
Molecular Characterization of the Complete Genome of Three Basal-BR Isolates of Turnip mosaic virus Infecting Raphanus sativus in China


Abstract
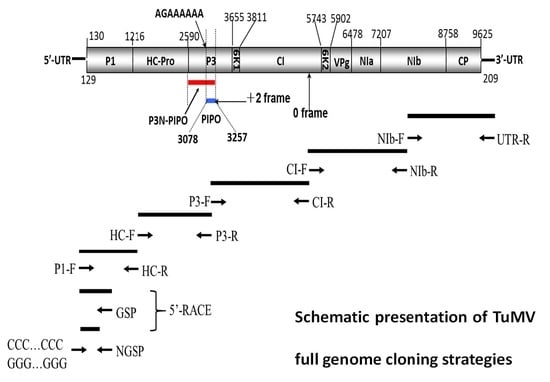
:
1. Introduction
2. Results
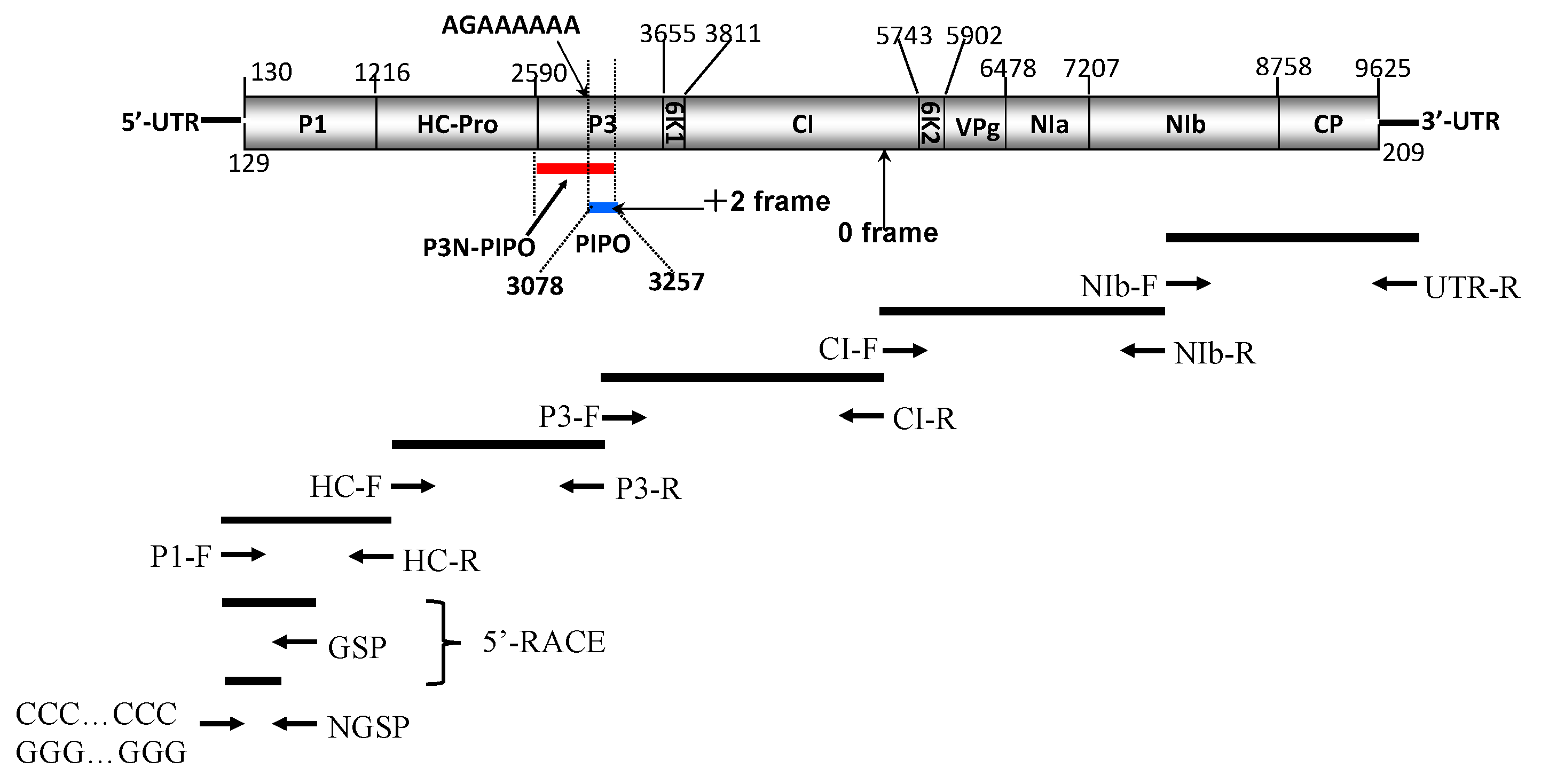
2.1. Genome Structure and Characterization of Putative Polyproteins
2.2. Percentage Identity
2.3. Phylogenetic Relationships
2.4. Recombination Analysis
2.5. Genetic Distance and Selection Pressure
3. Discussion
4. Materials and Methods
4.1. Virus Samples, RNA Extraction and Sequencing
4.2. Phylogenetic Analyses
4.3. Recombination Analyses
4.4. Genetic Distance and Selection Pressure
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Walsh, J.A.; Jenner, C.E. Turnip mosaic virus and the quest for durable resistance. Mol. Plant Pathol. 2002, 3, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Tomimura, K.; Gibbs, A.J.; Jenner, C.E.; Walsh, J.A.; Ohshima, K. The phylogeny of Turnip mosaic virus; comparisons of 38 genomic sequences reveal a Eurasian origin and a recent ”emergence” in East Asia. Mol. Ecol. 2003, 12, 2099–2111. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, J. Epidemiology and control of virus diseases of vegetables. Ann. Appl. Biol. 1987, 110, 661–681. [Google Scholar] [CrossRef]
- King, A.M.; Adams, M.J.; Lefkowitz, E.J. Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier Academic Press: San Diego, CA, USA, 2012; Volume 9. [Google Scholar]
- Vijayapalani, P.; Maeshima, M.; Nagasaki-Takekuchi, N.; Miller, W.A. Interaction of the trans-frame potyvirus protein P3N-PIPO with host protein PCaP1 facilitates Potyvirus movement. PLoS Pathog. 2012, 8, e1002639. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.Y.W.; Miller, W.A.; Atkins, J.F.; Firth, A.E. An overlapping essentiapgene in the Potyviridae. Proc. Natl. Acad. Sci. USA 2008, 105, 5897–5902. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.D.; Tomitaka, Y.; Ho, S.Y.; Duchêne, S.; Vetten, H.-J.; Lesemann, D.; Walsh, J.A.; Gibbs, A.J.; Ohshima, K. Turnip mosaic potyvirus probably first spread to Eurasian Brassica crops from wild orchids about 1000 years ago. PLoS ONE 2013, 8, e55336. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Liu, J.L.; Gao, R.; Chen, J.; Shao, Y.H.; Li, X.D. Complete genomic sequence analyses of Turnip mosaic virus basal-BR isolates from China. Virus Genes 2009, 38, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, A.J.; Nguyen, H.D.; Ohshima, K. The ”emergence” of Turnip mosaic virus was probably a ”gene-for-quasi-gene” event. Curr. Opin. Virol. 2015, 10, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K.; Akaishi, S.; Kajiyama, H.; Koga, R.; Gibbs, A.J. Evolutionary trajectory of Turnip mosaic virus populations adapting to a new host. J. Gen. Virol. 2010, 91, 788–801. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.D.; Tran, H.T.N.; Ohshima, K. Genetic variation of the Turnip mosaic virus population of Vietnam: A case study of founder, regional and local influences. Virus Res. 2013, 171, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K.; Yamaguchi, Y.; Hirota, R.; Hamamoto, T.; Tomimura, K.; Tan, Z.; Sano, T.; Azuhata, F.; Walsh, J.A.; Fletcher, J. Molecular evolution of Turnip mosaic virus: Evidence of host adaptation, genetic recombination and geographical spread. J. Gen. Virol. 2002, 83, 1511–1521. [Google Scholar] [CrossRef] [PubMed]
- Tomitaka, Y.; Ohshima, K. A phylogeographical study of the Turnip mosaic virus population in East Asia reveals an ”emergent” lineage in Japan. Mol. Ecol. 2006, 15, 4437–4457. [Google Scholar] [CrossRef] [PubMed]
- Yasaka, R.; Ohba, K.; Schwinghamer, M.W.; Fletcher, J.; Ochoa-Corona, F.M.; Thomas, J.E.; Ho, S.Y.; Gibbs, A.J.; Ohshima, K. Phylodynamic evidence of the migration of Turnip mosaic potyvirus from Europe to Australia and New Zealand. J. Gen. Virol. 2015, 96, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Tomimura, K.; Špak, J.; Katis, N.; Jenner, C.E.; Walsh, J.A.; Gibbs, A.J.; Ohshima, K. Comparisons of the genetic structure of populations of Turnip mosaic virus in West and East Eurasia. Virology 2004, 330, 408–423. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, A.; Ohshima, K. Potyviruses and the Digital Revolution. Annu. Rev. Phytopathol. 2010, 48, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.P.; Zhu, X.P.; Liu, J.L.; Yu, X.Q.; Du, J.; Kreuze, J.; Li, X.D. Molecular characterization of the 3′-terminal region of Turnip mosaic virus isolates from Eastern China. J. Phytopathol. 2007, 155, 333–341. [Google Scholar] [CrossRef]
- Song, Y.Z.; Li, L.L.; Zhu, C.X.; Wen, F.J.; Wen, F.K. Cloning and sequence analysis of coat protein genes of Turnip mosaic virus isolates obtained from Shandong. Scientia Agric. Sin. 2005, 38, 504–510. [Google Scholar]
- Shi, M.L. Cloning and sequence analysis of HC-Pro genes of Turnip mosaic virus Eurasian isolates. Acta Phytopathol. Sin. 2007, 37, 383–389. [Google Scholar]
- Turpen, T. Molecular cloning of a Potato virus Y genome: Nucleotide sequence homology in non-coding regions of Potyviruses. J. Gen. Virol. 1989, 70, 1951–1960. [Google Scholar] [CrossRef] [PubMed]
- Yukawa, Y.; Sugita, M.; Choisne, N.; Small, I.; Sugiura, M. The TATA motif, the CAA motif and the poly(T) transcription termination motif are all important for transcription re-initiation on plant tRNA genes. Plant J. 2000, 22, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Lütcke, H.; Chow, K.; Mickel, F.; Moss, K.; Kern, H.; Scheele, G. Selection of AUG initiation codons differs in plants and animals. EMBO J. 1987, 6, 43. [Google Scholar] [PubMed]
- Mlotshwa, S.; Verver, J.; Sithole-Niang, I.; Van Kampen, T.; Van Kammen, A.; Wellink, J. The genomic sequence of cowpea aphid-borne mosaic virus and its similarities with other potyviruses. Arch. Virol. 2002, 147, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.J.; Antoniw, J.F.; Beaudoin, F. Overview and analysis of the polyprotein cleavage sites in the family Potyviridae. Mol. Plant Pathol. 2005, 6, 471–487. [Google Scholar] [CrossRef] [PubMed]
- Atreya, C.D.; Pirone, T.P. Mutational analysis of the helper component-proteinase gene of a potyvirus: Effects of amino acid substitutions, deletions, and gene replacement on virulence and aphid transmissibility. Proc. Natl. Acad. Sci. USA 1993, 90, 11919–11923. [Google Scholar] [CrossRef] [PubMed]
- Gal-On, A. A point mutation in the FRNK motif of the potyvirus helper component-protease gene alters symptom expression in cucurbits and elicits protection against the severe homologous virus. Phytopathology 2000, 90, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Gal-On, A. Zucchini yellow mosaic virus: Insect transmission and pathogenicity—The tails of two proteins. Mol. Plant Pathol. 2007, 8, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Maia, I.G.; Bernardi, F. Nucleic acid-binding properties of a bacterially expressed Potato virus Y helper component-proteinase. J. Gen. Virol. 1996, 77, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Plisson, C.; Drucker, M.; Blanc, S.; German-Retana, S.; Le Gall, O.; Thomas, D.; Bron, P. Structural characterization of HC-Pro, a plant virus multifunctional protein. J. Biol. Chem. 2003, 278, 23753–23761. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Merits, A.; Saarma, M. Self-association and mapping of interaction domains of helper component-proteinase of potato A potyvirus. J. Gen. Virol. 1999, 80, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Kadaré, G.; Haenni, A.-L. Virus-encoded RNA helicases. J. Virol. 1997, 71, 2583. [Google Scholar] [PubMed]
- Liang, W.; Song, L.; Tian, G.; Li, H.; Fan, Z. The genomic sequence of Wisteria vein mosaic virus and its similarities with other potyviruses. Arch. Virol. 2006, 151, 2311–2319. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.F.; Klein, P.G.; Hunt, A.G.; Shaw, J.G. Replacement of the tyrosine residue that links a potyviral VPg to the viral RNA is lethal. Virology 1996, 220, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, W.G.; Parks, T.D.; Cary, S.M.; Bazan, J.F.; Fletterick, R.J. Characterization of the catalytic residues of the tobacco etch virus 49-kDa proteinase. Virology 1989, 172, 302–310. [Google Scholar] [CrossRef]
- Dujovny, G.; Sasaya, T.; Koganesawa, H.; Usugi, T.; Shohara, K.; Lenardon, S. Molecular characterization of a new potyvirus infecting sunflower. Arch. Virol. 2000, 145, 2249–2258. [Google Scholar] [CrossRef] [PubMed]
- Atreya, C.; Raccah, B.; Pirone, T. A point mutation in the coat protein abolishes aphid transmissibility of a potyvirus. Virology 1990, 178, 161–165. [Google Scholar] [CrossRef]
- Ohshima, K.; Tomitaka, Y.; Wood, J.T.; Minematsu, Y.; Kajiyama, H.; Tomimura, K.; Gibbs, A.J. Patterns of recombination in Turnip mosaic virus genomic sequences indicate hotspots of recombination. J. Gen. Virol. 2007, 88, 298–315. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Jenner, C.E.; Wang, X.; Tomimura, K.; Ohshima, K.; Ponz, F.; Walsh, J.A. The dual role of the potyvirus P3 protein of Turnip mosaic virus as a symptom and avirulence determinant in Brassicas. Mol. Plant Microbe Interact. 2003, 16, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Suehiro, N.; Natsuaki, T.; Watanabe, T.; Okuda, S. An important determinant of the ability of Turnip mosaic virus to infect Brassica spp. and/or Raphanus sativus is in its P3 protein. J. Gen. Virol. 2004, 85, 2087–2098. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Gibbs, A.J.; Tomitaka, Y.; Sánchez, F.; Ponz, F.; Ohshima, K. Mutations in Turnip mosaic virus genomes that have adapted to Raphanus sativus. J. Gen. Virol. 2005, 86, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Zhang, C.; Hong, J.; Xiong, R.; Kasschau, K.D.; Zhou, X.; Carrington, J.C.; Wang, A. Formation of complexes at plasmodesmata for potyvirus intercellular movement is mediated by the viral protein P3N-PIPO. PLoS Pathog. 2010, 6, e1000962. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Liu, J.; Zhang, C.; Liu, Y.; Wang, B.; Li, X.-D.; Guo, Z.; Valkonen, J. Genetic diversity of Potato virus Y infecting tobacco crops in China. Phytopathology 2011, 101, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Hillung, J.; Elena, S.F.; Cuevas, J.M. Intra-specific variability and biological relevance of P3N-PIPO protein length in potyviruses. BMC Evol. Biol. 2013, 13, 249–249. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.; Cong, Q.Q.; Li, X.D.; Mou, A.L.; Gao, R.; Liu, J.L.; Tian, Y.P. DEVELOPMENTALLY REGULATED PLASMA MEMBRANE PROTEIN of Nicotiana benthamiana contributes to potyvirus movement and transports to plasmodesmata via the early secretory pathway and the actomyosin system. Plant Physiol. 2015, 167, 394–410. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Hagiwara-Komoda, Y.; Nakahara, K.S.; Atsumi, G.; Shimada, R.; Hisa, Y.; Naito, S.; Uyeda, I. Quantitative and qualitative involvement of P3N-PIPO in overcoming recessive resistance against Clover yellow vein virus in pea carrying the cyv1 Gene. J. Virol. 2013, 87, 7326–7337. [Google Scholar] [CrossRef] [PubMed]
- Frohman, M.A. 5′-End cDNA amplification using classic RACE. CSH Protoc. 2005, 2006, 210–212. [Google Scholar]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Fuji, S.; Nakamae, H. Complete nucleotide sequence of the genomic RNA of a Japanese yam mosaic virus, a new potyvirus in Japan. Arch. Virol. 1999, 144, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Fuji, S.; Nakamae, H. Complete nucleotide sequence of the geonomic RNA of a mild strain of Japanese yam mosaic potyvirus. Rch. Virol. 2000, 145, 635–640. [Google Scholar] [CrossRef]
- Martin, D.P.; Lemey, P.; Lott, M.; Moulton, V.; Posada, D.; Lefeuvre, P. RDP3: A flexible and fast computer program for analyzing recombination. Bioinformatics 2010, 26, 2462–2463. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S. GENECONV: A Computer Package for the Statistical Detection of Gene Conversion; Washington University: St. Louis, MO, USA, 1999. [Google Scholar]
- Salminen, M.O.; CARR, J.K.; BURKE, D.S.; McCutchan, F.E. Identification of breakpoints in intergenotypic recombinants of HIV type 1 by bootscanning. AIDS Res. Hum. Retrovir. 1995, 11, 1423–1425. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.M. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Weiller, G.F. Phylogenetic profiles: A graphical method for detecting genetic recombinations in homologous sequences. Mol. Biol. Evol. 1998, 15, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-Scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
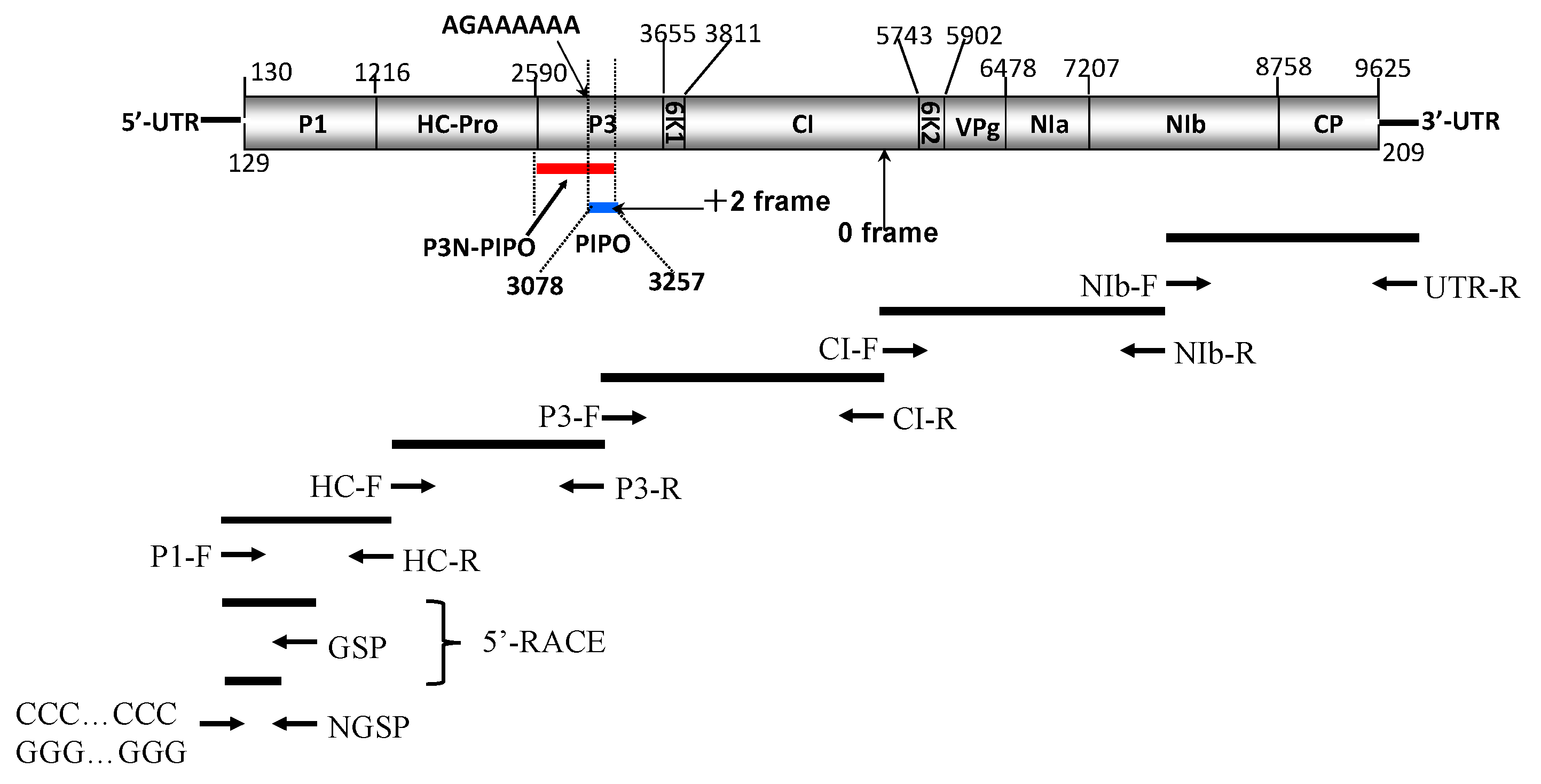


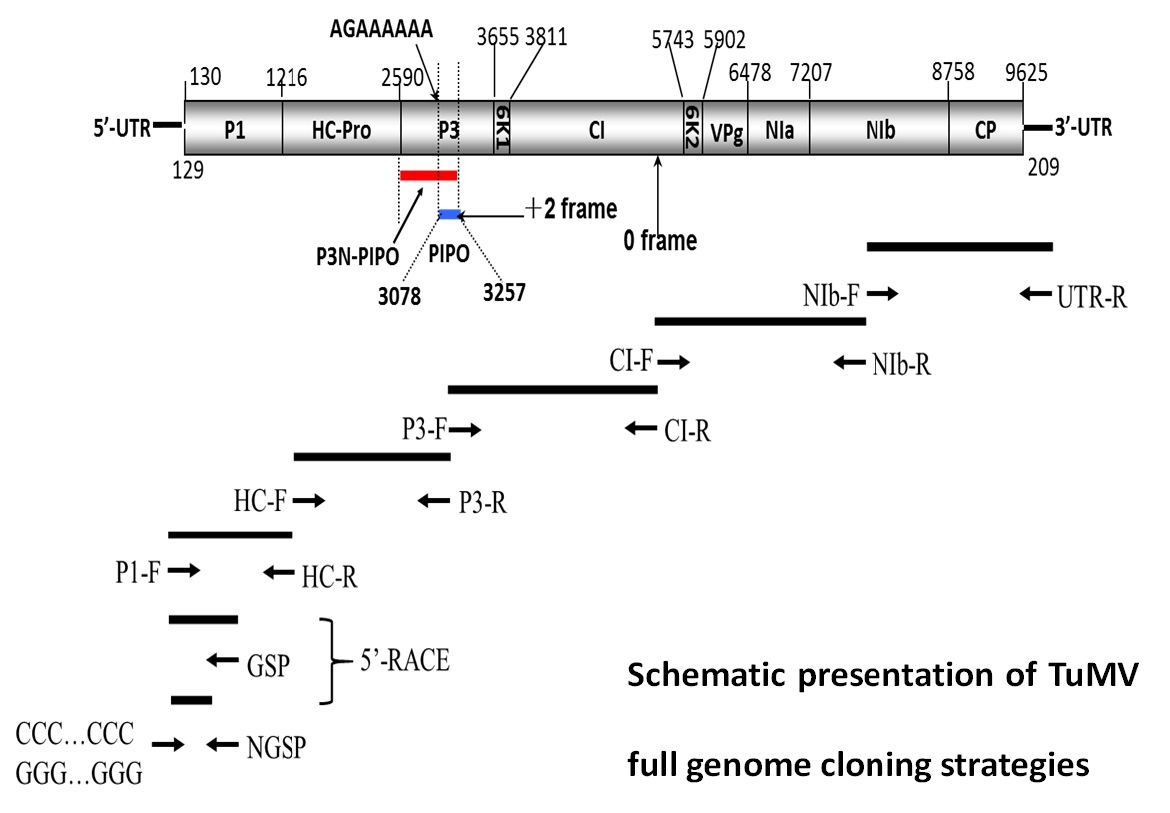{kind=link}
{kind=link}
{kind=link}
| Region (Gene) | Start–End Site | Size in nt/aa | Cleavage Site (C-Terminus) |
|---|---|---|---|
| 5′-UTR | 1–129 | 129/– | |
| P1 | 130–1215 | 1086/362 | Y/S |
| HC-Pro | 1216–2589 | 1374/458 | G/G |
| P3 | 2590–3654 | 1065/355 | Q/A |
| 6K1 | 3655–3810 | 156/52 | Q/T |
| CI | 3811–5742 | 1932/644 | Q/N |
| 6K2 | 5743–5901 | 159/53 | E/A |
| VPg | 5902–6477 | 576/192 | E/S |
| NIa-Pro | 6478–7206 | 729/243 | Q/T |
| NIb | 7207–8757 | 1551/517 | Q/A |
| CP | 8758–9621 | 864/288 | - |
| 3′-UTR | 9625–9833 | 209/- | - |
| Region (Gene) | CCLB | LWLB | WFLB14 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Isolate Name | nt (%) | aa (%) | Isolate Name | nt (%) | aa (%) | Isolate Name | nt (%) | aa (%) | |
| P1 | Cal1 | 95.3 | 94.2 | Cal1 | 94.7 | 93.4 | Cal1 | 95.5 | 94.5 |
| HC-Pro | DEU4 | 97.0 | 99.8 | DEU4 | 96.9 | 99.8 | DEU4 | 96.7 | 99.8 |
| PIPO | PV0104/DEU4 | 99.5 | 98.4 | PV0104/DEU4 | 99.5 | 98.4 | PV0104/DEU4 | 99.5 | 98.4 |
| P3 | PV0104/DEU4 | 97.1 | 98.6 | PV0104/DEU4 | 96.9 | 98.6 | PV0104/DEU4 | 96.8 | 98.6 |
| 6K1 | PV0104/DEU4 | 99.4 | 100.0 | PV0104/DEU4 | 98.1 | 100.0 | PV0104/DEU4 | 99.4 | 100.0 |
| CI | DEU4 | 98.7 | 99.8 | DEU4 | 98.6 | 99.8 | DEU4 | 98.3 | 99.8 |
| 6K2 | PV0104/DEU4 | 98.1 | 100.0 | PV0104/DEU4 | 96.9 | 96.2 | Cal1 | 98.1 | 100 |
| NIa-VPg | Cal1 | 95.5 | 99.0 | Cal1 | 95.7 | 99.0 | Cal1 | 95.8 | 99.5 |
| NIa-Pro | ITA8 | 91.5 | 98.8 | ITA8 | 90.5 | 98.8 | ITA8 | 90.9 | 98.8 |
| NIb | USA6 | 93.6 | 98.8 | USA6 | 93.3 | 98.6 | USA6 | 93.5 | 98.8 |
| CP | PV0104/DEU4 | 96.6 | 99.0 | PV0104/DEU4 | 96.5 | 99.0 | PV0104/DEU4 | 96.6 | 98.6 |
| Polyprotein | DEU4 | 94.9 | 98.1 | DEU4 | 94.6 | 97.9 | DEU4 | 94.7 | 98.0 |
| Genome Sequence | DEU4 | 95.0 | - | DEU4 | 94.7 | - | DEU4 | 94.8 | - |
| Accession Number | Isolates | Original Host | Location | Pathotype b | Year of Collection |
|---|---|---|---|---|---|
| AB093622 | 2J | Brassica pekinensis | Japan | BR | 1994 |
| AB093620 | 59J | Raphanus sativus | Japan | BR | 1996 |
| AB252097 | AD855J | Raphanus sativus | Japan | BR | 2002 |
| AB252099 | AKD161J | Raphanus sativus | Japan | BR | 1998 |
| AB252102 | AT181J | Eustoma russellianum | Japan | BR | <1998 |
| HQ446217 | BJ-C4 | cruciferous plants | China | Unknown | 1985–1987 |
| AB093601 | Cal1 | Calendula officinalis | Italy | BR | 1979 |
| AB252103 | CH6 | Raphanus sativus | China | BR | 1999 |
| AB252104 | CHK16 | Raphanus sativus | China | BR | 2000 |
| AB252105 | CHL13 | Raphanus sativus | China | BR | 1999 |
| AB093626 | CHN1 | Brassica sp. | China | BR | <1980 |
| AB093614 | CP845J | Calendula officinalis | Japan | BR | 1997 |
| AB701701 | DEU4 | Lactuca sativa | Germany | BR | 1986 |
| AB093623 | DMJ | Raphanus sativus | Japan | BR | 1996 |
| AB701705 | Eru1D | Eruca sativa | Italy | B | 1991 |
| AB252109 | FKD001J | Raphanus sativus | Japan | BR | 2000 |
| AB701696 | GK1 | Matthiola incana | Greece | B | <1989 |
| AB252118 | H1J | Raphanus sativus | Japan | BR | 1996 |
| AB093627 | HRD | Raphanus sativus | China | BR | 1998 |
| AB093602 | IS1 | Allium ampeloprasum | Israel | B | 1993 |
| AB701721 | ITA2 | Cheiranthus cheiri | Italy | BR | 1992 |
| AB701725 | ITA8 | Abutilon sp. | Italy | BR | 1993 |
| KM094174 | JPN 1 | Raphanus sativus | Japan | Unknown | 2014 |
| AB093605 | KEN1 | Brassica oleracea | Kenya | B | 1994 |
| AB252124 | KWB778J | Brassica oleracea | Japan | B | 2004 |
| AB252125 | KWB779J | Brassica rapa | Japan | BR | 2004 |
| AB252130 | ND10J | Raphanus sativus | Japan | BR | 1998 |
| AB701727 | NLD2 | Brassica oleracea | Netherlands | B | <1995 |
| AB701690 | OM | Orchis militaris | Germany | DI c | 1981 |
| AB701691 | OMA | Orchis militaris | Germany | DI | 1981 |
| AB701692 | ORM | Orchis morio | Germany | (B) | 1983 |
| AB701693 | OS | Orchis simia | Germany | DI | 1981 |
| AB093603 | PV0104 | Lactuca sativa | Germany | BR | 1993 |
| AY134473 | RC4 | Zantedeschia sp. | China | BR | 2000 |
| AB093615 | TD88J | Raphanus sativus | Japan | BR | 1998 |
| AB105134 | Tu-3 | Brassica oleracea | Japan | B | Unknown |
| AB362513 | TUR9 | Raphanus sativus | Turkey | B(R) | <2007 |
| AF169561 | UK1 | Brassica napus | UK | B | 1975 |
| AB701741 | USA6 | Raphanus sativus | USA | BR | 2002 |
| EU734434 | WFLB06 | Raphanus sativus | China | BR | 2006 |
| AF530055 | YC5 | Zantedeschia sp. | China | BR | 2000 |
| KF246570 | ZH1 | Phalaenopsis sp. | China | Unknown | 2012 |
| KR153038 | CCLB | Raphanus sativus | China | Unknown | 2014 |
| KR153039 | LWLB | Raphanus sativus | China | Unknown | 2014 |
| KR153040 | WFLB14 | Raphanus sativus | China | Unknown | 2014 |
| Isolate | Recombination Region | “Parential-Like” Isolate | Type of “Recombinant” | Recombination Detection | ||
|---|---|---|---|---|---|---|
| Major | Minor | Methods * | p-Value | |||
| CHN1 | nt 8872-9776 (5′-UTR-P1) | CH6 | 2J | Asian-BR × world-B | RGCS | 1.334 × 10−8 |
| ND10J | nt 143-723 (5′-UTR-P1) | CH6 | KWB778J | Asian-BR × world-B | RGMCS3 | 1.410 × 10−14 |
| nt 4598-5983 (VPg-CP) | 59J | DMJ | Asian-BR × world-B | RGBMCS3 | 1.608 × 10−25 | |
| nt 9133-9759 (VPg-CP) | CH6 | 2J | Asian-BR × world-B | RGCS | 1.334 × 10−8 | |
| 59J | nt 142-742 (HC-Pro-P3) | CH6 | KWB778J | Asian-BR × world-B | RGMCS3 | 1.410 × 10−14 |
| nt 9174-9759 (HC-Pro-P3) | CH6 | 2J | Asian-BR × world-B | RGCS | 1.334 × 10−8 | |
| Group | OMs | world-B | Asian-BR | basal-BR | basal-B |
|---|---|---|---|---|---|
| OMs | 0.003 ± 0.000 | ||||
| world-B | 0.278 ± 0.005 | 0.042 ± 0.001 | |||
| Asian-BR | 0.286 ± 0.005 | 0.175 ± 0.003 | 0.037 ± 0.001 | ||
| basal-BR | 0.281 ± 0.004 | 0.182 ± 0.003 | 0.158 ± 0.004 | 0.092 ± 0.002 | |
| basal-B | 0.283 ± 0.005 | 0.219 ± 0.004 | 0.222 ± 0.004 | 0.217 ± 0.003 | 0.185 ± 0.004 |
| Gene Name | China | Japan | ||||
|---|---|---|---|---|---|---|
| dN | dS | dN/dS | dN | dS | dN/dS | |
| P1 | 0.097 (±0.009) | 0.519 (±0.039) | 0.187 | 0.060 (±0.007) | 0.322 (±0.029) | 0.186 |
| HC-Pro | 0.016 (±0.003) | 0.854 (±0.083) | 0.019 | 0.015 (±0.003) | 0.716 (±0.094) | 0.021 |
| P3 | 0.065 (±0.008) | 0.705 (±0.057) | 0.092 | 0.064 (±0.007) | 0.519 (±0.055) | 0.123 |
| P3N-PIPO | 0.055 (±0.009) | 0.594 (±0.067) | 0.093 | 0.051 (±0.009) | 0.481 (±0.066) | 0.106 |
| PIPO | 0.047 (±0.013) | 0.105 (±0.038) | 0.448 | 0.050 (±0.016) | 0.133 (±0.045) | 0.375 |
| 6K1 | 0.015 (±0.008) | 0.769 (±0.151) | 0.020 | 0.017 (±0.009) | 0.801 (±0.211) | 0.021 |
| CI | 0.012 (±0.002) | 0.567 (±0.032) | 0.021 | 0.011 (±0.002) | 0.413 (±0.034) | 0.027 |
| 6K2 | 0.048 (±0.015) | 0.546 (±0.104) | 0.088 | 0.030 (±0.011) | 0.280 (±0.086) | 0.107 |
| VPg | 0.042 (±0.008) | 0.614 (±0.069) | 0.068 | 0.039 (±0.008) | 0.631 (±0.085) | 0.062 |
| NIa-Pro | 0.013 (±0.003) | 0.618 (±0.058) | 0.021 | 0.008 (±0.003) | 0.471 (±0.064) | 0.017 |
| NIb | 0.013 (±0.003) | 0.451 (±0.032) | 0.029 | 0.013 (±0.002) | 0.400 (±0.036) | 0.033 |
| CP | 0.017 (±0.004) | 0.240 (±0.027) | 0.071 | 0.012 (±0.003) | 0.175 (±0.024) | 0.069 |
| Primer | Sequence (5′→3′) |
|---|---|
| P1-F | AAAAATATAAAAACTCAACACAACATACACAAAACGA |
| HC-R | CTGTCGAAGCCTTTCCARAAGT |
| HC-F | ACTTYTGGAAAGGCTTCGACAG |
| P3-R | CGCTGTATCTGCCGCCTAAATCG |
| P3-F | CGATTTAGGCGGCAGATACAGCG |
| CI-R | TCCYTCAAGCACTGATATGTTCTC |
| CI-F | GAGAACATATCAGTGCTTGARGGA |
| NIb-R | TCTTCYTTCATCTCRGGTGTGAACTC |
| NIb-F | GAGTTCACACCYGAGATGAARGAAGA |
| UTR-R | TTTTTTTTTTTTTTTTTTGTCCCTTGCATCCTATCAAATG |
| 5′-RACE-QT | CCAGTGAGCAGAGTGACGAGGACTCGAGCTCAAGCTTTTTTTTTTTTTTTTT |
| 5′-RACE-QO | CCAGTGAGCAGAGTGACG |
| 5′-RACE-QI | GAGGACTCGAGCTCAAGC |
| GSP | AGCTGCGGCTTCCCTGAGGCTA |
| NGSP | TCCCAAATTGTACCATTCCGGTG |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, F.; Sun, Y.; Wang, Y.; Pan, H.; Wang, F.; Zhang, X.; Zhang, Y.; Liu, J. Molecular Characterization of the Complete Genome of Three Basal-BR Isolates of Turnip mosaic virus Infecting Raphanus sativus in China. Int. J. Mol. Sci. 2016, 17, 888. https://doi.org/10.3390/ijms17060888
Zhu F, Sun Y, Wang Y, Pan H, Wang F, Zhang X, Zhang Y, Liu J. Molecular Characterization of the Complete Genome of Three Basal-BR Isolates of Turnip mosaic virus Infecting Raphanus sativus in China. International Journal of Molecular Sciences. 2016; 17(6):888. https://doi.org/10.3390/ijms17060888
Chicago/Turabian StyleZhu, Fuxiang, Ying Sun, Yan Wang, Hongyu Pan, Fengting Wang, Xianghui Zhang, Yanhua Zhang, and Jinliang Liu. 2016. "Molecular Characterization of the Complete Genome of Three Basal-BR Isolates of Turnip mosaic virus Infecting Raphanus sativus in China" International Journal of Molecular Sciences 17, no. 6: 888. https://doi.org/10.3390/ijms17060888
APA StyleZhu, F., Sun, Y., Wang, Y., Pan, H., Wang, F., Zhang, X., Zhang, Y., & Liu, J. (2016). Molecular Characterization of the Complete Genome of Three Basal-BR Isolates of Turnip mosaic virus Infecting Raphanus sativus in China. International Journal of Molecular Sciences, 17(6), 888. https://doi.org/10.3390/ijms17060888