
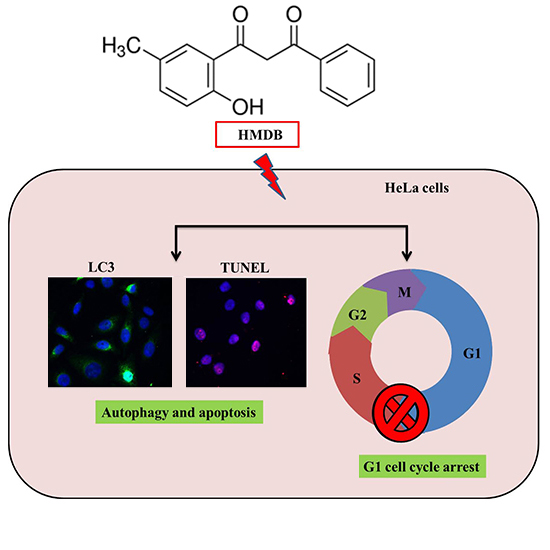
1-(2-Hydroxy-5-methylphenyl)-3-phenyl-1,3-propanedione Induces G1 Cell Cycle Arrest and Autophagy in HeLa Cervical Cancer Cells

 ,
,  ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
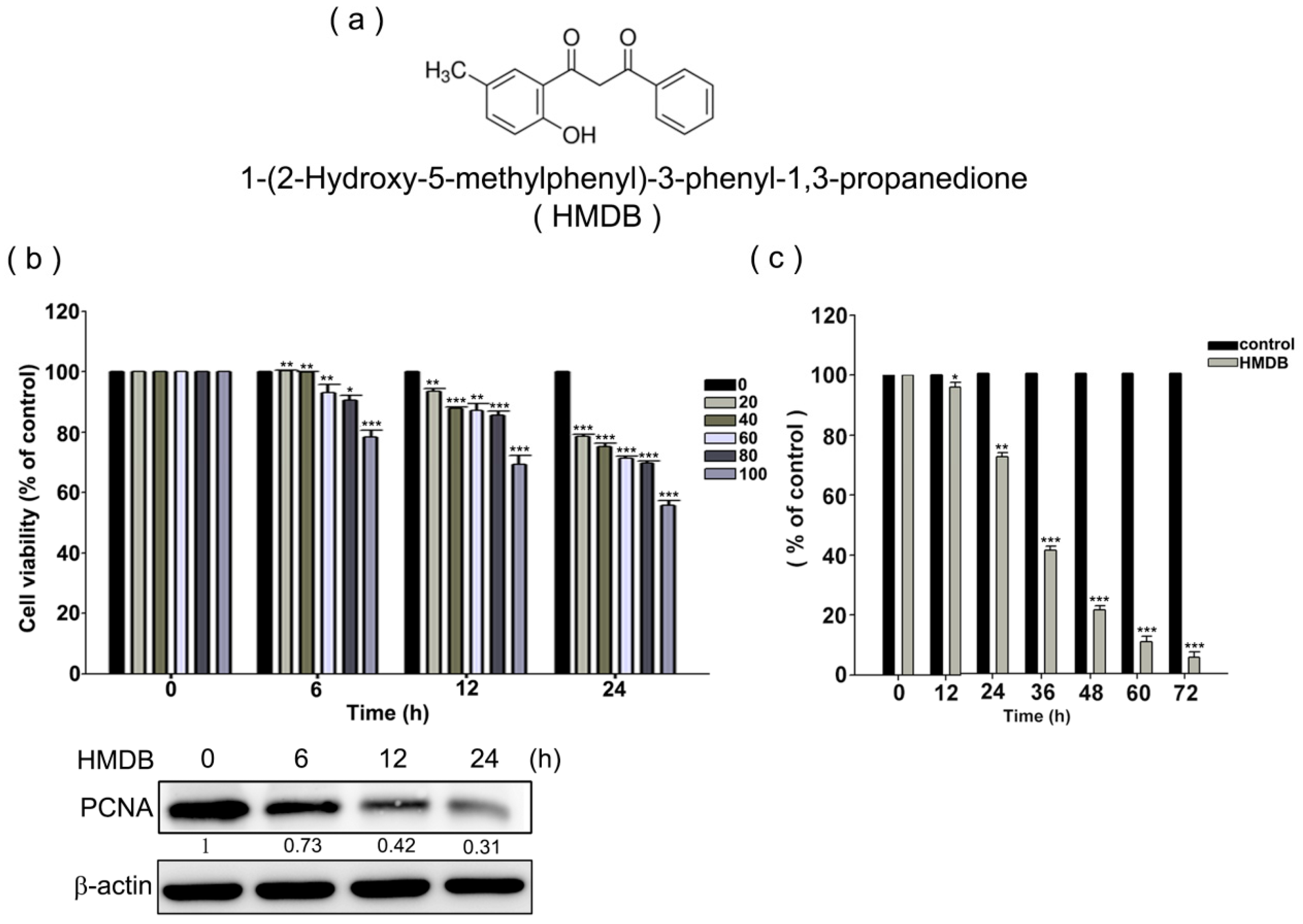
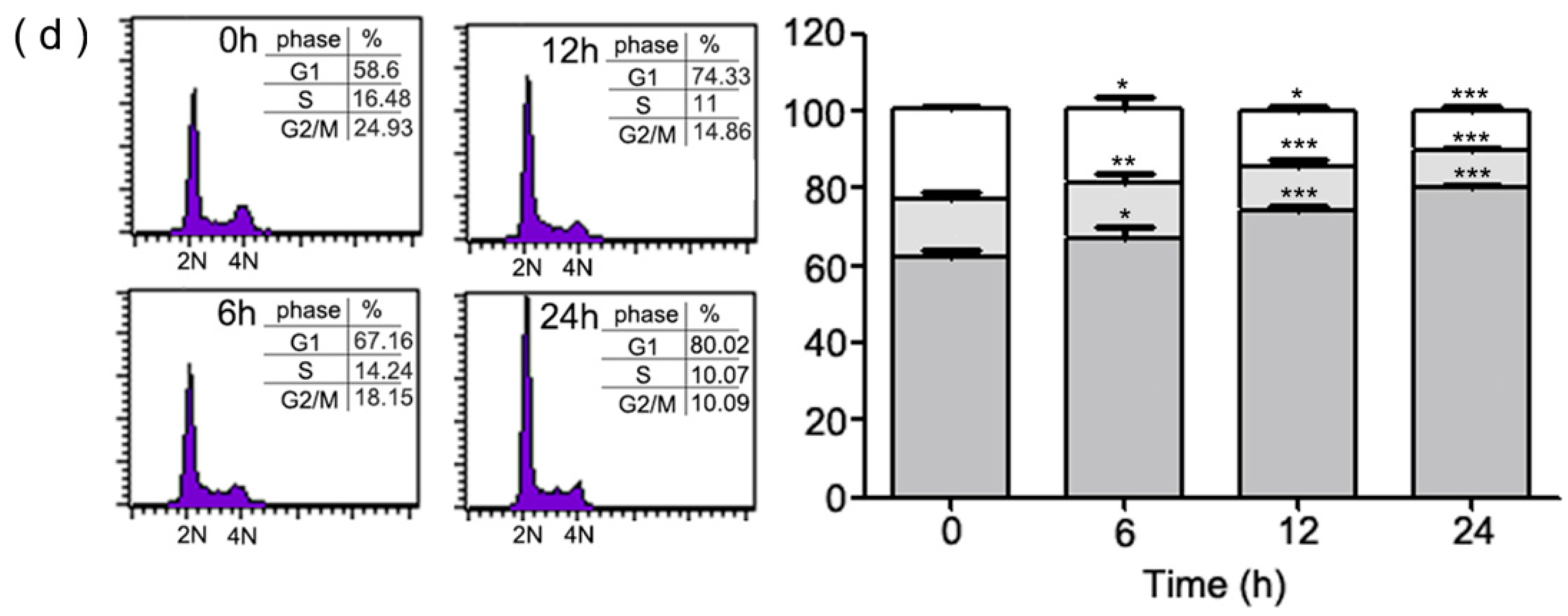
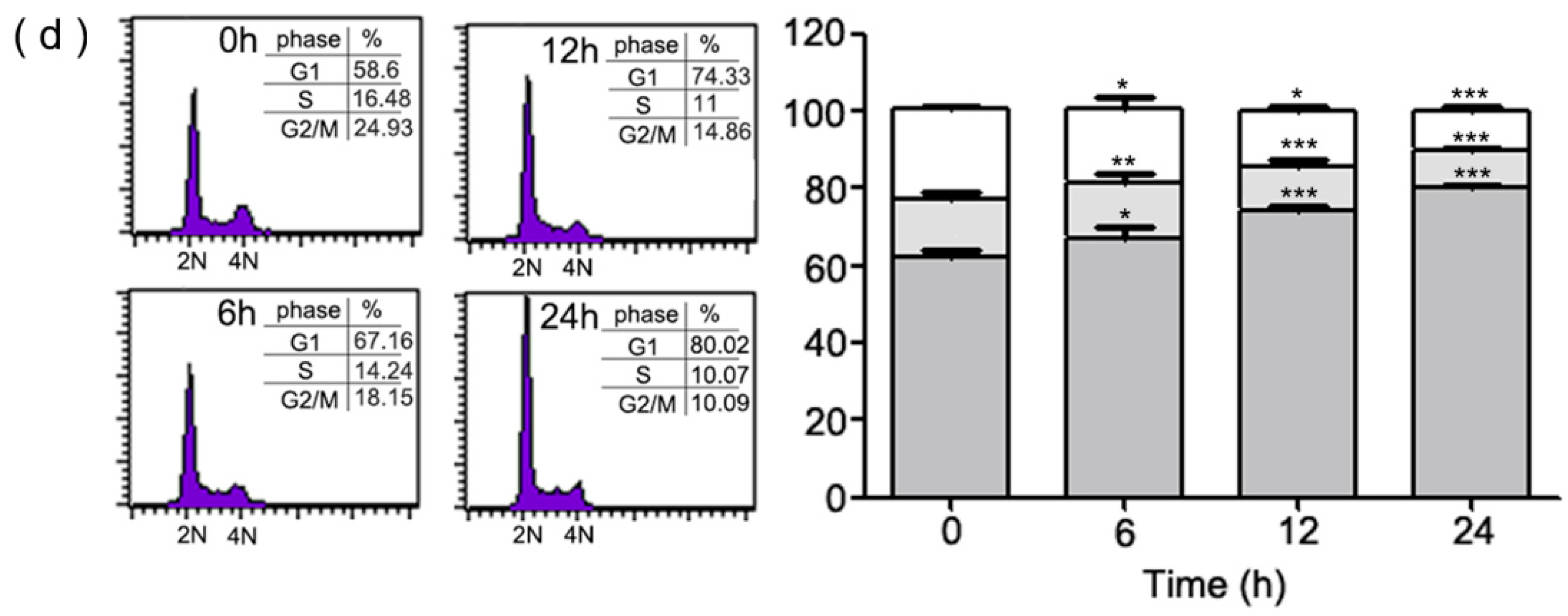
2.1. Inhibition of Cell Growth and Cell Cycle Progression at G1 Phase of HeLa Cells by 1-(2-Hydroxy-5-methylphenyl)-3-phenyl-1,3-propanedione (HMDB)
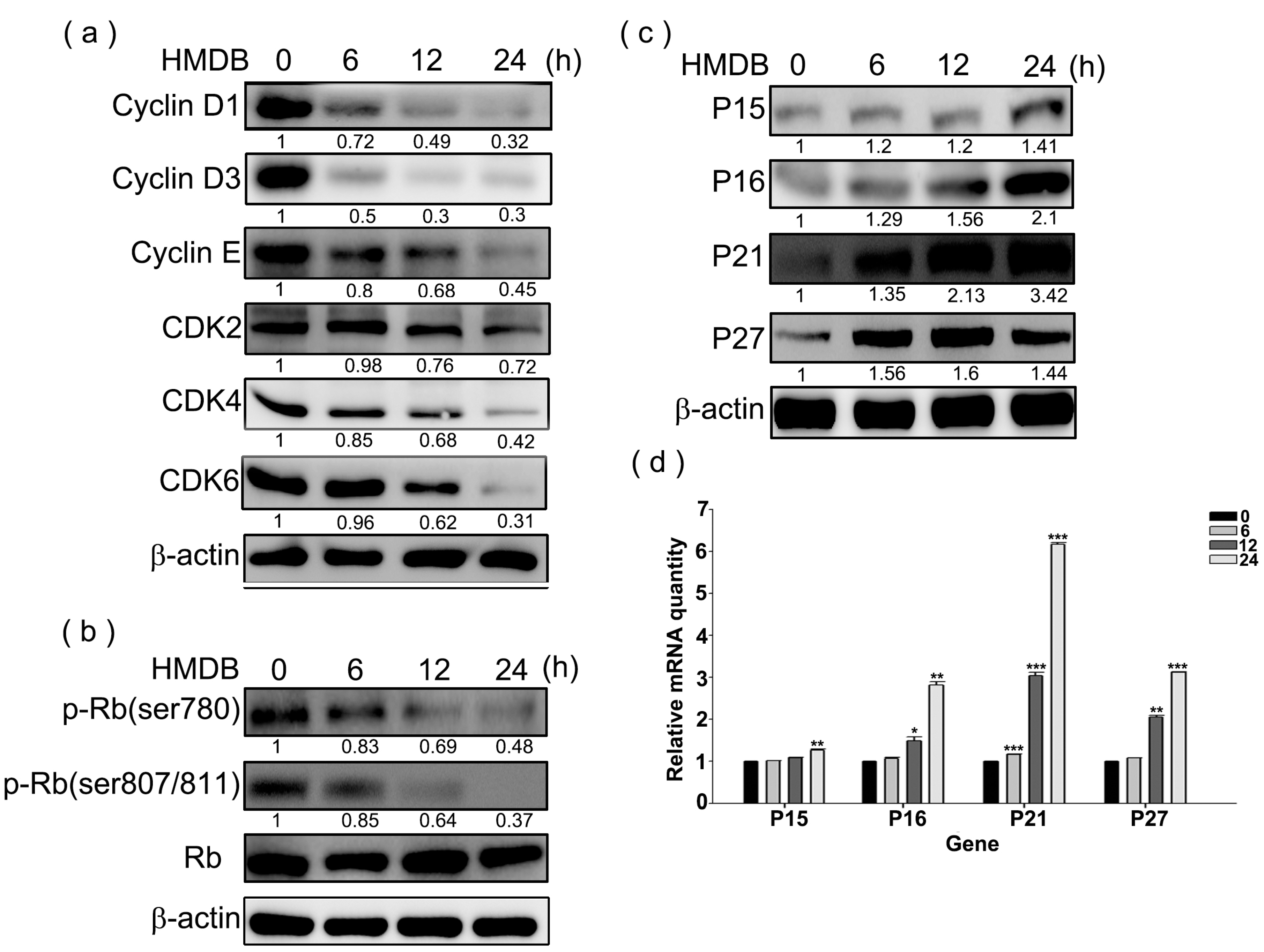
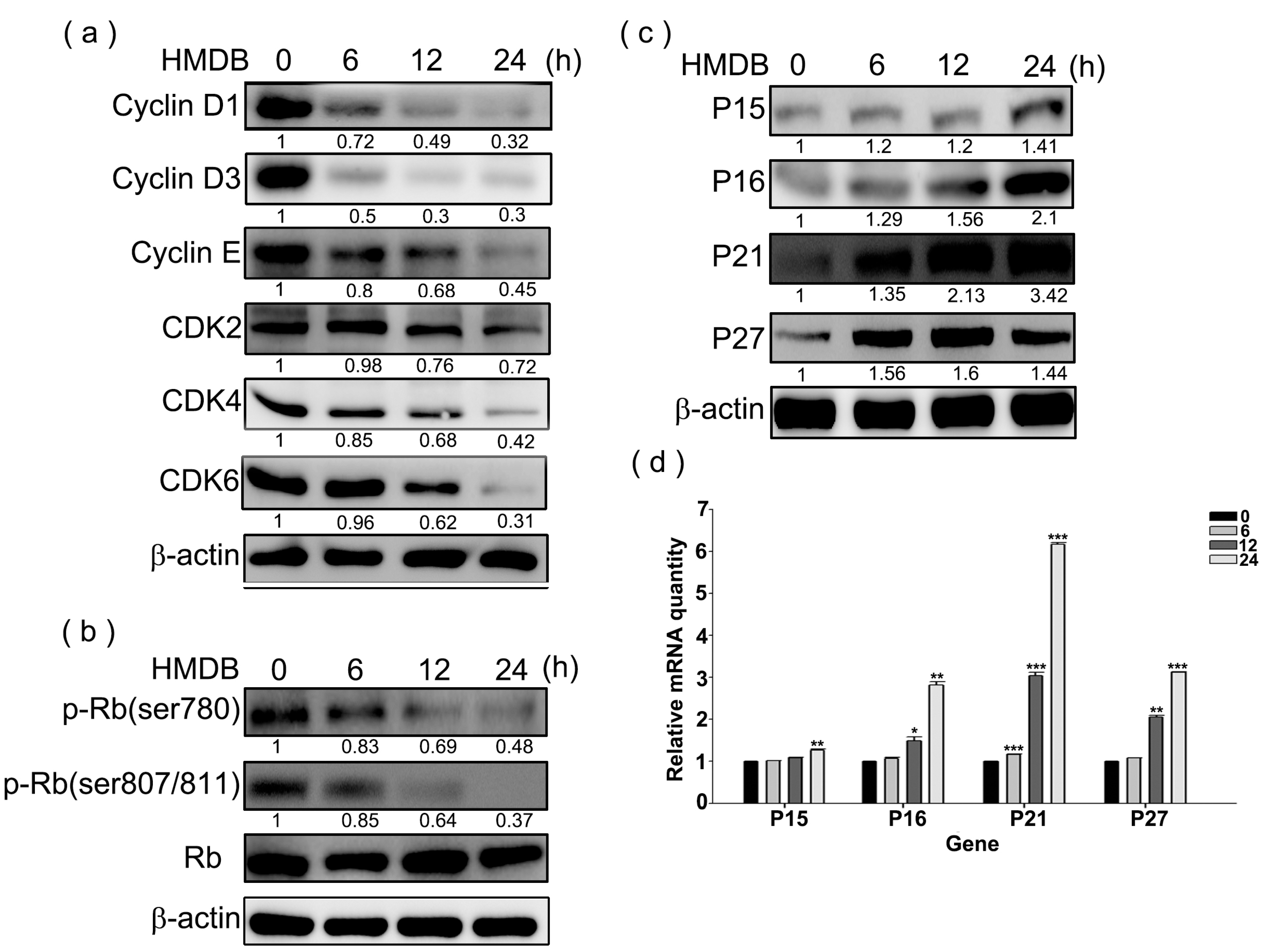
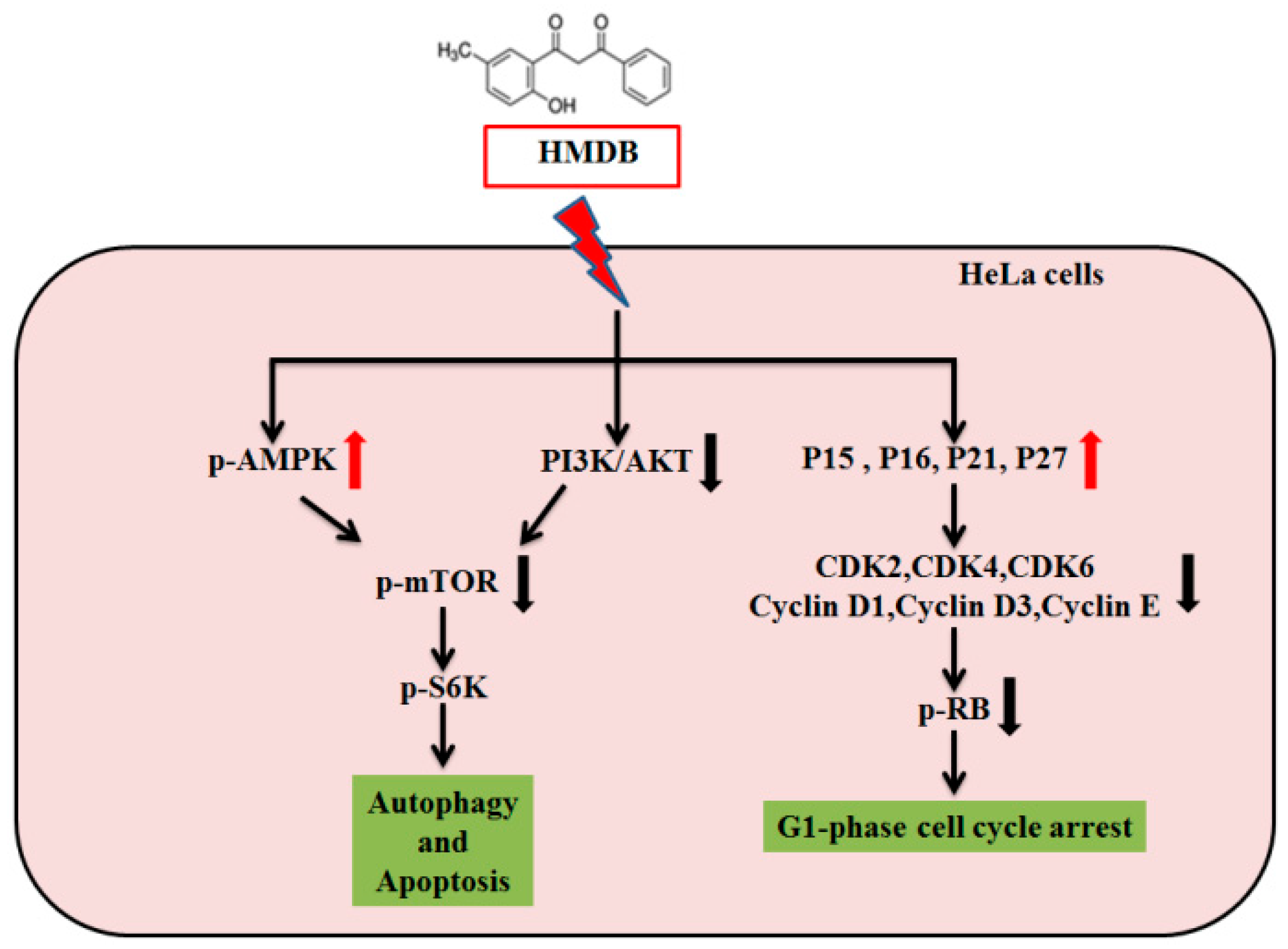
2.2. Modulation of the Expression of G1 Cell Cycle Checkpoint Regulators by HMDB in HeLa Cells
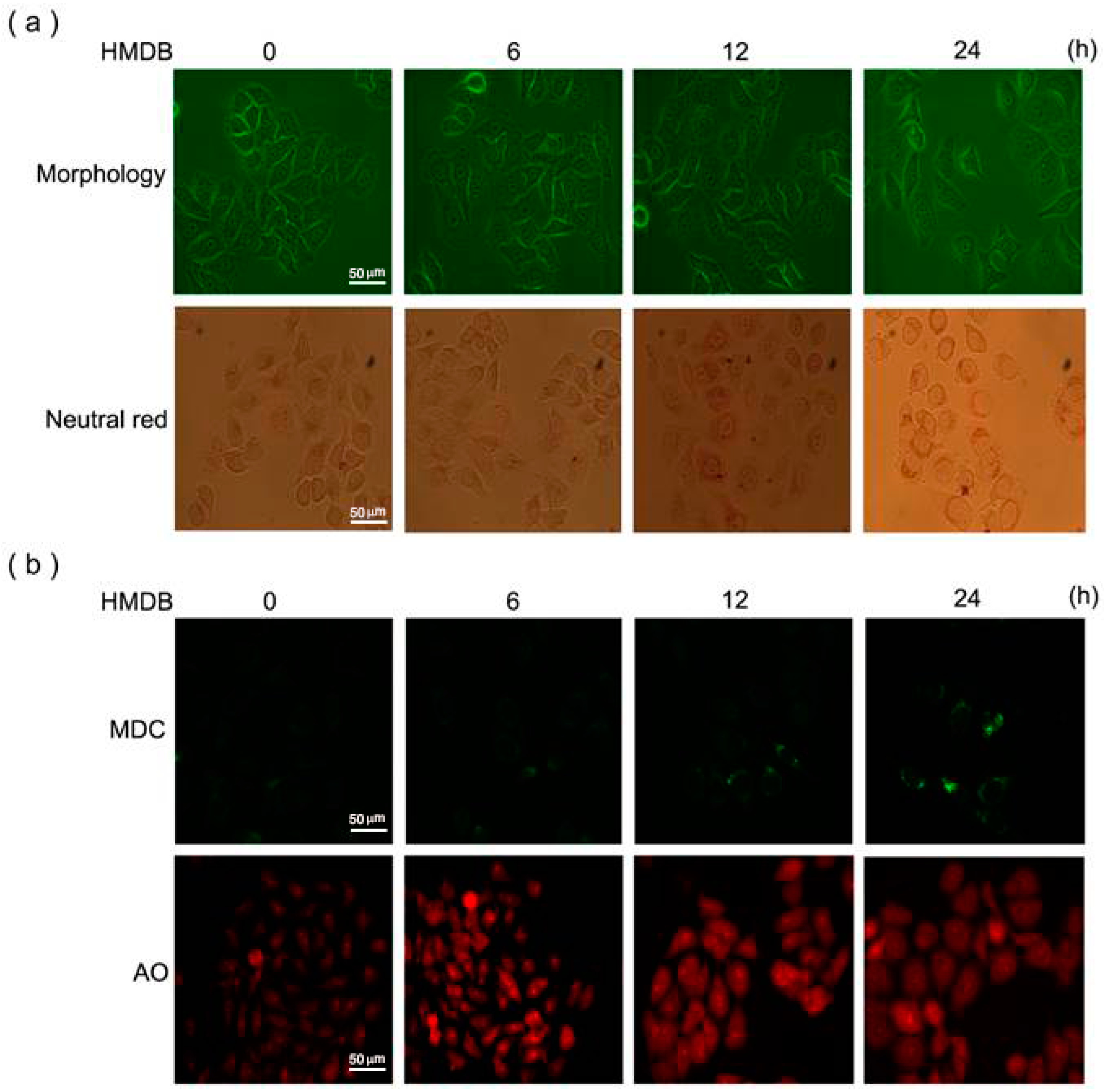
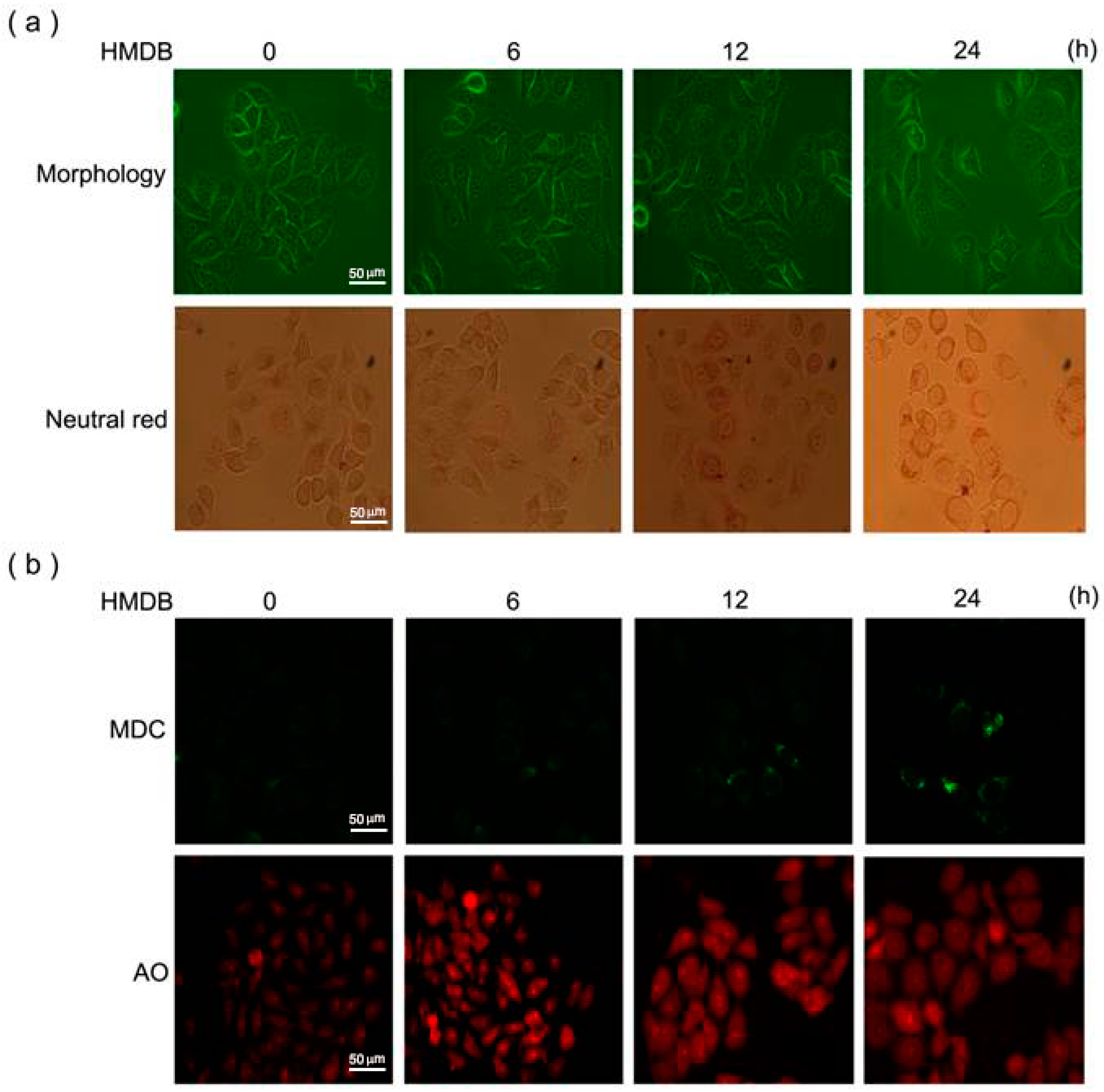
2.3. Induction of Cytoplasmic Vacuolation, Formation of Autolysosomes, and Accumulation of Acidic Vesicles in HMDB-Treated HeLa Cells
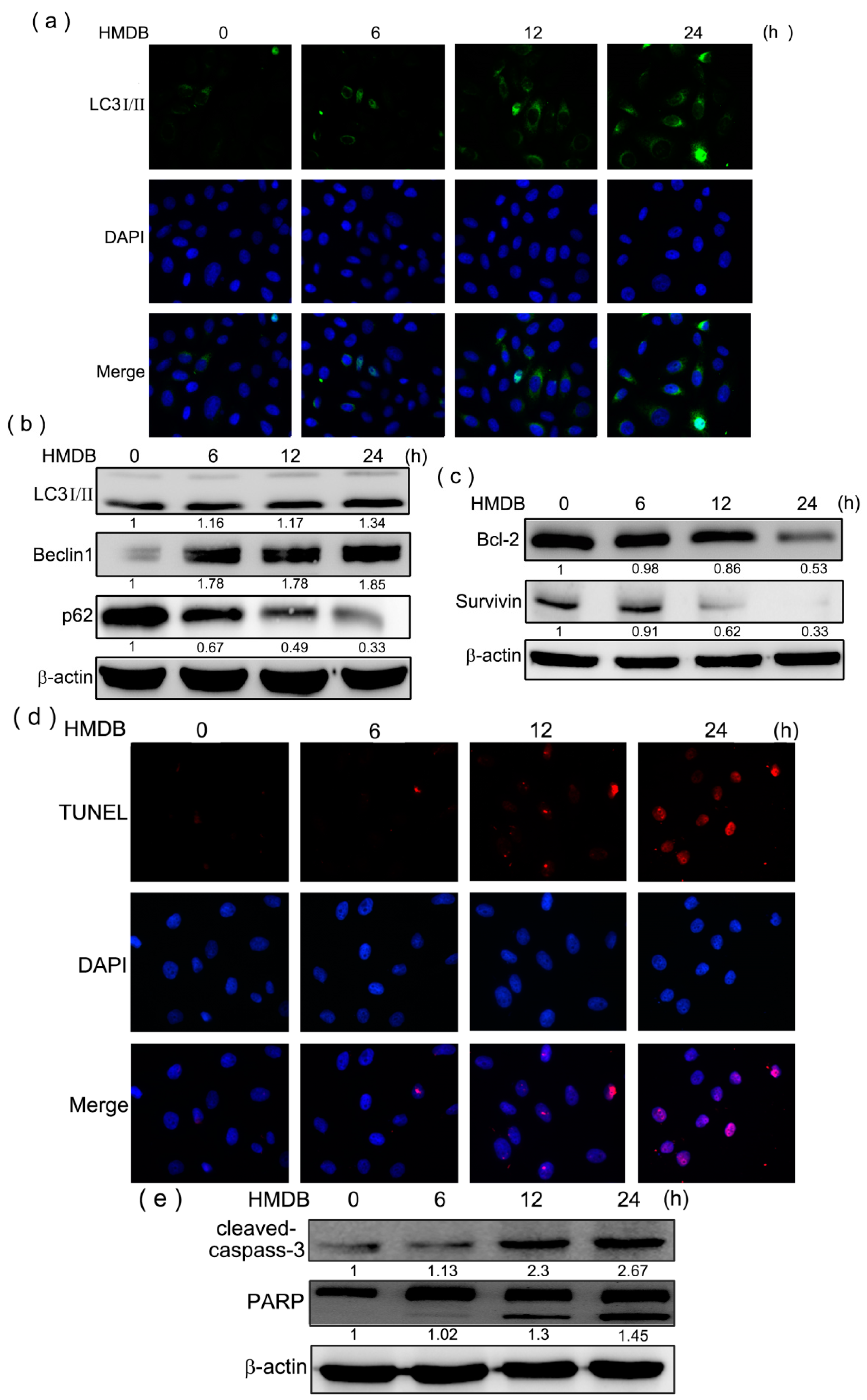
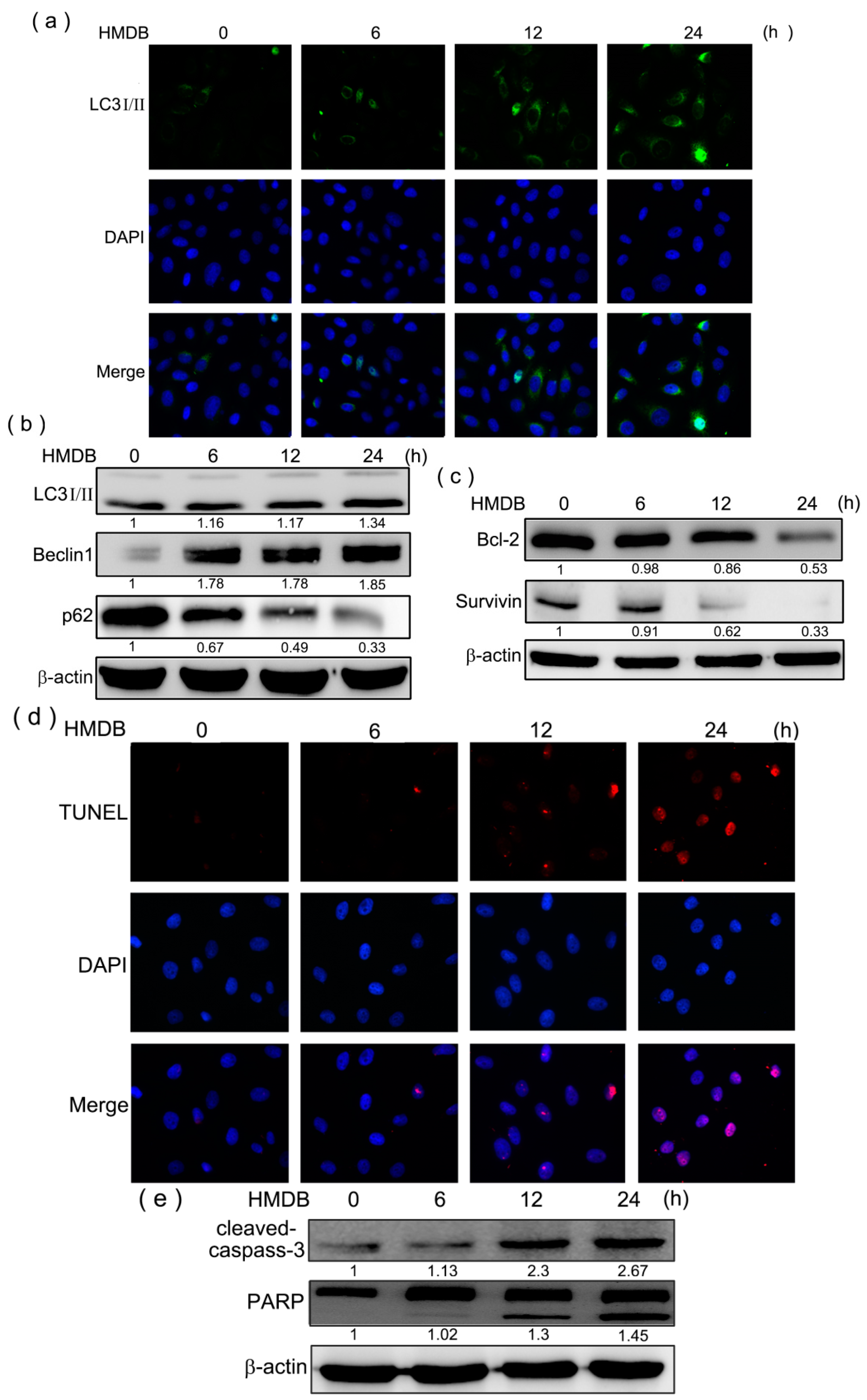
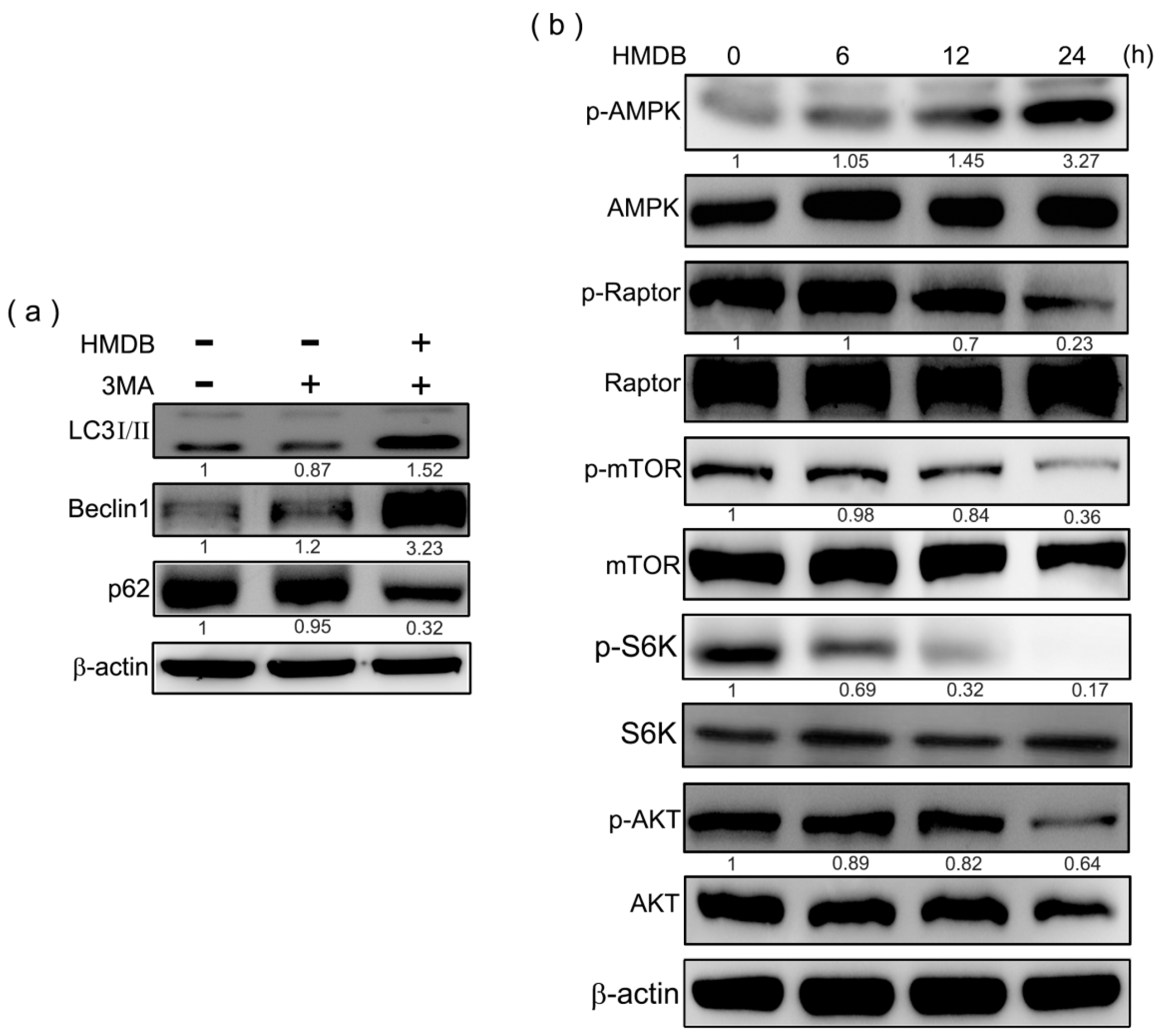
2.4. Formation of Autophagic Vacuoles with the Increases in Microtubule-Associated Protein 1 Light Chain 3 (LC3) and Beclin-1 Expression Induced by HMDB
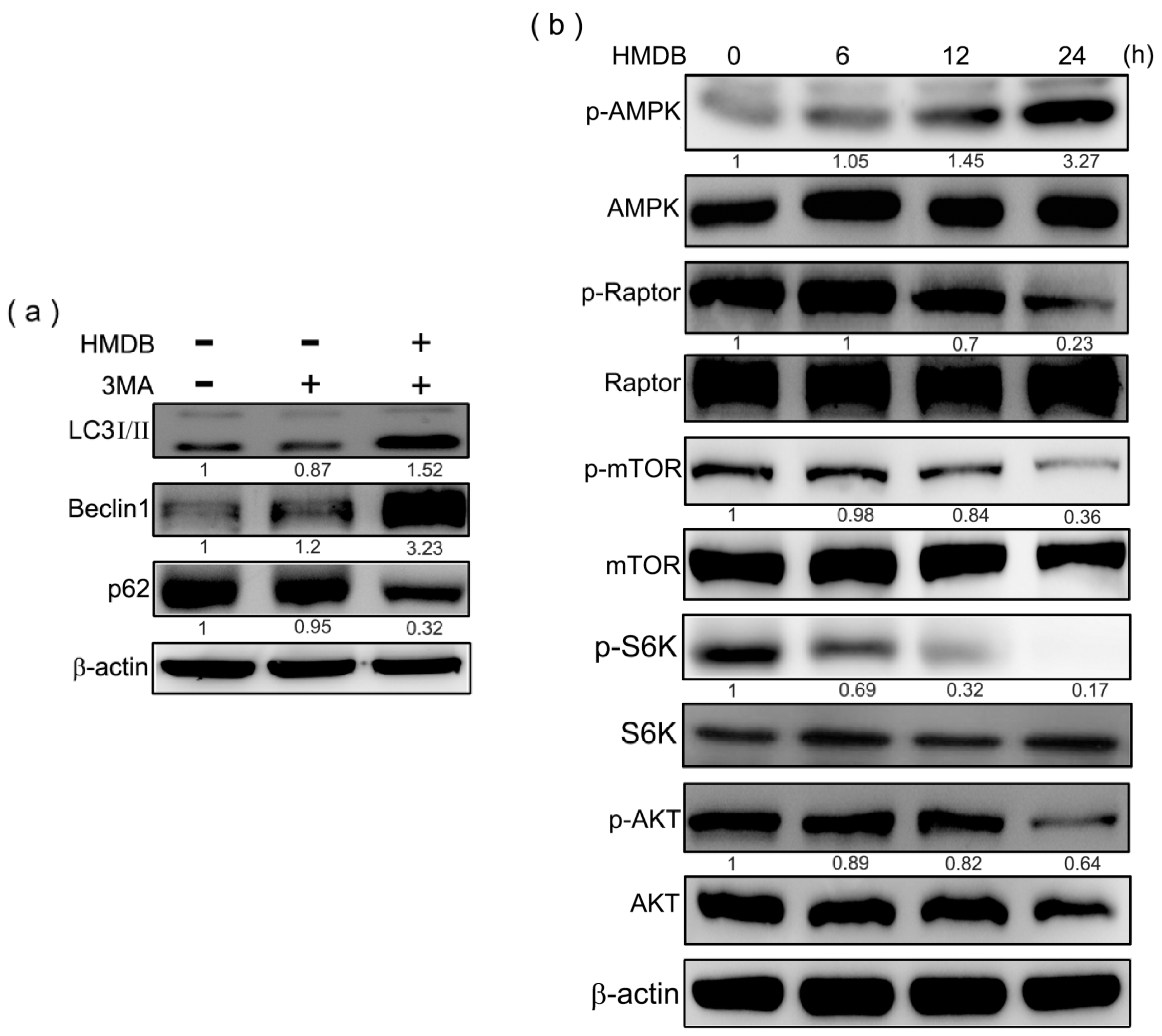
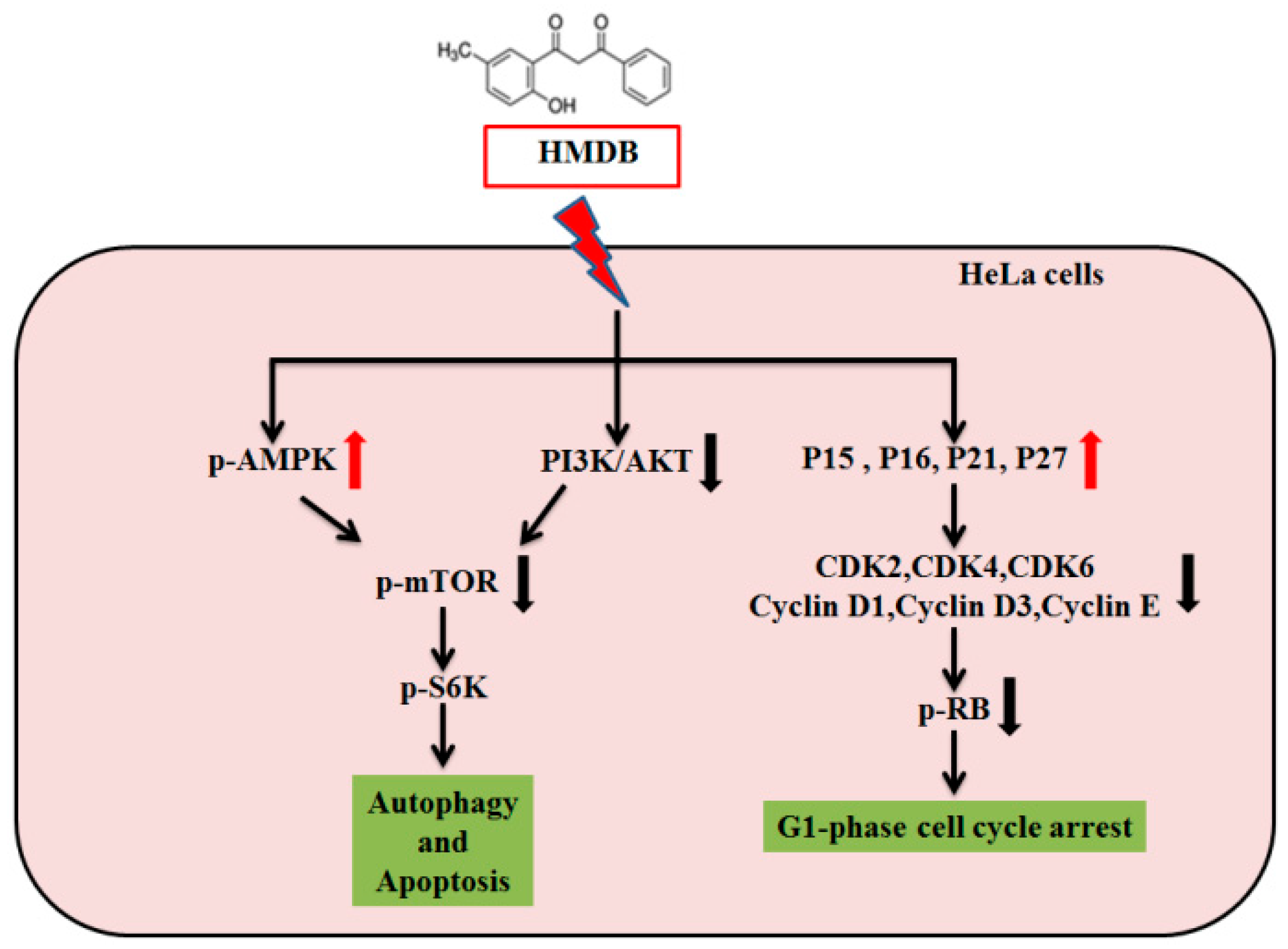
2.5. HMDB-Induced Autophagy Is Linked to the Mediation of AMPK/mTOR and Akt/mTOR Signaling
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture
4.3. MTT Assay
4.4. Trypan Blue Dye Exclusion Assay
4.5. Cell Cycle Analysis
4.6. Western Blotting
4.7. RNA Extraction, cDNA Synthesis, and qRT-PCR
4.8. Neutral Red Staining
4.9. Monodansylcadaverine and Acridine Orange Staining
4.10. Immunofluorescent Staining
4.11. TUNEL Assay
4.12. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.F.; Canfell, K.; Lew, J.B.; Qiao, Y.L. The burden of cervical cancer in China: Synthesis of the evidence. Int. J. Cancer 2012, 130, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Roa, J.C.; Garcia, P.; Gomez, J.; Fernández, W.; Gaete, F.; Espinoza, A.; Lepetic, A.; Suarez, E. HPV genotyping from invasive cervical cancer in Chile. Int. J. Gynecol. Obstet. 2009, 105, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Kwon, J.H.; Kim, E.H.; Kim, E.S.; Choi, K.Y. HMGB2 stabilizes p53 by interfering with E6/E6AP-mediated p53 degradation in human papillomavirus-positive HeLa cells. Cancer Lett. 2010, 292, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Cornelio, D.B.; Roesler, R.; Schwartsmann, G. Emerging therapeutic agents for cervical cancer. Recent Pat. Anticancer Drug Discov. 2009, 4, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Choudhari, A.S.; Suryavanshi, S.A.; Kaul-Ghanekar, R. The aqueous extract of Ficus religiosa induces cell cycle arrest in human cervical cancer cell lines SiHa (HPV-16 positive) and apoptosis in HeLa (HPV-18 positive). PLoS ONE 2013, 8, e70127. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Chen, J.J. Role of Cdk1 in DNA damage-induced G1 checkpoint abrogation by the human papillomavirus E7 oncogene. Cell Cycle 2014, 13, 3249–3259. [Google Scholar] [CrossRef] [PubMed]
- Perez de Castro, I.; de Carcer, G.; Malumbres, M. A census of mitotic cancer genes: New insights into tumor cell biology and cancer therapy. Carcinogenesis 2007, 28, 899–912. [Google Scholar] [CrossRef] [PubMed]
- Brenna, S.M.; Syrjanen, K.J. Regulation of cell cycles is of key importance in human papillomavirus (HPV)-associated cervical carcinogenesis. Sao Paulo Med. J. 2003, 121, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Kaldis, P. The cdk-activating kinase (CAK): From yeast to mammals. Cell. Mol. Life Sci. 1999, 55, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Capparelli, C.; Chiavarina, B.; Whitaker-Menezes, D.; Pestell, T.G.; Pestell, R.G.; Hulit, J.; Andò, S.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, ”fueling” tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle 2012, 11, 3599–3610. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Martin, V.; Gomez-Manzano, C.; Johnson, D.G.; Alonso, M.; White, E.; Xu, J.; McDonnell, T.J.; Shinojima, N.; Fueyo, J. The RB-E2F1 pathway regulates autophagy. Cancer Res. 2010, 70, 7882–7893. [Google Scholar] [CrossRef] [PubMed]
- Singletary, K.; Milner, J. Diet, autophagy, and cancer: A review. Cancer Epidemiol. Biomark. Prev. 2008, 17, 1596–1610. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lenardo, M.J. Reactive oxygen species regulate autophagy through redox-sensitive proteases. Dev. Cell 2007, 12, 484–485. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.P.; Yang, M.C.; Liu, H.S.; Lin, Y.S.; Lei, H.Y. Concanavalin A induces autophagy in hepatoma cells and has a therapeutic effect in a murine in situ hepatoma model. Hepatology 2007, 45, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Kanzawa, T.; Kondo, Y.; Ito, H.; Kondo, S.; Germano, I. Induction of autophagic cell death in malignant glioma cells by arsenic trioxide. Cancer Res. 2003, 63, 2103–2108. [Google Scholar] [PubMed]
- Eskelinen, E.L.; Saftig, P. Autophagy: A lysosomal degradation pathway with a central role in health and disease. Biochim. Biophys. Acta 2009, 1793, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Miracco, C.; Cevenini, G.; Franchi, A.; Luzi, P.; Cosci, E.; Mourmouras, V.; Monciatti, I.; Mannucci, S.; Biagioli, M.; Toscano, M.; et al. Beclin 1 and LC3 autophagic gene expression in cutaneous melanocytic lesions. Hum. Pathol. 2010, 41, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.H.; Li, L.; Peng, Z.L.; Duan, Z.L. Effect of autophagy gene Beclin 1 on the growth of cervical cancer HeLa cells in vitro and vivo. Zhonghua Zhong Liu Za Zhi 2011, 33, 804–809. [Google Scholar] [PubMed]
- Klionsky, D.J.; Abeliovich, H.; Agostinis, P.; Agrawal, D.K.; Aliev, G.; Askew, D.S.; Baba, M.; Baehrecke, E.H.; Bahr, B.A.; Ballabio, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008, 4, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Pan, X.; Li, F.; Zhang, Y.; Lu, X. Expression of Beclin 1 and LC3 in FIGO stage I-II cervical squamous cell carcinoma and relationship to survival. Tumour Biol. 2012, 33, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.H.; Huang, M.C.; Wang, Y.J.; Lin, J.K.; Lin, C.H. Induction of apoptosis by hydroxydibenzoylmethane through coordinative modulation of cyclin D3, Bcl-XL, and Bax, release of cytochrome c, and sequential activation of caspases in human colorectal carcinoma cells. J. Agric. Food Chem. 2003, 51, 3977–3984. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.H.; Sin, Y.H.; Lai, C.S.; Wang, Y.J.; Lin, J.K.; Wang, M.; Ho, C.T. Induction of apoptosis by 1-(2-hydroxy-5-methylphenyl)-3-phenyl-1,3-propanedione through reactive oxygen species production, GADD153 expression, and caspases activation in human epidermoid carcinoma cells. J. Agric. Food Chem. 2005, 53, 9039–9049. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.C.; Li, C.F.; Ko, C.Y.; Pan, M.H.; Chen, P.J.; Tseng, J.T.; Wu, W.C.; Chang, W.C.; Huang, A.M.; Sterneck, E.; et al. CEBPD reverses RB/E2F1-mediated gene repression and participates in HMDB-induced apoptosis of cancer cells. Clin. Cancer Res. 2010, 16, 5770–5780. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, M.; Grogan, M. Guidelines for the treatment of recurrent and metastatic cervical cancer. Oncologist 2002, 7, 342–347. [Google Scholar] [PubMed]
- Suh, D.H.; Kim, J.W.; Kim, K.; Kim, H.J.; Lee, K.H. Major clinical research advances in gynecologic cancer in 2012. J. Gynecol. Oncol. 2013, 24, 66–82. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liu, J.H.; Jin, L.; Lin, S.M.; Yang, Y.; Sui, Y.X.; Shi, H. Over-expression of the Beclin1 gene upregulates chemosensitivity to anti-cancer drugs by enhancing therapy-induced apoptosis in cervix squamous carcinoma CaSki cells. Cancer Lett. 2010, 294, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 2005, 30, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, E.S.; Wang, J.Y. Targeting the RB-pathway in cancer therapy. Clin. Cancer Res. 2010, 16, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavaré, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Lei, Y.; Liu, R.; Li, J.; Yuan, K.; Li, Y.; Chen, Y.; Liu, Y.; Lu, Y.; Edwards, C.K., III; et al. Pyrvinium targets autophagy addiction to promote cancer cell death. Cell Death Dis. 2013, 4, e614. [Google Scholar]
- Kelekar, A. Autophagy. Ann. N. Y. Acad. Sci. 2005, 1066, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Hoyer-Hansen, M.; Bastholm, L.; Mathiasen, I.S.; Elling, F.; Jäättelä, M. Vitamin D analog EB1089 triggers dramatic lysosomal changes and Beclin 1-mediated autophagic cell death. Cell Death Differ. 2005, 12, 1297–1309. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.H.; Peng, Z.L.; Duan, Z.L.; Liu, H. Expression and clinical significance of autophagy gene Beclin 1 in cervical squamous cell carcinoma. Sichuan Da Xue Xue Bao Yi Xue Ban 2006, 37, 860–863. [Google Scholar] [PubMed]
- Cheng, H.Y.; Zhang, Y.N.; Wu, Q.L.; Sun, X.M.; Sun, J.R.; Huang, X. Expression of beclin 1, an autophagy-related protein, in human cervical carcinoma and its clinical significance. Eur. J. Gynaecol. Oncol. 2012, 33, 15–20. [Google Scholar] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Cornu, M.; Albert, V.; Hall, M.N. mTOR in aging, metabolism, and cancer. Curr. Opin. Genet. Dev. 2013, 23, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, C.; Chen, S.; Chen, S.; Ye, Y.; Guo, M.; Ren, Q.; Liu, L.; Zhang, H.; Xu, C.; et al. Activation of AMPK and inactivation of Akt result in suppression of mTOR-mediated S6K1 and 4E-BP1 pathways leading to neuronal cell death in in vitro models of Parkinson’s disease. Cell Signal. 2014, 26, 1680–1689. [Google Scholar] [CrossRef] [PubMed]
- Slomovitz, B.M.; Coleman, R.L. The PI3K/AKT/mTOR pathway as a therapeutic target in endometrial cancer. Clin. Cancer Res. 2012, 18, 5856–5864. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.; Afaq, F.; Khan, N.; Johnson, J.J.; Khusro, F.H.; Mukhtar, H. Fisetin induces autophagic cell death through suppression of mTOR signaling pathway in prostate cancer cells. Carcinogenesis 2010, 31, 1424–1433. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, J.-H.; Hsu, L.-S.; Huang, H.-C.; Lin, C.-L.; Pan, M.-H.; Hong, H.-M.; Chen, W.-J. 1-(2-Hydroxy-5-methylphenyl)-3-phenyl-1,3-propanedione Induces G1 Cell Cycle Arrest and Autophagy in HeLa Cervical Cancer Cells. Int. J. Mol. Sci. 2016, 17, 1274. https://doi.org/10.3390/ijms17081274
Tsai J-H, Hsu L-S, Huang H-C, Lin C-L, Pan M-H, Hong H-M, Chen W-J. 1-(2-Hydroxy-5-methylphenyl)-3-phenyl-1,3-propanedione Induces G1 Cell Cycle Arrest and Autophagy in HeLa Cervical Cancer Cells. International Journal of Molecular Sciences. 2016; 17(8):1274. https://doi.org/10.3390/ijms17081274
Chicago/Turabian StyleTsai, Jie-Heng, Li-Sung Hsu, Hsiu-Chen Huang, Chih-Li Lin, Min-Hsiung Pan, Hui-Mei Hong, and Wei-Jen Chen. 2016. "1-(2-Hydroxy-5-methylphenyl)-3-phenyl-1,3-propanedione Induces G1 Cell Cycle Arrest and Autophagy in HeLa Cervical Cancer Cells" International Journal of Molecular Sciences 17, no. 8: 1274. https://doi.org/10.3390/ijms17081274
APA StyleTsai, J.-H., Hsu, L.-S., Huang, H.-C., Lin, C.-L., Pan, M.-H., Hong, H.-M., & Chen, W.-J. (2016). 1-(2-Hydroxy-5-methylphenyl)-3-phenyl-1,3-propanedione Induces G1 Cell Cycle Arrest and Autophagy in HeLa Cervical Cancer Cells. International Journal of Molecular Sciences, 17(8), 1274. https://doi.org/10.3390/ijms17081274