
Site-Specific Integration of Exogenous Genes Using Genome Editing Technologies in Zebrafish

Abstract
: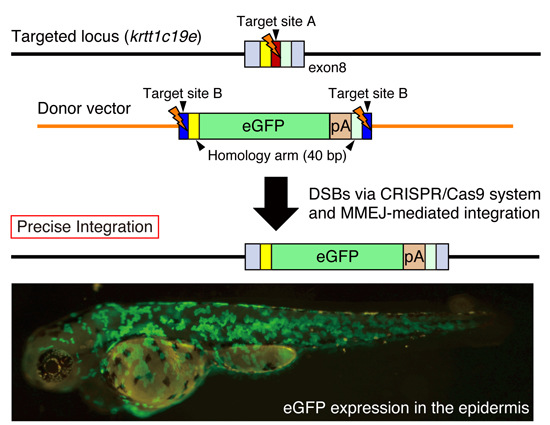
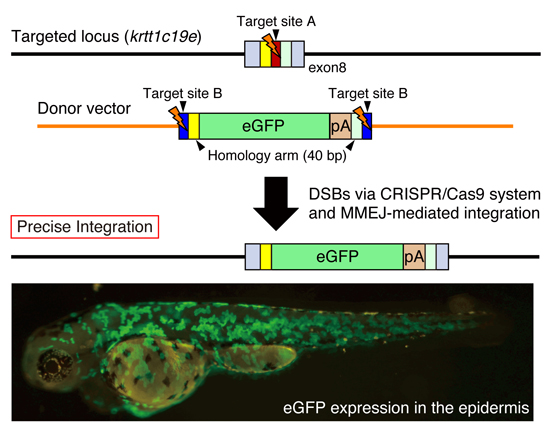{kind=link}
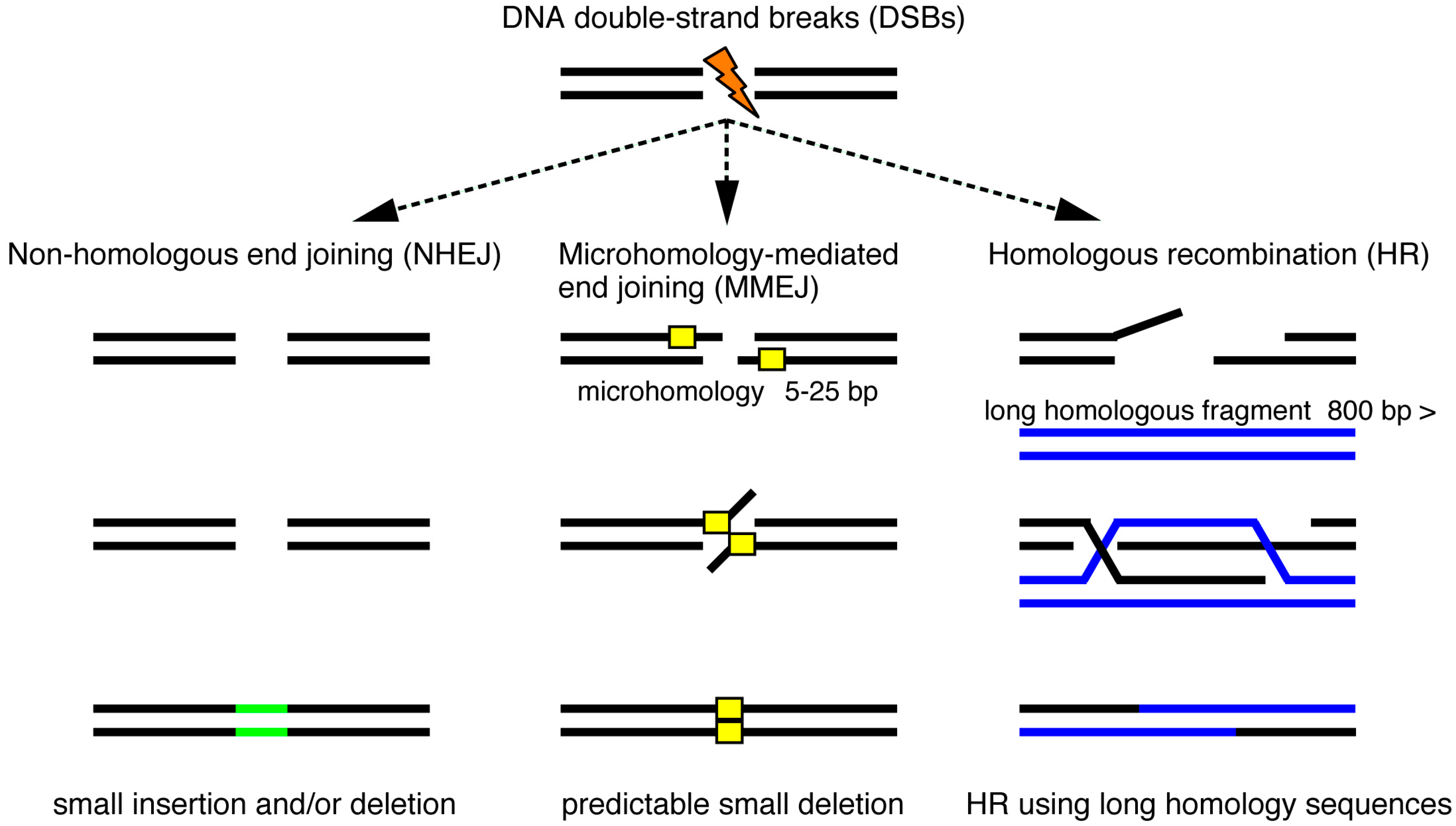{kind=link}
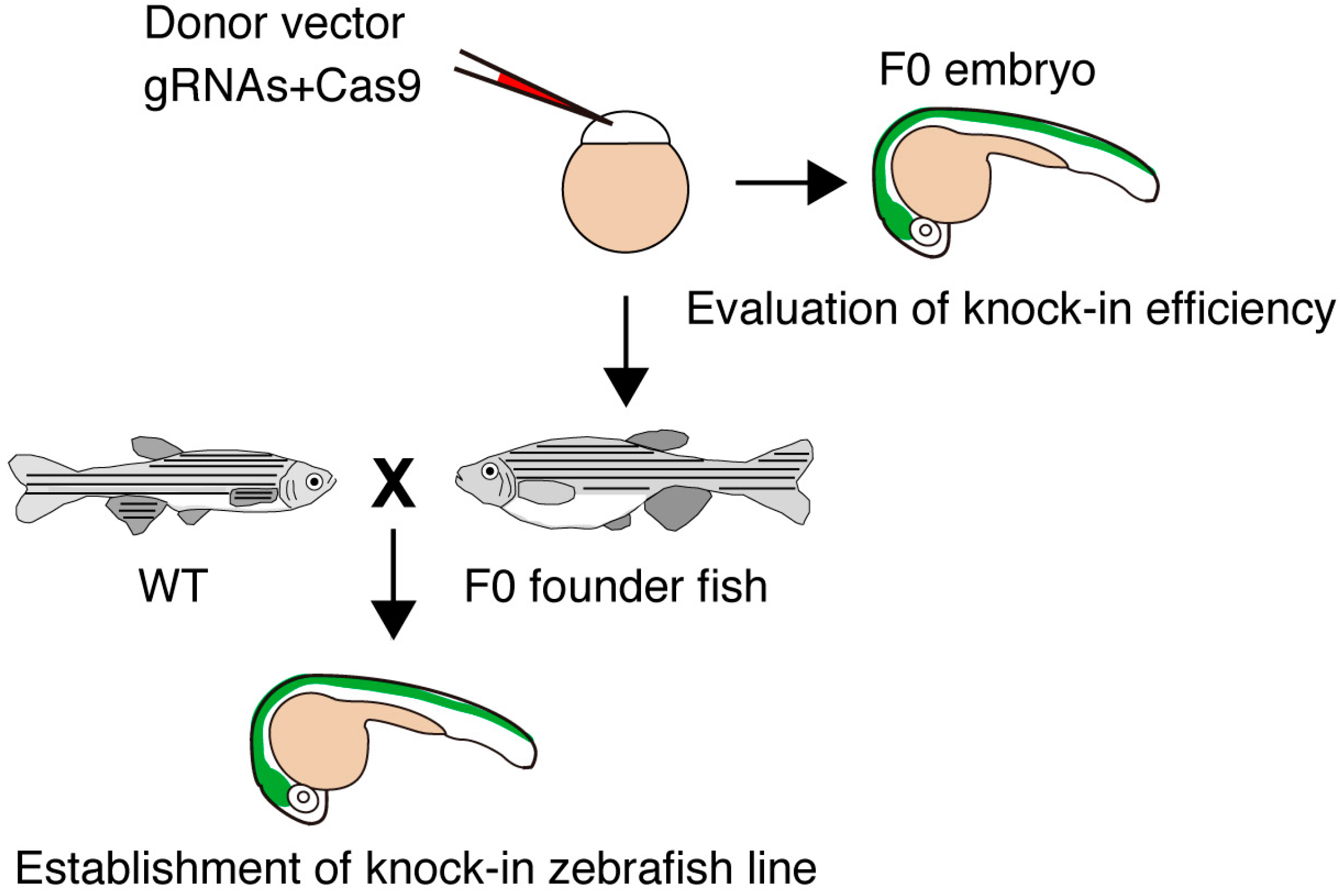{kind=link}
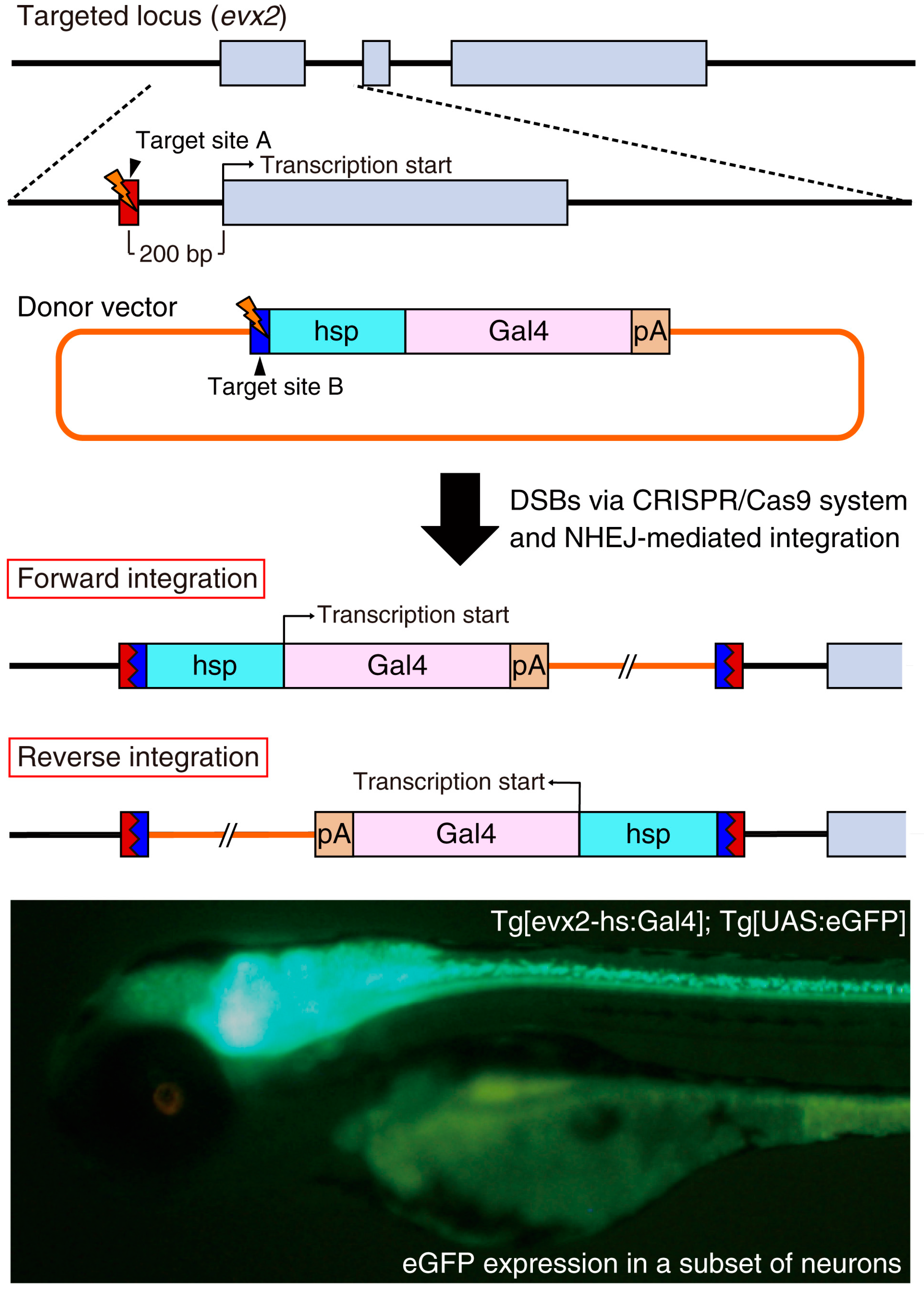
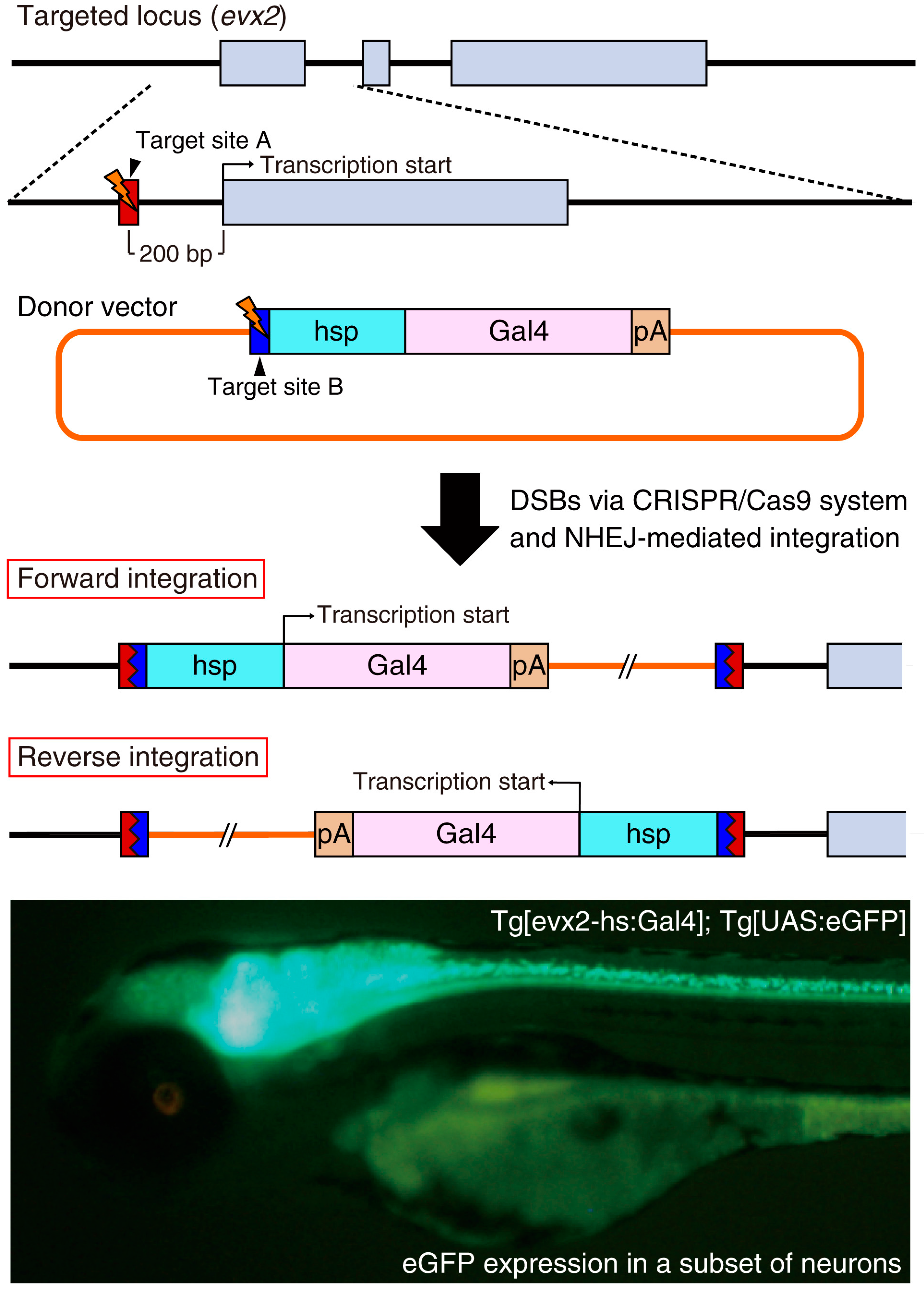{kind=link}
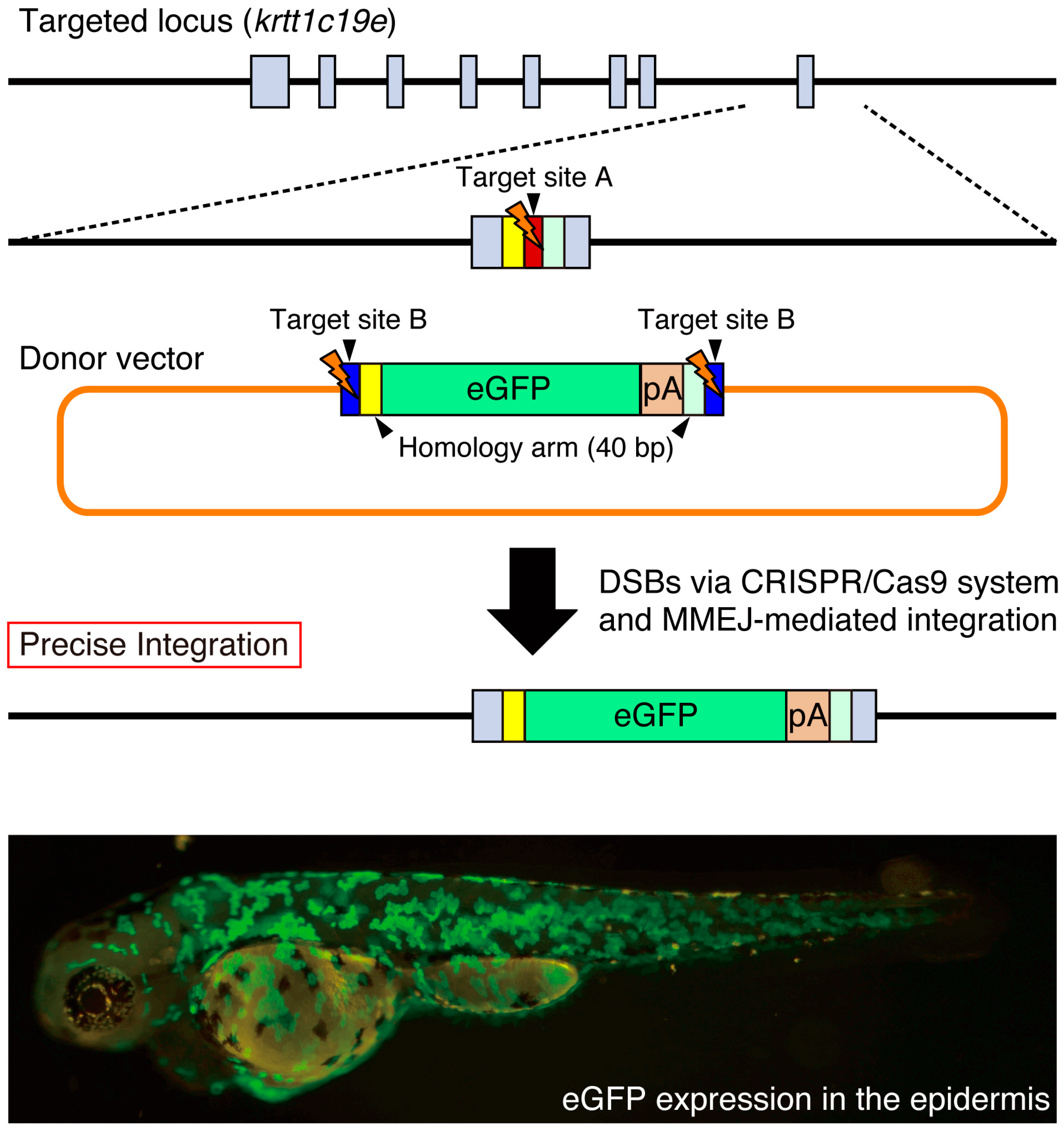{kind=link}
1. Introduction
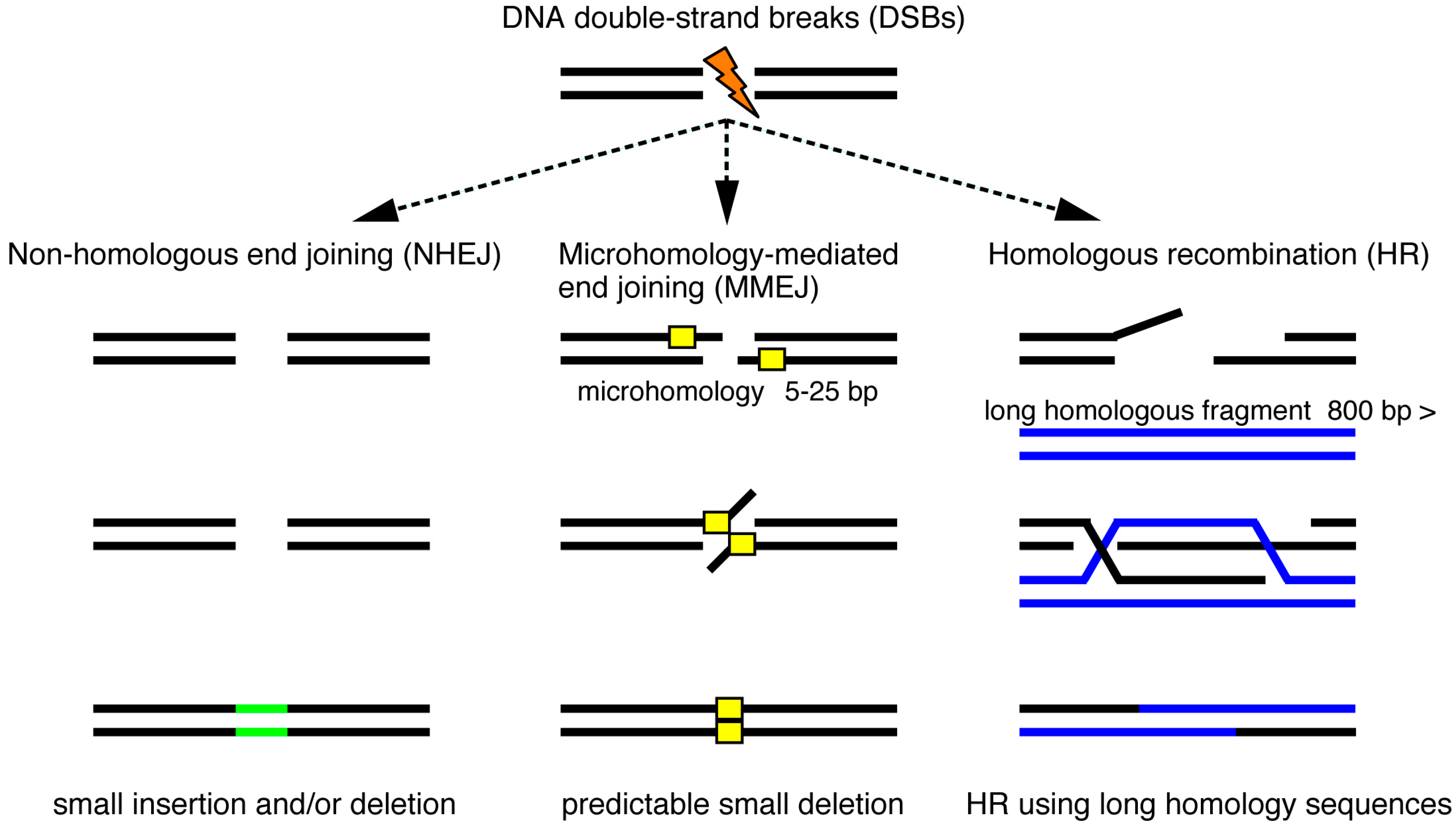
2. Genomic Insertion of Single-Stranded Oligodeoxynucleotides (ssODNs)
3. Precise Site-Specific Integration of Donor DNA by Homologous Recombination (HR)
4. Genomic Insertion of Donor DNA by Non-Homologous End Joining (NHEJ)
5. Precise Targeted Integration of Donor DNA by Microhomology-Mediated End Joining (MMEJ)
6. Concluding Remarks and Future Perspective
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ZFN | zinc finger nuclease |
| TALEN | transcription activator-like effector nuclease |
| CRISPR | clustered regularly interspaced short palindromic repeats |
| MO | morpholino oligonucleotide |
| RVD | repeat-variable di-residue |
| crRNA | CRISPR RNA |
| tracrRNA | trans-activating crRNA |
| PAM | protospacer-adjacent motif |
| gRNA | guide RNA |
| DSB | double-strand break |
| NHEJ | non-homologous end joining |
| MMEJ | microhomology-mediated end joining |
| HR | homologous recombination |
| indel | insertion and/or deletion |
| ssODN | single-stranded oligodeoxynucleotide |
| HA | hemagglutinin |
| BAC | bacterial artificial chromosome |
| iPS | induced pluripotent stem |
References
- Ota, S.; Kawahara, A. Zebrafish: A model vertebrate suitable for the analysis of human genetic disorders. Congenit. Anom. (Kyoto) 2014, 54, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Chiang, C.Y.; Tsai, H.-J. Zebrafish and Medaka: New model organisms for modern biomedical research. J. Biomed. Sci. 2016, 23, 19. [Google Scholar] [CrossRef] [PubMed]
- Nasevicius, A.; Ekker, S.C. Effective targeted gene “knockdown” in zebrafish. Nat. Genet. 2000, 26, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Robu, M.E.; Larson, J.D.; Nasevicius, A.; Beiraghi, S.; Brenner, C.; Farber, S.A.; Ekker, S.C. p53 activation by knockdown technologies. PLoS Genet. 2007, 3, 787–801. [Google Scholar] [CrossRef] [PubMed]
- Hisano, Y.; Ota, S.; Kawahara, A. Genome editing using artificial site-specific nucleases in zebrafish. Dev. Growth Differ. 2014, 56, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Sung, Y.H.; Kim, J.M.; Kim, H.T.; Lee, J.; Jeon, J.; Jin, Y.; Choi, J.H.; Ban, Y.H.; Ha, S.J.; Kim, C.H.; et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Res. 2014, 24, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Noyes, M.B.; Zhu, L.J.; Lawson, N.D.; Wolfe, S.A. Targeted gene inactivation in zebrafish using engineered zinc-finger nucleases. Nat. Biotechnol. 2008, 26, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Doyon, Y.; Mccammon, J.M.; Miller, J.C.; Faraji, F.; Katibah, G.E.; Amora, R.; Hocking, T.D.; Zhang, L.; Rebar, E.J.; Gregory, P.D.; et al. Heritable Targeted Gene Disruption in Zebrafish Using Designed Zinc Finger Nucleases. Nat. Biotechnol. 2009, 26, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Van Impel, A.; Zhao, Z.; Hermkens, D.M.; Roukens, M.G.; Fischer, J.C.; Peterson-Maduro, J.; Duckers, H.; Ober, E.A.; Ingham, P.W.; Schulte-Merker, S. Divergence of zebrafish and mouse lymphatic cell fate specification pathways. Development 2014, 141, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Kok, F.O.; Shin, M.; Ni, C.W.; Gupta, A.; Grosse, A.S.; VanImpel, A.; Kirchmaier, B.C.; Peterson-Maduro, J.; Kourkoulis, G.; Male, I.; et al. Reverse genetic screening reveals poor correlation between morpholino-induced and mutant phenotypes in zebrafish. Dev. Cell 2015, 32, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Hisano, Y.; Inoue, A.; Taimatsu, K.; Ota, S.; Ohga, R.; Kotani, H.; Muraki, M.; Aoki, J.; Kawahara, A. Comprehensive analysis of sphingosine-1-phosphate receptor mutants during zebrafish embryogenesis. Genes Cells 2015, 20, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Hisano, Y.; Inoue, A.; Okudaira, M.; Taimatsu, K.; Matsumoto, H.; Kotani, H.; Ohga, R.; Aoki, J.; Kawahara, A. Maternal and Zygotic Sphingosine Kinase 2 are Indispensable for Cardiac Development in Zebrafish. J. Biol. Chem. 2015, 290, 14841–14851. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, K.; Lan, Y.; Hla, T.; Evans, T. Maternal or zygotic sphingosine kinase is required to regulate zebrafish cardiogenesis. Dev. Dyn. 2015, 244, 948–954. [Google Scholar] [CrossRef] [PubMed]
- Carroll, D. Genome engineering with zinc-finger nucleases. Genetics 2011, 188, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Ansai, S.; Kinoshita, M. Targeted mutagenesis using CRISPR/Cas system in medaka. Biol. Open 2014, 3, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Carroll, D.; Segal, D.J.; Trautman, J.K.; Smith, J.; Kim, Y.G.; Chandrasegaran, S. Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Mol. Cell. Biol. 2001, 21, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Cermak, T.; Doyle, E.L.; Christian, M.; Wang, L.; Zhang, Y.; Schmidt, C.; Baller, J.A.; Somia, N.V.; Bogdanove, A.J.; Voytas, D.F. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011, 39, e82. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, T.; Hosoi, S.; Woltjen, K.; Suzuki, K.I.; Kashiwagi, K.; Wada, H.; Ochiai, H.; Miyamoto, T.; Kawai, N.; Sasakura, Y.; et al. Efficient TALEN construction and evaluation methods for human cell and animal applications. Genes Cells 2013, 18, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Maggio, I.; Gonçalves, M.A.F.V. Genome editing at the crossroads of delivery, specificity, and fidelity. Trends Biotechnol. 2015, 33, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Streubel, J.; Blücher, C.; Landgraf, A.; Boch, J. TAL effector RVD specificities and efficiencies. Nat. Biotechnol. 2012, 30, 593–595. [Google Scholar] [CrossRef] [PubMed]
- Hisano, Y.; Ota, S.; Arakawa, K.; Muraki, M.; Kono, N.; Oshita, K.; Sakuma, T.; Tomita, M.; Yamamoto, T.; Okada, Y.; et al. Quantitative assay for TALEN activity at endogenous genomic loci. Biol. Open 2013, 2, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ann Ran, F.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; Zhang, F. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Aida, T.; Chiyo, K.; Usami, T.; Ishikubo, H.; Imahashi, R.; Wada, Y.; Tanaka, K.F.; Sakuma, T.; Yamamoto, T.; Tanaka, K. Cloning-free CRISPR/Cas system facilitates functional cassette knock-in in mice. Genome Biol. 2015, 16, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kotani, H.; Taimatsu, K.; Ohga, R.; Ota, S.; Kawahara, A. Efficient multiple genome modifications induced by the crRNAs, tracrRNA and Cas9 Protein complex in zebrafish. PLoS ONE 2015, 10, e0128319. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55. [Google Scholar] [CrossRef] [PubMed]
- McVey, M.; Lee, S.E. MMEJ repair of double-strand breaks (director’s cut): Deleted sequences and alternative endings. Trends Genet. 2008, 24, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Bedell, V.M.; Wang, Y.; Campbell, J.M.; Poshusta, T.L.; Starker, C.G.; Krug, R.G.; Tan, W.; Penheiter, S.G.; Ma, A.C.; Leung, A.Y.H.; et al. In vivo genome editing using a high-efficiency TALEN system. Nature 2012, 491, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Yoshimi, K.; Kunihiro, Y.; Kaneko, T.; Nagahora, H.; Voigt, B.; Mashimo, T. ssODN-mediated knock-in with CRISPR-Cas for large genomic regions in zygotes. Nat. Commun. 2016, 7, 10431. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Kaini, P.; Sander, J.D.; Joung, J.K.; Peterson, R.T.; Yeh, J.R.J. Heritable and Precise Zebrafish Genome Editing Using a CRISPR-Cas System. PLoS ONE 2013, 8, e68708. [Google Scholar] [CrossRef] [PubMed]
- Hruscha, A.; Krawitz, P.; Rechenberg, A.; Heinrich, V.; Hecht, J.; Haass, C.; Schmid, B. Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish. Development 2013, 140, 4982–4987. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.; Sun, C.; Gao, L.; Zhu, D.; Xu, X.; Zhu, X.; Xiong, J.W.; Xi, J.J. Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res. 2013, 23, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Zu, Y.; Tong, X.; Wang, Z.; Liu, D.; Pan, R.; Li, Z.; Hu, Y.; Luo, Z.; Huang, P.; Wu, Q.; et al. TALEN-mediated precise genome modification by homologous recombination in zebrafish. Nat. Methods 2013, 10, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Chen, J.; Solnica-Krezel, L. Efficient homologous recombination-mediated genome engineering in zebrafish using TALE nucleases. Development 2014, 141, 3807–3818. [Google Scholar] [CrossRef] [PubMed]
- Irion, U.; Krauss, J.; Nusslein-Volhard, C. Precise and efficient genome editing in zebrafish using the CRISPR/Cas9 system. Development 2014, 141, 4827–4830. [Google Scholar] [CrossRef] [PubMed]
- Cristea, S.; Freyvert, Y.; Santiago, Y.; Holmes, M.C.; Urnov, F.D.; Gregory, P.D.; Cost, G.J. In vivo cleavage of transgene donors promotes nuclease-mediated targeted integration. Biotechnol. Bioeng. 2013, 110, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Maresca, M.; Lin, V.G.; Guo, N.; Yang, Y. Obligate ligation-gated recombination (ObLiGaRe): Custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Res. 2013, 23, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Auer, T.O.; Duroure, K.; De Cian, A.; Concordet, J.P.; Del Bene, F. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome Res. 2014, 24, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Hisano, Y.; Kawahara, A.; Higashijima, S.-I. Efficient generation of knock-in transgenic zebrafish carrying reporter/driver genes by CRISPR/Cas9-mediated genome engineering. Sci. Rep. 2014, 4, 6545. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Okamura, Y.; Higashijima, S.I. alx, a zebrafish homolog of Chx10, marks ipsilateral descending excitatory interneurons that participate in the regulation of spinal locomotor circuits. J. Neurosci. 2006, 26, 5684–5697. [Google Scholar] [CrossRef] [PubMed]
- Nakade, S.; Tsubota, T.; Sakane, Y.; Kume, S.; Sakamoto, N.; Obara, M.; Daimon, T.; Sezutsu, H.; Yamamoto, T.; Sakuma, T.; et al. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nat. Commun. 2014, 5, 5560. [Google Scholar] [CrossRef] [PubMed]
- Hisano, Y.; Sakuma, T.; Nakade, S.; Ohga, R.; Ota, S.; Okamoto, H.; Yamamoto, T.; Kawahara, A. Precise in-frame integration of exogenous DNA mediated by CRISPR/Cas9 system in zebrafish. Sci. Rep. 2015, 5, 8841. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, T.; Takenaga, M.; Kawabe, Y.; Nakamura, T.; Kamihira, M.; Yamamoto, T. Homologous recombination-independent large gene cassette knock-in in CHO cells using TALEN and MMEJ-directed donor plasmids. Int. J. Mol. Sci. 2015, 16, 23849–23866. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawahara, A.; Hisano, Y.; Ota, S.; Taimatsu, K. Site-Specific Integration of Exogenous Genes Using Genome Editing Technologies in Zebrafish. Int. J. Mol. Sci. 2016, 17, 727. https://doi.org/10.3390/ijms17050727
Kawahara A, Hisano Y, Ota S, Taimatsu K. Site-Specific Integration of Exogenous Genes Using Genome Editing Technologies in Zebrafish. International Journal of Molecular Sciences. 2016; 17(5):727. https://doi.org/10.3390/ijms17050727
Chicago/Turabian StyleKawahara, Atsuo, Yu Hisano, Satoshi Ota, and Kiyohito Taimatsu. 2016. "Site-Specific Integration of Exogenous Genes Using Genome Editing Technologies in Zebrafish" International Journal of Molecular Sciences 17, no. 5: 727. https://doi.org/10.3390/ijms17050727
APA StyleKawahara, A., Hisano, Y., Ota, S., & Taimatsu, K. (2016). Site-Specific Integration of Exogenous Genes Using Genome Editing Technologies in Zebrafish. International Journal of Molecular Sciences, 17(5), 727. https://doi.org/10.3390/ijms17050727