
Less Is More: Substrate Reduction Therapy for Lysosomal Storage Disorders




Abstract
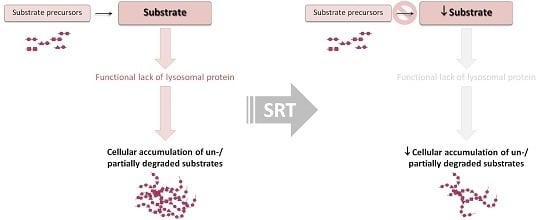
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. From Concept to Clinics
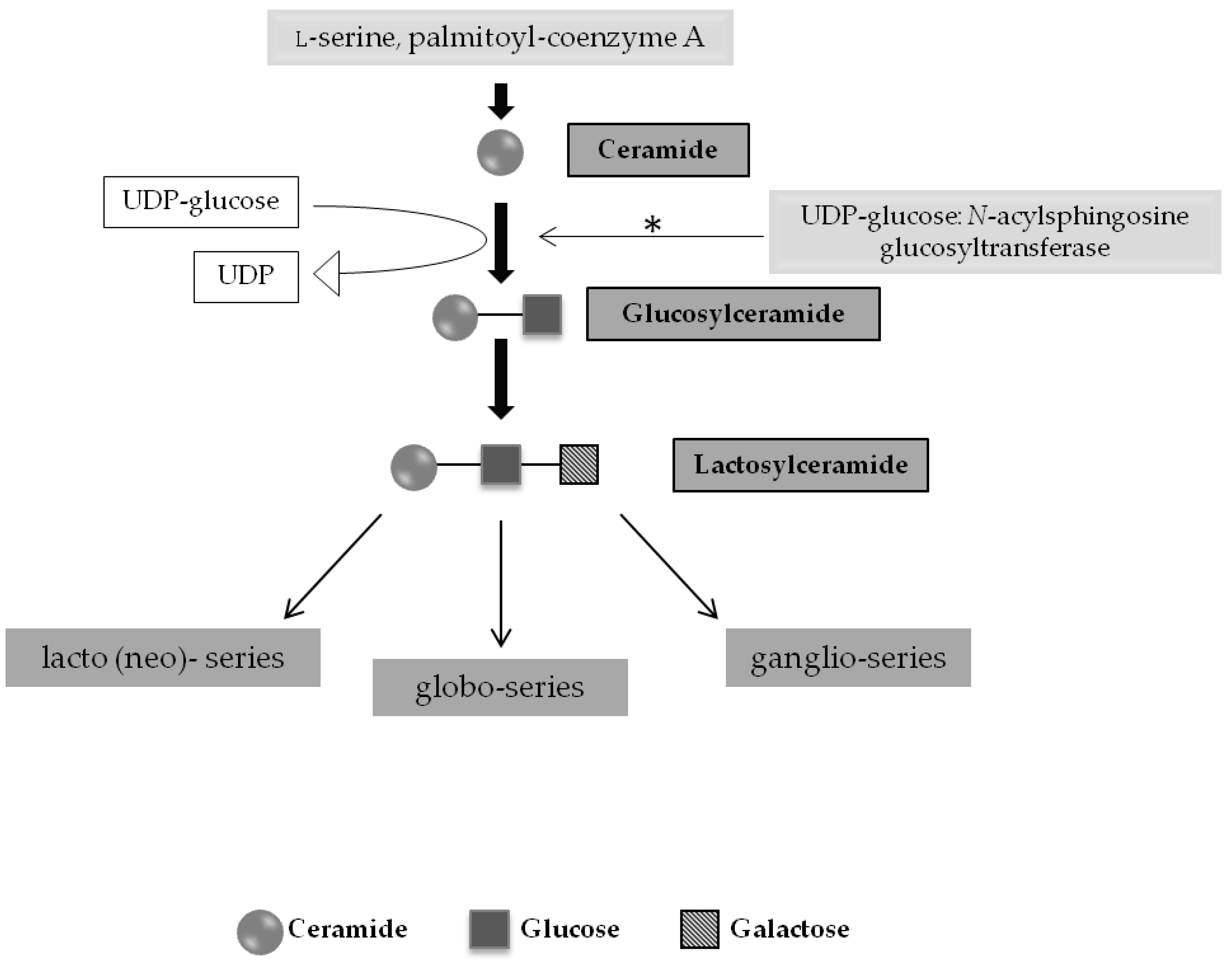
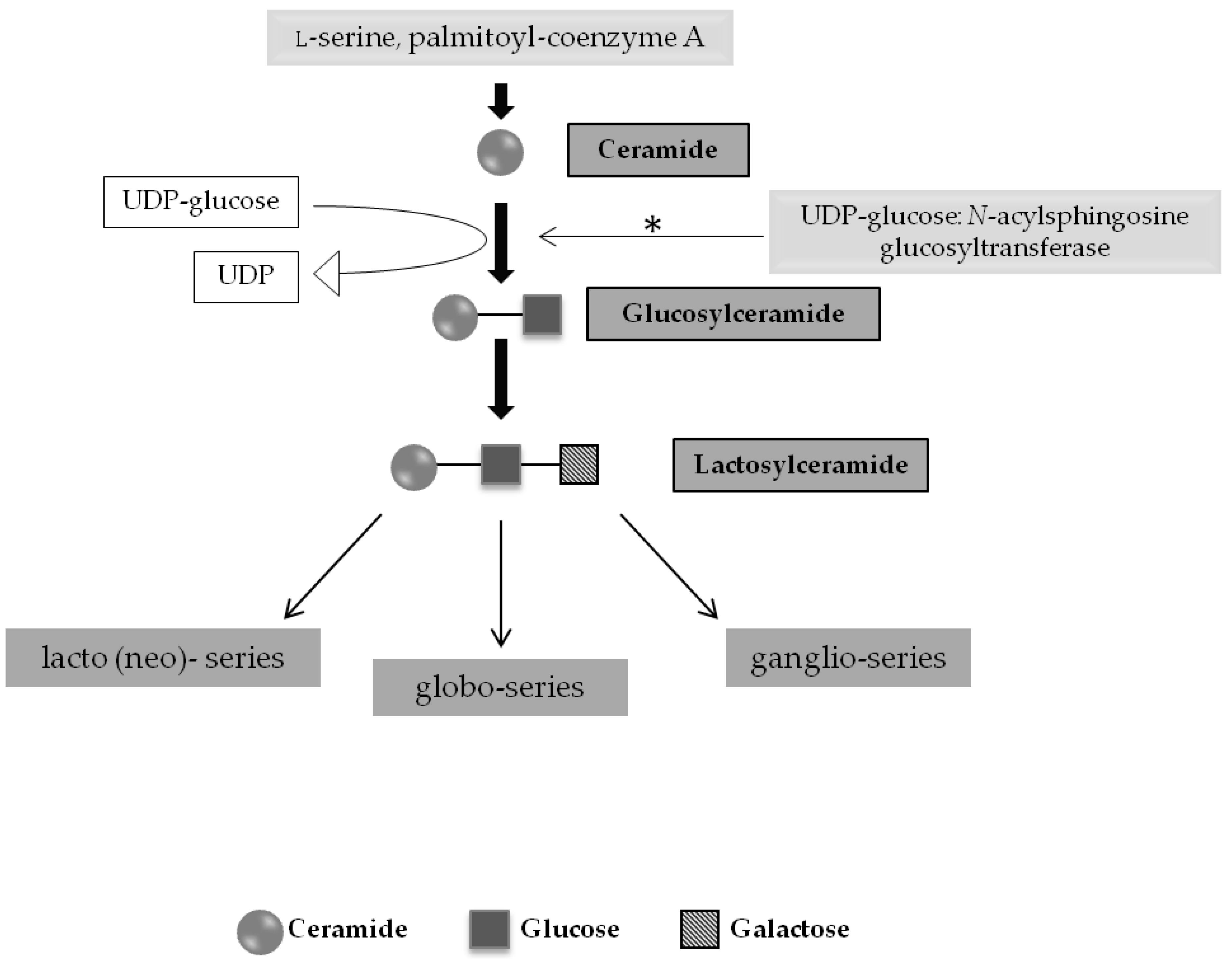
2.1. SRT for Glycosphingolipidoses and Related Disorders
2.1.1. Gaucher Disease and Other Glycosphingolipidoses
2.1.2. Niemann-Pick Type C
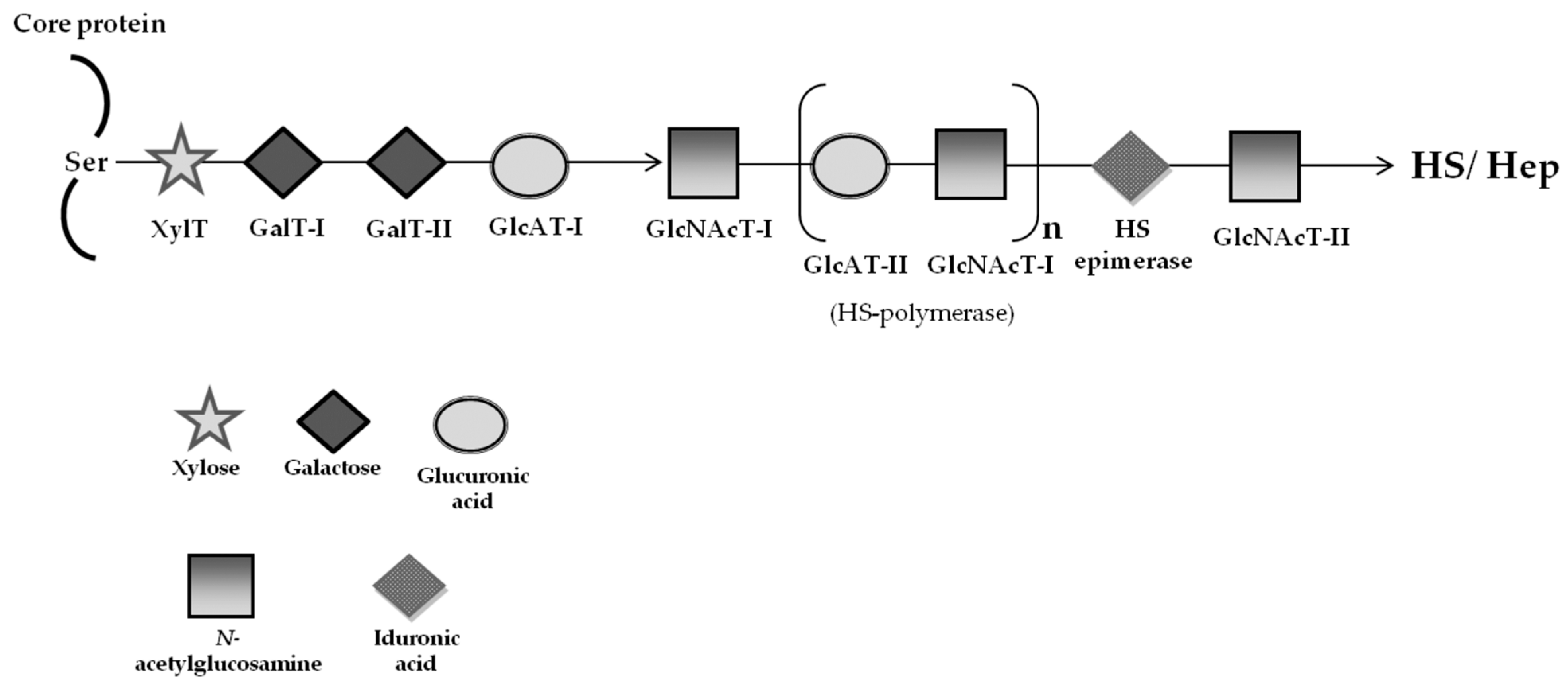
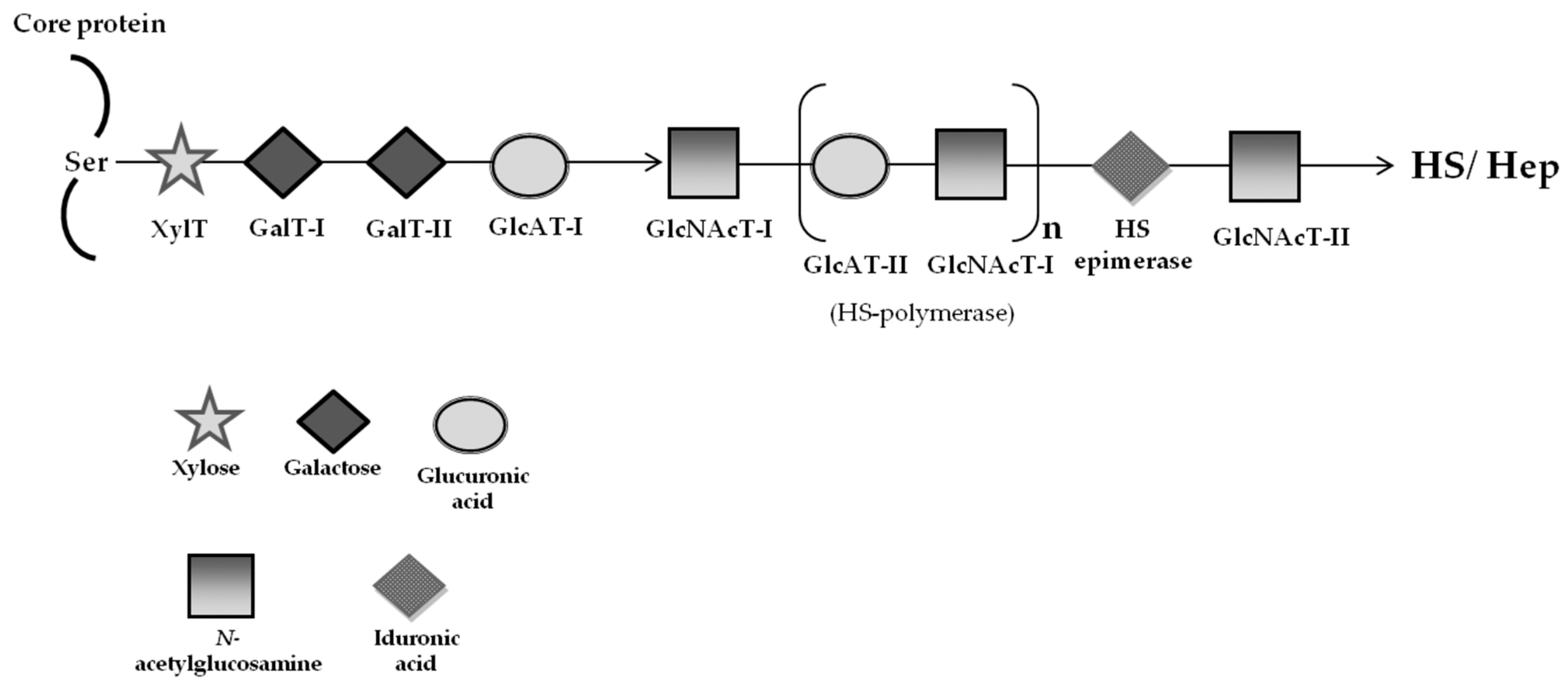
2.2. Mucopolysaccharidoses—Special Focus on MPS Type III (Sanfilippo Syndrome)
3. A Step Forward: Second Generation Compounds and Genetic SRT
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A
A1. Further Reading
A1.1. On the Individual SRT Drugs
A1.2. On LSD and Their Therapeutic Possibilities
References
- Hers, H.G. Inborn lysosomal diseases. Gastroenterology 1965, 48, 625–633. [Google Scholar] [PubMed]
- De Duve, C. From lysosomes to storage diseases and back: A personal reminiscence. In Lysosomal Storage Disorders; Barranger, J.A., Cabrera-Salazar, M., Eds.; Springer US: New York, NY, USA, 2007; pp. 1–5. [Google Scholar]
- Deduve, C. From cytases to lysosomes. Fed. Proc. 1964, 23, 1045–1049. [Google Scholar] [PubMed]
- Fratantoni, J.C.; Hall, C.W.; Neufeld, E.F. The defect in hurler and hunter syndromes. II. Deficiency of specific factors involved in mucopolysaccharide degradation. Proc. Natl. Acad. Sci. USA 1969, 64, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, M.F.; Prata, M.J.; Alves, S. Mannose-6-phosphate pathway: A review on its role in lysosomal function and dysfunction. Mol. Genet. Metab. 2012, 105, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Desnick, R.J.; Schuchman, E.H. Enzyme replacement therapy for lysosomal diseases: Lessons from 20 years of experience and remaining challenges. Annu. Rev. Genom. Hum. Genet. 2012, 13, 307–335. [Google Scholar] [CrossRef] [PubMed]
- Brady, R.O.; Pentchev, P.G.; Gal, A.E.; Hibbert, S.R.; Dekaban, A.S. Replacement therapy for inherited enzyme deficiency. Use of purified glucocerebrosidase in Gaucher’s disease. N. Engl. J. Med. 1974, 291, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Barton, N.W.; Furbish, F.S.; Murray, G.J.; Garfield, M.; Brady, R.O. Therapeutic response to intravenous infusions of glucocerebrosidase in a patient with Gaucher disease. Proc. Natl. Acad. Sci. USA 1990, 87, 1913–1916. [Google Scholar] [CrossRef] [PubMed]
- Barton, N.W.; Brady, R.O.; Dambrosia, J.M.; Di Bisceglie, A.M.; Doppelt, S.H.; Hill, S.C.; Mankin, H.J.; Murray, G.J.; Parker, R.I.; Argoff, C.E.; et al. Replacement therapy for inherited enzyme deficiency—Macrophage-targeted glucocerebrosidase for Gaucher’s disease. N. Engl. J. Med. 1991, 324, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Jakobkiewicz-Banecka, J.; Wegrzyn, A.; Wegrzyn, G. Substrate deprivation therapy: A new hope for patients suffering from neuronopathic forms of inherited lysosomal storage diseases. J. Appl. Genet. 2007, 48, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Wraith, J.E.; Beck, M.; Lane, R.; van der Ploeg, A.; Shapiro, E.; Xue, Y.; Kakkis, E.D.; Guffon, N. Enzyme replacement therapy in patients who have Mucopolysaccharidosis I and are younger than 5 years: Results of a multinational study of recombinant human α-l-iduronidase (laronidase). Pediatrics 2007, 120, e37–e46. [Google Scholar] [CrossRef] [PubMed]
- Wraith, J.E.; Clarke, L.A.; Beck, M.; Kolodny, E.H.; Pastores, G.M.; Muenzer, J.; Rapoport, D.M.; Berger, K.I.; Swiedler, S.J.; Kakkis, E.D.; et al. Enzyme replacement therapy for Mucopolysaccharidosis I: A randomized, double-blinded, placebo-controlled, multinational study of recombinant human α-l-iduronidase (laronidase). J. Pediatr. 2004, 144, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Corzo, D.; Leslie, N.D.; Gruskin, D.; Van der Ploeg, A.; Clancy, J.P.; Parini, R.; Morin, G.; Beck, M.; Bauer, M.S.; et al. Early treatment with alglucosidase α prolongs long-term survival of infants with pompe disease. Pediatr. Res. 2009, 66, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Deegan, P.B. Fabry disease, enzyme replacement therapy and the significance of antibody responses. J. Inherit. Metab. Dis. 2012, 35, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Pastores, G.M.; Rosenbloom, B.; Weinreb, N.; Goker-Alpan, O.; Grabowski, G.; Cohn, G.M.; Zahrieh, D. A multicenter open-label treatment protocol (HGT-GCB-058) of velaglucerase alfa enzyme replacement therapy in patients with Gaucher disease type 1: Safety and tolerability. Genet. Med. Off. J. Am. Coll. Med. Genet. 2014, 16, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Broomfield, A.; Jones, S.A.; Hughes, S.M.; Bigger, B.W. The impact of the immune system on the safety and efficiency of enzyme replacement therapy in lysosomal storage disorders. J. Inherit. Metab. Dis. 2016, 39, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.Q.; Ishii, S. Active-site-specific chaperone therapy for fabry disease. Yin and yang of enzyme inhibitors. FEBS J. 2007, 274, 4962–4971. [Google Scholar] [CrossRef] [PubMed]
- Feldhammer, M.; Durand, S.; Pshezhetsky, A.V. Protein misfolding as an underlying molecular defect in Mucopolysaccharidosis III type C. PLoS ONE 2009, 4, e7434. [Google Scholar] [CrossRef] [PubMed]
- Pastores, G.M.; Sathe, S. A chaperone-mediated approach to enzyme enhancement as a therapeutic option for the lysosomal storage disorders. Drugs R D 2006, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Keeling, K.M.; Brooks, D.A.; Hopwood, J.J.; Li, P.; Thompson, J.N.; Bedwell, D.M. Gentamicin-mediated suppression of hurler syndrome stop mutations restores a low level of α-l-iduronidase activity and reduces lysosomal glycosaminoglycan accumulation. Hum. Mol. Genet. 2001, 10, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Hein, L.K.; Bawden, M.; Muller, V.J.; Sillence, D.; Hopwood, J.J.; Brooks, D.A. α-l-iduronidase premature stop codons and potential read-through in Mucopolysaccharidosis type I patients. J. Mol. Biol. 2004, 338, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.A.; Muller, V.J.; Hopwood, J.J. Stop-codon read-through for patients affected by a lysosomal storage disorder. Trends Mol. Med. 2006, 12, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Matos, L.; Canals, I.; Dridi, L.; Choi, Y.; Prata, M.J.; Jordan, P.; Desviat, L.R.; Perez, B.; Pshezhetsky, A.V.; Grinberg, D.; et al. Therapeutic strategies based on modified U1 snRNAs and chaperones for sanfilippo C splicing mutations. Orphanet J. Rare Dis. 2014, 9, 180. [Google Scholar] [CrossRef] [PubMed]
- Matos, L.; Gonçalves, V.; Pinto, E.; Laranjeira, F.; Prata, M.J.; Jordan, P.; Desviat, L.R.; Pérez, B.; Alves, S. Functional analysis of splicing mutations in the IDS gene and the use of antisense oligonucleotides to exploit an alternative therapy for MPS II. Biochim. Biophys. Acta 2015, 1852, 2712–2721. [Google Scholar] [CrossRef] [PubMed]
- Warnock, D.G.; Bichet, D.G.; Holida, M.; Goker-Alpan, O.; Nicholls, K.; Thomas, M.; Eyskens, F.; Shankar, S.; Adera, M.; Sitaraman, S.; et al. Oral migalastat HCl leads to greater systemic exposure and tissue levels of active α-galactosidase a in fabry patients when co-administered with infused agalsidase. PLoS ONE 2015, 10, e0134341. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Lun, Y.; Brignol, N.; Hamler, R.; Schilling, A.; Frascella, M.; Sullivan, S.; Boyd, R.E.; Chang, K.; Soska, R.; et al. Coformulation of a novel human α-galactosidase a with the pharmacological chaperone AT1001 leads to improved substrate reduction in fabry mice. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Radin, N.S. Treatment of Gaucher disease with an enzyme inhibitor. Glycoconj. J. 1996, 13, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; Jeyakumar, M.; Andersson, U.; Heare, T.; Dwek, R.A.; Butters, T.D. Substrate reduction therapy in mouse models of the glycosphingolipidoses. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2003, 358, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, S.; Hirabayashi, Y. Glucosylceramide synthase and glycosphingolipid synthesis. Trends Cell Biol. 1998, 8, 198–202. [Google Scholar] [CrossRef]
- Sandhoff, K.; Kolter, T. Biosynthesis and degradation of mammalian glycosphingolipids. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2003, 358, 847–861. [Google Scholar] [CrossRef] [PubMed]
- Schapiro, F.B.; Lingwood, C.; Furuya, W.; Grinstein, S. pH-independent retrograde targeting of glycolipids to the golgi complex. Am. J. Physiol. 1998, 274, C319–C332. [Google Scholar] [PubMed]
- Van Echten, G.; Sandhoff, K. Ganglioside metabolism. Enzymology, topology, and regulation. J. Biol. Chem. 1993, 268, 5341–5344. [Google Scholar] [PubMed]
- Sandhoff, K.; Kolter, T. Biochemistry of glycosphingolipid degradation. Clin. Chim. Acta Int. J. Clin. Chem. 1997, 266, 51–61. [Google Scholar] [CrossRef]
- Sandhoff, K.; Klein, A. Intracellular trafficking of glycosphingolipids: Role of sphingolipid activator proteins in the topology of endocytosis and lysosomal digestion. FEBS Lett. 1994, 346, 103–107. [Google Scholar] [CrossRef]
- Sandhoff, K.; Kolter, T. Topology of glycosphingolipid degradation. Trends Cell Biol. 1996, 6, 98–103. [Google Scholar] [CrossRef]
- Neufeld, E.F. Lysosomal storage diseases. Annu. Rev. Biochem. 1991, 60, 257–280. [Google Scholar] [CrossRef] [PubMed]
- Butters, T.D.; Dwek, R.A.; Platt, F.M. Imino sugar inhibitors for treating the lysosomal glycosphingolipidoses. Glycobiology 2005, 15, 43R–52R. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; Neises, G.R.; Dwek, R.A.; Butters, T.D. N-butyldeoxynojirimycin is a novel inhibitor of glycolipid biosynthesis. J. Biol. Chem. 1994, 269, 8362–8365. [Google Scholar] [PubMed]
- Platt, F.M.; Neises, G.R.; Karlsson, G.B.; Dwek, R.A.; Butters, T.D. N-butyldeoxygalactonojirimycin inhibits glycolipid biosynthesis but does not affect N-linked oligosaccharide processing. J. Biol. Chem. 1994, 269, 27108–27114. [Google Scholar] [PubMed]
- Aerts, J.M.; Hollak, C.E.; Boot, R.G.; Groener, J.E.; Maas, M. Substrate reduction therapy of glycosphingolipid storage disorders. J. Inherit. Metab. Dis. 2006, 29, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Dwek, R.A.; Butters, T.D.; Platt, F.M.; Zitzmann, N. Targeting glycosylation as a therapeutic approach. Rev. Drug Discov. 2002, 1, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Butters, T.D.; Dwek, R.A.; Platt, F.M. Therapeutic applications of imino sugars in lysosomal storage disorders. Curr. Top. Med. Chem. 2003, 3, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Butters, T.D.; Mellor, H.R.; Narita, K.; Dwek, R.A.; Platt, F.M. Small-molecule therapeutics for the treatment of glycolipid lysosomal storage disorders. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2003, 358, 927–945. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; Butters, T.D. Substrate reduction therapy. In Lysosomal Storage Disorders; Barranger, J.A., Cabrera-Salazar, M.A., Eds.; Springer US: New York, NY, USA, 2007; pp. 153–168. [Google Scholar]
- Yamanaka, S.; Johnson, M.D.; Grinberg, A.; Westphal, H.; Crawley, J.N.; Taniike, M.; Suzuki, K.; Proia, R.L. Targeted disruption of the hexa gene results in mice with biochemical and pathologic features of tay-sachs disease. Proc. Natl. Acad. Sci. USA 1994, 91, 9975–9979. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; Neises, G.R.; Reinkensmeier, G.; Townsend, M.J.; Perry, V.H.; Proia, R.L.; Winchester, B.; Dwek, R.A.; Butters, T.D. Prevention of lysosomal storage in tay-sachs mice treated with N-butyldeoxynojirimycin. Science 1997, 276, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Sango, K.; Yamanaka, S.; Hoffmann, A.; Okuda, Y.; Grinberg, A.; Westphal, H.; McDonald, M.P.; Crawley, J.N.; Sandhoff, K.; Suzuki, K.; et al. Mouse models of tay-sachs and sandhoff diseases differ in neurologic phenotype and ganglioside metabolism. Nat. Genet. 1995, 11, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Ashe, K.M.; Bangari, D.; Li, L.; Cabrera-Salazar, M.A.; Bercury, S.D.; Nietupski, J.B.; Cooper, C.G.; Aerts, J.M.; Lee, E.R.; Copeland, D.P.; et al. Iminosugar-based inhibitors of glucosylceramide synthase increase brain glycosphingolipids and survival in a mouse model of sandhoff disease. PLoS ONE 2011, 6, e21758. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.; Lachmann, R.; Hollak, C.; Aerts, J.; van Weely, S.; Hrebicek, M.; Platt, F.; Butters, T.; Dwek, R.; Moyses, C.; et al. Novel oral treatment of Gaucher’s disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet 2000, 355, 1481–1485. [Google Scholar] [CrossRef]
- Elstein, D.; Hollak, C.; Aerts, J.M.; van Weely, S.; Maas, M.; Cox, T.M.; Lachmann, R.H.; Hrebicek, M.; Platt, F.M.; Butters, T.D.; et al. Sustained therapeutic effects of oral miglustat (zavesca, N-butyldeoxynojirimycin, OGT 918) in type I Gaucher disease. J. Inherit. Metab. Dis. 2004, 27, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, R.; Fitzgibbon, E.J.; Harris, C.; DeVile, C.; Davies, E.H.; Abel, L.; van Schaik, I.N.; Benko, W.; Timmons, M.; Ries, M.; et al. Randomized, controlled trial of miglustat in Gaucher’s disease type 3. Ann. Neurol. 2008, 64, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Bennett, L.L.; Mohan, D. Gaucher disease and its treatment options. Ann. Pharmacother. 2013, 47, 1182–1193. [Google Scholar] [CrossRef] [PubMed]
- Nietupski, J.B.; Pacheco, J.J.; Chuang, W.L.; Maratea, K.; Li, L.; Foley, J.; Ashe, K.M.; Cooper, C.G.; Aerts, J.M.; Copeland, D.P.; et al. Iminosugar-based inhibitors of glucosylceramide synthase prolong survival but paradoxically increase brain glucosylceramide levels in Niemann-Pick C mice. Mol. Genet. Metab. 2012, 105, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Ridley, C.M.; Thur, K.E.; Shanahan, J.; Thillaiappan, N.B.; Shen, A.; Uhl, K.; Walden, C.M.; Rahim, A.A.; Waddington, S.N.; Platt, F.M.; et al. β-Glucosidase 2 (GBA2) activity and imino sugar pharmacology. J. Biol. Chem. 2013, 288, 26052–26066. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.K.; Liu, J.; Sun, L.; Chuang, W.L.; Yuen, T.; Yang, R.; Lu, P.; Zhang, K.; Li, J.; Keutzer, J.; et al. Glucocerebrosidase 2 gene deletion rescues type 1 Gaucher disease. Proc. Natl. Acad. Sci. USA 2014, 111, 4934–4939. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.R.; Aten, J.; Ottenhoff, R.; van Roomen, C.P.; Herrera Moro, D.; Claessen, N.; Vinueza Veloz, M.F.; Zhou, K.; Lin, Z.; Mirzaian, M.; et al. Reducing GBA2 activity ameliorates neuropathology in Niemann-Pick type C mice. PLoS ONE 2015, 10, e0135889. [Google Scholar] [CrossRef] [PubMed]
- Alfonso, P.; Pampin, S.; Estrada, J.; Rodriguez-Rey, J.C.; Giraldo, P.; Sancho, J.; Pocovi, M. Miglustat (NB-DNJ) works as a chaperone for mutated acid β-glucosidase in cells transfected with several Gaucher disease mutations. Blood Cells Mol. Dis. 2005, 35, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Gabig-Ciminska, M.; Jakobkiewicz-Banecka, J.; Malinowska, M.; Kloska, A.; Piotrowska, E.; Chmielarz, I.; Moskot, M.; Wegrzyn, A.; Wegrzyn, G. Combined therapies for lysosomal storage diseases. Curr. Mol. Med. 2015, 15, 746–771. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, S.; Sakiyama, H.; Suzuki, G.; Hidari, K.I.; Hirabayashi, Y. Expression cloning of a cDNA for human ceramide glucosyltransferase that catalyzes the first glycosylation step of glycosphingolipid synthesis. Proc. Natl. Acad. Sci. USA 1996, 93, 4638–4693. [Google Scholar] [CrossRef] [PubMed]
- Vunnam, R.R.; Radin, N.S. Analogs of ceramide that inhibit glucocerebroside synthetase in mouse brain. Chem. Phys. Lipids 1980, 26, 265–278. [Google Scholar] [CrossRef]
- Shayman, J.A. The design and clinical development of inhibitors of glycosphingolipid synthesis: Will invention be the mother of necessity? Trans. Am. Clin. Climatol. Assoc. 2013, 124, 46–60. [Google Scholar] [PubMed]
- Lee, K.; Jung, W.H.; Hwang, S.Y.; Lee, S.H. Fluorobenzamidrazone thrombin inhibitors: Influence of fluorine on enhancing oral absorption. Bioorg. Med. Chem. Lett. 1999, 9, 2483–2486. [Google Scholar] [CrossRef]
- Abe, A.; Arend, L.J.; Lee, L.; Lingwood, C.; Brady, R.O.; Shayman, J.A. Glycosphingolipid depletion in fabry disease lymphoblasts with potent inhibitors of glucosylceramide synthase. Kidney Int. 2000, 57, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Abe, A.; Gregory, S.; Lee, L.; Killen, P.D.; Brady, R.O.; Kulkarni, A.; Shayman, J.A. Reduction of globotriaosylceramide in fabry disease mice by substrate deprivation. J. Clin. Investig. 2000, 105, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Shayman, J.A. Eliglustat tartrate: Glucosylceramide synthase inhibitor treatment of type 1 Gaucher disease. Drugs Future 2010, 35, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Brumshtein, B.; Greenblatt, H.M.; Butters, T.D.; Shaaltiel, Y.; Aviezer, D.; Silman, I.; Futerman, A.H.; Sussman, J.L. Crystal structures of complexes of N-butyl- and N-nonyl-deoxynojirimycin bound to acid β-glucosidase: Insights into the mechanism of chemical chaperone action in Gaucher disease. J. Biol. Chem. 2007, 282, 29052–29058. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Ollé, G.; Duque, J.; Egido-Gabás, M.; Casas, J.; Lluch, M.; Chabás, A.; Grinberg, D.; Vilageliu, L. Promising results of the chaperone effect caused by imino sugars and aminocyclitol derivatives on mutant glucocerebrosidases causing Gaucher disease. Blood Cells Mol. Dis. 2009, 42, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Peterschmitt, M.J.; Burke, A.; Blankstein, L.; Smith, S.E.; Puga, A.C.; Kramer, W.G.; Harris, J.A.; Mathews, D.; Bonate, P.L. Safety, tolerability, and pharmacokinetics of eliglustat tartrate (genz-112638) after single doses, multiple doses, and food in healthy volunteers. J. Clin. Pharmacol. 2011, 51, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Lukina, E.; Watman, N.; Arreguin, E.A.; Banikazemi, M.; Dragosky, M.; Iastrebner, M.; Rosenbaum, H.; Phillips, M.; Pastores, G.M.; Rosenthal, D.I.; et al. A phase 2 study of eliglustat tartrate (genz-112638), an oral substrate reduction therapy for Gaucher disease type 1. Blood 2010, 116, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Lukina, E.; Watman, N.; Arreguin, E.A.; Dragosky, M.; Iastrebner, M.; Rosenbaum, H.; Phillips, M.; Pastores, G.M.; Kamath, R.S.; Rosenthal, D.I.; et al. Improvement in hematological, visceral, and skeletal manifestations of Gaucher disease type 1 with oral eliglustat tartrate (genz-112638) treatment: 2-year results of a phase 2 study. Blood 2010, 116, 4095–4098. [Google Scholar] [CrossRef] [PubMed]
- Lukina, E.; Watman, N.; Dragosky, M.; Pastores, G.M.; Arreguin, E.A.; Rosenbaum, H.; Zimran, A.; Angell, J.; Ross, L.; Puga, A.C.; et al. Eliglustat, an investigational oral therapy for Gaucher disease type 1: Phase 2 trial results after 4 years of treatment. Blood Cells Mol. Dis. 2014, 53, 274–276. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.M. Eliglustat tartrate, an orally active glucocerebroside synthase inhibitor for the potential treatment of Gaucher disease and other lysosomal storage diseases. Curr. Opin. Investig. Drugs 2010, 11, 1169–1181. [Google Scholar] [PubMed]
- Cox, T.M. Innovative treatments for lysosomal diseases. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 275–311. [Google Scholar] [CrossRef] [PubMed]
- Kamath, R.S.; Lukina, E.; Watman, N.; Dragosky, M.; Pastores, G.M.; Arreguin, E.A.; Rosenbaum, H.; Zimran, A.; Aguzzi, R.; Puga, A.C.; et al. Skeletal improvement in patients with Gaucher disease type 1: A phase 2 trial of oral eliglustat. Skelet. Radiol. 2014, 43, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.K.; Lukina, E.; Ben Turkia, H.; Amato, D.; Baris, H.; Dasouki, M.; Ghosn, M.; Mehta, A.; Packman, S.; Pastores, G.; et al. Effect of oral eliglustat on splenomegaly in patients with Gaucher disease type 1: The engage randomized clinical trial. JAMA 2015, 313, 695–706. [Google Scholar] [CrossRef] [PubMed]
- CerdelgaTM. Highlights of Prescribing Information. Available online: http://www.cerdelga.com/pdf/cerdelga_prescribing_information.pdf (accessed on 26 October 2016).
- Belmatoug, N.; Di Rocco, M.; Fraga, C.; Giraldo, P.; Hughes, D.; Lukina, E.; Maison-Blanche, P.; Merkel, M.; Niederau, C.; Plckinger, U.; et al. Management and monitoring recommendations for the use of eliglustat in adults with type 1 gaucher disease in europe. Eur. J. Intern. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M. Sphingolipid lysosomal storage disorders. Nature 2014, 510, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Salazar, M.A.; Deriso, M.; Bercury, S.D.; Li, L.; Lydon, J.T.; Weber, W.; Pande, N.; Cromwell, M.A.; Copeland, D.; Leonard, J.; et al. Systemic delivery of a glucosylceramide synthase inhibitor reduces CNS substrates and increases lifespan in a mouse model of type 2 Gaucher disease. PLoS ONE 2012, 7, e43310. [Google Scholar] [CrossRef] [PubMed]
- Kanfer, J.N.; Legler, G.; Sullivan, J.; Raghavan, S.S.; Mumford, R.A. The Gaucher mouse. Biochem. Biophys. Res. Commun. 1975, 67, 85–90. [Google Scholar] [CrossRef]
- Sun, Y.; Liou, B.; Ran, H.; Skelton, M.R.; Williams, M.T.; Vorhees, C.V.; Kitatani, K.; Hannun, Y.A.; Witte, D.P.; Xu, Y.H.; et al. Neuronopathic Gaucher disease in the mouse: Viable combined selective saposin C deficiency and mutant glucocerebrosidase (V394L) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits. Hum. Mol. Genet. 2010, 19, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.; Sun, Y.; Bangari, D.S.; Budman, E.; Park, H.; Nietupski, J.B.; Allaire, A.; Cromwell, M.A.; Wang, B.; Grabowski, G.A.; et al. CNS-accessible inhibitor of glucosylceramide synthase for substrate reduction therapy of neuronopathic Gaucher disease. Mol. Ther. J. A Soc. Gene Ther. 2016, 24, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Puga, A.C. Crossing the barrier and meaning it: Evaluation of a novel substrate reduction therapy in Gaucher disease type 3. Available online: http://www.brains4brain.eu/wp-content/uploads/2016/01/Brains-For-Brain-2016.pdf (accessed on 29 June 2016).
- Elliot-Smith, E.; Speak, A.O.; Lloyd-Evans, E.; Smith, D.A.; van der Spoel, A.C.; Jeyakumar, M.; Butters, T.D.; Dwek, R.A.; d’Azzo, A.; Platt, F.M. Beneficial effects of substrate reduction therapy in a mouse model of GM1 gangliosidosis. Mol. Genet. Metab. 2008, 94, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Kasperzyk, J.L.; d’Azzo, A.; Platt, F.M.; Alroy, J.; Seyfried, T.N. Substrate reduction reduces gangliosides in postnatal cerebrum-brainstem and cerebellum in GM1 gangliosidosis mice. J. Lipid Res. 2005, 46, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Jeyakumar, M.; Butters, T.D.; Cortina-Borja, M.; Hunnam, V.; Proia, R.L.; Perry, V.H.; Dwek, R.A.; Platt, F.M. Delayed symptom onset and increased life expectancy in sandhoff disease mice treated with N-butyldeoxynojirimycin. Proc. Natl. Acad. Sci. USA 1999, 96, 6388–6393. [Google Scholar] [CrossRef] [PubMed]
- Baek, R.C.; Kasperzyk, J.L.; Platt, F.M.; Seyfried, T.N. N-butyldeoxygalactonojirimycin reduces brain ganglioside and GM2 content in neonatal sandhoff disease mice. Neurochem. Int. 2008, 52, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Masciullo, M.; Santoro, M.; Modoni, A.; Ricci, E.; Guitton, J.; Tonali, P.; Silvestri, G. Substrate reduction therapy with miglustat in chronic GM2 gangliosidosis type sandhoff: Results of a 3-year follow-up. J. Inherit. Metab. Dis. 2010, 33 (Suppl. S3), S355–S361. [Google Scholar] [CrossRef] [PubMed]
- Burrow, T.A.; Grabowski, G.A. Emerging treatments and future outcomes. In Lysosomal Storage Disorders—A Practical Guide, 1th ed.; John Wiley & Sons, Ltd.: Oxford, UK, 2012; pp. 174–180. [Google Scholar]
- Zervas, M.; Somers, K.L.; Thrall, M.A.; Walkley, S.U. Critical role for glycosphingolipids in Niemann-Pick disease type c. Curr. Biol. 2001, 11, 1283–1287. [Google Scholar] [CrossRef]
- Patterson, M.C.; Vecchio, D.; Prady, H.; Abel, L.; Wraith, J.E. Miglustat for treatment of Niemann-Pick C disease: A randomised controlled study. Lancet Neurol. 2007, 6, 765–772. [Google Scholar] [CrossRef]
- Wraith, J.E.; Vecchio, D.; Jacklin, E.; Abel, L.; Chadha-Boreham, H.; Luzy, C.; Giorgino, R.; Patterson, M.C. Miglustat in adult and juvenile patients with Niemann-Pick disease type C: Long-term data from a clinical trial. Mol. Genet. Metab. 2010, 99, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Pineda, M.; Perez-Poyato, M.S.; O’Callaghan, M.; Vilaseca, M.A.; Pocovi, M.; Domingo, R.; Portal, L.R.; Perez, A.V.; Temudo, T.; Gaspar, A.; et al. Clinical experience with miglustat therapy in pediatric patients with Niemann-Pick disease type C: A case series. Mol. Genet. Metab. 2010, 99, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.H.; Peng, S.F.; Yang, C.C.; Lee, N.C.; Tsai, L.K.; Huang, A.C.; Su, S.C.; Tseng, C.C.; Hwu, W.L. Long-term efficacy of miglustat in paediatric patients with Niemann-Pick disease type C. J. Inherit. Metab. Dis. 2013, 36, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Fecarotta, S.; Romano, A.; Della Casa, R.; Del Giudice, E.; Bruschini, D.; Mansi, G.; Bembi, B.; Dardis, A.; Fiumara, A.; Di Rocco, M.; et al. Long term follow-up to evaluate the efficacy of miglustat treatment in italian patients with Niemann-Pick disease type C. Orphanet J. Rare Dis. 2015, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Santos-Lozano, A.; Villamandos Garcia, D.; Sanchis-Gomar, F.; Fiuza-Luces, C.; Pareja-Galeano, H.; Garatachea, N.; Nogales Gadea, G.; Lucia, A. Niemann-Pick disease treatment: A systematic review of clinical trials. Ann. Transl. Med. 2015, 3, 360. [Google Scholar] [PubMed]
- Wegrzyn, G.; Jakobkiewicz-Banecka, J.; Gabig-Ciminska, M.; Piotrowska, E.; Narajczyk, M.; Kloska, A.; Malinowska, M.; Dziedzic, D.; Golebiewska, I.; Moskot, M.; et al. Genistein: A natural isoflavone with a potential for treatment of genetic diseases. Biochem. Soc. Trans. 2010, 38, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.F.; Muenzer, J. The mucopolysaccharidoses. In The Metabolic Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill Professional: New York, NY, USA, 2001; Volume 3. [Google Scholar]
- Coutinho, M.F.; Lacerda, L.; Alves, S. Glycosaminoglycan storage disorders: A review. Biochem. Res. Int. 2012, 2012, 471325. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, E.; Jakobkiewicz-Banecka, J.; Baranska, S.; Tylki-Szymanska, A.; Czartoryska, B.; Wegrzyn, A.; Wegrzyn, G. Genistein-mediated inhibition of glycosaminoglycan synthesis as a basis for gene expression-targeted isoflavone therapy for mucopolysaccharidoses. Eur. J. Hum. Genet. 2006, 14, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Friso, A.; Tomanin, R.; Salvalaio, M.; Scarpa, M. Genistein reduces glycosaminoglycan levels in a mouse model of Mucopolysaccharidosis type II. Br. J. Pharmacol. 2010, 159, 1082–1091. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.L.; Rees, M.H.; Klebe, S.; Fletcher, J.M.; Byers, S. Improvement in behaviour after substrate deprivation therapy with rhodamine B in a mouse model of MPS IIIA. Mol. Genet. Metab. 2007, 92, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.L.; Thomas, B.J.; Wilkinson, A.S.; Fletcher, J.M.; Byers, S. Inhibition of glycosaminoglycan synthesis using rhodamine B in a mouse model of Mucopolysaccharidosis type IIIA. Pediatr. Res. 2006, 60, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.L.; Fletcher, J.M.; Moore, L.; Byers, S. Trans-generational exposure to low levels of rhodamine B does not adversely affect litter size or liver function in murine Mucopolysaccharidosis type IIIA. Mol. Genet. Metab. 2010, 101, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, E.; Jakobkiewicz-Banecka, J.; Tylki-Szymanska, A.; Liberek, A.; Maryniak, A.; Malinowska, M.; Czartoryska, B.; Puk, E.; Kloska, A.; Liberek, T.; et al. Genistin-rich soy isoflavone extract in substrate reduction therapy for sanfilippo syndrome: An open-label, pilot study in 10 pediatric patients. Curr. Ther. Res. Clin. Exp. 2008, 69, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, K.; Baranowska-Bosiacka, I.; Marchlewicz, M.; Gutowska, I.; Nocen, I.; Zawislak, M.; Chlubek, D.; Wiszniewska, B. Changes in male reproductive system and mineral metabolism induced by soy isoflavones administered to rats from prenatal life until sexual maturity. Nutrition 2011, 27, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Marucha, J.; Tylki-Szymanska, A.; Jakobkiewicz-Banecka, J.; Piotrowska, E.; Kloska, A.; Czartoryska, B.; Wegrzyn, G. Improvement in the range of joint motion in seven patients with Mucopolysaccharidosis type II during experimental gene expression-targeted isoflavone therapy (get it). Am. J. Med. Genet. Part A 2011, 155A, 2257–2262. [Google Scholar] [CrossRef] [PubMed]
- Malinowska, M.; Wilkinson, F.L.; Bennett, W.; Langford-Smith, K.J.; O’Leary, H.A.; Jakobkiewicz-Banecka, J.; Wynn, R.; Wraith, J.E.; Wegrzyn, G.; Bigger, B.W. Genistein reduces lysosomal storage in peripheral tissues of mucopolysaccharide iiib mice. Mol. Genet. Metab. 2009, 98, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Malinowska, M.; Wilkinson, F.L.; Langford-Smith, K.J.; Langford-Smith, A.; Brown, J.R.; Crawford, B.E.; Vanier, M.T.; Grynkiewicz, G.; Wynn, R.F.; Wraith, J.E.; et al. Genistein improves neuropathology and corrects behaviour in a mouse model of neurodegenerative metabolic disease. PLoS ONE 2010, 5, e14192. [Google Scholar] [CrossRef] [PubMed]
- The European Union Clinical Trials. High Dose Genistein in Sanfilippo Syndrome. Available online: https://www.clinicaltrialsregister.eu/ctr-search/trial/2013-001479-18/GB (accessed on 26 October 2016).
- Mizumoto, S.; Ikegawa, S.; Sugahara, K. Human genetic disorders caused by mutations in genes encoding biosynthetic enzymes for sulfated glycosaminoglycans. J. Biol. Chem. 2013, 288, 10953–10961. [Google Scholar] [CrossRef] [PubMed]
- Dire, D.J.; Wilkinson, J.A. Acute exposure to rhodamine B. J. Toxicol. Clin. Toxicol. 1987, 25, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Jakobkiewicz-Banecka, J.; Piotrowska, E.; Narajczyk, M.; Baranska, S.; Wegrzyn, G. Genistein-mediated inhibition of glycosaminoglycan synthesis, which corrects storage in cells of patients suffering from mucopolysaccharidoses, acts by influencing an epidermal growth factor-dependent pathway. J. Biomed. Sci. 2009, 16, 26. [Google Scholar] [CrossRef] [PubMed]
- Moskot, M.; Montefusco, S.; Jakobkiewicz-Banecka, J.; Mozolewski, P.; Wegrzyn, A.; di Bernardo, D.; Wegrzyn, G.; Medina, D.L.; Ballabio, A.; Gabig-Ciminska, M. The phytoestrogen genistein modulates lysosomal metabolism and transcription factor EB (TFEB) activation. J. Biol. Chem. 2014, 289, 17054–17069. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.; Lee, J.I. Current and potential therapeutic strategies for mucopolysaccharidoses. J. Clin. Pharm. Ther. 2014, 39, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Smith, D.; Jeyakumar, M.; Butters, T.D.; Borja, M.C.; Dwek, R.A.; Platt, F.M. Improved outcome of N-butyldeoxygalactonojirimycin-mediated substrate reduction therapy in a mouse model of sandhoff disease. Neurobiol. Dis. 2004, 16, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Butters, T.D.; Dwek, R.A.; Platt, F.M. N-butyldeoxygalactonojirimycin: A more selective inhibitor of glycosphingolipid biosynthesis than N-butyldeoxynojirimycin, in vitro and in vivo. Biochem. Pharmacol. 2000, 59, 821–829. [Google Scholar] [CrossRef]
- Ramachandran, P.V.; Ignacimuthu, S. Rna interference—A silent but an efficient therapeutic tool. Appl. Biochem. Biotechnol. 2013, 169, 1774–1789. [Google Scholar] [CrossRef] [PubMed]
- Diallo, M.; Arenz, C.; Schmitz, K.; Sandhoff, K.; Schepers, U. Long endogenous dsrnas can induce complete gene silencing in mammalian cells and primary cultures. Oligonucleotides 2003, 13, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Font, A.; Chabas, A.; Grinberg, D.; Vilageliu, L. Rnai-mediated inhibition of the glucosylceramide synthase (GCS) gene: A preliminary study towards a therapeutic strategy for Gaucher disease and other glycosphingolipid storage diseases. Blood Cells Mol. Dis. 2006, 37, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Dziedzic, D.; Wegrzyn, G.; Jakobkiewicz-Banecka, J. Impairment of glycosaminoglycan synthesis in Mucopolysaccharidosis type IIIA cells by using siRNA: A potential therapeutic approach for sanfilippo disease. Eur. J. Hum. Genet. 2010, 18, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Canals, I.; Beneto, N.; Cozar, M.; Vilageliu, L.; Grinberg, D. EXTL2 and EXTL3 inhibition with siRNAs as a promising substrate reduction therapy for sanfilippo C syndrome. Sci. Rep. 2015, 5, 13654. [Google Scholar] [CrossRef] [PubMed]
- Haskins, M.E.; Giger, U.; Patterson, D.F. “Animal models of lysosomal storage diseases: Their development and clinical relevance” in fabry disease: Perspectives from 5 years of fos. In Oxford Pharmagenesis; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford Pharmagenesis: Oxford, UK, 2006. [Google Scholar]
- Burrow, T.A.; Grabowski, G.A. “Emerging Treatments and Future Outcomes” in Lysosomal Storage Disorders—A Practical Guide, 1st ed.; Wiley: New York, NY, USA, 2012. [Google Scholar]
- Capablo, J.L.; Franco, R.; de Cabezon, A.S.; Alfonso, P.; Pocovi, M.; Giraldo, P. Neurologic improvement in a type 3 Gaucher disease patient treated with imiglucerase/miglustat combination. Epilepsia 2007, 48, 1406–1408. [Google Scholar] [CrossRef] [PubMed]
- Cox-Brinkman, J.; van Breemen, M.J.; van Maldegem, B.T.; Bour, L.; Donker, W.E.; Hollak, C.E.; Wijburg, F.A.; Aerts, J.M. Potential efficacy of enzyme replacement and substrate reduction therapy in three siblings with Gaucher disease type III. J. Inherit. Metab. Dis. 2008, 31, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.; McEachern, K.A.; Chuang, W.L.; Hutto, E.; Siegel, C.S.; Shayman, J.A.; Grabowski, G.A.; Scheule, R.K.; Copeland, D.P.; Cheng, S.H. Improved management of lysosomal glucosylceramide levels in a mouse model of type 1 Gaucher disease using enzyme and substrate reduction therapy. J. Inherit. Metab. Dis. 2010, 33, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.; Ashe, K.M.; Bangari, D.; McEachern, K.; Chuang, W.L.; Pacheco, J.; Copeland, D.P.; Desnick, R.J.; Shayman, J.A.; Scheule, R.K.; et al. Substrate reduction augments the efficacy of enzyme therapy in a mouse model of fabry disease. PLoS ONE 2010, 5, e15033. [Google Scholar] [CrossRef] [PubMed]
- Jeyakumar, M.; Norflus, F.; Tifft, C.J.; Cortina-Borja, M.; Butters, T.D.; Proia, R.L.; Perry, V.H.; Dwek, R.A.; Platt, F.M. Enhanced survival in sandhoff disease mice receiving a combination of substrate deprivation therapy and bone marrow transplantation. Blood 2001, 97, 327–329. [Google Scholar] [CrossRef] [PubMed]
- Villamizar-Schiller, I.T.; Pabon, L.A.; Hufnagel, S.B.; Serrano, N.C.; Karl, G.; Jefferies, J.L.; Hopkin, R.J.; Prada, C.E. Neurological and cardiac responses after treatment with miglustat and a ketogenic diet in a patient with sandhoff disease. Eur. J. Med. Genet. 2015, 58, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, B.E.; Pastores, G.M.; Gianutsos, J.; Luzy, C.; Kolodny, E.H. Miglustat in late-onset tay-sachs disease: A 12-month, randomized, controlled clinical study with 24 months of extended treatment. Genet. Med. Off. J. Am. Coll. Med. Genet. 2009, 11, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Denny, C.A.; Heinecke, K.A.; Kim, Y.P.; Baek, R.C.; Loh, K.S.; Butters, T.D.; Bronson, R.T.; Platt, F.M.; Seyfried, T.N. Restricted ketogenic diet enhances the therapeutic action of N-butyldeoxynojirimycin towards brain GM2 accumulation in adult sandhoff disease mice. J. Neurochem. 2010, 113, 1525–1535. [Google Scholar] [CrossRef] [PubMed]
- Williams, I.M.; Wallom, K.L.; Smith, D.A.; Al Eisa, N.; Smith, C.; Platt, F.M. Improved neuroprotection using miglustat, curcumin and ibuprofen as a triple combination therapy in Niemann-Pick disease type C1 mice. Neurobiol. Dis. 2014, 67, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Narajczyk, M.; Moskot, M.; Konieczna, A. Quantitative estimation of lysosomal storage in mucopolysaccharidoses by electron microscopy analysis. Acta Biochim. Pol. 2012, 59, 693–696. [Google Scholar] [PubMed]
- Jakobkiewicz-Banecka, J.; Piotrowska, E.; Gabig-Ciminska, M.; Borysiewicz, E.; Slominska-Wojewodzka, M.; Narajczyk, M.; Wegrzyn, A.; Wegrzyn, G. Substrate reduction therapies for mucopolysaccharidoses. Curr. Pharm. Biotechnol. 2011, 12, 1860–1865. [Google Scholar] [CrossRef] [PubMed]
- Venier, R.E.; Igdoura, S.A. Miglustat as a therapeutic agent: Prospects and caveats. J. Med. Genet. 2012, 49, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Banecka-Majkutewicz, Z.; Jakobkiewicz-Banecka, J.; Gabig-Ciminska, M.; Wegrzyn, A.; Wegrzyn, G. Putative biological mechanisms of efficiency of substrate reduction therapies for mucopolysaccharidoses. Arch. Immunol. Ther. Exp. 2012, 60, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.B.; Winchester, B. (Eds.) Lysosomal Storage Disorders: A Practical Guide; Wiley: Malden, MA, USA, 2012; p. 208.
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coutinho, M.F.; Santos, J.I.; Alves, S. Less Is More: Substrate Reduction Therapy for Lysosomal Storage Disorders. Int. J. Mol. Sci. 2016, 17, 1065. https://doi.org/10.3390/ijms17071065
Coutinho MF, Santos JI, Alves S. Less Is More: Substrate Reduction Therapy for Lysosomal Storage Disorders. International Journal of Molecular Sciences. 2016; 17(7):1065. https://doi.org/10.3390/ijms17071065
Chicago/Turabian StyleCoutinho, Maria Francisca, Juliana Inês Santos, and Sandra Alves. 2016. "Less Is More: Substrate Reduction Therapy for Lysosomal Storage Disorders" International Journal of Molecular Sciences 17, no. 7: 1065. https://doi.org/10.3390/ijms17071065
APA StyleCoutinho, M. F., Santos, J. I., & Alves, S. (2016). Less Is More: Substrate Reduction Therapy for Lysosomal Storage Disorders. International Journal of Molecular Sciences, 17(7), 1065. https://doi.org/10.3390/ijms17071065