
Genome-Wide Analysis of Codon Usage Bias in Epichloë festucae



Abstract
:
1. Introduction
2. Results and Discussion
2.1. Base Composition of E. festucae
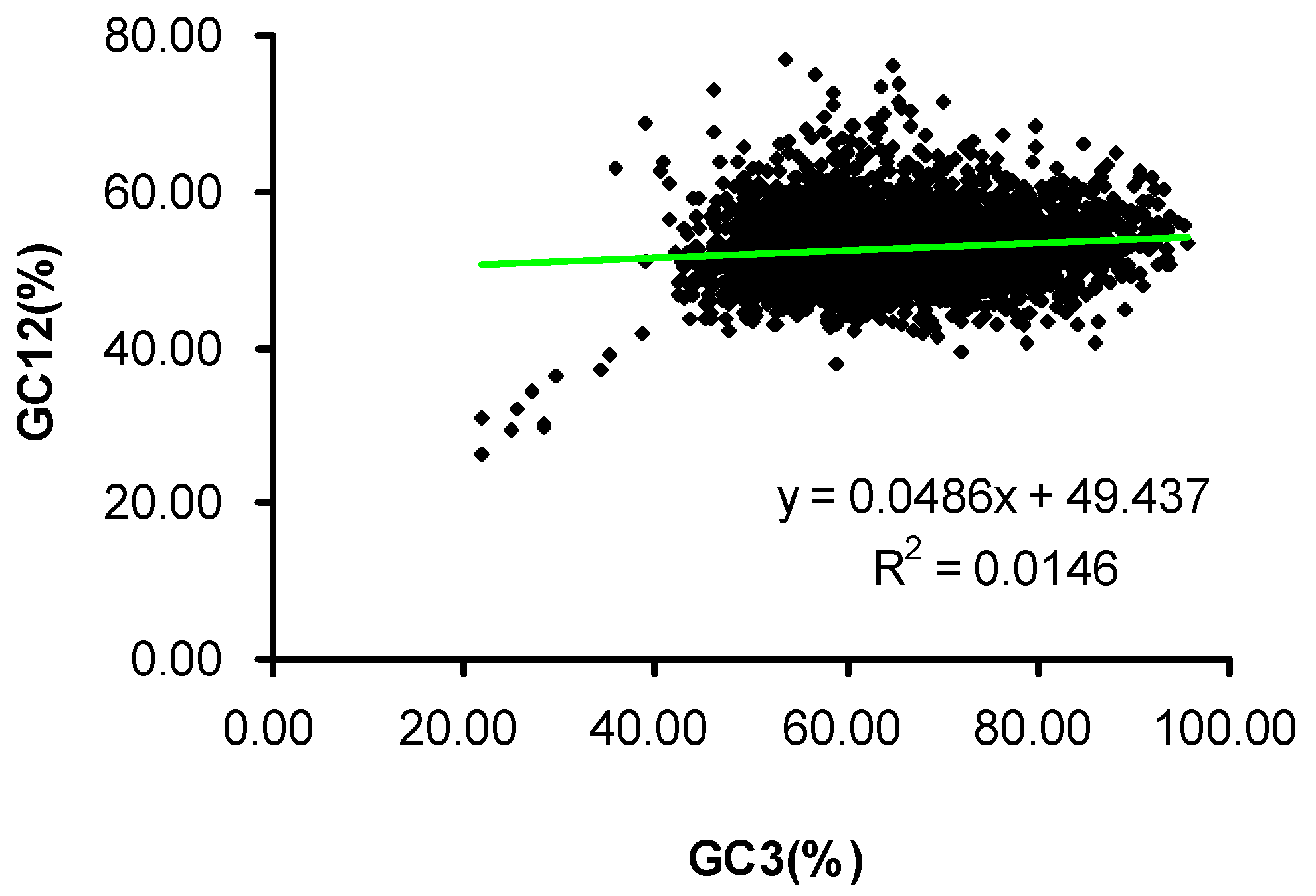
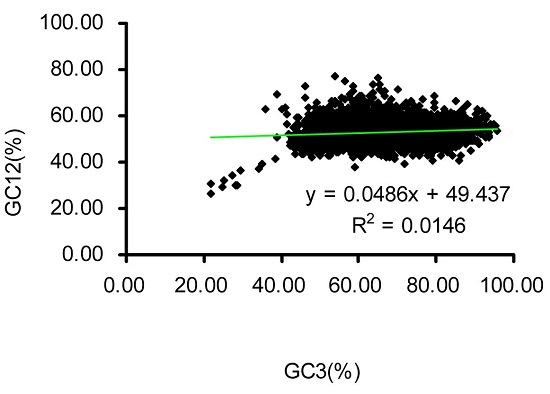
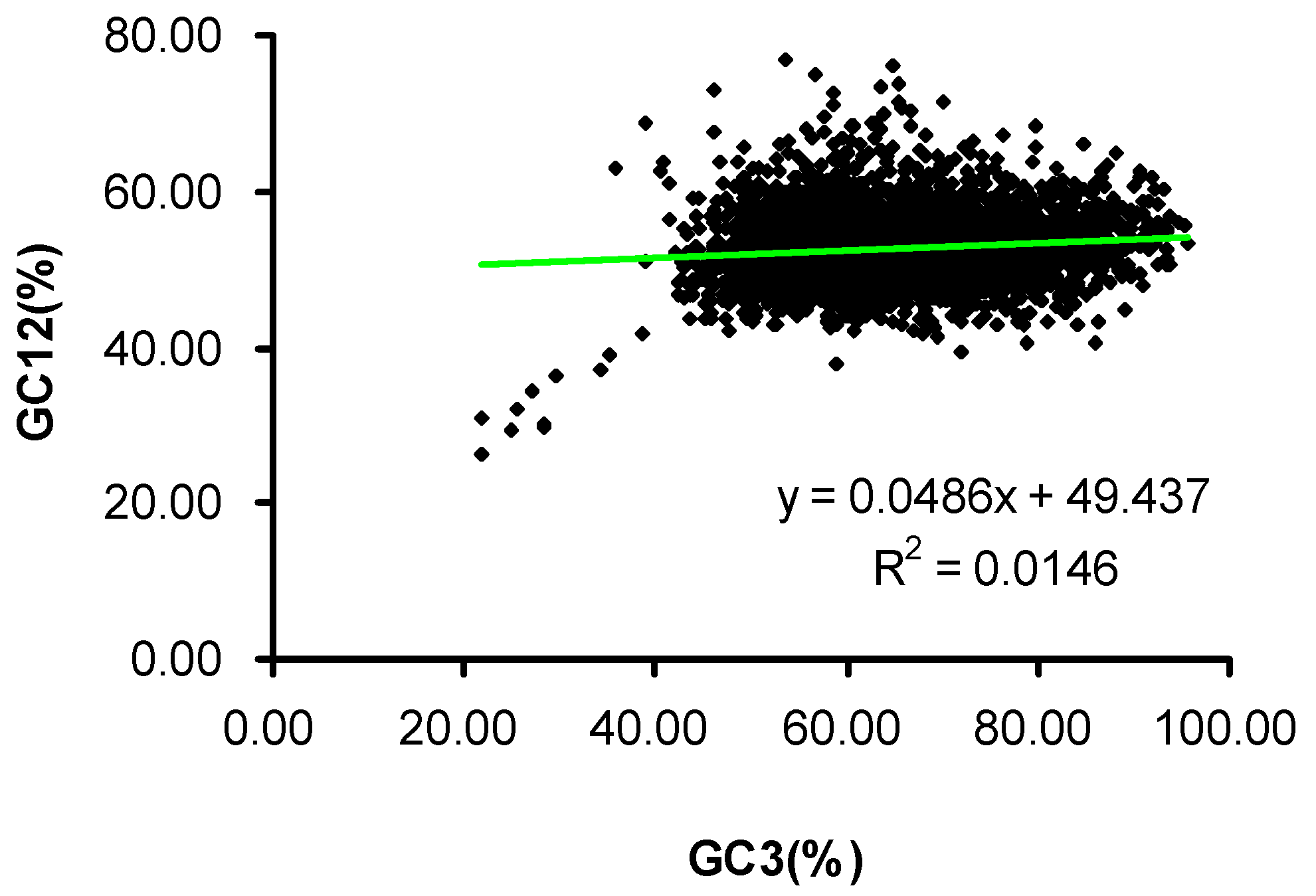
2.2. Neutrality Plot
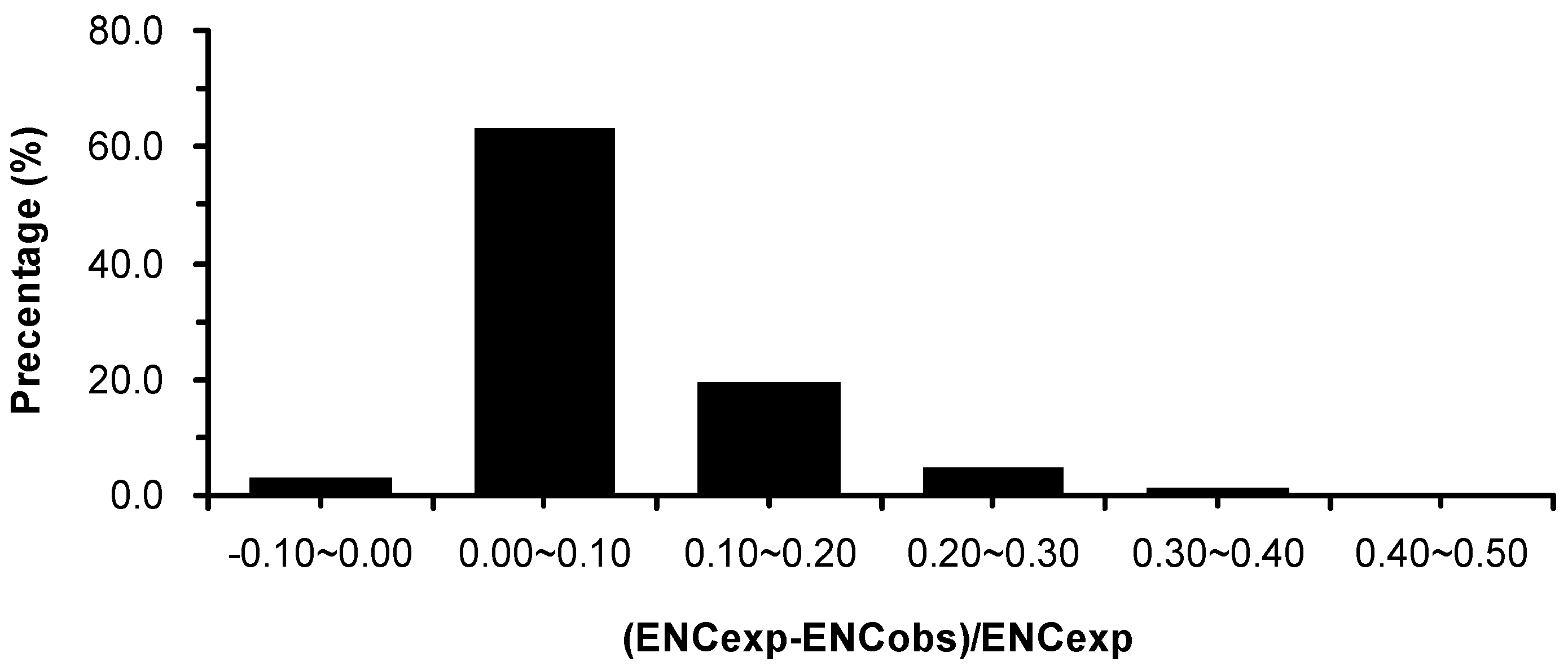
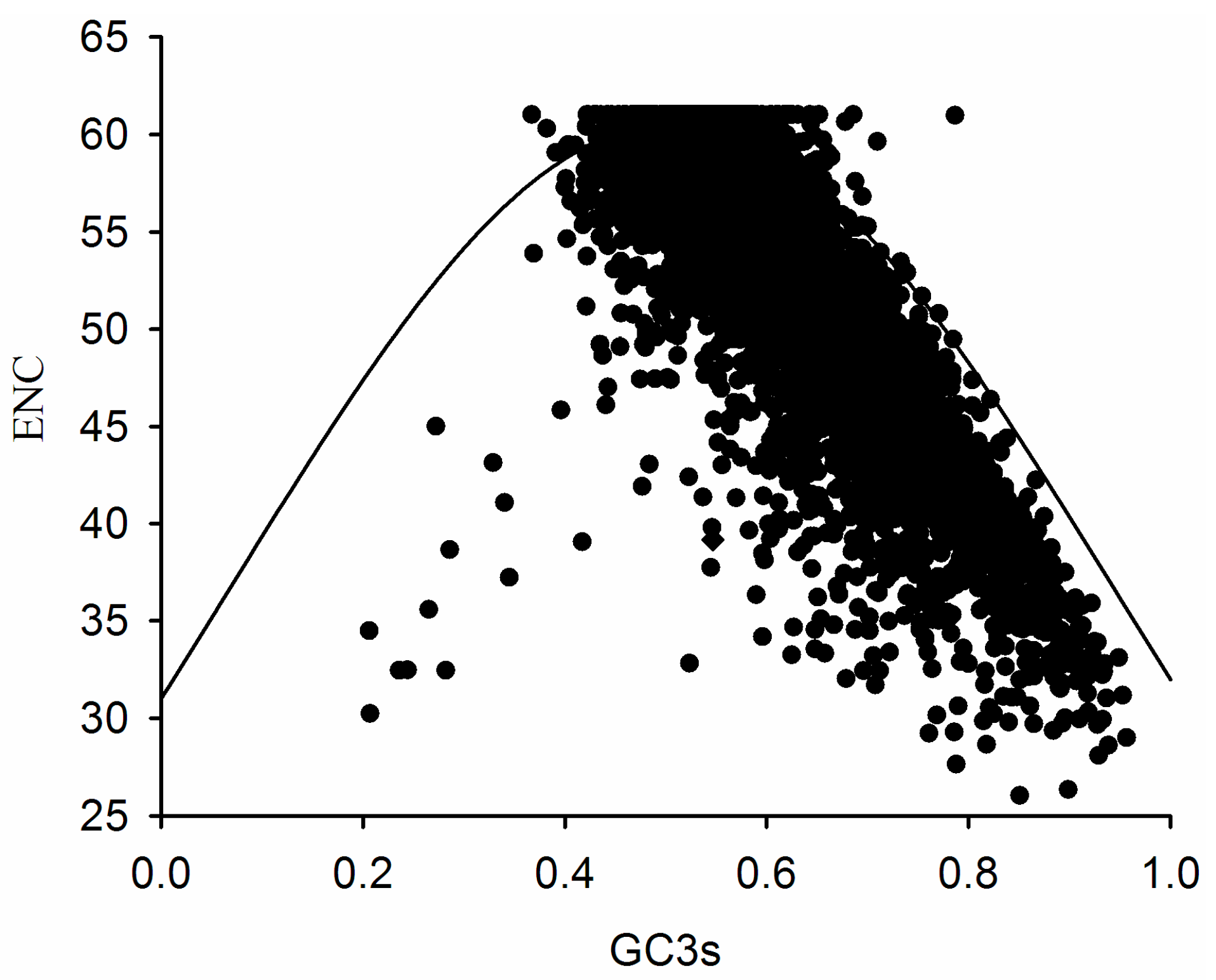
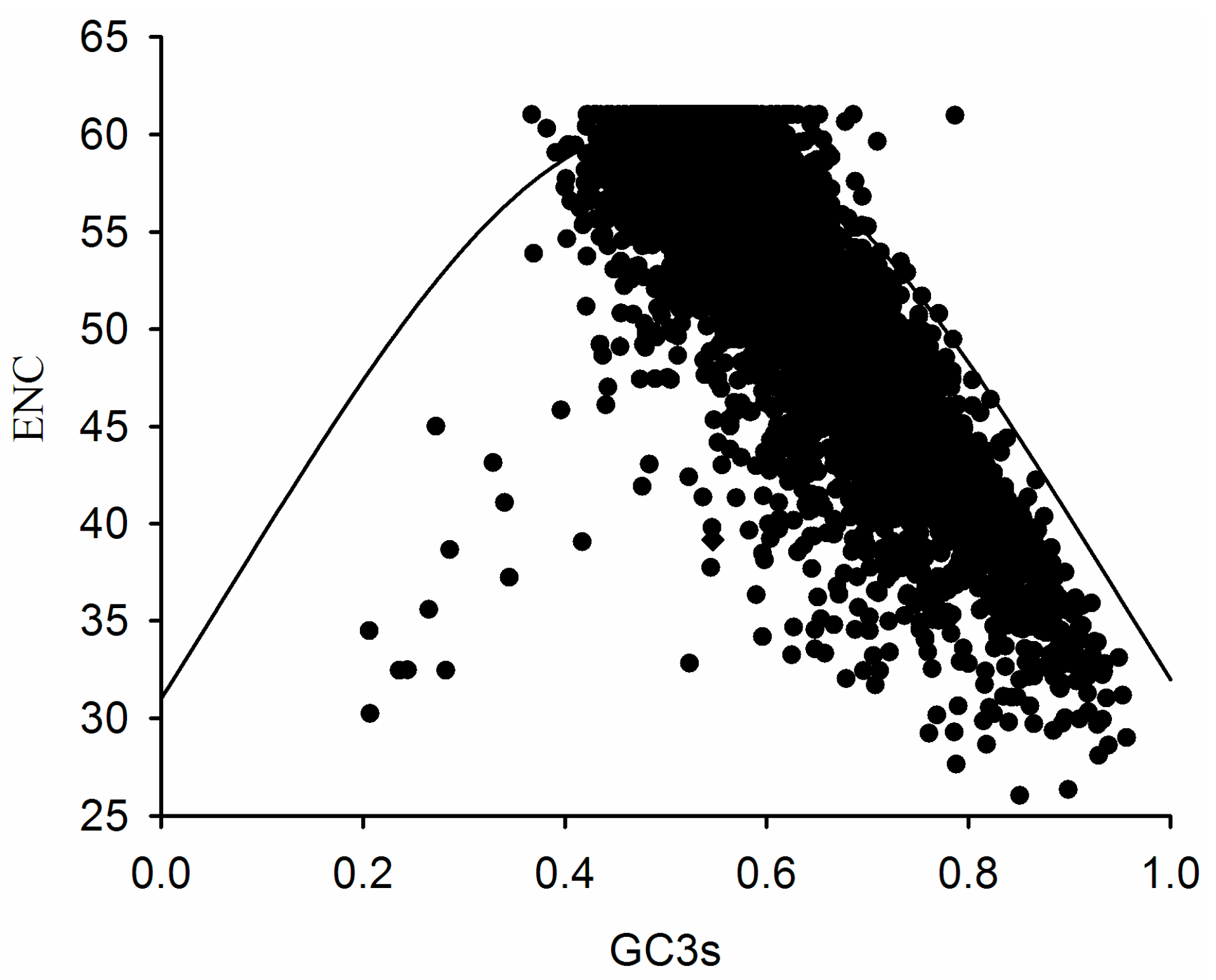
2.3. Association between Effective Number of Codons (ENC) and GC3s
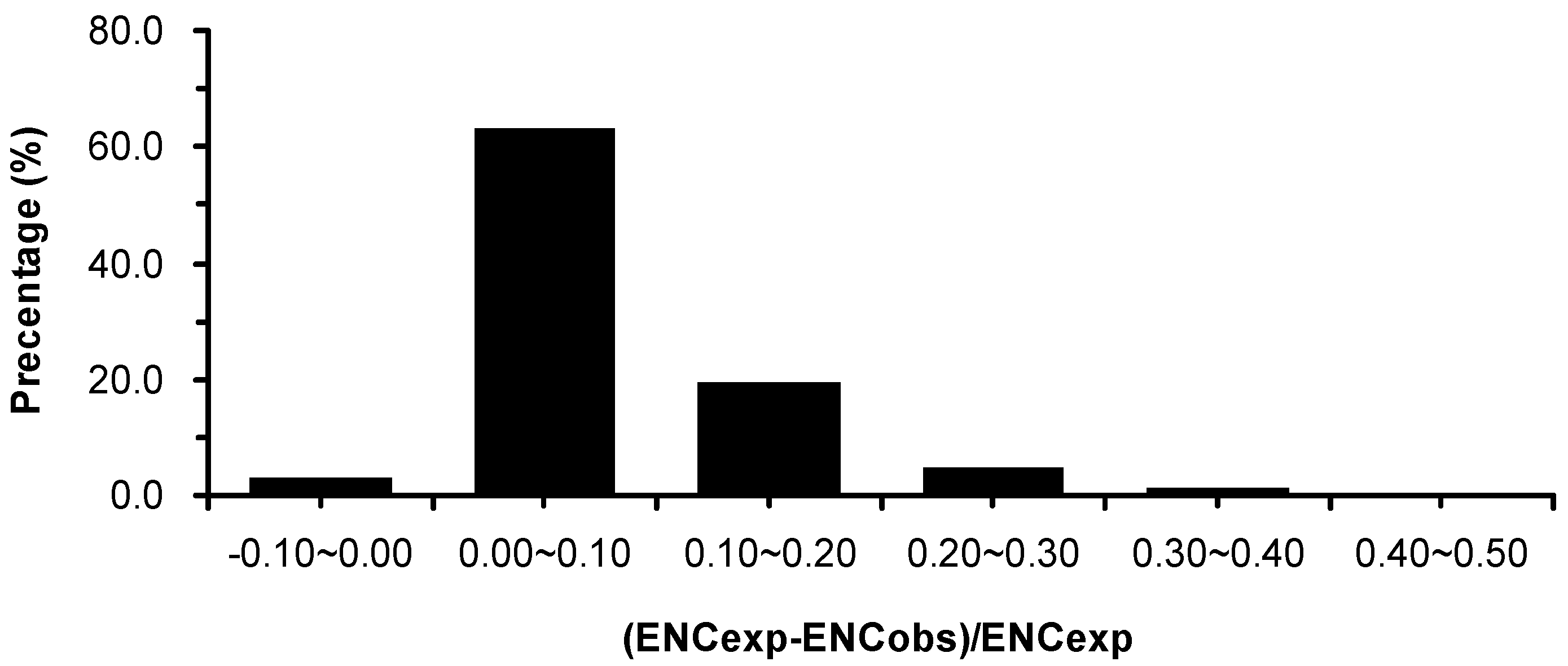
2.4. Correlations between Codon Usage Bias, Hydrophobicity, Aromaticity, and Gene Length in E. festucae
2.5. Optimal Codons in E. festucae
3. Materials and Methods
3.1. Indices of Codon Usage and Synonymous Codon Usage Bias
3.2. ENC Plot
3.3. Neutrality Plot
3.4. Determination of Optimal Codons
3.5. Correspondence Analysis of RSCU
3.6. Software Used
3.7. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Grantham, R.; Gautier, C.; Gouy, M.; Mercier, R.; Pave, A. Codon catalog usage and the genome hypothesis. Nucleic Acids Res. 1980, 8, 197–197. [Google Scholar]
- Martin, A.; Bertranpetit, J.; Oliver, J.; Medina, J. Variation in g + c-content and codon choice: Differences among synonymous codon groups in vertebrate genes. Nucleic Acids Res. 1989, 17, 6181–6189. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.H. Codon usage in regulatory genes in Escherichia coli does not reflect selection for ‘rare’codons. Nucleic Acids Res. 1986, 14, 7737–7749. [Google Scholar] [CrossRef] [PubMed]
- Duret, L.; Mouchiroud, D. Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis. Proc. Natl. Acad. Sci. USA 1999, 96, 4482–4487. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Zhou, T.; Ma, J.; Sun, X.; Lu, Z. Analysis of synonymous codon usage in sars coronavirus and other viruses in the nidovirales. Virus Res. 2004, 101, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Van der, L.; Marx, G.; de Farias, S.T. Correlation between codon usage and thermostability. Extremophiles 2006, 10, 479–481. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Cowe, E.; Higgins, D.G.; Shields, D.C.; Wolfe, K.H.; Wright, F. Codon usage patterns in Escherichia coli, Bacillus subtilis, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Drosophila melanogaster and Homo sapiens; a review of the considerable within-species diversity. Nucleic Acids Res. 1988, 16, 8207–8211. [Google Scholar] [CrossRef] [PubMed]
- Chiapello, H.; Lisacek, F.; Caboche, M.; Henaut, A. Codon usage and gene function are related in sequences of Arabidopsis thaliana. Gene 1998, 209, GC1–GC38. [Google Scholar] [CrossRef]
- Moriyama, E.N.; Powell, J.R. Gene length and codon usage bias in Drosophila melanogaster, Saccharomyces cerevisiae and Escherichia coli. Nucleic Acids Res. 1998, 26, 3188–3193. [Google Scholar] [CrossRef] [PubMed]
- Sueoka, N.; Kawanishi, Y. DNA g + c content of the third codon position and codon usage biases of human genes. Gene 2000, 261, 53–62. [Google Scholar] [CrossRef]
- Marais, G.; Mouchiroud, D.; Duret, L. Does recombination improve selection on codon usage? Lessons from nematode and fly complete genomes. Proc. Natl. Acad. Sci. USA 2001, 98, 5688–5692. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Gu, W.; Ma, J.; Sun, X.; Lu, Z. Analysis of synonymous codon usage in h5n1 virus and other influenza a viruses. Biosystems 2005, 81, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.T.; Gu, Y.X.; Liu, Y.S. Analysis of synonymous codon usage in dengue viruses. J. Anim. Vet. Adv. 2013, 12, 88–98. [Google Scholar]
- Butt, A.M.; Nasrullah, I.; Tong, Y. Genome-wide analysis of codon usage and influencing factors in chikungunya viruses. PLoS ONE 2014, 9, e90905. [Google Scholar] [CrossRef] [PubMed]
- Schardl, C.L.; Balestrini, R.; Florea, S.; Zhang, D.X.; Scott, B. Epichloë endophytes: Clavicipitaceous symbionts of grasses. In Plant Relationships; Springer: London, UK, 2009; pp. 275–306. [Google Scholar]
- Leuchtmann, A.; Bacon, C.W.; Schardl, C.L.; White, J.F.; Tadych, M. Nomenclatural realignment of neotyphodium species with genus Epichloë. Mycologia 2014, 106, 202–215. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Takemoto, D.; Chujo, T.; Scott, B. Fungal endophytes of grasses. Curr. Opin. Plant Biol. 2012, 15, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Clay, K.; Schardl, C. Evolutionary origins and ecological consequences of endophyte symbiosis with grasses. Am. Nat. 2002, 160, S99–S127. [Google Scholar] [CrossRef] [PubMed]
- Hahn, H.; McManus, M.T.; Warnstorff, K.; Monahan, B.J.; Young, C.A.; Davies, E.; Tapper, B.A.; Scott, B. Neotyphodium fungal endophytes confer physiological protection to perennial ryegrass (Lolium perenne L.) subjected to a water deficit. Environ. Exp. Bot. 2008, 63, 183–199. [Google Scholar] [CrossRef]
- Schardl, C.L.; Leuchtmann, A.; Spiering, M.J. Symbioses of grasses with seedborne fungal endophytes. Annu. Rev. Plant Biol. 2004, 55, 315–340. [Google Scholar] [CrossRef] [PubMed]
- Schardl, C.L.; Grossman, R.B.; Nagabhyru, P.; Faulkner, J.R.; Mallik, U.P. Loline alkaloids: Currencies of mutualism. Phytochemistry 2007, 68, 980–996. [Google Scholar] [CrossRef] [PubMed]
- Latch, G. An overview of Neotyphodium-grass interactions. In Neotyphodium/Grass Interactions; Springer: London, UK, 1997; pp. 1–11. [Google Scholar]
- Leuchtmann, A.; Schardl, C.L.; Siegel, M.R. Sexual compatibility and taxonomy of a new species of Epichloë symbiotic with fine fescue grasses. Mycologia 1994, 86, 802–812. [Google Scholar] [CrossRef]
- Schardl, C.L. Epichloë festucae and related mutualistic symbionts of grasses. Fungal Genet. Biol. 2001, 33, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.J.; Bennett, R.J.; Schmid, J. Growth of Epichloë/Neotyphodium and p-endophytes in leaves of Lolium and Festuca grasses. Mycol. Res. 2002, 106, 93–106. [Google Scholar] [CrossRef]
- Scott, B.; Becker, Y.; Becker, M.; Cartwright, G. Morphogenesis, growth, and development of the grass symbiont Epichlöe festucae. In Morphogenesis and Pathogenicity in Fungi; Springer: London, UK, 2012; pp. 243–264. [Google Scholar]
- Schardl, C.L.; Young, C.A.; Uljana, H.; Amyotte, S.G.; Kalina, A.; Calie, P.J.; Fleetwood, D.J.; Haws, D.C.; Neil, M.; Birgitt, O. Plant-symbiotic fungi as chemical engineers: Multi-genome analysis of the clavicipitaceae reveals dynamics of alkaloid loci. PLoS Genet. 2013, 9, e1003323. [Google Scholar] [CrossRef] [PubMed]
- Bulmer, M. The selection-mutation-drift theory of synonymous codon usage. Genetics 1991, 129, 897. [Google Scholar] [PubMed]
- Nakamura, Y.; Gojobori, T.; Ikemura, T. Codon usage tabulated from the international DNA sequence databases; its status 1999. Nucleic Acids Res. 1999, 27, 292. [Google Scholar] [CrossRef] [PubMed]
- Wright, F. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Fuglsang, A. The ‘effective number of codons’ revisited. Biochem. Bioph. Res. Commun. 2004, 317, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Shields, D.C.; Sharp, P.M. Synonymous codon usage in bacillus subtilis reflects both translational selection and mutational biases. Nucleic Acids Res. 1987, 15, 8023–8040. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.J.; Zhou, J.; Li, Z.F.; Wang, L.; Gu, X.; Zhong, Y. Comparative analysis of codon usage patterns among mitochondrion, chloroplast and nuclear genes in Triticum aestivum L. J. Integr. Plant Biol. 2007, 49, 246–254. [Google Scholar] [CrossRef]
- Kawabe, A.; Miyashita, N.T. Patterns of codon usage bias in three dicot and four monocot plant species. Genes Genet. Syst. 2003, 78, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Ikemura, T. Correlation between the abundance of Escherichia coli transfer rnas and the occurrence of the respective codons in its protein genes. J. Mol. Biol. 1981, 146, 1–21. [Google Scholar] [CrossRef]
- Varenne, S.; Baty, D.; Verheij, H.; Shire, D.; Lazdunski, C. The maximum rate of gene expression is dependent in the downstream context of unfavourable codons. Biochimie 1989, 71, 1221–1229. [Google Scholar] [CrossRef]
- Li, H.; Luo, L.F. The relations of gene expression level with codon usage and its prediction. J. Inner Mong. Univ. (Nat. Sci.) 1995, 26, 544–561. [Google Scholar]
- Perrière, G.; Thioulouse, J. Use and misuse of correspondence analysis in codon usage studies. Nucleic Acids Res. 2002, 30, 4548–4555. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Brown, C.J.; Forney, L.J.; Top, E.M. Comparison of correspondence analysis methods for synonymous codon usage in bacteria. DNA Res. 2008, 15, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, A.T.; Sharp, P.M. Codon usage in Aspergillus nidulans. Mol. Genet. Genom. 1991, 230, 288–294. [Google Scholar] [CrossRef]
- Liu, Q.P.; Feng, Y.; Zhao, X.A.; Dong, H.; Xue, Q.Z. Synonymous codon usage bias in Oryza sativa. Plant Sci. 2004, 167, 101–105. [Google Scholar] [CrossRef]
- Liu, H.; He, R.; Zhang, H.; Huang, Y.; Tian, M.; Zhang, J. Analysis of synonymous codon usage in Zea mays. Mol. Biol. Rep. 2010, 37, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Cowe, E. Synonymous codon usage in Saccharomyces cerevisiae. Yeast 1991, 7, 657–678. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Liu, S.Y.; Zheng, H.; Li, B.; Qi, Q.; Wei, L.; Zhao, T.Y.; He, J.; Sun, J.C. Non-uniqueness of factors constraint on the codon usage in bombyx mori. BMC Genom. 2015, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Genome Projects at University of Kentucky. Available online: http://csbio-l.csr.uky.edu/endophyte/ (accessed on 14 July 2016).
- GenBank. Available online: http://www.ncbi.nlm.nih.gov (accessed on 14 July 2016).
- Jansen, R.; Bussemaker, H.J.; Gerstein, M. Revisiting the codon adaptation index from a whole-genome perspective: Analyzing the relationship between gene expression and codon occurrence in yeast using a variety of models. Nucleic Acids Res. 2003, 31, 2242–2251. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.H. The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef]
- Sueoka, N. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. USA 1988, 85, 2653–2657. [Google Scholar] [CrossRef] [PubMed]
- Mobyle server. Available online: http://mobyle.pasteur.fr (accessed on 14 July 2016).
- Codon W. Available online: http://codonw.sourceforge.net/culong.html#CodonW (accessed on 14 July 2016).
- CHIPS. Available online: http://mobyle.pasteur.fr/cgi-bin/portal.py#forms::chips (accessed on 14 July 2016).
- CUSP. Available online: http://mobyle.pasteur.fr/cgi-bin/portal.py#forms::cusp (accessed on 14 July 2016).
- Kramer, C.Y. Extension of multiple range tests to group means with unequal numbers of replications. Biometrics 1956, 12, 307–310. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Genes | Codons | GC (%) | GC1 (%) | GC2 (%) | GC3 (%) | GC3s (%) | T3s (%) | C3s (%) | A3s (%) | G3s (%) | Gravy | Aro | ENC | CAI |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | 4870 | 2498255 | 56.41 ± 4.60 | 58.68 ± 5.30 | 46.43 ± 5.80 | 64.11 ± 10.16 | 62.86 ± 10.52 | 23.91 ± 7.41 | 43.85 ± 9.99 | 21.13 ± 7.5 | 33.66 ± 7.37 | −0.41 ± 0.37 | 0.07 ± 0.02 | 51.58 ± 7.14 | 0.22 ± 0.04 |
| Length | GC | GC1 | GC2 | GC3 | GC3S | A3S | T3S | C3S | G3S | GRAVY | AROMO | ENC | CAI | AXIS1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GC | −0.191 ** | ||||||||||||||
| GC1 | −0.041 ** | 0.568 ** | |||||||||||||
| GC2 | −0.069 ** | 0.443 ** | 0.085 ** | ||||||||||||
| GC3 | −0.199 ** | 0.808 ** | 0.201 ** | −0.013 | |||||||||||
| GC3S | −0.200 ** | 0.816 ** | 0.214 ** | −0.004 | 0.998 ** | ||||||||||
| A3S | 0.189 ** | −0.710 ** | −0.181 ** | −0.028 * | −0.854 ** | −0.857 ** | |||||||||
| T3S | 0.160 ** | −0.753 ** | −0.196 ** | −0.092 ** | −0.868 ** | −0.868 ** | 0.508 ** | ||||||||
| C3S | −0.152 ** | 0.635 ** | 0.120 ** | −0.063 ** | 0.835 ** | 0.838 ** | −0.760 ** | −0.671 ** | |||||||
| G3S | −0.117 ** | 0.393 ** | 0.168 ** | −0.194 ** | 0.557 ** | 0.555 ** | −0.383 ** | −0.530 ** | 0.060 ** | ||||||
| GRAVY | −0.065 ** | 0.033 * | −0.180 ** | −0.138 ** | 0.217 ** | 0.207 ** | −0.336 ** | −0.114 ** | 0.198 ** | −0.063 ** | |||||
| AROMO | −0.040 ** | −0.195 ** | −0.412 ** | −0.317 ** | 0.130 ** | 0.106 ** | −0.129 ** | −0.028 | 0.163 ** | 0.004 | 0.420 ** | ||||
| ENC | 0.227 ** | −0.673 ** | −0.211 ** | −0.008 | −0.799 ** | −0.808 ** | 0.786 ** | 0.608 ** | −0.807 ** | −0.252 ** | −0.164 ** | −0.034 * | |||
| CAI | −0.024 | 0.108 ** | −0.015 | −0.193 ** | 0.265 ** | 0.266 ** | −0.455 ** | 0.045 ** | 0.526 ** | −0.176 ** | 0.080 ** | 0.144 ** | −0.402 ** | ||
| AXIS1 | 0.195 ** | −0.813 ** | −0.231 ** | −0.023 | −0.970 ** | −0.972 ** | 0.850 ** | 0.830 ** | −0.884 ** | −0.436 ** | −0.182 ** | −0.087 ** | 0.836 ** | −0.328 ** | |
| AXIS2 | −0.050 ** | 0.119 ** | 0.034 * | 0.042 ** | 0.120 ** | 0.115 ** | 0.177 ** | −0.374 ** | −0.345 ** | 0.690 ** | −0.054 ** | −0.025 | 0.193 ** | −0.613 ** | 0.000 |
| CAI | GRAVY | Aromo | Axis1 | Axis2 | Axis3 | |
|---|---|---|---|---|---|---|
| Gravy | 0.080 ** | |||||
| Aromo | 0.144 ** | 0.420 ** | ||||
| Axis1 | −0.328 ** | −0.182 ** | −0.087 ** | |||
| Axis2 | −0.613 ** | −0.054 ** | −0.025 * | 0.000 | ||
| Axis3 | −0.159 ** | −0.056 ** | 0.007 | 0.000 | 0.001 | |
| Axis4 | 0.005 | −0.008 | −0.036 ** | −0.003 | −0.001 | 0.002 |
| Amino Acid | Codon | Total Count | RSCU |
|---|---|---|---|
| Phe | UUU | 31,361 | 0.71 |
| UUC | 53,807 | 1.28 | |
| Leu | UUA | 8539 | 0.22 |
| UUG | 39,289 | 1.05 | |
| CUU | 32,567 | 0.86 | |
| CUC | 63,009 | 1.84 | |
| CUA | 15,908 | 0.42 | |
| CUG | 57,144 | 1.61 | |
| Ile | AUU | 36,953 | 0.97 |
| AUC | 56,383 | 1.61 | |
| AUA | 14,980 | 0.42 | |
| Met | AUG | 56,564 | 1.00 |
| Val | GUU | 33,011 | 0.83 |
| GUC | 63,583 | 1.69 | |
| GUA | 15,230 | 0.39 | |
| GUG | 41,264 | 1.08 | |
| Tyr | UAU | 21,165 | 0.68 |
| UAC | 39,027 | 1.29 | |
| Cys | UGU | 10,231 | 0.59 |
| UGC | 20,393 | 1.25 | |
| His | CAU | 27,393 | 0.81 |
| CAC | 36,289 | 1.16 | |
| Gln | CAA | 44,072 | 0.80 |
| CAG | 59,569 | 1.19 | |
| Asn | AAU | 32,251 | 0.70 |
| AAC | 55,829 | 1.29 | |
| Lys | AAA | 37,695 | 0.63 |
| AAG | 80,172 | 1.37 | |
| Asp | GAU | 60,666 | 0.79 |
| GAC | 87,756 | 1.21 | |
| Glu | GAA | 60,978 | 0.76 |
| GAG | 89,320 | 1.23 | |
| Ser | UCU | 33,326 | 0.87 |
| UCC | 44,291 | 1.33 | |
| UCA | 30,242 | 0.76 | |
| UCG | 39,274 | 1.14 | |
| Pro | CCU | 35,400 | 0.86 |
| CCC | 49,710 | 1.37 | |
| CCA | 35,372 | 0.83 | |
| CCG | 36,167 | 0.93 | |
| Thr | ACU | 27,362 | 0.71 |
| ACC | 46,502 | 1.30 | |
| ACA | 31,612 | 0.81 | |
| ACG | 40,791 | 1.17 | |
| Ala | GCU | 49,281 | 0.84 |
| GCC | 88,269 | 1.58 | |
| GCA | 44,142 | 0.75 | |
| GCG | 45,406 | 0.83 | |
| TER | UGA | 2445 | 1.39 |
| UAA | 1271 | 0.74 | |
| UAG | 1529 | 0.87 | |
| Trp | UGG | 34,101 | 0.94 |
| Arg | CGU | 20,731 | 0.73 |
| CGC | 40,880 | 1.55 | |
| CGA | 33,836 | 1.13 | |
| CGG | 23,806 | 0.86 | |
| AGA | 24,225 | 0.84 | |
| AGG | 23,489 | 0.89 | |
| Gly | GGU | 35,358 | 0.75 |
| GGC | 79,015 | 1.79 | |
| GGA | 35,677 | 0.78 | |
| GGG | 28,001 | 0.67 | |
| Ser | AGU | 21,367 | 0.57 |
| AGC | 46,172 | 1.32 |
| Amino Acid | Codon | High RSCU | N | Low RSCU | N |
|---|---|---|---|---|---|
| Phe | UUU | 0.41 | 1246 | 0.97 | 4176 |
| UUC * | 1.59 | 4874 | 1.03 | 4414 | |
| Leu | UUA | 0.03 | 86 | 0.54 | 2081 |
| UUG | 0.55 | 1375 | 1.27 | 4893 | |
| CUU | 0.29 | 744 | 1.22 | 4713 | |
| CUC * | 2.90 | 7320 | 1.10 | 4257 | |
| CUA | 0.14 | 358 | 0.71 | 2725 | |
| CUG * | 2.08 | 5252 | 1.16 | 4456 | |
| Ile | AUU | 0.57 | 1339 | 1.20 | 4799 |
| AUC * | 2.24 | 5277 | 1.13 | 4511 | |
| AUA | 0.19 | 442 | 0.68 | 2704 | |
| Met | AUG | 1.00 | 3940 | 1.00 | 5668 |
| Val | GUU | 0.35 | 1025 | 1.18 | 4457 |
| GUC * | 2.39 | 7011 | 1.21 | 4561 | |
| GUA | 0.11 | 322 | 0.63 | 2379 | |
| GUG * | 1.16 | 3396 | 0.97 | 3649 | |
| Tyr | UAU | 0.29 | 651 | 1.03 | 3182 |
| UAC * | 1.71 | 3910 | 0.97 | 2979 | |
| Ser | AGU | 0.24 | 488 | 0.76 | 3234 |
| AGC * | 1.61 | 3266 | 1.05 | 4495 | |
| His | CAU | 0.42 | 875 | 1.11 | 3946 |
| CAC * | 1.58 | 3322 | 0.89 | 3180 | |
| Gln | CAA | 0.42 | 1184 | 1.07 | 6735 |
| CAG * | 1.58 | 4457 | 0.93 | 5867 | |
| Asn | AAU | 0.30 | 808 | 0.98 | 4824 |
| AAC * | 1.70 | 4557 | 1.02 | 4978 | |
| Lys | AAA | 0.29 | 1091 | 0.94 | 5763 |
| AAG * | 1.71 | 6306 | 1.06 | 6539 | |
| Asp | GAU | 0.40 | 1941 | 1.03 | 8086 |
| GAC * | 1.60 | 7765 | 0.97 | 7678 | |
| Glu | GAA | 0.39 | 1802 | 1.03 | 8278 |
| GAG * | 1.61 | 7396 | 0.97 | 7846 | |
| Ser | UCU | 0.40 | 818 | 1.22 | 5215 |
| UCC * | 1.96 | 3989 | 0.89 | 3811 | |
| UCA | 0.31 | 635 | 1.17 | 5018 | |
| UCG * | 1.48 | 2998 | 0.91 | 3878 | |
| Pro | CCU | 0.43 | 1030 | 1.10 | 4937 |
| CCC * | 2.13 | 5100 | 0.81 | 3633 | |
| CCA | 0.32 | 763 | 1.27 | 5704 | |
| CCG * | 1.12 | 2689 | 0.81 | 3651 | |
| Thr | ACU | 0.28 | 685 | 1.04 | 4130 |
| ACC * | 1.80 | 4428 | 0.91 | 3615 | |
| ACA | 0.38 | 941 | 1.20 | 4774 | |
| ACG * | 1.54 | 3796 | 0.86 | 3422 | |
| Ala | GCU | 0.38 | 1665 | 1.14 | 6365 |
| GCC * | 2.29 | 9960 | 1.05 | 5840 | |
| GCA | 0.34 | 1462 | 1.10 | 6141 | |
| GCG * | 0.99 | 4333 | 0.70 | 3903 | |
| Cys | UGU | 0.29 | 322 | 0.95 | 1626 |
| UGC * | 1.71 | 1869 | 1.05 | 1790 | |
| Trp | UGG | 1.00 | 2561 | 1.00 | 3365 |
| Gly | GGU | 0.39 | 1322 | 0.96 | 4175 |
| GGC * | 2.55 | 8641 | 1.35 | 5864 | |
| GGA | 0.37 | 1263 | 1.03 | 4480 | |
| GGG | 0.69 | 2353 | 0.65 | 2813 | |
| Arg | AGA | 0.45 | 825 | 1.17 | 3566 |
| AGG * | 1.29 | 2390 | 0.79 | 2427 | |
| CGU | 0.37 | 680 | 0.82 | 2510 | |
| CGC * | 2.34 | 4329 | 1.02 | 3113 | |
| CGA | 0.50 | 932 | 1.41 | 4306 | |
| CGG * | 1.05 | 1934 | 0.80 | 2440 | |
| TER | UAA | 0.62 | 101 | 0.79 | 169 |
| UAG | 0.92 | 150 | 0.74 | 158 | |
| UGA | 1.47 | 240 | 1.48 | 317 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Song, H.; Kuang, Y.; Chen, S.; Tian, P.; Li, C.; Nan, Z. Genome-Wide Analysis of Codon Usage Bias in Epichloë festucae. Int. J. Mol. Sci. 2016, 17, 1138. https://doi.org/10.3390/ijms17071138
Li X, Song H, Kuang Y, Chen S, Tian P, Li C, Nan Z. Genome-Wide Analysis of Codon Usage Bias in Epichloë festucae. International Journal of Molecular Sciences. 2016; 17(7):1138. https://doi.org/10.3390/ijms17071138
Chicago/Turabian StyleLi, Xiuzhang, Hui Song, Yu Kuang, Shuihong Chen, Pei Tian, Chunjie Li, and Zhibiao Nan. 2016. "Genome-Wide Analysis of Codon Usage Bias in Epichloë festucae" International Journal of Molecular Sciences 17, no. 7: 1138. https://doi.org/10.3390/ijms17071138
APA StyleLi, X., Song, H., Kuang, Y., Chen, S., Tian, P., Li, C., & Nan, Z. (2016). Genome-Wide Analysis of Codon Usage Bias in Epichloë festucae. International Journal of Molecular Sciences, 17(7), 1138. https://doi.org/10.3390/ijms17071138