [Google Scholar] [CrossRef] [PubMed]
- Riker, A.I.; Enkemann, S.A.; Fodstad, O.; Liu, S.; Ren, S.; Morris, C.; Xi, Y.; Howell, P.; Metge, B.; Samant, R.S.; et al. The gene expression profiles of primary and metastatic melanoma yields a transition point of tumor progression and metastasis. BMC Med. Genom. 2008, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Brastianos, P.K.; Carter, S.L.; Santagata, S.; Cahill, D.P.; Taylor-Weiner, A.; Jones, R.T.; van Allen, E.M.; Lawrence, M.S.; Horowitz, P.M.; Cibulskis, K.; et al. Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discov. 2015, 5, 1164–1177. [Google Scholar] [CrossRef]
- Gavrilovic, I.T.; Posner, J.B. Brain metastases: Epidemiology and pathophysiology. J. Neuro-Oncol. 2005, 75, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Kienast, Y.; von Baumgarten, L.; Fuhrmann, M.; Klinkert, W.E.F.; Goldbrunner, R.; Herms, J.; Winkler, F. Real-time imaging reveals the single steps of brain metastasis formation. Nat. Med. 2010, 16, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Repesh, L.A.; Fitzgerald, T.J. Interactions of tumor cells with intact capillaries: A model for intravasation. Clin. Exp. Metastasis 1984, 2, 139–150. [Google Scholar] [CrossRef]
- Winkelhake, J.L.; Nicolson, G.L. Determination of adhesive properties of variant metastatic melanoma cells to BALB/3T3 cells and their virus-transformed derivatives by a monolayer attachment assay. J. Natl. Cancer Inst. 1976, 56, 285–291. [Google Scholar] [PubMed]
- Molnar, J.; Fazakas, C.; Hasko, J.; Sipos, O.; Nagy, K.; Nyul-Toth, A.; Farkas, A.E.; Vegh, A.G.; Varo, G.; Galajda, P.; et al. Transmigration characteristics of breast cancer and melanoma cells through the brain endothelium: Role of Rac and PI3K. Cell Adhes. Migr. 2016, 10, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, I.; Molnar, J.; Fazakas, C.; Hasko, J.; Krizbai, I.A. Role of the blood-brain barrier in the formation of brain metastases. Int. J. Mol. Sci. 2013, 14, 1383–1411. [Google Scholar] [CrossRef] [PubMed]
- Monteagudo, C.; Pellin-Carcelen, A.; Martin, J.M.; Ramos, D. Role of chemokines in melanoma progression. Actas Dermo-Sifiliogr. 2011, 102, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1889, 133, 571–573. [Google Scholar] [CrossRef]
- Murakami, T.; Cardones, A.R.; Hwang, S.T. Chemokine receptors and melanoma metastasis. J. Dermatol. Sci. 2004, 36, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Borrull, A.; Allard, B.; Wijkhuisen, A.; Herbet, A.; Lamourette, P.; Birouk, W.; Leiber, D.; Tanfin, Z.; Ducancel, F.; Boquet, D.; et al. Rendomab B4, a monoclonal antibody that discriminates the human endothelin B receptor of melanoma cells and inhibits their migration. mAbs 2016. [Google Scholar] [CrossRef] [PubMed]
- Izraely, S.; Klein, A.; Sagi-Assif, O.; Meshel, T.; Tsarfaty, G.; Hoon, D.S.B.; Witz, I.P. Chemokine-chemokine receptor axes in melanoma brain metastasis. Immunol. Lett. 2010, 130, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Ou, B.; Zhao, J.; Guan, S.; Feng, H.; Wangpu, X.; Zhu, C.; Zong, Y.; Ma, J.; Sun, J.; Shen, X.; et al. CCR4 promotes metastasis via ERK/NF-kappaB/MMP13 pathway and acts downstream of TNF-α in colorectal cancer. Oncotarget 2016, 7, 47637–47649. [Google Scholar]
- Yang, Y.; Du, L.; Yang, X.; Qu, A.; Zhang, X.; Zhou, C.; Wang, C. Aberrant CCR4 expression is involved in tumor invasion of lymph node-negative human gastric cancer. PLoS ONE 2015, 10, e0120059. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Ou, Z.L.; Yu, S.J.; Gu, X.L.; Yang, C.; Chen, A.X.; Di, G.H.; Shen, Z.Z.; Shao, Z.M. The chemokine receptor CCR4 promotes tumor growth and lung metastasis in breast cancer. Breast Cancer Res. Treat. 2012, 131, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.; Sagi-Assif, O.; Izraely, S.; Meshel, T.; Pasmanik-Chor, M.; Nahmias, C.; Couraud, P.-O.; Erez, N.; Hoon, D.S.B.; Witz, I.P. The metastatic microenvironment: Brain-derived soluble factors alter the malignant phenotype of cutaneous and brain-metastasizing melanoma cells. Int. J. Cancer 2012, 131, 2509–2518. [Google Scholar] [CrossRef] [PubMed]
- Lok, E.; Chung, A.S.; Swanson, K.D.; Wong, E.T. Melanoma brain metastasis globally reconfigures chemokine and cytokine profiles in patient cerebrospinal fluid. Melanoma Res. 2014, 24, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Saldana-Caboverde, A.; Kos, L. Roles of endothelin signaling in melanocyte development and melanoma. Pigment Cell Melanoma Res. 2010, 23, 160–170. [Google Scholar] [CrossRef]
- Cruz-Munoz, W.; Jaramillo, M.L.; Man, S.; Xu, P.; Banville, M.; Collins, C.; Nantel, A.; Francia, G.; Morgan, S.S.; Cranmer, L.D.; et al. Roles for endothelin receptor B and BCL2A1 in spontaneous CNS metastasis of melanoma. Cancer Res. 2012, 72, 4909–4919. [Google Scholar] [CrossRef] [PubMed]
- Firth, J.D.; Ratcliffe, P.J. Organ distribution of the three rat endothelin messenger RNAs and the effects of ischemia on renal gene expression. J. Clin. Investig. 1992, 90, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Wouters, J.; Hunger, R.E.; Garrod, T.; Dubuis, B.; Hunziker, T.; van den Oord, J.J.; Lahav-le Coutre, R. First-in-human proof-of-concept study: Intralesional administration of BQ788, an endothelin receptor B antagonist, to melanoma skin metastases. Oncologist 2015, 20, 1121–1122. [Google Scholar] [CrossRef] [PubMed]
- Fabricant, R.N.; de Larco, J.E.; Todaro, G.J. Nerve growth factor receptors on human melanoma cells in culture. Proc. Natl. Acad. Sci. USA 1977, 74, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Herlyn, M.; Thurin, J.; Balaban, G.; Bennicelli, J.L.; Herlyn, D.; Elder, D.E.; Bondi, E.; Guerry, D.; Nowell, P.; Clark, W.H.; et al. Characteristics of Cultured Human Melanocytes Isolated from Different Stages of Tumor Progression. Cancer Res. 1985, 45, 5670–5676. [Google Scholar]
- Herrmann, J.L.; Menter, D.G.; Hamada, J.; Marchetti, D.; Nakajima, M.; Nicolson, G.L. Mediation of NGF-stimulated extracellular matrix invasion by the human melanoma low-affinity p75 neurotrophin receptor: Melanoma p75 functions independently of trkA. Mol. Biol. Cell 1993, 4, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, D.; Murry, B.; Galjour, J.; Wilke-Greiter, A. Human melanoma TrkC: Its association with a purine-analog-sensitive kinase activity. J. Cell. Biochem. 2003, 88, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, D.; McCutcheon, I.; Ross, M.; Nicolson, G. Inverse expression of neurotrophins and neurotrophin receptros at the invasion front of human-melanoma brain metastases. Int. J. Oncol. 1995, 7, 87–94. [Google Scholar] [PubMed]
- Yoshida, K.; Gage, F.H. Cooperative regulation of nerve growth factor synthesis and secretion in fibroblasts and astrocytes by fibroblast growth factor and other cytokines. Brain Res. 1992, 569, 14–25. [Google Scholar] [CrossRef]
- Komai, T.; Okamura, T.; Yamamoto, K.; Fujio, K. The effects of TGF-β on immune responses. Jpn J. Clin. Immunol. 2016, 39, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, F.; Tsan, R.; Fidler, I.J. Transforming growth factor-β2 is a molecular determinant for site-specific melanoma metastasis in the brain. Cancer Res. 2009, 69, 828–835. [Google Scholar] [CrossRef] [PubMed]
- Li, J.R.; Wang, J.Q.; Gong, Q.; Fang, R.H.; Guo, Y.L. MicroRNA-328 inhibits proliferation of human melanoma cells by targeting TGFβ2. Asian Pac. J. Cancer Prev. 2015, 16, 1575–1579. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood–brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Brightman, M.W.; Reese, T.S. Junctions between intimately apposed cell membranes in the vertebrate brain. J. Cell Biol. 1969, 40, 648–677. [Google Scholar] [CrossRef] [PubMed]
- Vlodavsky, I.; Goldshmidt, O.; Zcharia, E.; Atzmon, R.; Rangini-Guatta, Z.; Elkin, M.; Peretz, T.; Friedmann, Y. Mammalian heparanase: Involvement in cancer metastasis, angiogenesis and normal development. Semin. Cancer Biol. 2002, 12, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, D. Heparanase: A target for therapy of brain invasive tumors? Exp. Rev. Neurother. 2002, 2, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, D.; Li, J.; Shen, R. Astrocytes contribute to the metastatic brain specificity of melanoma cells by producing heparanase. Cancer Res. 2000, 60, 4767–4770. [Google Scholar]
- Murry, B.P.; Blust, B.E.; Singh, A.; Foster, T.P.; Marchetti, D. Heparanase brain: Mechanisms of melanoma metastasis to the development and use of a brain slice model. J. Cell. Biochem. 2006, 97, 217–225. [Google Scholar] [CrossRef]
- Marchetti, D.; Menter, D.; Jin, L.; Nakajima, M.; Nicolson, G.L. Nerve growth factor effects on human and mouse melanoma cell invasion and heparanase production. Int. J. Cancer 1993, 55, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Fang, H.; Chen, H.; Jiang, X.; Fang, D.; Wang, Y.; Zhu, D. An artificial miRNA against HPSE suppresses melanoma invasion properties, correlating with a down-regulation of chemokines and MAPK phosphorylation. PLoS ONE 2012, 7, e38659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sullivan, P.S.; Goodman, J.C.; Gunaratne, P.H.; Marchetti, D. MicroRNA-1258 suppresses breast cancer brain metastasis by targeting heparanase. Cancer Res. 2011, 71, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Fazakas, C.; Wilhelm, I.; Nagyoszi, P.; Farkas, A.E.; Hasko, J.; Molnar, J.; Bauer, H.; Bauer, H.C.; Ayaydin, F.; Dung, N.T.; et al. Transmigration of melanoma cells through the blood-brain barrier: Role of endothelial tight junctions and melanoma-released serine proteases. PLoS ONE 2011, 6, e20758. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.; Andl, T.; Li, G.; Meinkoth, J.L.; Herlyn, M. Cadherin repertoire determines partner-specific gap junctional communication during melanoma progression. J. Cell Sci. 2000, 113, 1535–1542. [Google Scholar] [PubMed]
- Ito, A.; Katoh, F.; Kataoka, T.R.; Okada, M.; Tsubota, N.; Asada, H.; Yoshikawa, K.; Maeda, S.; Kitamura, Y.; Yamasaki, H.; et al. A role for heterologous gap junctions between melanoma and endothelial cells in metastasis. J. Clin. Investig. 2000, 105, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Stoletov, K.; Strnadel, J.; Zardouzian, E.; Momiyama, M.; Park, F.D.; Kelber, J.A.; Pizzo, D.P.; Hoffman, R.; VandenBerg, S.R.; Klemke, R.L. Role of connexins in metastatic breast cancer and melanoma brain colonization. J. Cell Sci. 2013, 126, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Sargen, M.R.; Gormley, R.H.; Pasha, T.L.; Yum, S.; Acs, G.; Xu, X.; Zhang, P.J. Melanocytic tumors express connexin 43 but not 26: Immunohistochemical analysis with potential significance in melanocytic oncogenesis. Am. J. Dermatopathol. 2013, 35, 813–817. [Google Scholar] [CrossRef]
- Ableser, M.J.; Penuela, S.; Lee, J.; Shao, Q.; Laird, D.W. Connexin43 reduces melanoma growth within a keratinocyte microenvironment and during tumorigenesis in vivo. J. Biol. Chem. 2014, 289, 1592–1603. [Google Scholar] [CrossRef] [PubMed]
- Tittarelli, A.; Guerrero, I.; Tempio, F.; Gleisner, M.A.; Avalos, I.; Sabanegh, S.; Ortiz, C.; Michea, L.; Lopez, M.N.; Mendoza-Naranjo, A.; et al. Overexpression of connexin 43 reduces melanoma proliferative and metastatic capacity. Br. J. Cancer 2015, 113, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Herwig, N.; Belter, B.; Pietzsch, J. Extracellular S100A4 affects endothelial cell integrity and stimulates transmigration of A375 melanoma cells. Biochem. Biophys. Res. Commun. 2016, 477, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Herwig, N.; Belter, B.; Wolf, S.; Haase-Kohn, C.; Pietzsch, J. Interaction of extracellular S100A4 with RAGE prompts prometastatic activation of A375 melanoma cells. J. Cell. Mol. Med. 2016, 20, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Medapati, M.R.; Dahlmann, M.; Ghavami, S.; Pathak, K.A.; Lucman, L.; Klonisch, T.; Hoang-Vu, C.; Stein, U.; Hombach-Klonisch, S. RAGE mediates the pro-migratory response of extracellular S100A4 in human thyroid cancer cells. Thyroid 2015, 25, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Dahlmann, M.; Okhrimenko, A.; Marcinkowski, P.; Osterland, M.; Herrmann, P.; Smith, J.; Heizmann, C.W.; Schlag, P.M.; Stein, U. RAGE mediates S100A4-induced cell motility via MAPK/ERK and hypoxia signaling and is a prognostic biomarker for human colorectal cancer metastasis. Oncotarget 2014, 5, 3220–3233. [Google Scholar] [CrossRef] [PubMed]
- Siddique, H.R.; Adhami, V.M.; Parray, A.; Johnson, J.J.; Siddiqui, I.A.; Shekhani, M.T.; Murtaza, I.; Ambartsumian, N.; Konety, B.R.; Mukhtar, H.; et al. The S100A4 oncoprotein promotes prostate tumorigenesis in a transgenic mouse model: Regulating NFκB through the rage receptor. Genes Cancer 2013, 4, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, J.L.; Padilla, L.; Dakhel, S.; Coll, T.; Hervas, R.; Adan, J.; Masa, M.; Mitjans, F.; Martinez, J.M.; Coma, S.; et al. Therapeutic targeting of tumor growth and angiogenesis with a novel anti-S100A4 monoclonal antibody. PLoS ONE 2013, 8, e72480. [Google Scholar] [CrossRef] [PubMed]
- Jilaveanu, L.B.; Parisi, F.; Barr, M.L.; Zito, C.R.; Cruz-Munoz, W.; Kerbel, R.S.; Rimm, D.L.; Bosenberg, M.W.; Halaban, R.; Kluger, Y.; et al. PLEKHA5 as a Biomarker and Potential Mediator of Melanoma Brain Metastasis. Clin. Cancer Res. 2015, 21, 2138–2147. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.; Schwartz, H.; Sagi-Assif, O.; Meshel, T.; Izraely, S.; Ben Menachem, S.; Bengaiev, R.; Ben-Shmuel, A.; Nahmias, C.; Couraud, P.O.; et al. Astrocytes facilitate melanoma brain metastasis via secretion of IL-23. J. Pathol. 2015, 236, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Li, J.; Zhu, H.; Li, P.; Zou, Z.; Xiao, Y. Hmgb1-IL-23-IL-17-IL-6-Stat3 axis promotes tumor growth in murine models of melanoma. Med. Inflamm. 2013, 2013, 713859. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.X.; Huang, F.J.; Aldape, K.D.; Kang, S.H.; Liu, M.; Gershenwald, J.E.; Xie, K.; Sawaya, R.; Huang, S. Activation of stat3 in human melanoma promotes brain metastasis. Cancer Res. 2006, 66, 3188–3196. [Google Scholar] [CrossRef] [PubMed]
- Redondo, P.; Lloret, P.; Idoate, M.; Inoges, S. Expression and serum levels of MMP-2 and MMP-9 during human melanoma progression. Clin. Exp. Dermatol. 2005, 30, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Rotte, A.; Martinka, M.; Li, G. MMP2 expression is a prognostic marker for primary melanoma patients. Cell. Oncol. 2012, 35, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.; Vasco, C.; Girgenti, V.; Fugnanesi, V.; Calatozzolo, C.; Canazza, A.; Salmaggi, A.; Rivoltini, L.; Morbin, M.; Ciusani, E. Melanoma cells homing to the brain: An in vitro model. BioMed Res. Int. 2015, 2015, 476069. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, H.; Kubota, H.; Lindberg, R.L.; Leppert, D.; Gloor, S.M.; Errede, M.; Virgintino, D.; Fontana, A.; Yonekawa, Y.; Frei, K. Endothelial cell barrier impairment induced by glioblastomas and transforming growth factor β2 involves matrix metalloproteinases and tight junction proteins. J. Neuropathol. Exp. Neurol. 2008, 67, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.; Antfolk, M.; Brodin, B.; Tenje, M. In vitro blood-brain barrier models—An overview of established models and new microfluidic approaches. J. Pharm. Sci. 2015, 104, 2727–2746. [Google Scholar] [CrossRef] [PubMed]
- Berghoff, A.S.; Rajky, O.; Winkler, F.; Bartsch, R.; Furtner, J.; Hainfellner, J.A.; Goodman, S.L.; Weller, M.; Schittenhelm, J.; Preusser, M. Invasion patterns in brain metastases of solid cancers. Neuro-Oncology 2013, 15, 1664–1672. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.J.; Steeg, P.S.; Price, J.E.; Chiu, W.T.; Chou, P.C.; Xie, K.; Sawaya, R.; Huang, S. Molecular basis for the critical role of suppressor of cytokine signaling-1 in melanoma brain metastasis. Cancer Res. 2008, 68, 9634–9642. [Google Scholar] [CrossRef] [PubMed]
- Ilhan-Mutlu, A.; Siehs, C.; Berghoff, A.S.; Ricken, G.; Widhalm, G.; Wagner, L.; Preusser, M. Expression profiling of angiogenesis-related genes in brain metastases of lung cancer and melanoma. Tumour Biol. 2016, 37, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Ju, R.; Zhuang, Z.W.; Zhang, J.; Lanahan, A.A.; Kyriakides, T.; Sessa, W.C.; Simons, M. Angiopoietin-2 Secretion by Endothelial Cell Exosomes: Regulation by the Phosphatidylinositol 3-kinase (PI3K)/Akt/Endothelial nitric oxide synthase (eNOS) and Syndecan-4/Synthenin pathways. J. Biol. Chem. 2014, 289, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Karar, J.; Maity, A. PI3K/AKT/mTOR pathway in Angiogenesis. Front. Mol. Neurosci. 2011. [Google Scholar] [CrossRef] [PubMed]
- Phung, T.L.; Du, W.; Xue, Q.; Ayyaswamy, S.; Gerald, D.; Antonello, Z.; Nhek, S.; Perruzzi, C.A.; Acevedo, I.; Ramanna-Valmiki, R.; et al. Akt1 and Akt3 exert opposing roles in the regulation of vascular tumor growth. Cancer Res. 2015, 75, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Phung, T.L.; Ziv, K.; Dabydeen, D.; Eyiah-Mensah, G.; Riveros, M.; Perruzzi, C.; Sun, J.; Monahan-Earley, R.A.; Shiojima, I.; Nagy, J.A.; et al. Pathological angiogenesis is induced by sustained Akt signaling and inhibited by rapamycin. Cancer Cell 2006, 10, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.L.; Martinka, M.; Li, G. Prognostic significance of activated Akt expression in melanoma: A clinicopathologic study of 292 cases. J. Clin. Oncol. 2005, 23, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.A.; Stemke-Hale, K.; Lin, E.; Tellez, C.; Deng, W.; Gopal, Y.N.; Woodman, S.E.; Calderone, T.C.; Ju, Z.; Lazar, A.J.; et al. Integrated molecular and clinical analysis of Akt activation in metastatic melanoma. Clin. Cancer Res. 2009, 15, 7538–7546. [Google Scholar] [CrossRef] [PubMed]
- Smalley, K.S.M. Understanding melanoma signaling networks as the basis for molecular targeted therapy. J. Investig. Dermatol. 2009, 130, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Altomare, D.A.; Testa, J.R. Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar] [CrossRef] [PubMed]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.A.; Stemke-Hale, K.; Tellez, C.; Calderone, T.L.; Deng, W.; Prieto, V.G.; Lazar, A.J.F.; Gershenwald, J.E.; Mills, G.B. A novel AKT3 mutation in melanoma tumours and cell lines. Br. J. Cancer 2008, 99, 1265–1268. [Google Scholar] [CrossRef] [PubMed]
- Manca, A.; Lissia, A.; Capone, M.; Ascierto, P.A.; Botti, G.; Caraco, C.; Stanganelli, I.; Colombino, M.; Sini, M.; Cossu, A.; et al. Activating PIK3CA mutations coexist with BRAF or NRAS mutations in a limited fraction of melanomas. J. Transl. Med. 2015, 13, 37. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [Google Scholar]
- Bunney, T.D.; Katan, M. Phosphoinositide signalling in cancer: Beyond PI3K and PTEN. Nat. Rev. Cancer 2010, 10, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Glitza, I.C.; Davies, M.A. Genotyping of cutaneous melanoma. Chin. Clin. Oncol. 2014, 3, 27. [Google Scholar] [PubMed]
- Davies, M.A. The role of the PI3K-AKT pathway in melanoma. Cancer J. 2012, 18, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, D.C.; Zhou, X.P.; Cummings, M.C.; Pavey, S.; Hayward, N.K.; Eng, C. Nuclear PTEN expression and clinicopathologic features in a population-based series of primary cutaneous melanoma. Int. J. Cancer 2002, 99, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Poetsch, M.; Dittberner, T.; Woenckhaus, C. PTEN/MMAC1 in malignant melanoma and its importance for tumor progression. Cancer Genet. Cytogenet. 2001, 125, 21–26. [Google Scholar] [CrossRef]
- Stahl, J.M.; Sharma, A.; Cheung, M.; Zimmerman, M.; Cheng, J.Q.; Bosenberg, M.W.; Kester, M.; Sandirasegarane, L.; Robertson, G.P. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 2004, 64, 7002–7010. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, B.; Sligh, J.E.; Vincent, B.J.; Li, M.; Canter, J.A.; Nickoloff, B.J.; Rodenburg, R.J.; Smeitink, J.A.; Oberley, L.; Zhang, Y.; et al. Overexpression of Akt converts radial growth melanoma to vertical growth melanoma. J. Clin. Investig. 2007, 117, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Bucheit, A.D.; Chen, G.; Siroy, A.; Tetzlaff, M.; Broaddus, R.; Milton, D.; Fox, P.; Bassett, R.; Hwu, P.; Gershenwald, J.E.; et al. Complete loss of PTEN protein expression correlates with shorter time to brain metastasis and survival in stage IIIB/C melanoma patients with BRAFV600 mutations. Clin. Cancer Res. 2014, 20, 5527–5536. [Google Scholar] [CrossRef] [PubMed]
- Niessner, H.; Forschner, A.; Klumpp, B.; Honegger, J.B.; Witte, M.; Bornemann, A.; Dummer, R.; Adam, A.; Bauer, J.; Tabatabai, G.; et al. Targeting hyperactivation of the AKT survival pathway to overcome therapy resistance of melanoma brain metastases. Cancer Med. 2013, 2, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Chakravarti, N.; Aardalen, K.; Lazar, A.J.; Tetzlaff, M.T.; Wubbenhorst, B.; Kim, S.-B.; Kopetz, S.; Ledoux, A.A.; Gopal, Y.N.V.; et al. Molecular profiling of patient-matched brain and extracranial melanoma metastases implicates the PI3K pathway as a therapeutic target. Clin. Cancer Res. 2014, 20, 5537–5546. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Robinson, J.P.; Arave, R.A.; Burnett, W.J.; Kircher, D.A.; Chen, G.; Davies, M.A.; Grossmann, A.H.; VanBrocklin, M.W.; McMahon, M.; et al. AKT1 Activation promotes development of melanoma metastases. Cell Rep. 2015, 13, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Zetser, A.; Bashenko, Y.; Edovitsky, E.; Levy-Adam, F.; Vlodavsky, I.; Ilan, N. Heparanase induces vascular endothelial growth factor expression: Correlation with p38 phosphorylation levels and Src activation. Cancer Res. 2006, 66, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Park, D.J.; Wallick, C.J.; Martyn, K.D.; Lau, A.F.; Jin, C.; Warn-Cramer, B.J. Akt phosphorylates Connexin43 on Ser373, a “mode-1” binding site for 14–3-3. Cell Commun. Adhes. 2007, 14, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Vogt, P.K.; Hart, J.R. PI3K and STAT3: A new alliance. Cancer Discov. 2011, 1, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Park, E.S.; Kim, S.J.; Kim, S.W.; Yoon, S.L.; Leem, S.H.; Kim, S.B.; Kim, S.M.; Park, Y.Y.; Cheong, J.H.; Woo, H.G.; et al. Cross-species hybridization of microarrays for studying tumor transcriptome of brain metastasis. Proc. Natl. Acad. Sci. USA 2011, 108, 17456–17461. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Q.; Jiang, S.; Li, C.; Zhang, B.; Li, Q.J. miR-17–92 cluster targets phosphatase and tensin homology and Ikaros Family Zinc Finger 4 to promote TH17-mediated inflammation. J. Biol. Chem. 2014, 289, 12446–12456. [Google Scholar] [CrossRef] [PubMed]
- Olive, V.; Bennett, M.J.; Walker, J.C.; Ma, C.; Jiang, I.; Cordon-Cardo, C.; Li, Q.J.; Lowe, S.W.; Hannon, G.J.; He, L. miR-19 is a key oncogenic component of mir-17–92. Genes Dev. 2009, 23, 2839–2849. [Google Scholar] [CrossRef] [PubMed]
- Olive, V.; Jiang, I.; He, L. mir-17–92, a cluster of miRNAs in the midst of the cancer network. Int. J. Biochem. Cell Biol. 2010, 42, 1348–1354. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Han, C.; Lu, D.; Wu, T. miR-17–92 cluster promotes cholangiocarcinoma growth: Evidence for PTEN as downstream target and IL-6/Stat3 as upstream activator. Am. J. Pathol. 2014, 184, 2828–2839. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, S.; Yao, J.; Lowery, F.J.; Zhang, Q.; Huang, W.C.; Li, P.; Li, M.; Wang, X.; Zhang, C.; et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 2015, 527, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Munoz, W.; Man, S.; Xu, P.; Kerbel, R.S. Development of a Preclinical Model of Spontaneous Human Melanoma Central Nervous System Metastasis. Cancer Res. 2008, 68, 4500–4505. [Google Scholar] [CrossRef] [PubMed]
- Niessner, H.; Schmitz, J.; Tabatabai, G.; Schmid, A.; Calaminus, C.; Sinnberg, T.; Weide, B.; Eigentler, T.K.; Garbe, C.; Schittek, B.; et al. PI3K pathway inhibition achieves potent antitumor activity in melanoma brain metastases in vitro and in vivo. Clin. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Sundstrom, T.; Daphu, I.; Wendelbo, I.; Hodneland, E.; Lundervold, A.; Immervoll, H.; Skaftnesmo, K.O.; Babic, M.; Jendelova, P.; Sykova, E.; et al. Automated tracking of nanoparticle-labeled melanoma cells improves the predictive power of a brain metastasis model. Cancer Res. 2013, 73, 2445–2456. [Google Scholar] [CrossRef]
- Izraely, S.; Sagi-Assif, O.; Klein, A.; Meshel, T.; Tsarfaty, G.; Pasmanik-Chor, M.; Nahmias, C.; Couraud, P.O.; Ateh, E.; Bryant, J.L.; et al. The metastatic microenvironment: Brain-residing melanoma metastasis and dormant micrometastasis. Int. J. Cancer 2012, 131, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Shinohara, H.; Herbst, R.S.; Kuniyasu, H.; Bucana, C.D.; Ellis, L.M.; Davis, D.W.; McConkey, D.J.; Fidler, I.J. Expression of vascular endothelial growth factor is necessary but not sufficient for production and growth of brain metastasis. Cancer Res. 2000, 60, 4959–4967. [Google Scholar]
- Fujimaki, T.; Price, J.E.; Fan, D.; Bucana, C.D.; Itoh, K.; Kirino, T.; Fidler, I.J. Selective growth of human melanoma cells in the brain parenchyma of nude mice. Melanoma Res. 1996, 6, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Takahashi, M.; Akhand, A.A.; Liu, W.; Dai, Y.; Shimizu, S.; Iwamoto, T.; Suzuki, H.; Nakashima, I. Transgenic mouse model for skin malignant melanoma. Oncogene 1998, 17, 1885–1888. [Google Scholar] [CrossRef] [PubMed]
- Umansky, V.; Abschuetz, O.; Osen, W.; Ramacher, M.; Zhao, F.; Kato, M.; Schadendorf, D. Melanoma-specific memory T cells are functionally active in Ret transgenic mice without macroscopic tumors. Cancer Res. 2008, 68, 9451–9458. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, H.; Blacher, E.; Amer, M.; Livneh, N.; Abramovitz, L.; Klein, A.; Ben-Shushan, D.; Soffer, S.; Blazquez, R.; Barrantes-Freer, A.; et al. Incipient melanoma brain metastases instigate astrogliosis and neuroinflammation. Cancer Res. 2016, 76, 4359–4371. [Google Scholar] [CrossRef] [PubMed]
- Morsi, A.; Gaziel-Sovran, A.; Cruz-Munoz, W.; Kerbel, R.S.; Golfinos, J.G.; Hernando, E.; Wadghiri, Y.Z. Development and characterization of a clinically relevant mouse model of melanoma brain metastasis. Pigment Cell Melanoma Res. 2013, 26, 743–745. [Google Scholar] [CrossRef] [PubMed]
- Heilmann, S.; Ratnakumar, K.; Langdon, E.M.; Kansler, E.R.; Kim, I.S.; Campbell, N.R.; Perry, E.B.; McMahon, A.J.; Kaufman, C.K.; van Rooijen, E.; et al. A Quantitative system for studying metastasis using transparent zebrafish. Cancer Res. 2015, 75, 4272–4282. [Google Scholar] [CrossRef] [PubMed]
- Budman, D.R.; Camacho, E.; Wittes, R.E. The current causes of death in patients with malignant melanoma. Eur. J. Cancer 1978, 14, 327–330. [Google Scholar] [CrossRef]
- Davies, M.A.; Liu, P.; McIntyre, S.; Kim, K.B.; Papadopoulos, N.; Hwu, W.-J.; Hwu, P.; Bedikian, A. Prognostic factors for survival in melanoma patients with brain metastases. Cancer 2011, 117, 1687–1696. [Google Scholar] [CrossRef] [PubMed]
- Sandru, A.; Voinea, S.; Panaitescu, E.; Blidaru, A. Survival rates of patients with metastatic malignant melanoma. J. Med. Life 2014, 7, 572–576. [Google Scholar] [PubMed]
- Sampson, J.H.; Carter, J.H., Jr.; Friedman, A.H.; Seigler, H.F. Demographics, prognosis, and therapy in 702 patients with brain metastases from malignant melanoma. J. Neurosurg. 1998, 88, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, D.; Denkins, Y.; Reiland, J.; Greiter-Wilke, A.; Galjour, J.; Murry, B.; Blust, J.; Roy, M. Brain-metastatic melanoma: A neurotrophic perspective. Pathol. Oncol. Res. 2003, 9, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Steeg, P.S.; Camphausen, K.A.; Smith, Q.R. Brain metastases as preventive and therapeutic targets. Nat. Rev. Cancer 2011, 11, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Amer, M.H.; Al-Sarraf, M.; Baker, L.H.; Vaitkevicius, V.K. Malignant melanoma and central nervous system metastases. Incidence, diagnosis, treatment and survival. Cancer 1978, 42, 660–668. [Google Scholar] [CrossRef]
- Chaichana, K.K.; Chaichana, K.L. Diagnosis and Treatment Options for Brain Metastasis of Melanoma; INTECH Open Access Publisher: Vienna, Austria, 2011. [Google Scholar]
- Tas, F. Metastatic behavior in melanoma: Timing, pattern, survival, and influencing factors. J. Oncol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Fife, K.M.; Colman, M.H.; Stevens, G.N.; Firth, I.C.; Moon, D.; Shannon, K.F.; Harman, R.; Petersen-Schaefer, K.; Zacest, A.C.; Besser, M.; et al. Determinants of outcome in melanoma patients with cerebral metastases. J. Clin. Oncol. 2004, 22, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, S.; Spagnolo, F.; Merlo, D.F.; Signori, A.; Acquati, M.; Pronzato, P.; Queirolo, P. The treatment of melanoma brain metastases before the advent of targeted therapies: Associations between therapeutic choice, clinical symptoms and outcome with survival. Melanoma Res. 2014, 24, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Sperduto, P.W.; Berkey, B.; Gaspar, L.E.; Mehta, M.; Curran, W. A new prognostic index and comparison to three other indices for patients with brain metastases: An analysis of 1,960 patients in the RTOG database. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Sperduto, P.W.; Kased, N.; Roberge, D.; Xu, Z.; Shanley, R.; Luo, X.; Sneed, P.K.; Chao, S.T.; Weil, R.J.; Suh, J.; et al. Summary report on the graded prognostic assessment: An accurate and facile diagnosis-specific tool to estimate survival for patients with brain metastases. J. Clin. Oncol. 2012, 30, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, A.; Furness, A.; Corbett, R.W.; Bloomfield, A.; Porta, N.; Morris, S.; Ali, Z.; Larkin, J.; Harrington, K. The melanoma-specific graded prognostic assessment does not adequately discriminate prognosis in a modern population with brain metastases from malignant melanoma. Br. J. Cancer 2015, 113, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Ewend, M.G.; Carey, L.A.; Brem, H. Treatment of melanoma metastases in the brain. Semin. Surg. Oncol. 1996, 12, 429–435. [Google Scholar] [CrossRef]
- Marek, W.; Ehud, A. Surgical treatment of brain metastases from melanoma: A retrospective study of 91 patients. J. Neurosurg. 2000, 93, 9–18. [Google Scholar]
- Gorantla, V.; Kirkwood, J.; Tawbi, H. Melanoma brain metastases: An unmet challenge in the era of active therapy. Curr. Oncol. Rep. 2013, 15, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Eichler, A.F.; Loeffler, J.S. Multidisciplinary management of brain metastases. Oncologist 2007, 12, 884–898. [Google Scholar] [CrossRef]
- Flanigan, J.C.; Jilaveanu, L.B.; Faries, M.; Sznol, M.; Ariyan, S.; Yu, J.B.; Knisely, J.P.S.; Chiang, V.L.; Kluger, H.M. Melanoma Brain Metastases: Is It Time to Reassess the Bias? Curr. Probl. Cancer 2011, 35, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Chukwueke, U.; Batchelor, T.; Brastianos, P. Management of brain metastases in patients with melanoma. J. Oncol. Pract. 2016, 12, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, F.; Picasso, V.; Lambertini, M.; Ottaviano, V.; Dozin, B.; Queirolo, P. Survival of patients with metastatic melanoma and brain metastases in the era of MAP-kinase inhibitors and immunologic checkpoint blockade antibodies: A systematic review. Cancer Treat. Rev. 2016, 45, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Goulart, C.R.; Mattei, T.A.; Ramina, R. Cerebral melanoma metastases: A critical review on diagnostic methods and therapeutic options. ISRN Surg. 2011. [Google Scholar] [CrossRef] [PubMed]
- Sperduto, P.W.; Chao, S.T.; Sneed, P.K.; Luo, X.; Suh, J.; Roberge, D.; Bhatt, A.; Jensen, A.W.; Brown, P.D.; Shih, H.; et al. Diagnosis-specific prognostic factors, indexes, and treatment outcomes for patients with newly diagnosed brain metastases: A multi-institutional analysis of 4259 patients. Int. J. Radiat. Oncol. Biol. Phys. 2010, 77, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.J.; Dicker, A.P. On the Merits and limitations of whole-brain radiation therapy. J. Clin. Oncol. 2013, 31, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Hatiboglu, M.A.; Wildrick, D.M.; Sawaya, R. The role of surgical resection in patients with brain metastases. Ecancermedicalscience 2013, 7, 308. [Google Scholar] [PubMed]
- Barranco, S.C.; Romsdahl, M.M.; Humphrey, R.M. The Radiation response of human malignant melanoma cells grown in vitro. Cancer Res. 1971, 31, 830–833. [Google Scholar] [PubMed]
- Goldinger, S.M.; Panje, C.; Nathan, P. Treatment of melanoma brain metastases. Curr. Opin. Oncol. 2016, 28, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Tsao, M.N.; Rades, D.; Wirth, A.; Lo, S.S.; Danielson, B.L.; Gaspar, L.E.; Sperduto, P.W.; Vogelbaum, M.A.; Radawski, J.D.; Wang, J.Z.; et al. Radiotherapeutic and surgical management for newly diagnosed brain metastasis(es): An American Society for Radiation Oncology evidence-based guideline. Pract. Radiat. Oncol. 2012, 2, 210–225. [Google Scholar] [CrossRef] [PubMed]
- Staudt, M.; Lasithiotakis, K.; Leiter, U.; Meier, F.; Eigentler, T.; Bamberg, M.; Tatagiba, M.; Brossart, P.; Garbe, C. Determinants of survival in patients with brain metastases from cutaneous melanoma. Br. J. Cancer 2010, 102, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Skibber, J.M.; Soong, S.J.; Austin, L.; Balch, C.M.; Sawaya, R.E. Cranial irradiation after surgical excision of brain metastases in melanoma patients. Ann. Surg. Oncol. 1996, 3, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Hagen, N.A.; Cirrincione, C.; Thaler, H.T.; DeAngelis, L.M. The role of radiation therapy following resection of single brain metastasis from melanoma. Neurology 1990, 40, 158–160. [Google Scholar] [CrossRef] [PubMed]
- Nieder, C.; Grosu, A.; Gaspar, L. Stereotactic radiosurgery (SRS) for brain metastases: A systematic review. Radiat. Oncol. 2014, 9, 155. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, H.; Shirato, H.; Tago, M.; Nakagawa, K.; Toyoda, T.; Hatano, K.; Kenjyo, M.; Oya, N.; Hirota, S.; Shioura, H.; et al. Stereotactic radiosurgery plus whole-brain radiation therapy vs stereotactic radiosurgery alone for treatment of brain metastases: A randomized controlled trial. JAMA 2006, 295, 2483–2491. [Google Scholar] [CrossRef] [PubMed]
- Sahgal, A.; Aoyama, H.; Kocher, M.; Neupane, B.; Collette, S.; Tago, M.; Shaw, P.; Beyene, J.; Chang, E.L. Phase 3 trials of stereotactic radiosurgery with or without whole-brain radiation therapy for 1 to 4 brain metastases: Individual patient data meta-analysis. Int. J. Radiat. Oncol. Biol. Phys. 2015, 91, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, I.H.; Ojerholm, E.; McMillan, M.T.; Miller, D.; Kolker, J.D.; Kurtz, G.; Dorsey, J.F.; Nagda, S.N.; Geiger, G.A.; Brem, S.; et al. Novel risk scores for survival and intracranial failure in patients treated with radiosurgery alone to melanoma brain metastases. Radiat. Oncol. 2015, 10, 248. [Google Scholar] [CrossRef] [PubMed]
- DeAngelis, L.M.; Delattre, J.Y.; Posner, J.B. Radiation-induced dementia in patients cured of brain metastases. Neurology 1989, 39, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Goyal, S.; Silk, A.W.; Tian, S.; Mehnert, J.; Danish, S.; Ranjan, S.; Kaufman, H.L. Clinical management of multiple melanoma brain metastases: A systematic review. JAMA Oncol. 2015, 1, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Agarwala, S.S.; Kirkwood, J.M.; Gore, M.; Dreno, B.; Thatcher, N.; Czarnetski, B.; Atkins, M.; Buzaid, A.; Skarlos, D.; Rankin, E.M. Temozolomide for the treatment of brain metastases associated with metastatic melanoma: A phase II study. J. Clin. Oncol. 2004, 22, 2101–2107. [Google Scholar] [CrossRef] [PubMed]
- Avril, M.F.; Aamdal, S.; Grob, J.J.; Hauschild, A.; Mohr, P.; Bonerandi, J.J.; Weichenthal, M.; Neuber, K.; Bieber, T.; Gilde, K.; et al. Fotemustine compared with dacarbazine in patients with disseminated malignant melanoma: A phase III study. J. Clin. Oncol. 2004, 22, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Fisher, D.E.; Garbe, C.; Gershenwald, J.E.; Grob, J.J.; Halpern, A.; Herlyn, M.; Marchetti, M.A.; McArthur, G.; Ribas, A.; et al. Melanoma. Nat. Rev. Dis. Primers 2015, 1, 15003. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Trefzer, U.; Davies, M.A.; Kefford, R.F.; Ascierto, P.A.; Chapman, P.B.; Puzanov, I.; Hauschild, A.; Robert, C.; Algazi, A.; et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): A multicentre, open-label, phase 2 trial. Lancet Oncol. 2012, 13, 1087–1095. [Google Scholar] [CrossRef]
- Kefford, R.F.; Maio, M.; Arance, A.; Nathan, P.; Blank, C.; Avril, M.F. Vemurafenib in metastatic melanoma patients with brain metastases: An open-label, singlearm, phase 2, multicenter study. Pigment Cell Melanoma Res. 2013, 26, 965. [Google Scholar]
- Maxwell, R.; Garzon-Muvdi, T.; Lipson, E.J.; Sharfman, W.H.; Bettegowda, C.; Redmond, K.J.; Kleinberg, L.R.; Ye, X.; Lim, M. BRAF-V600 mutational status affects recurrence patterns of melanoma brain metastasis. Int. J. Cancer 2016. [Google Scholar] [CrossRef] [PubMed]
- Gummadi, T.; Zhang, B.Y.; Valpione, S.; Kim, C.; Kottschade, L.A.; Mittapalli, R.K.; Chiarion-Sileni, V.; Pigozzo, J.; Elmquist, W.F.; Dudek, A.Z. Impact of BRAF mutation and BRAF inhibition on melanoma brain metastases. Melanoma Res. 2015, 25, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAFV600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Goldinger, S.M.; Turtschi, C.P.; Eggmann, N.B.; Michielin, O.; Mitchell, L.; Veronese, L.; Hilfiker, P.R.; Felderer, L.; Rinderknecht, J.D. Vemurafenib in patients with BRAF (V600) mutation-positive melanoma with symptomatic brain metastases: Final results of an open-label pilot study. Eur. J. Cancer 2014, 50, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.J.; Catalanotti, F.; Munhoz, R.R.; Cheng, D.T.; Yaqubie, A.; Kelly, N.; McDermott, G.C.; Kersellius, R.; Merghoub, T.; Lacouture, M.E.; et al. A retrospective evaluation of vemurafenib as treatment for BRAF-mutant melanoma brain metastases. Oncologist 2015, 20, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Sakji-Dupre, L.; Le Rhun, E.; Templier, C.; Desmedt, E.; Blanchet, B.; Mortier, L. Cerebrospinal fluid concentrations of vemurafenib in patients treated for brain metastatic BRAF-V600 mutated melanoma. Melanoma Res. 2015, 25, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.S.; Long, G.V.; Kurzrock, R.; Kim, K.B.; Arkenau, T.H.; Brown, M.P.; Hamid, O.; Infante, J.R.; Millward, M.; Pavlick, A.C.; et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: A phase 1 dose-escalation trial. Lancet 2012, 379, 1893–1901. [Google Scholar] [CrossRef]
- Azer, M.W.; Menzies, A.M.; Haydu, L.E.; Kefford, R.F.; Long, G.V. Patterns of response and progression in patients with BRAF-mutant melanoma metastatic to the brain who were treated with dabrafenib. Cancer 2014, 120, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Mittapalli, R.K.; Vaidhyanathan, S.; Dudek, A.Z.; Elmquist, W.F. Mechanisms limiting distribution of the threonine-protein kinase BRAFV600E inhibitor dabrafenib to the brain: Implications for the treatment of melanoma brain metastases. J. Pharmacol. Exp. Ther. 2013, 344, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Wong, M.K.; Daniels, G.A.; McDermott, D.F.; Aung, S.; Lowder, J.N.; Morse, M.A. The use of registries to improve cancer treatment: A national database for patients treated with interleukin-2 (IL-2). J. Personal. Med. 2014, 4, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.; Dudek, A.Z. Single-institution outcome of high-dose interleukin-2 (HD IL-2) therapy for metastatic melanoma and analysis of favorable response in brain metastases. Anticancer Res. 2009, 29, 4189–4193. [Google Scholar] [PubMed]
- Chu, M.B.; Fesler, M.J.; Armbrecht, E.S.; Fosko, S.W.; Hsueh, E.; Richart, J.M. High-dose interleukin-2 (HD IL-2) therapy should be considered for treatment of patients with melanoma brain Metastases. Chemother. Res. Pract. 2013, 2013, 726925. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H.; June, C.H. Engineering T cells for cancer: Our synthetic future. Immunol. Rev. 2014, 257, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.J.; Rosenberg, S.A.; Dudley, M.E.; Yang, J.C.; White, D.E.; Butman, J.A.; Sherry, R.M. Successful Treatment of melanoma brain metastases with adoptive cell therapy. Clin. Cancer Res. 2010, 16, 4892–4898. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Azvolinsky, A. One size does not fit all: Personalized immune therapies poised to take center stage. J. Natl. Cancer Inst. 2013, 105, 583–584. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hodi, F.S.; Robert, C. CTLA-4 and PD-1/PD-L1 Blockade: New Immunotherapeutic Modalities with Durable Clinical Benefit in Melanoma Patients. Clin. Cancer Res. 2013, 19, 5300–5309. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.M. The immune checkpoint inhibitors: Where are we now? Nat. Rev. Drug Discov. 2014, 13, 883–884. [Google Scholar] [CrossRef] [PubMed]
- Margolin, K.; Ernstoff, M.S.; Hamid, O.; Lawrence, D.; McDermott, D.; Puzanov, I.; Wolchok, J.D.; Clark, J.I.; Sznol, M.; Logan, T.F.; et al. Ipilimumab in patients with melanoma and brain metastases: An open-label, phase 2 trial. Lancet Oncol. 2012, 13, 459–465. [Google Scholar] [CrossRef]
- Di Giacomo, A.M.; Margolin, K. Immune checkpoint blockade in patients with melanoma metastatic to the brain. Semin. Oncol. 2015, 42, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Knisely, J.P.; Yu, J.B.; Flanigan, J.C.; Sznol, M.; Kluger, H.M.; Chiang, V.L. Radiosurgery for melanoma brain metastases in the ipilimumab era and the possibility of longer survival. J. Neurosurg. 2012, 117, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Kiess, A.P.; Wolchok, J.D.; Barker, C.A.; Postow, M.A.; Tabar, V.; Huse, J.T.; Chan, T.A.; Yamada, Y.; Beal, K. Stereotactic radiosurgery for melanoma brain metastases in patients receiving ipilimumab: Safety profile and efficacy of combined treatment. Int. J. Radiat. Oncol. Biol. Phys. 2015, 92, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Mathew, M.; Tam, M.; Ott, P.A.; Pavlick, A.C.; Rush, S.C.; Donahue, B.R.; Golfinos, J.G.; Parker, E.C.; Huang, P.P.; Narayana, A. Ipilimumab in melanoma with limited brain metastases treated with stereotactic radiosurgery. Melanoma Res. 2013, 23, 191–195. [Google Scholar] [CrossRef]
- Patel, K.R.; Shoukat, S.; Oliver, D.E.; Chowdhary, M.; Rizzo, M.; Lawson, D.H.; Khosa, F.; Liu, Y.; Khan, M.K. Ipilimumab and Stereotactic Radiosurgery Versus Stereotactic Radiosurgery Alone for Newly Diagnosed Melanoma Brain Metastases. Am. J. Clin. Oncol. 2015. [Google Scholar] [CrossRef]
- Qian, J.M.; Yu, J.B.; Kluger, H.M.; Chiang, V.L. Timing and type of immune checkpoint therapy affect the early radiographic response of melanoma brain metastases to stereotactic radiosurgery. Cancer 2016. [Google Scholar] [CrossRef]
- Schartz, N.E.C.; Farges, C.; Madelaine, I.; Bruzzoni, H.; Calvo, F.; Hoos, A.; Lebbe, C. Complete regression of a previously untreated melanoma brain metastasis with ipilimumab. Melanoma Res. 2010, 20, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Silk, A.W.; Bassetti, M.F.; West, B.T.; Tsien, C.I.; Lao, C.D. Ipilimumab and radiation therapy for melanoma brain metastases. Cancer Med. 2013, 2, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Tazi, K.; Hathaway, A.; Chiuzan, C.; Shirai, K. Survival of melanoma patients with brain metastases treated with ipilimumab and stereotactic radiosurgery. Cancer Med. 2015, 4, 1–6. [Google Scholar] [CrossRef]
- Weber, J.S.; Amin, A.; Minor, D.; Siegel, J.; Berman, D.; O’Day, S.J. Safety and clinical activity of ipilimumab in melanoma patients with brain metastases: Retrospective analysis of data from a phase 2 trial. Melanoma Res. 2011, 21, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, S.B.; Gettinger, S.N.; Mahajan, A.; Chiang, A.C.; Herbst, R.S.; Sznol, M.; Tsiouris, A.J.; Cohen, J.; Vortmeyer, A.; Jilaveanu, L.; et al. Pembrolizumab for patients with melanoma or non-small-cell lung cancer and untreated brain metastases: Early analysis of a non-randomised, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 976–983. [Google Scholar] [CrossRef]
- Kluger, H.M.; Zito, C.R.; Barr, M.L.; Baine, M.K.; Chiang, V.L.; Sznol, M.; Rimm, D.L.; Chen, L.; Jilaveanu, L.B. Characterization of PD-L1 expression and associated T-cell infiltrates in metastatic melanoma samples from variable anatomic sites. Clin. Cancer Res. 2015, 21, 3052–3060. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.A.; Stallworth, D.G.; Kim, Y.; Johnstone, P.A.; Harrison, L.B.; Caudell, J.J.; Yu, H.H.; Etame, A.B.; Weber, J.S.; Gibney, G.T. Clinical outcomes of melanoma brain metastases treated with stereotactic radiation and anti-PD-1 therapy. Ann. Oncol. 2016, 27, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Trial | Phase | Status | Primary Outcome Measured | Title |
|---|---|---|---|---|
| NCT01721603 | II | Active, not recruiting | Safety/efficacy | A Phase 2 Prospective Trial of Dabrafenib with Stereotactic Radiosurgery in BRAFV600E Melanoma Brain Metastases |
| NCT02115139 | II | Recruiting | 1-year survival rate | GEM STUDY: Radiation and Yervoy in Patients with Melanoma and Brain Metastases (GRAY-B) |
| NCT01703507 | I | Active, not recruiting | Maximum tolerated dose ipilimumab | Phase I Study of Ipilimumab Combined with Whole Brain Radiation Therapy or Radiosurgery for Melanoma |
| NCT02085070 | II | Recruiting | Response rate | MK-3475 in Melanoma and NSCLC Patients with Brain Metastases |
| NCT02097732 | II | Recruiting | Local control rate | Ipilimumab Induction in Patients with Melanoma Brain Metastases Receiving Stereotactic Radiosurgery |
| NCT02039947 | II | Recruiting | Intracranial response rate | Study to Evaluate Treatment of Dabrafenib Plus Trametinib in Subjects with BRAF Mutation-Positive Melanoma That Has Metastasized to the Brain |
| NCT01378975 | II | Completed, no posts | Best overall response rate | A Study of Vemurafenib in Metastatic Melanoma Patients with Brain Metastases |
| NCT01978236 | II | Recruiting | Concentrations of dabrafenib & trametinib in metastases | Dabrafenib/Trametinib, BRAF or BRAF AND MEK Pre-op with BRAF and MEK Post-op, Phase IIB, Melanoma with Brain Mets, Biomarkers and Metabolites |
| NCT02320058 | II | Recruiting | Clinical benefit rate | A Multi-Center Phase 2 Open-Label Study to Evaluate Safety and Efficacy in Subjects with Melanoma Metastatic to the Brain Treated with Nivolumab in Combination with Ipilimumab Followed by Nivolumab Monotherapy (CheckMate 204) |
| NCT01503827 | III | Recruiting | Distant intracranial failure | Whole Brain Radiotherapy Following Local Treatment of Intracranial Metastases of Melanoma (WBRTMel) |
| NCT01644591 | III | Active, not recruiting | Time to local failure | Trial to Compare Local Control and Neurocognitive Preservation after Initial Treatment with Stereotactic Radiosurgery (SRS) versus Whole Brain Radiation Therapy (WBRT) for Patients with >3 Brain Metastases from Melanoma |
| NCT02460068 | III | Recruiting | Overall survival rate | A Study of Fotemustine(FTM) vs. FTM and Ipilimumab (IPI) or IPI and Nivolumab in Melanoma Brain Metastasis (NIBIT-M2) |
| NCT02374242 | II | Recruiting | Intracranial response rate | Anti-PD 1 Brain Collaboration for Patients with Melanoma Brain Metastases (ABC) |
| NCT02662725 | II | Completed, no posts | Overall survival rate | Ipilimumab Combined with a Stereotactic Radiosurgery in Melanoma Patients with Brain Metastases (IPI + RTS) |
| NCT02308020 | II | Recruiting | Complete response, partial response, objective intracranial response rates | A Study of Abemaciclib (LY2835219) in Participants with Breast Cancer, Non-small Cell Lung Cancer, or Melanoma That Has Spread to the Brain |
| NCT02681549 | II | Recruiting | Brain metastasis response rate | Pembrolizumab Plus Bevacizumab for Treatment of Brain Metastases in Metastatic Melanoma or Non-small Cell Lung Cancer |
| NCT02621515 | II | Recruiting | Best overall response rate | Nivolumab in Symptomatic Brain Metastases (CA209-322) |
| NCT02716948 | I | Recruiting | Incidence of serious adverse events | Stereotactic Radiosurgery and Nivolumab in Treating Patients with Newly Diagnosed Melanoma Metastases in the Brain or Spine |
| NCT01904123 | I | Not yet recruiting | Maximum tolerated dose WP1066 | A Phase I Trial of WP1066 in Patients with Central Nervous System (CNS) Melanoma and Recurrent Glioblastoma Multiforme (GBM) |
| NCT02452294 | II | Recruting | Intracranial disease control rate | Buparlisib in Melanoma Patients Suffering from Brain Metastases (BUMPER) |
| NCT02537600 | II | Recruiting | Complete or partial intracranial response rate | Vemurafenib and Cobimetinib Combination in BRAF Mutated Melanoma with Brain Metastasis (CONVERCE) |
| NCT02107755 | II | Recruiting | Progression-free survival rate | Stereotactic Radiation Therapy and Ipilimumab in Treating Patients with Metastatic Melanoma |
| NCT01983124 | II | Completed, no posts | Progression-free survival rate | Vemurafenib + Fotemustine to Treat Advanced Melanoma Patients with V600BRAF Mutation Recurred While on Vemurafenib (BeyPro1) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kircher, D.A.; Silvis, M.R.; Cho, J.H.; Holmen, S.L. Melanoma Brain Metastasis: Mechanisms, Models, and Medicine. Int. J. Mol. Sci. 2016, 17, 1468. https://doi.org/10.3390/ijms17091468
Kircher DA, Silvis MR, Cho JH, Holmen SL. Melanoma Brain Metastasis: Mechanisms, Models, and Medicine. International Journal of Molecular Sciences. 2016; 17(9):1468. https://doi.org/10.3390/ijms17091468
Chicago/Turabian StyleKircher, David A., Mark R. Silvis, Joseph H. Cho, and Sheri L. Holmen. 2016. "Melanoma Brain Metastasis: Mechanisms, Models, and Medicine" International Journal of Molecular Sciences 17, no. 9: 1468. https://doi.org/10.3390/ijms17091468
APA StyleKircher, D. A., Silvis, M. R., Cho, J. H., & Holmen, S. L. (2016). Melanoma Brain Metastasis: Mechanisms, Models, and Medicine. International Journal of Molecular Sciences, 17(9), 1468. https://doi.org/10.3390/ijms17091468