
Understanding the Functions of Long Non-Coding RNAs through Their Higher-Order Structures



Abstract
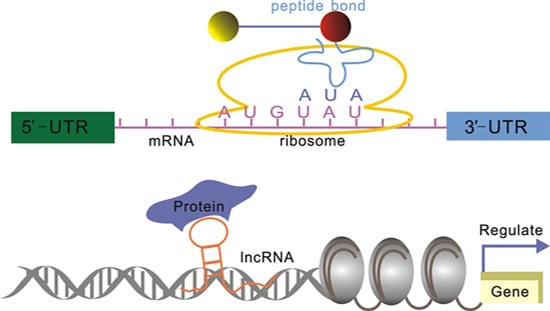
:
1. Introduction
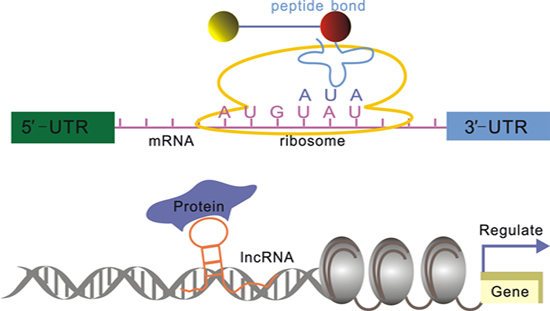
2. lncRNA Structure and Biological Function Relationships
2.1. lncRNAs in Animals
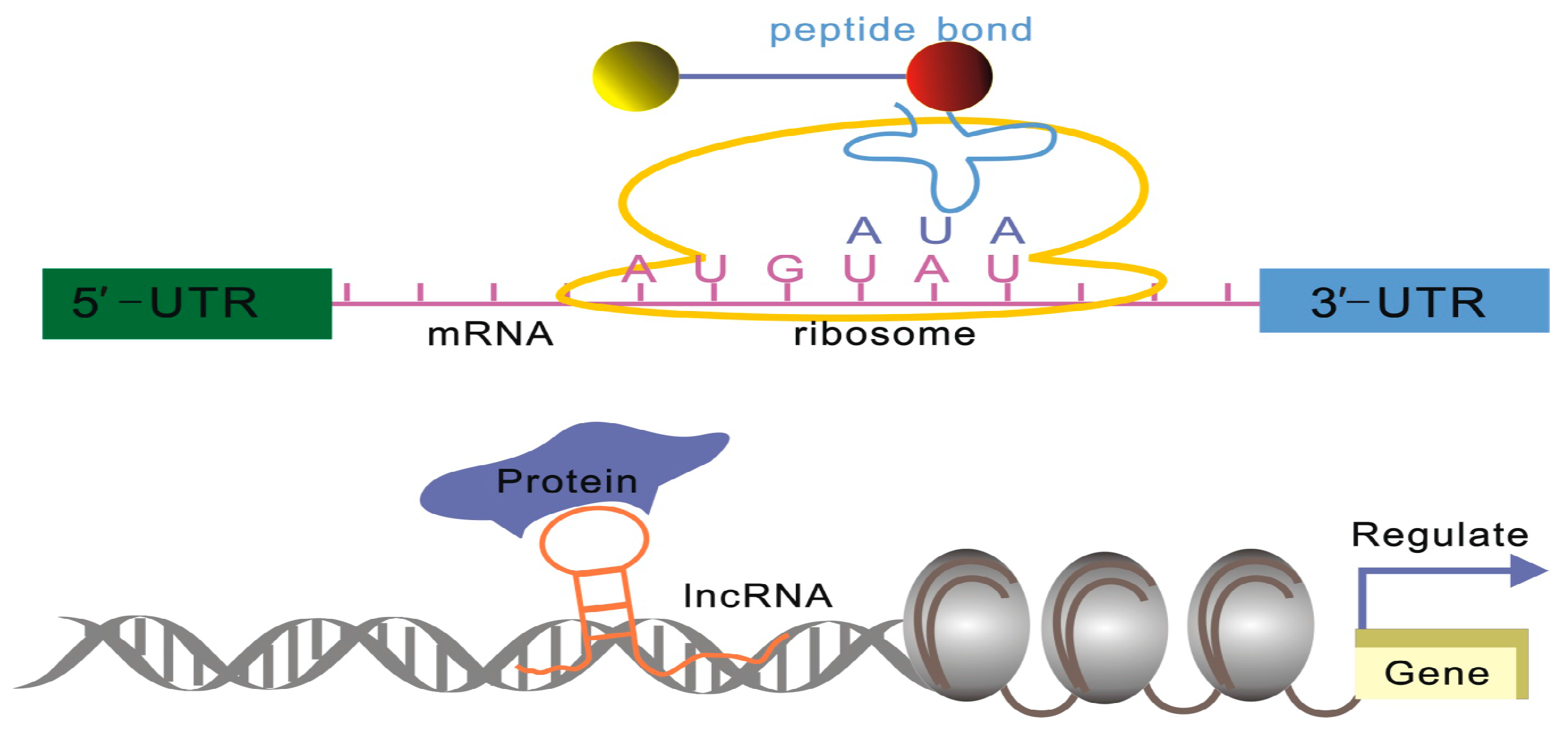
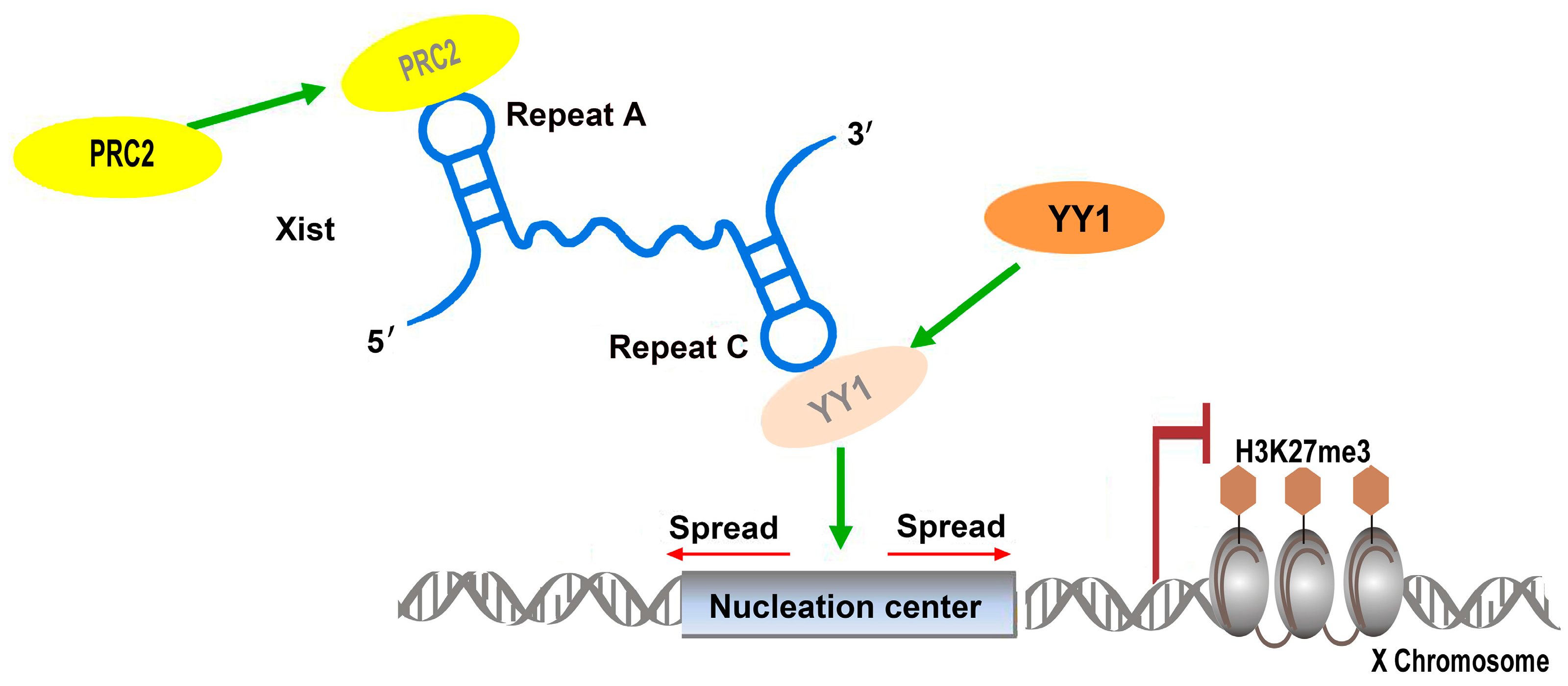
2.1.1. Xist: Repetitive Elements Involved in Protein Complex Recruitment
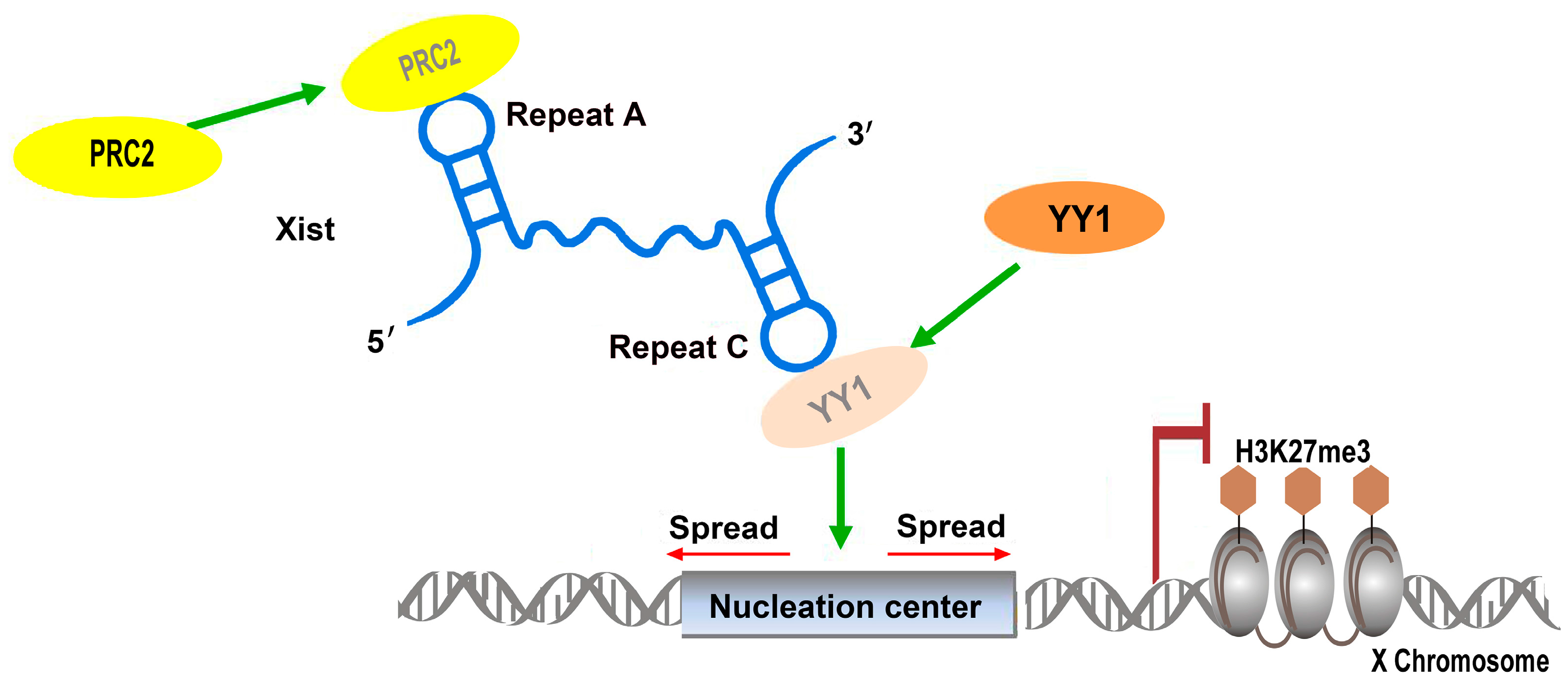
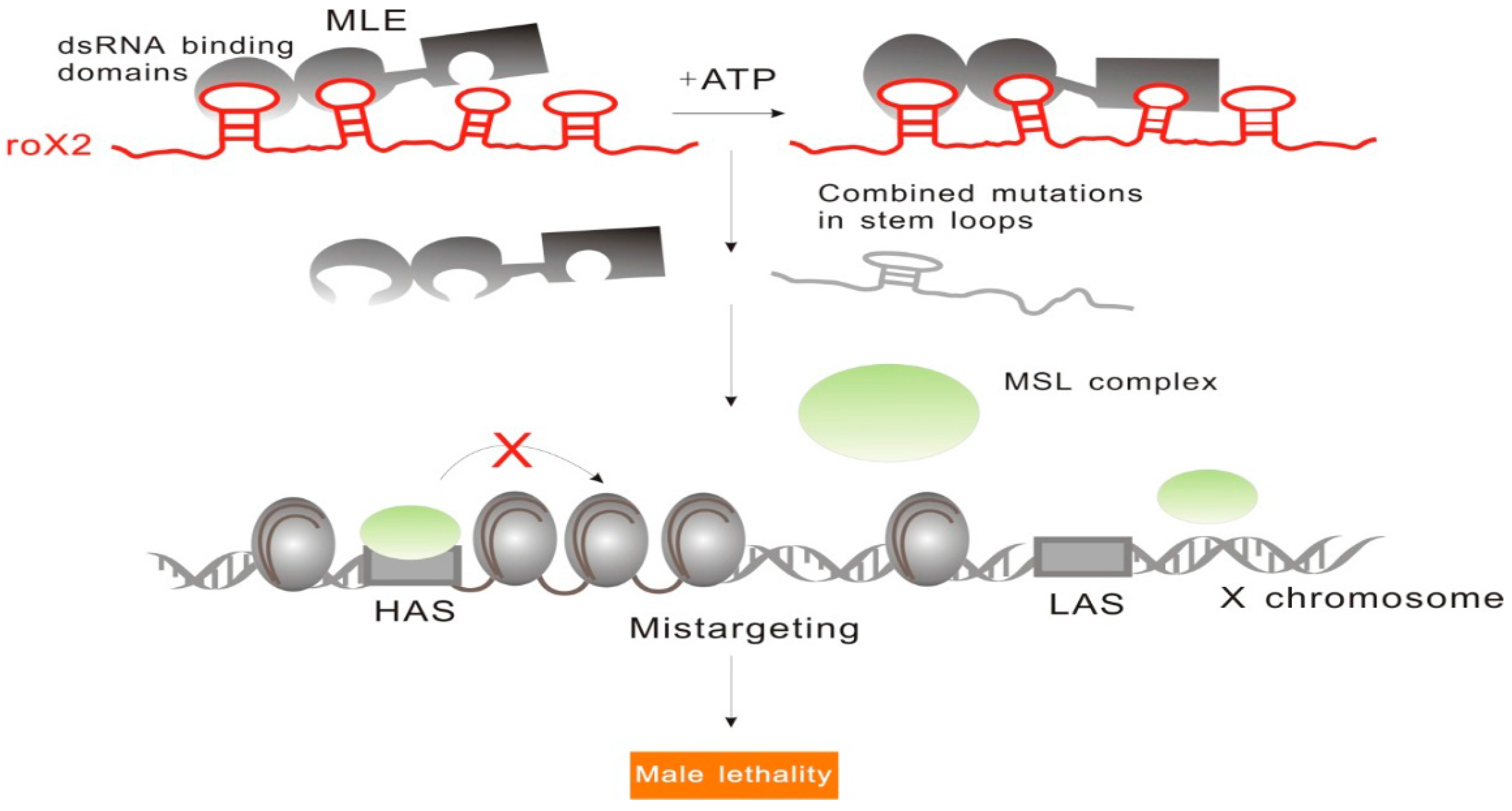
2.1.2. RoX: Tandem Stem-Loops Direct MSL Complex Assembly
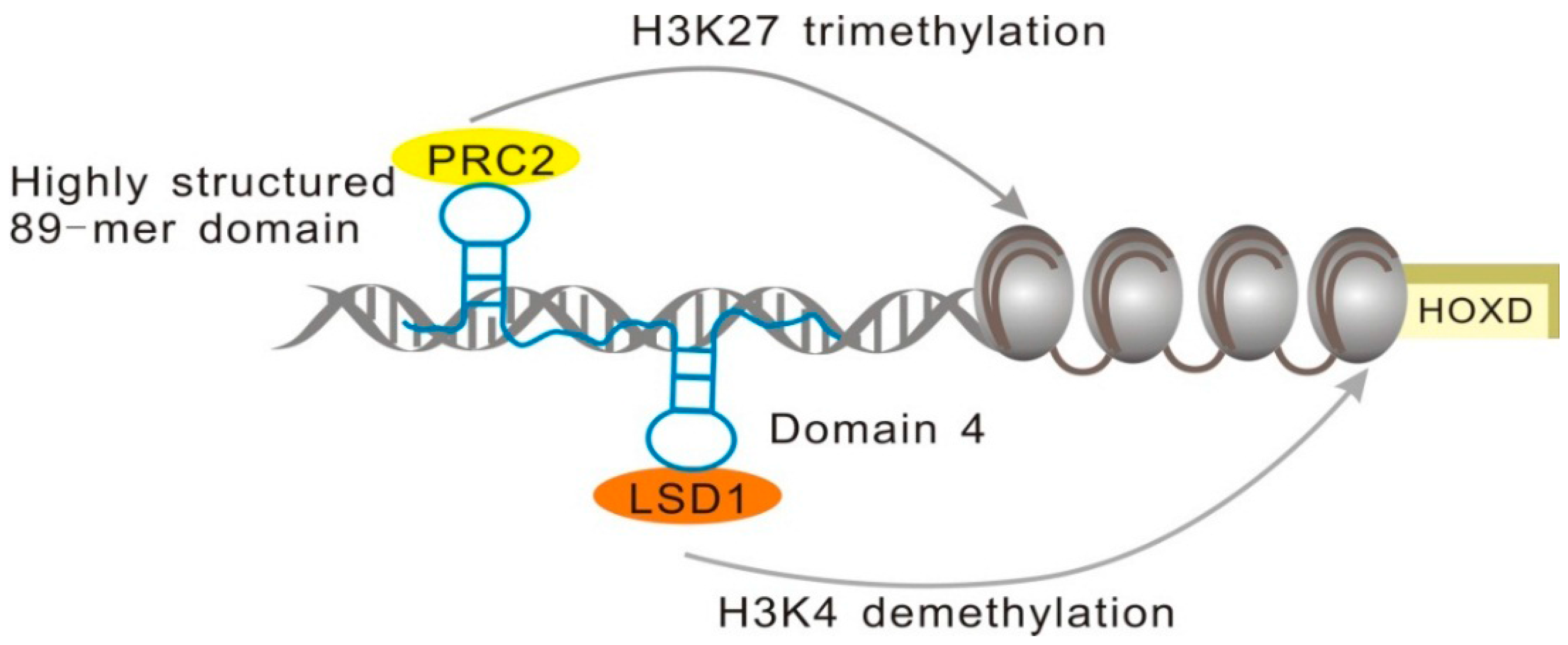
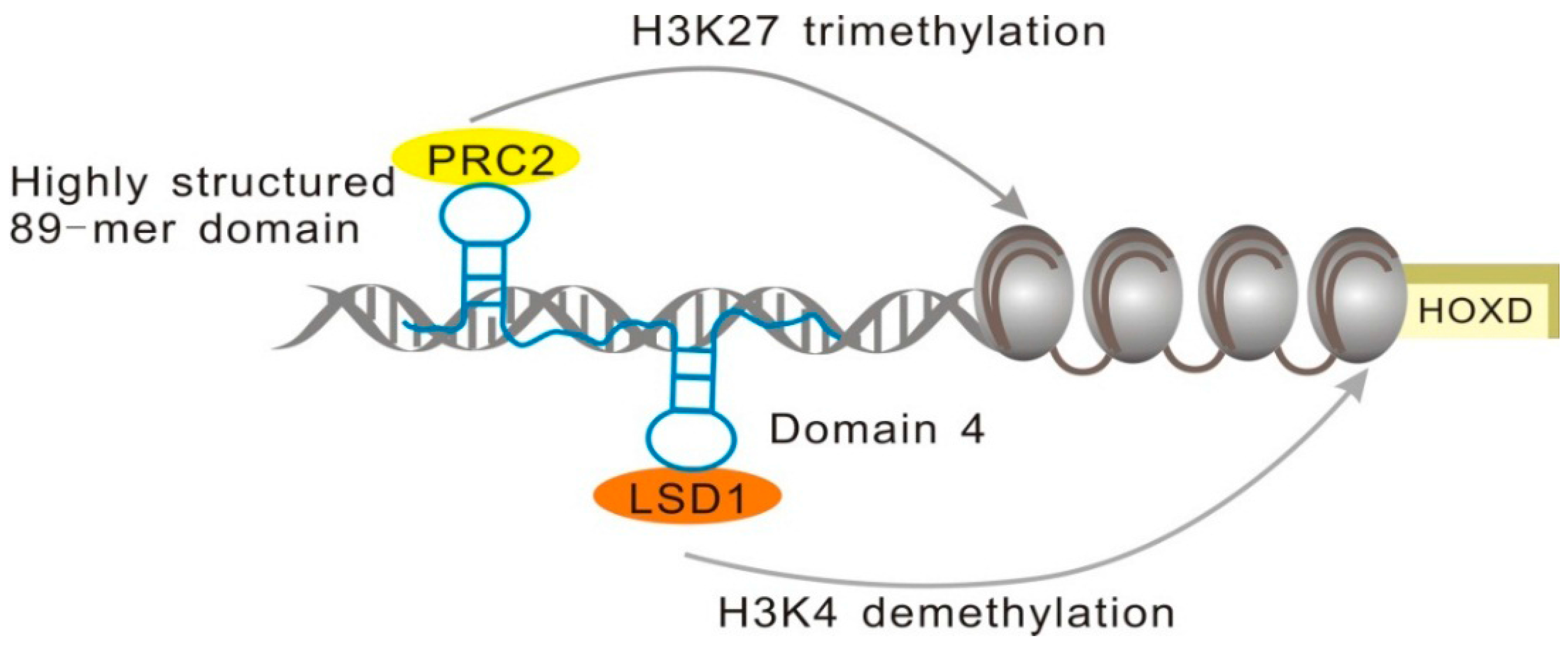
2.1.3. minHOTAIR Binds PRC2, while D4 Domain Recruits the LSD1 Complex
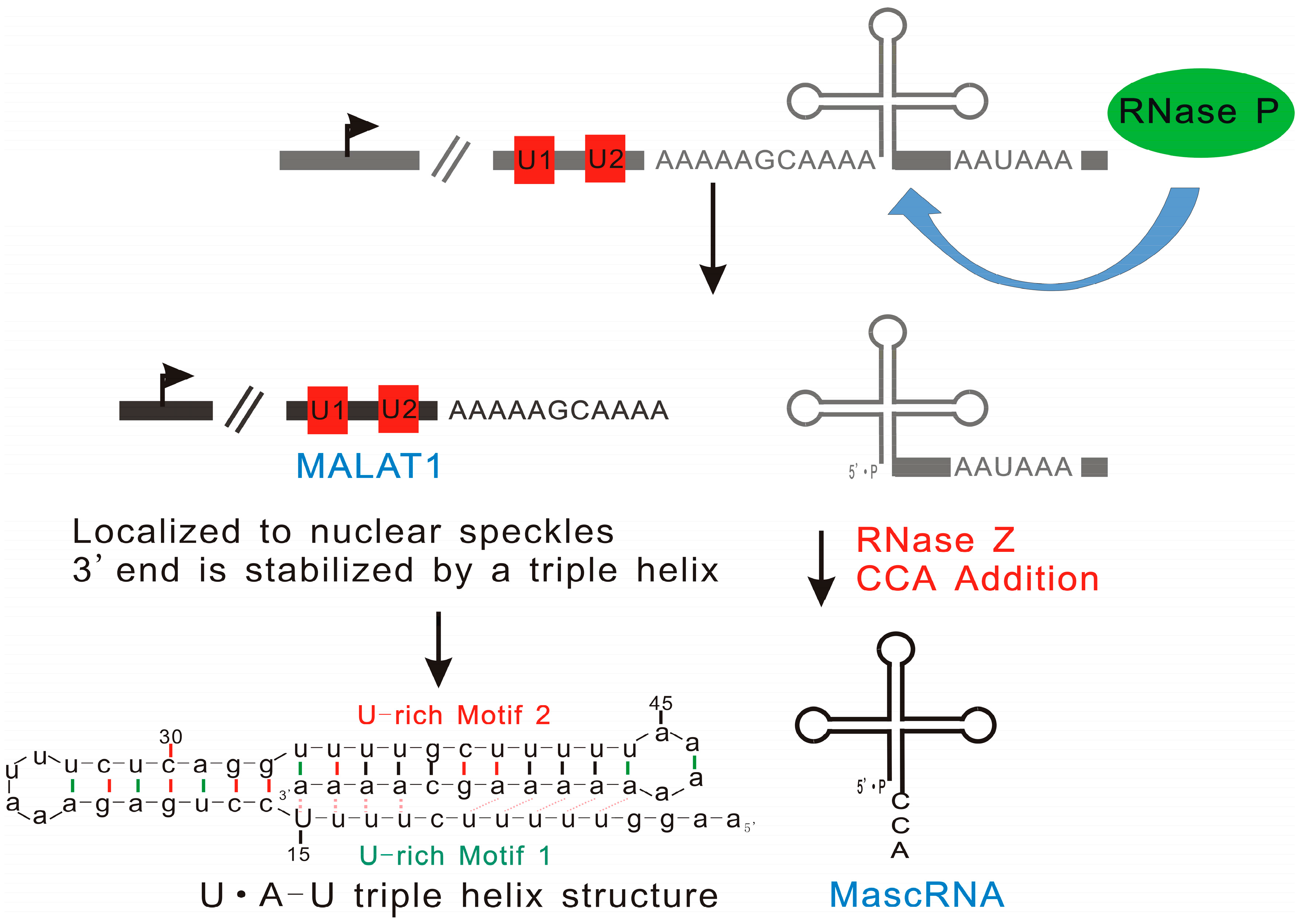
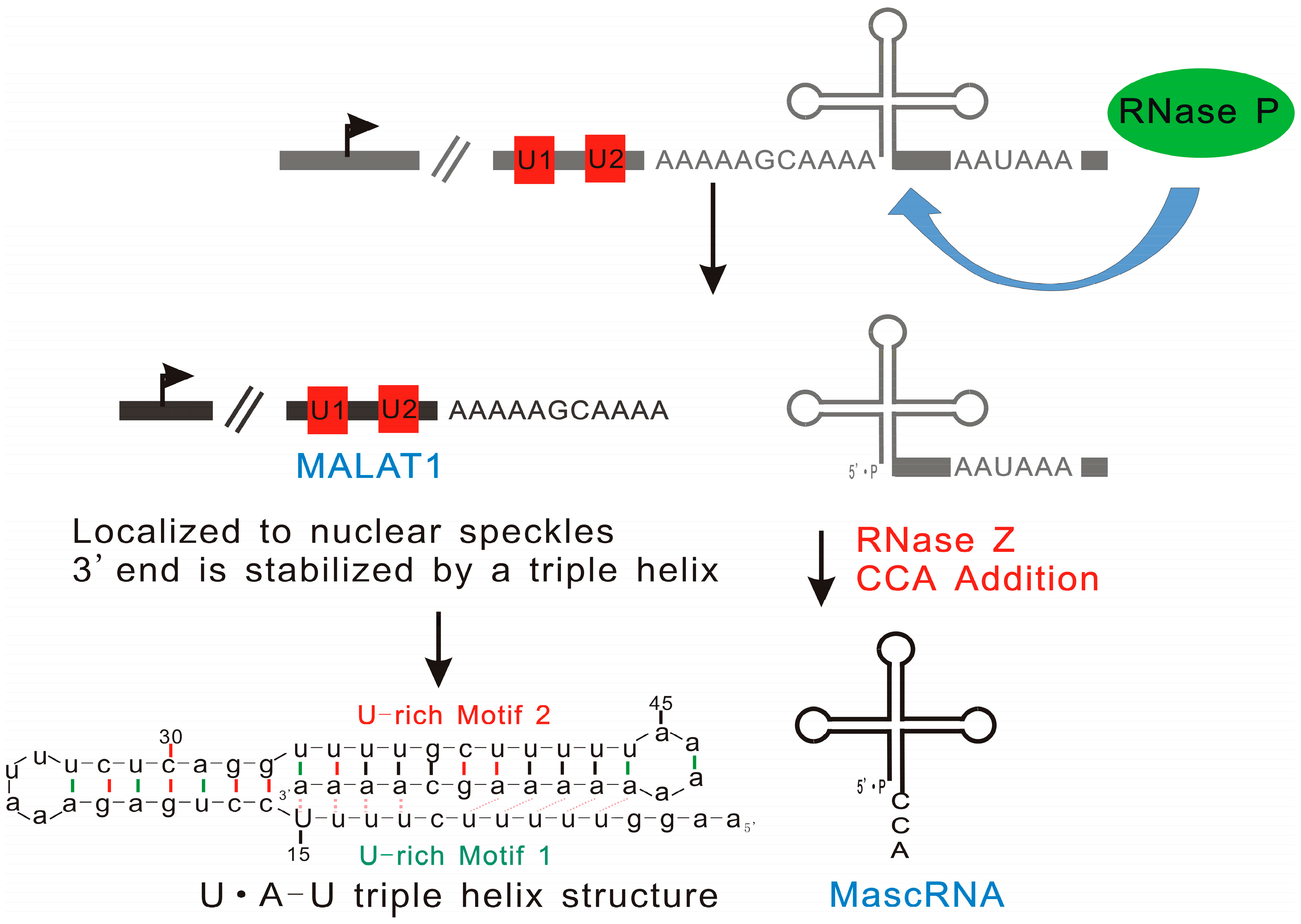
2.1.4. MALAT1: Triple Helix Structure Explains the High Stability of Long Nuclear-Retained Transcripts
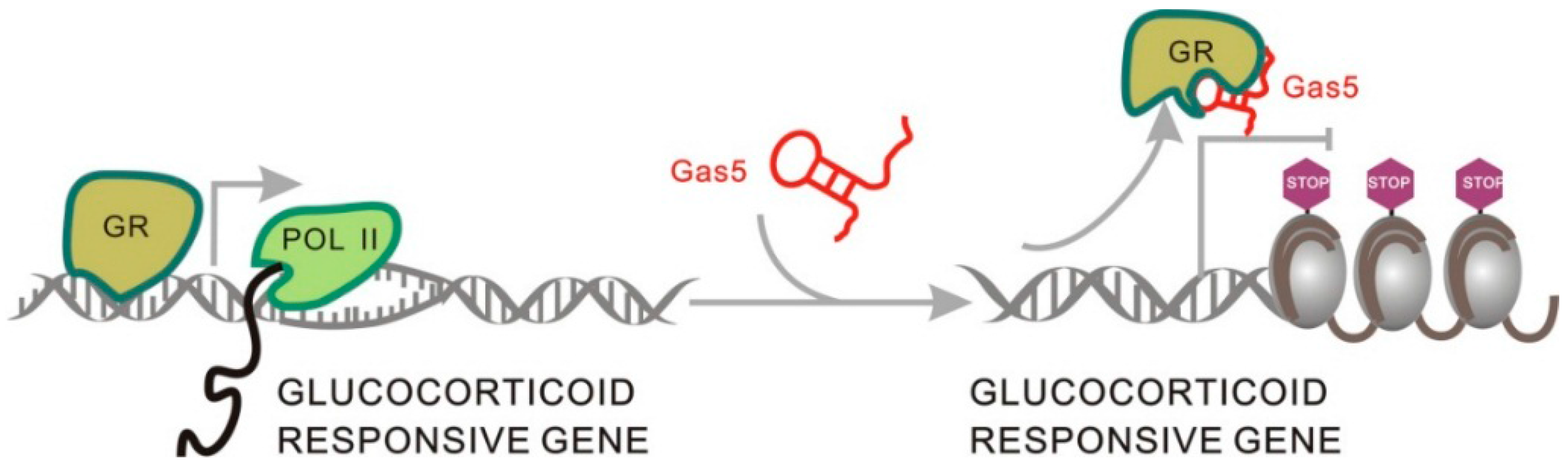
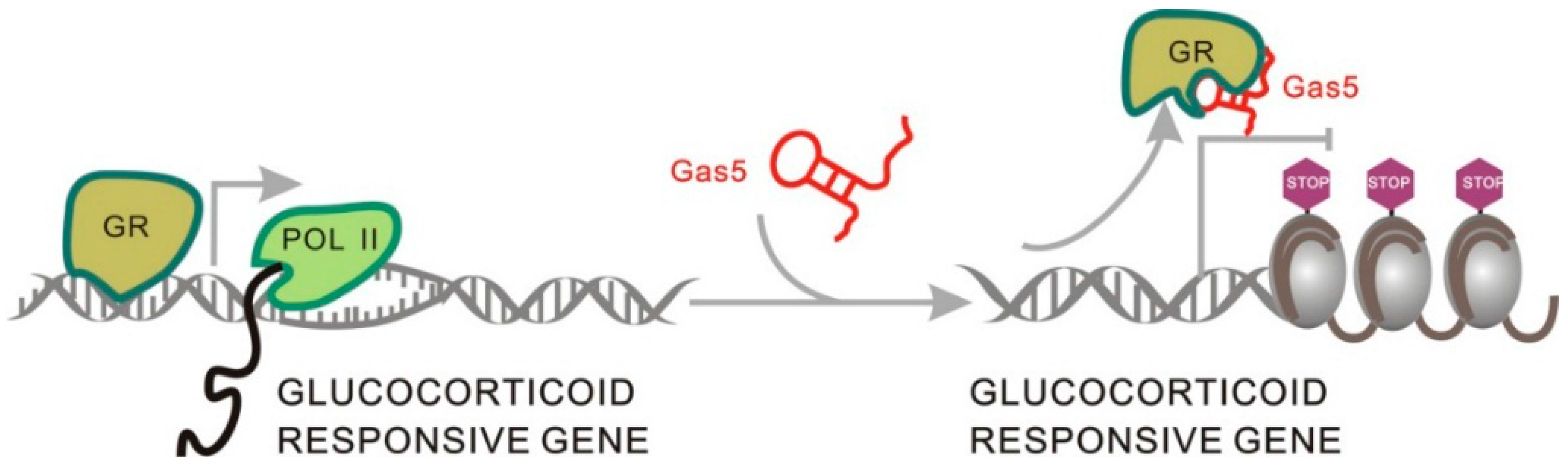
2.1.5. Gas5 Acts as a Decoy for the Glucocorticoid Receptor through Structure Transformation
2.2. lncRNAs in Plants
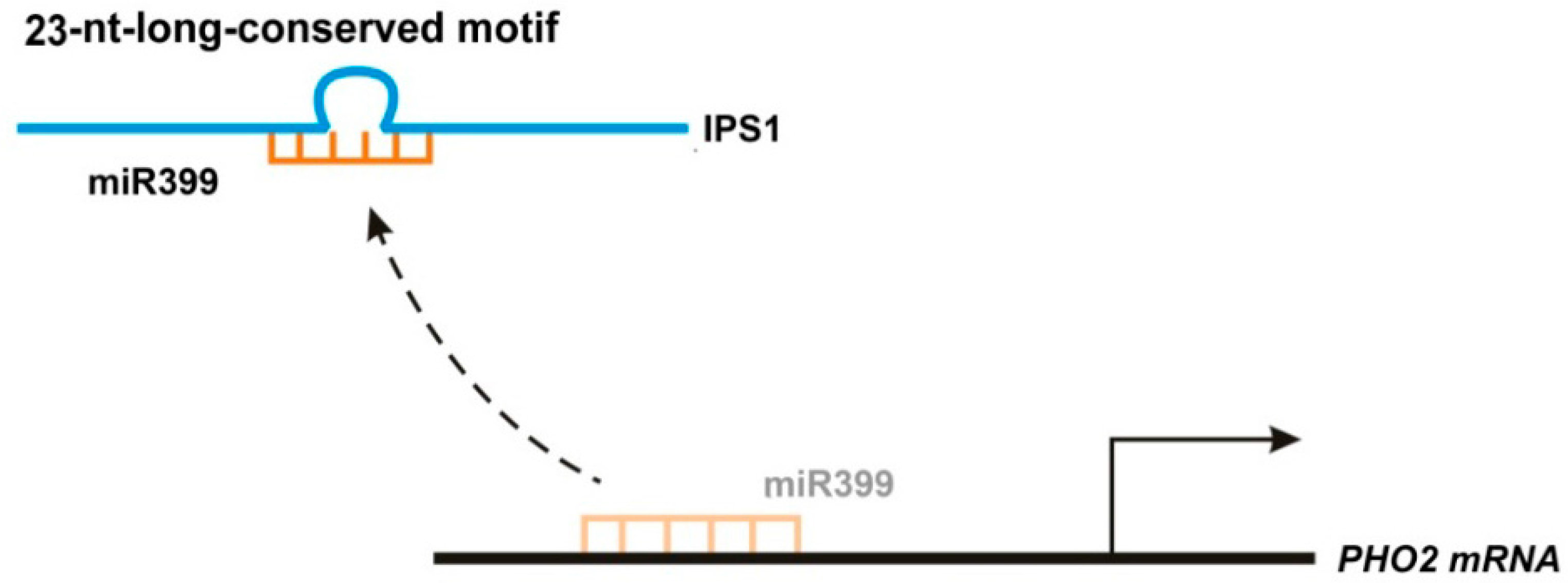
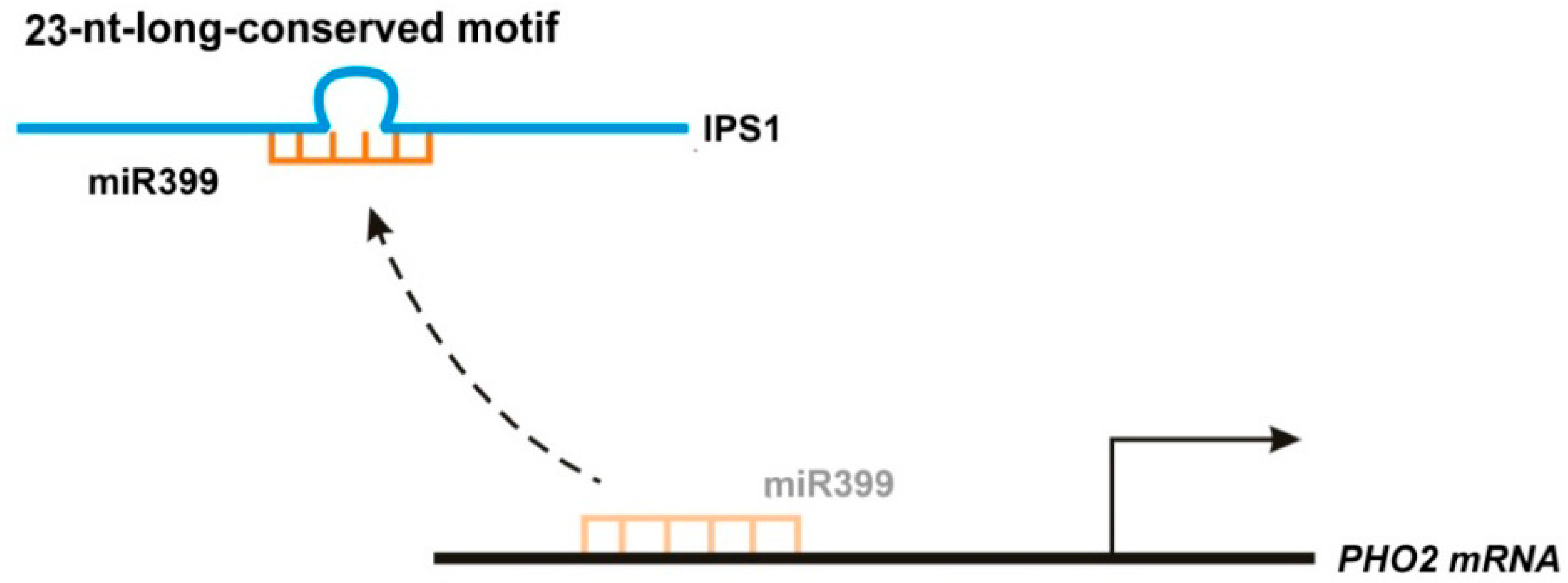
2.2.1. IPS1 Functions as an Endogenous Target Mimic Using Its 23-Nucleotide Conserved Motif
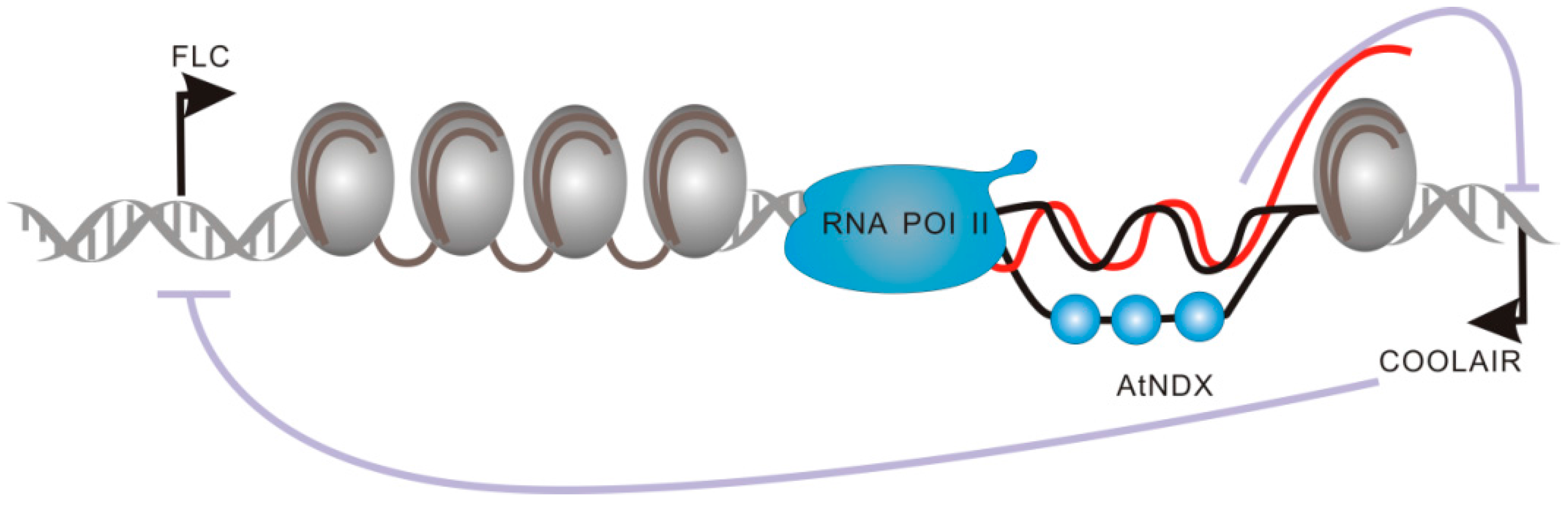
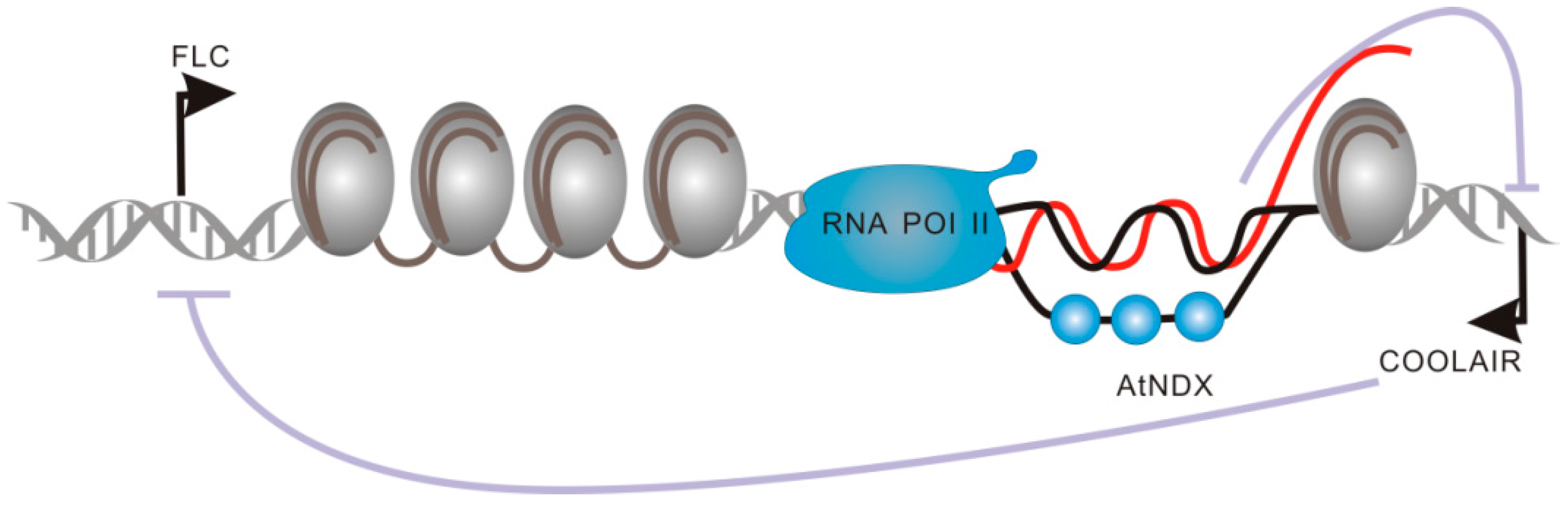
2.2.2. Functional Domains of COOLAIR and COLDAIR Are Involved in the Repression of Flowering Locus C (FLC)
2.2.3. LDMAR: lncRNA Structural Integrity Is Required in Order to Exert Biological Functions
2.2.4. ENOD40 Highly Structured Motif Is Involved in MtRBP1 Binding and Trafficking
3. Technologies Used in the Structural Studies of RNAs
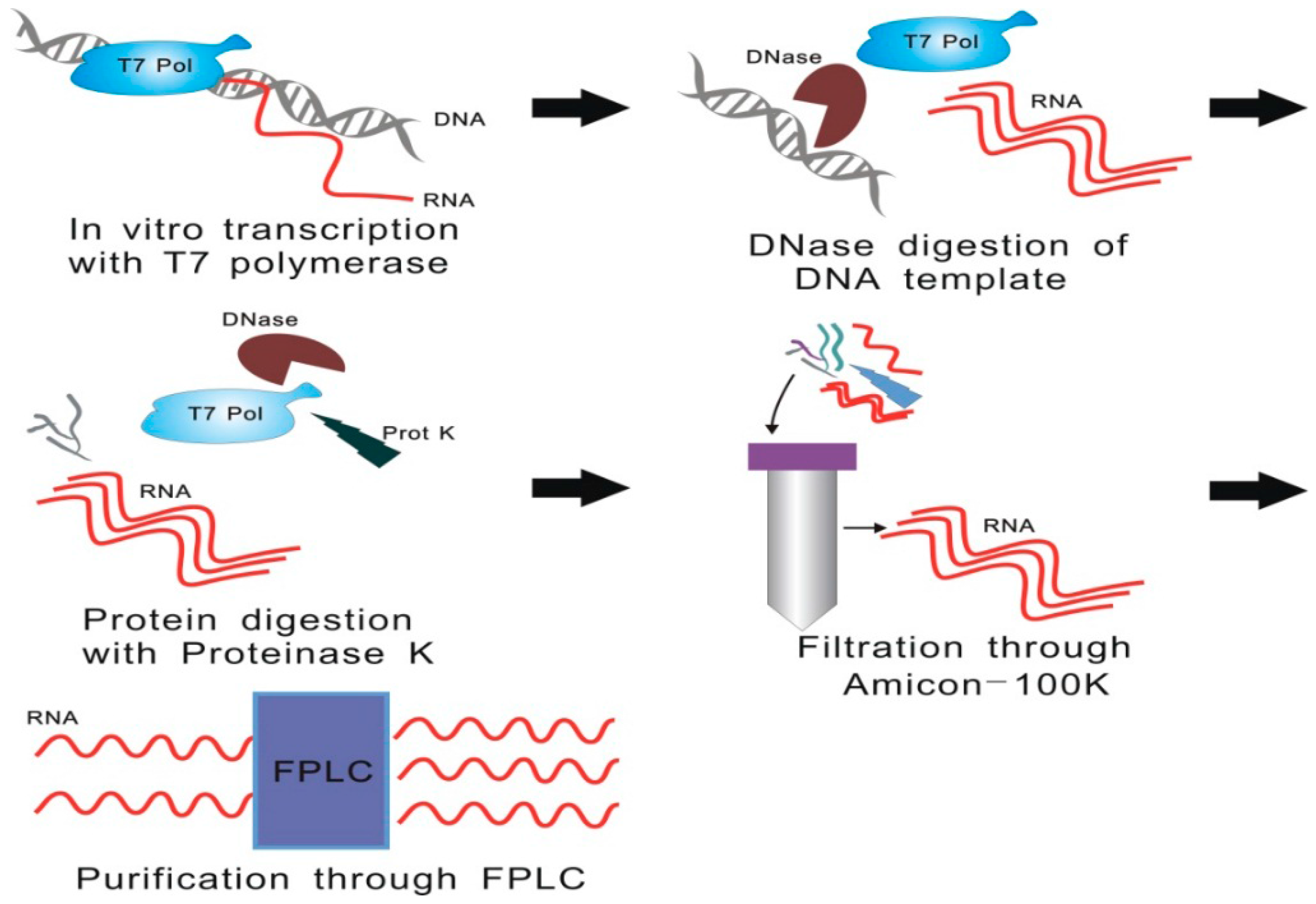
3.1. Methodologies of lncRNA Purification for Motif Determination
3.2. Methodologies of RNA Structure Probing in Vitro
3.2.1. SHAPE-seq, SHAPE-MAP, and RING-Map
3.2.2. PARS and FragSeq
3.2.3. ss/dsRNA-seq Techniques
3.3. RNA Structure Probing in Vivo
3.3.1. DMS-seq, Structural-seq, and Mod-seq
3.3.2. icSHAPE
3.3.3. CLASH and hiCLIP
3.3.4. RNA Proximity Ligation (RPL)
4. Conclusions and Future Direction
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Shabalina, S.A.; Ogurtsov, A.Y.; Spiridonov, N.A. A periodic pattern of mRNA secondary structure created by the genetic code. Nucleic Acids Res. 2006, 34, 2428–2437. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Belgard, T.G. Transcribed dark matter: Meaning or myth? Hum. Mol. Genet. 2010, 19, R162–R168. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.D.; Sung, S. Long noncoding RNA: Unveiling hidden layer of gene regulatory networks. Trends Plant Sci. 2012, 17, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Simon, S.A.; Meyers, B.C. Small RNA-mediated epigenetic modifications in plants. Curr. Opin. Plant Biol. 2011, 14, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wen, L.; Zhu, H. Unveiling the hidden function of long non-coding RNA by identifying its major partner-protein. Cell Biosci. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; Carmichael, G.G. Decoding the function of nuclear long non-coding RNAs. Curr. Opin. Cell Biol. 2010, 22, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Mattick, J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 2013, 20, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Johnsson, P.; Lipovich, L.; Grander, D.; Morris, K.V. Evolutionary conservation of long non-coding RNAs; sequence, structure, function. Biochim. Biophys. Acta 2014, 1840, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Novikova, I.V.; Hennelly, S.P.; Sanbonmatsu, K.Y. Structural architecture of the human long non-coding RNA, steroid receptor RNA activator. Nucleic Acids Res. 2012, 40, 5034–5051. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, X.; Wang, C.; Xu, Z.; Wang, Y.; Liu, X.; Kang, Z.; Ji, W. Long non-coding genes implicated in response to stripe rust pathogen stress in wheat (Triticum aestivum L.). Mol. Biol. Rep. 2013, 40, 6245–6253. [Google Scholar] [CrossRef] [PubMed]
- Di, C.; Yuan, J.; Wu, Y.; Li, J.; Lin, H.; Hu, L.; Zhang, T.; Qi, Y.; Gerstein, M.B.; Guo, Y.; et al. Characterization of stress-responsive lncRNAs in Arabidopsis thaliana by integrating expression, epigenetic and structural features. Plant J. 2014, 80, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Schubert, M.; Duss, O.; Ravindranathan, S.; Allain, F.H. Structure determination and dynamics of protein-RNA complexes by NMR spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 2011, 58, 1–61. [Google Scholar] [CrossRef] [PubMed]
- Kwok, C.K.; Tang, Y.; Assmann, S.M.; Bevilacqua, P.C. The RNA structurome: Transcriptome-wide structure probing with next-generation sequencing. Trends Biochem. Sci. 2015, 40, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Novikova, I.V.; Hennelly, S.P.; Tung, C.S.; Sanbonmatsu, K.Y. Rise of the RNA machines: Exploring the structure of long non-coding RNAs. J. Mol. Biol. 2013, 425, 3731–3746. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The gencode v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.D.; Pinter, S.F.; Fang, R.; Sarma, K.; Rutenberg-Schoenberg, M.; Bowman, S.K.; Kesner, B.A.; Maier, V.K.; Kingston, R.E.; Lee, J.T. High-resolution Xist binding maps reveal two-step spreading during X-chromosome inactivation. Nature 2013, 504, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Pontier, D.B.; Gribnau, J. Xist regulation and function explored. Hum. Genet. 2011, 130, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Froberg, J.E.; Yang, L.; Lee, J.T. Guided by RNAs: X-inactivation as a model for lncRNA function. J. Mol. Biol. 2013, 425, 3698–3706. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chapman, A.G.; Kelsey, A.D.; Minks, J.; Cotton, A.M.; Brown, C.J. X-chromosome inactivation: Molecular mechanisms from the human perspective. Hum. Genet. 2011, 130, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Morey, C.; Arnaud, D.; Avner, P.; Clerc, P. Tsix-mediated repression of Xist accumulation is not sufficent for normal random X inactivation. Hum. Mol. Genet. 2001, 10, 1403–1411. [Google Scholar] [CrossRef] [PubMed]
- Migeon, B.R.; Lee, C.H.; Chowdhury, A.K.; Carpenter, H. Species differences in Tsix/Tsix reveal the roles of these genes in X-chromosome inactivation. Am. J. Hum. Genet. 2002, 71, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Maenner, S.; Blaud, M.; Fouillen, L.; Savoye, A.; Marchand, V.; Dubois, A.; Sanglier-Cianferani, S.; van Dorsselaer, A.; Clerc, P.; Avner, P.; et al. 2-D structure of the a region of Xist RNA and its implication for PRC2 association. PLoS Biol. 2010, 8, e1000276. [Google Scholar] [CrossRef] [PubMed]
- Duszczyk, M.M.; Wutz, A.; Rybin, V.; Sattler, M. The Xist RNA A-repeat comprises a novel AUCG tetraloop fold and a platform for multimerization. RNA 2011, 17, 1973–1982. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.; Lee, J.T. Yy1 tethers Xist RNA to the inactive X nucleation center. Cell 2011, 146, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.; Moss, W.N.; Rutenberg-Schoenberg, M.; Simon, M.D. Probing Xist RNA structure in cells using targeted structure-seq. PLoS Genet. 2015, 11, e1005668. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Yuan, L.; Song, Y.; Sui, T.; Li, Z.; Lai, L. D-repeat in the Xist gene is required for X chromosome inactivation. RNA Biol. 2016, 13, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Flintoft, L. Non-coding RNA: Structure and function for lncRNAs. Nat. Rev. Genet. 2013, 14, 598. [Google Scholar] [CrossRef] [PubMed]
- Wutz, A. Noncoding RoX RNA remodeling triggers fly dosage compensation complex assembly. Mol. Cell 2013, 51, 131–132. [Google Scholar] [CrossRef] [PubMed]
- Maenner, S.; Muller, M.; Frohlich, J.; Langer, D.; Becker, P.B. ATP-dependent RoX RNA remodeling by the helicase maleless enables specific association of MSL proteins. Mol. Cell 2013, 51, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Ilik, I.A.; Quinn, J.J.; Georgiev, P.; Tavares-Cadete, F.; Maticzka, D.; Toscano, S.; Wan, Y.; Spitale, R.C.; Luscombe, N.; Backofen, R.; et al. Tandem stem-loops in RoX RNAs act together to mediate X chromosome dosage compensation in drosophila. Mol. Cell 2013, 51, 156–173. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOT AIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Arfat, Y.; Li, D.; Zhao, F.; Chen, Z.; Yin, C.; Sun, Y.; Hu, L.; Yang, T.; Qian, A. Structure prediction: New insights into decrypting long noncoding RNAs. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Loewen, G.; Jayawickramarajah, J.; Zhuo, Y.; Shan, B. Functions of lncRNA HOT AIR in lung cancer. J. Hematol. Oncol. 2014, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Su, Y.; Yang, Q.; Lv, D.; Zhang, W.; Tang, K.; Wang, H.; Zhang, R.; Liu, Y. Overexpression of long non-coding RNA HOT AIR promotes tumor growth and metastasis in human osteosarcoma. Mol. Cells 2015, 38, 432–440. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Liu, S.; Zhu, H. The sequence, structure and evolutionary features of HOTAIR in mammals. BMC Evol. Biol. 2011, 11. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Murat, P.; Matak-Vinkovic, D.; Murrell, A.; Balasubramanian, S. Binding interactions between long noncoding RNAHOTAIR and PRC2 proteins. Biochemistry 2013, 52, 9519–9527. [Google Scholar] [CrossRef] [PubMed]
- Somarowthu, S.; Legiewicz, M.; Chillon, I.; Marcia, M.; Liu, F.; Pyle, A.M. HOTAIR forms an intricate and modular secondary structure. Mol. Cell 2015, 58, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, R.; Mayeda, A.; Yoshida, M.; Nakagawa, S. MALAT1 long non-coding RNA in cancer. Biochim. Biophys. Acta 2016, 1859, 192–199. [Google Scholar] [CrossRef] [PubMed]
- West, J.A.; Davis, C.P.; Sunwoo, H.; Simon, M.D.; Sadreyev, R.I.; Wang, P.I.; Tolstorukov, M.Y.; Kingston, R.E. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol. Cell 2014, 55, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, B.; Wang, T.; Wang, H. LncRNA malat1 overexpression is an unfavorable prognostic factor in human cancer: Evidence from a meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 5499–5505. [Google Scholar] [PubMed]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating Sr splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E.; Freier, S.M.; Spector, D.L. 3ʹ end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell 2008, 135, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E.; JnBaptiste, C.K.; Lu, L.Y.; Kuhn, C.D.; Joshua-Tor, L.; Sharp, P.A. A triple helix stabilizes the 3ʹ ends of long noncoding RNAs that lack poly(A) tails. Genes Dev. 2012, 26, 2392–2407. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Valenstein, M.L.; Yario, T.A.; Tycowski, K.T.; Steitz, J.A. Formation of triple-helical structures by the 3’-endsequences of MALAT1 and MENβ noncoding RNAs. Proc. Natl. Acad. Sci. USA 2012, 109, 19202–19207. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Bulkley, D.; Wang, J.; Valenstein, M.L.; Yario, T.A.; Steitz, T.A.; Steitz, J.A. Structural insights into the stabilization of MALAT1 noncoding RNA by a bipartite triple helix. Nat. Struct. Mol. Biol. 2014, 21, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Pickard, M.R.; Williams, G.T. Molecular and cellular mechanisms of action of tumour suppressor Gas5 lncRNA. Genes (Basel) 2015, 6, 484–499. [Google Scholar] [CrossRef] [PubMed]
- Kino, T.; Hurt, D.E.; Ichijo, T.; Nader, N.; Chrousos, G.P. Noncoding RNAGas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci. Signal. 2010, 3. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, H.; Chua, N.H. Long noncoding RNA transcriptome of plants. Plant Biotechnol. J. 2015, 13, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, Z.; Fu, X.; Han, W. LncRNAs: Insights into their function and mechanics in underlying disorders. Mutat. Res. Rev. Mutat. Res. 2014, 762, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicki, A.T. The role of long non-coding RNA in transcriptional gene silencing. Curr. Opin. Plant Biol. 2012, 15, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.C.; Chen, Y.Q. Long noncoding RNAs: New regulators in plant development. Biochem. Biophys. Res. Commun. 2013, 436, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Franco-Zorrilla, J.M.; Valli, A.; Todesco, M.; Mateos, I.; Puga, M.I.; Rubio-Somoza, I.; Leyva, A.; Weigel, D.; Garcia, J.A.; Paz-Ares, J. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat. Genet. 2007, 39, 1033–1037. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.B.; Lee, Y.S.; Sung, S. Epigenetic regulation by long noncoding RNAs in plants. Chromosome Res. 2013, 21, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Guil, S.; Esteller, M. RNA–RNA interactions in gene regulation: The coding and noncoding players. Trends Biochem. Sci. 2015, 40, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, A.; Abe, M. Regulation of reproductive development by non-coding RNA in Arabidopsis: To flower or not to flower. J. Plant Res. 2012, 125, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Csorba, T.; Questa, J.I.; Sun, Q.; Dean, C. Antisense coolair mediates the coordinated switching of chromatin states at FLC during vernalization. Proc. Natl. Acad. Sci. USA 2014, 111, 16160–16165. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sung, S. Environmentally coordinated epigenetic silencing of FLC by protein and long noncoding RNA components. Curr. Opin. Plant Biol. 2012, 15, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.W.; Wu, Z.; Raitskin, O.; Sun, Q.; Dean, C. Antisense-mediated FLC transcriptional repression requires the P-TEFb transcription elongation factor. Proc. Natl. Acad. Sci. USA 2014, 111, 7468–7473. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Csorba, T.; Skourti-Stathaki, K.; Proudfoot, N.J.; Dean, C. R-loop stabilization represses antisense transcription at the Arabidopsis FLC locus. Science 2013, 340, 619–621. [Google Scholar] [CrossRef] [PubMed]
- Chekanova, J.A. Long non-coding RNAs and their functions in plants. Curr. Opin. Plant Biol. 2015, 27, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T. Lessons from X-chromosome inactivation: Long ncRNA as guides and tethers to the epigenome. Genes Dev. 2009, 23, 1831–1842. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ohsumi, T.K.; Kung, J.T.; Ogawa, Y.; Grau, D.J.; Sarma, K.; Song, J.J.; Kingston, R.E.; Borowsky, M.; Lee, J.T. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell 2010, 40, 939–953. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Lu, Q.; Ouyang, Y.; Mao, H.; Zhang, P.; Yao, J.; Xu, C.; Li, X.; Xiao, J.; Zhang, Q. A long noncoding RNA regulates photoperiod-sensitive male sterility, an essential component of hybrid rice. Proc. Natl. Acad. Sci. USA 2012, 109, 2654–2659. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Shen, J.; Mao, H.; Xie, W.; Li, X.; Zhang, Q. RNA-directed DNA methylation is involved in regulating photoperiod-sensitive male sterility in rice. Mol. Plant 2012, 5, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Mujahid, H.; Hou, Y.; Nallamilli, B.R.; Peng, Z. Plant long ncRNAs: A new frontier for gene regulatory control. Am. J. Plant Sci. 2013, 4, 1038–1045. [Google Scholar] [CrossRef]
- Bardou, F.; Merchan, F.; Ariel, F.; Crespi, M. Dual RNAs in plants. Biochimie 2011, 93, 1950–1954. [Google Scholar] [CrossRef] [PubMed]
- Gultyaev, A.P.; Roussis, A. Identification of conserved secondary structures and expansion segments in ENOD40 RNAs reveals new ENOD40 homologues in plants. Nucleic Acids Res. 2007, 35, 3144–3152. [Google Scholar] [CrossRef] [PubMed]
- Ariel, F.; Romero-Barrios, N.; Jegu, T.; Benhamed, M.; Crespi, M. Battles and hijacks: Noncoding transcription in plants. Trends Plant Sci. 2015, 20, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Rohrig, H.; Schmidt, J.; Miklashevichs, E.; Schell, J.; John, M. Soybean ENOD40 encodes two peptides that bind to sucrose synthase. Proc. Natl. Acad. Sci. USA 2002, 99, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Girard, G.; Roussis, A.; Gultyaev, A.P.; Pleij, C.W.; Spaink, H.P. Structural motifs in the RNA encoded by the early nodulation gene enod40 of soybean. Nucleic Acids Res. 2003, 31, 5003–5015. [Google Scholar] [CrossRef] [PubMed]
- Campalans, A.; Kondorosi, A.; Crespi, M. Enod40, a short open reading frame-containing mRNA, induces cytoplasmic localization of a nuclear RNA binding protein in medicago truncatula. Plant Cell 2004, 16, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.M.; Anderson, K.M.; Chang, C.L.; Makarewich, C.A.; Nelson, B.R.; McAnally, J.R.; Kasaragod, P.; Shelton, J.M.; Liou, J.; Bassel-Duby, R.; et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 2015, 160, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Bardou, F.; Ariel, F.; Simpson, C.G.; Romero-Barrios, N.; Laporte, P.; Balzergue, S.; Brown, J.W.; Crespi, M. Long noncoding RNA modulates alternative splicing regulators in Arabidopsis. Dev. Cell 2014, 30, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Ziehler, W.A.; Engelke, D.R. Probing RNA structure with chemical reagents and enzymes. Curr. Protoc. Nucleic Acid Chem. 2001, 6. [Google Scholar] [CrossRef]
- Cheong, H.K.; Hwang, E.; Lee, C.; Choi, B.S.; Cheong, C. Rapid preparation of RNA samples for NMR spectroscopy and X-ray crystallography. Nucleic Acids Res. 2004, 32, e84. [Google Scholar] [CrossRef] [PubMed]
- Batey, R.T.; Kieft, J.S. Improved native affinity purification of RNA. RNA 2007, 13, 1384–1389. [Google Scholar] [CrossRef] [PubMed]
- Said, N.; Rieder, R.; Hurwitz, R.; Deckert, J.; Urlaub, H.; Vogel, J. In vivo expression and purification of aptamer-tagged small RNA regulators. Nucleic Acids Res. 2009, 37, e133. [Google Scholar] [CrossRef] [PubMed]
- Chillon, I.; Marcia, M.; Legiewicz, M.; Liu, F.; Somarowthu, S.; Pyle, A.M. Native purification and analysis of long RNAs. Methods Enzymol. 2015, 558, 3–37. [Google Scholar] [PubMed]
- Poulsen, L.D.; Kielpinski, L.J.; Salama, S.R.; Krogh, A.; Vinther, J. SHAPE selection (SHAPES) enrich for RNA structure signal in SHAPE sequencing-based probing data. RNA 2015, 21, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Spitale, R.C.; Crisalli, P.; Flynn, R.A.; Torre, E.A.; Kool, E.T.; Chang, H.Y. RNA SHAPE analysis in living cells. Nat. Chem. Biol. 2013, 9, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Lucks, J.B.; Mortimer, S.A.; Trapnell, C.; Luo, S.; Aviran, S.; Schroth, G.P.; Pachter, L.; Doudna, J.A.; Arkin, A.P. Multiplexed RNA structure characterization with selective 2′-hydroxyl acylation analyzed by primer extension sequencing (SHAPE-seq). Proc. Natl. Acad. Sci. USA 2011, 108, 11063–11068. [Google Scholar] [CrossRef] [PubMed]
- Siegfried, N.A.; Busan, S.; Rice, G.M.; Nelson, J.A.; Weeks, K.M. RNA motif discovery by shape and mutational profiling (SHAPE-MAP). Nat. Methods 2014, 11, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Homan, P.J.; Favorov, O.V.; Lavender, C.A.; Kursun, O.; Ge, X.; Busan, S.; Dokholyan, N.V.; Weeks, K.M. Single-molecule correlated chemical probing of RNA. Proc. Natl. Acad. Sci. USA 2014, 111, 13858–13863. [Google Scholar] [CrossRef] [PubMed]
- Foley, S.W.; Vandivier, L.E.; Kuksa, P.P.; Gregory, B.D. Transcriptome-wide measurement of plant RNA secondary structure. Curr. Opin. Plant Biol. 2015, 27, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Kertesz, M.; Wan, Y.; Mazor, E.; Rinn, J.L.; Nutter, R.C.; Chang, H.Y.; Segal, E. Genome-wide measurement of RNA secondary structure in yeast. Nature 2010, 467, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Qu, K.; Ouyang, Z.; Chang, H.Y. Genome-wide mapping of RNA structure using nuclease digestion and high-throughput sequencing. Nat. Protoc. 2013, 8, 849–869. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Qu, K.; Zhang, Q.C.; Flynn, R.A.; Manor, O.; Ouyang, Z.; Zhang, J.; Spitale, R.C.; Snyder, M.P.; Segal, E.; et al. Landscape and variation of RNA secondary structure across the human transcriptome. Nature 2014, 505, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Novikova, I.V.; Hennelly, S.P.; Sanbonmatsu, K.Y. Tackling structures of long noncoding RNAs. Int. J. Mol. Sci. 2013, 14, 23672–23684. [Google Scholar] [CrossRef] [PubMed]
- Underwood, J.G.; Uzilov, A.V.; Katzman, S.; Onodera, C.S.; Mainzer, J.E.; Mathews, D.H.; Lowe, T.M.; Salama, S.R.; Haussler, D. Fragseq: Transcriptome-wide RNA structure probing using high-throughput sequencing. Nat. Methods 2010, 7, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Kashi, K.; Henderson, L.; Bonetti, A.; Carninci, P. Discovery and functional analysis of lncRNAs: Methodologies to investigate an uncharacterized transcriptome. Biochim. Biophys. Acta 2016, 1859, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Ryvkin, P.; Li, F.; Dragomir, I.; Valladares, O.; Yang, J.; Cao, K.; Wang, L.S.; Gregory, B.D. Genome-wide double-stranded RNA sequencing reveals the functional significance of base-paired RNAs in Arabidopsis. PLoS Genet. 2010, 6, e1001141. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zheng, Q.; Ryvkin, P.; Dragomir, I.; Desai, Y.; Aiyer, S.; Valladares, O.; Yang, J.; Bambina, S.; Sabin, L.R.; et al. Global analysis of RNA secondary structure in two metazoans. Cell Rep. 2012, 1, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zheng, Q.; Vandivier, L.E.; Willmann, M.R.; Chen, Y.; Gregory, B.D. Regulatory impact of RNA secondary structure across the Arabidopsis transcriptome. Plant Cell 2012, 24, 4346–4359. [Google Scholar] [CrossRef] [PubMed]
- Cordero, P.; Kladwang, W.; VanLang, C.C.; Das, R. Quantitative dimethyl sulfate mapping for automated RNA secondary structure inference. Biochemistry 2012, 51, 7037–7039. [Google Scholar] [CrossRef] [PubMed]
- Kubota, M.; Tran, C.; Spitale, R.C. Progress and challenges for chemical probing of RNA structure inside living cells. Nat. Chem. Biol. 2015, 11, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Talkish, J.; May, G.; Lin, Y.; Woolford, J.L., Jr.; McManus, C.J. Mod-seq: High-throughput sequencing for chemical probing of RNA structure. RNA 2014, 20, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; May, G.E.; Joel McManus, C. Mod-seq: A high-throughput method for probing RNA secondary structure. Methods Enzymol. 2015, 558, 125–152. [Google Scholar] [PubMed]
- Rouskin, S.; Zubradt, M.; Washietl, S.; Kellis, M.; Weissman, J.S. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 2014, 505, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Tang, Y.; Kwok, C.K.; Zhang, Y.; Bevilacqua, P.C.; Assmann, S.M. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 2014, 505, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Chang, H.Y. Decoding the RNA structurome. Curr. Opin. Struct. Biol. 2016, 36, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.A.; Zhang, Q.C.; Spitale, R.C.; Lee, B.; Mumbach, M.R.; Chang, H.Y. Transcriptome-wide interrogation of RNA secondary structure in living cells with icSHAPE. Nat. Protoc. 2016, 11, 273–290. [Google Scholar] [CrossRef] [PubMed]
- Spitale, R.C.; Flynn, R.A.; Zhang, Q.C.; Crisalli, P.; Lee, B.; Jung, J.W.; Kuchelmeister, H.Y.; Batista, P.J.; Torre, E.A.; Kool, E.T.; et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature 2015, 519, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the human miRNA interactome by clash reveals frequent noncanonical binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Kudla, G.; Granneman, S.; Hahn, D.; Beggs, J.D.; Tollervey, D. Cross-linking, ligation, and sequencing of hybrids reveals RNA–RNA interactions in yeast. Proc. Natl. Acad. Sci. USA 2011, 108, 10010–10015. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, Y.; Vigilante, A.; Darbo, E.; Zirra, A.; Militti, C.; D′Ambrogio, A.; Luscombe, N.M.; Ule, J. Hiclip reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1. Nature 2015, 519, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Ramani, V.; Qiu, R.; Shendure, J. High-throughput determination of RNA structure by proximity ligation. Nat. Biotechnol. 2015, 33, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Cao, J. The functional role of long non-coding RNAs and epigenetics. Biol. Proced. Online 2014, 16, 11. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Abdelmohsen, K.; Gorospe, M. Posttranscriptional gene regulation by long noncoding RNA. J. Mol. Biol. 2013, 425, 3723–3730. [Google Scholar] [CrossRef] [PubMed]
- Novikova, I.V.; Hennelly, S.P.; Sanbonmatsu, K.Y. Sizing up long non-coding RNAs do lncRNAs have secondary and tertiary structure. Bioarchitecture 2012, 2, 189–199. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Application to Date | Probe | Data | References |
|---|---|---|---|---|
| SHAPE-seq | In vitro | 1M7, NMIA | 1D | [87] |
| SHAPE-MAP | In vitro | 1M7 | 1D | [88] |
| RING-Map | In vitro | DMS | 1D | [89] |
| PARS | In vitro | RNase S1 (ssRNA); RNase V1 (dsRNA) | 1D | [91] |
| Fragseq | In vitro | RNase P1 (ssRNA) | 1D | [95] |
| ss/dsRNA-seq | In vitro | RNase I (ssRNA); RNase V1 (dsRNA) | 1D | [90,97] |
| Mod-seq | In vivo | DMS | 1D | [102,103] |
| DMS-seq | In vivo | DMS | 1D | [104] |
| Structural-seq | In vivo | DMS | 1D | [105] |
| icSHAPE | In vivo | NAI-N3 | 1D | [107,108] |
| CLASH | In vivo | UV crosslinking | 2D | [109,110] |
| hiCLIP | In vivo | UV crosslinking | 2D | [111] |
| RPL | In vivo | No crosslinking | 2D | [112] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Zhu, H.; Luo, Y. Understanding the Functions of Long Non-Coding RNAs through Their Higher-Order Structures. Int. J. Mol. Sci. 2016, 17, 702. https://doi.org/10.3390/ijms17050702
Li R, Zhu H, Luo Y. Understanding the Functions of Long Non-Coding RNAs through Their Higher-Order Structures. International Journal of Molecular Sciences. 2016; 17(5):702. https://doi.org/10.3390/ijms17050702
Chicago/Turabian StyleLi, Rui, Hongliang Zhu, and Yunbo Luo. 2016. "Understanding the Functions of Long Non-Coding RNAs through Their Higher-Order Structures" International Journal of Molecular Sciences 17, no. 5: 702. https://doi.org/10.3390/ijms17050702
APA StyleLi, R., Zhu, H., & Luo, Y. (2016). Understanding the Functions of Long Non-Coding RNAs through Their Higher-Order Structures. International Journal of Molecular Sciences, 17(5), 702. https://doi.org/10.3390/ijms17050702