
Optimizing Hybrid de Novo Transcriptome Assembly and Extending Genomic Resources for Giant Freshwater Prawns (Macrobrachium rosenbergii): The Identification of Genes and Markers Associated with Reproduction

 ,
,  ,
, 
Abstract
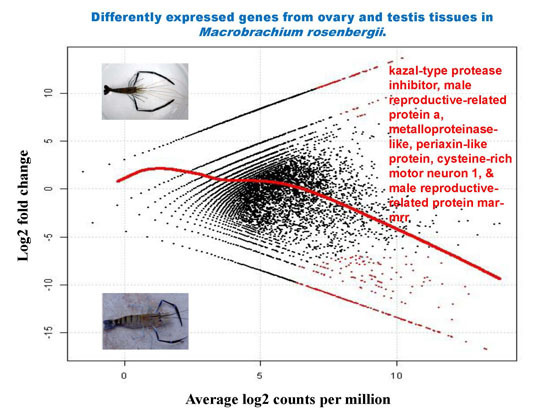
:
1. Introduction
2. Results
2.1. Transcriptome Sequencing
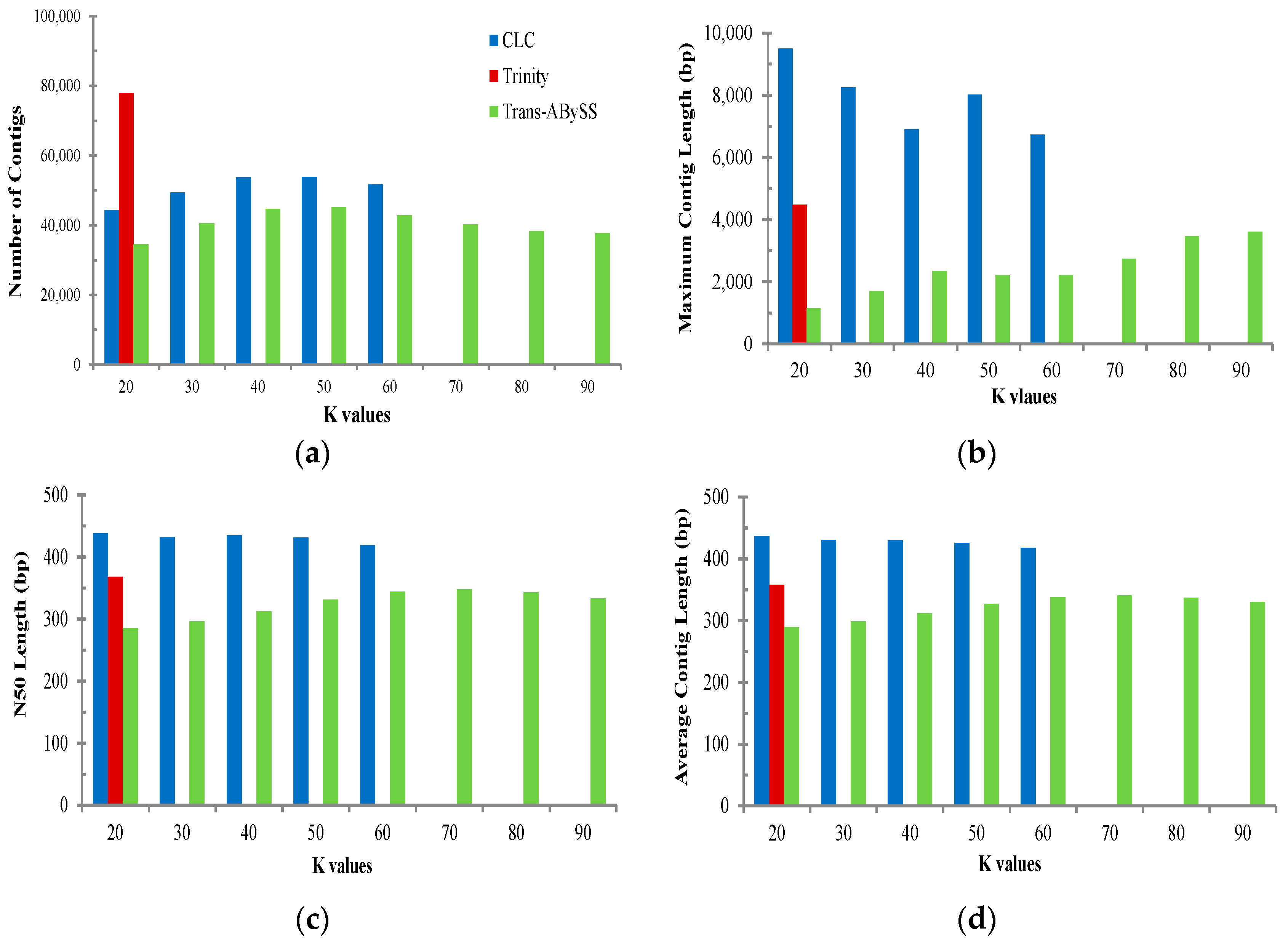
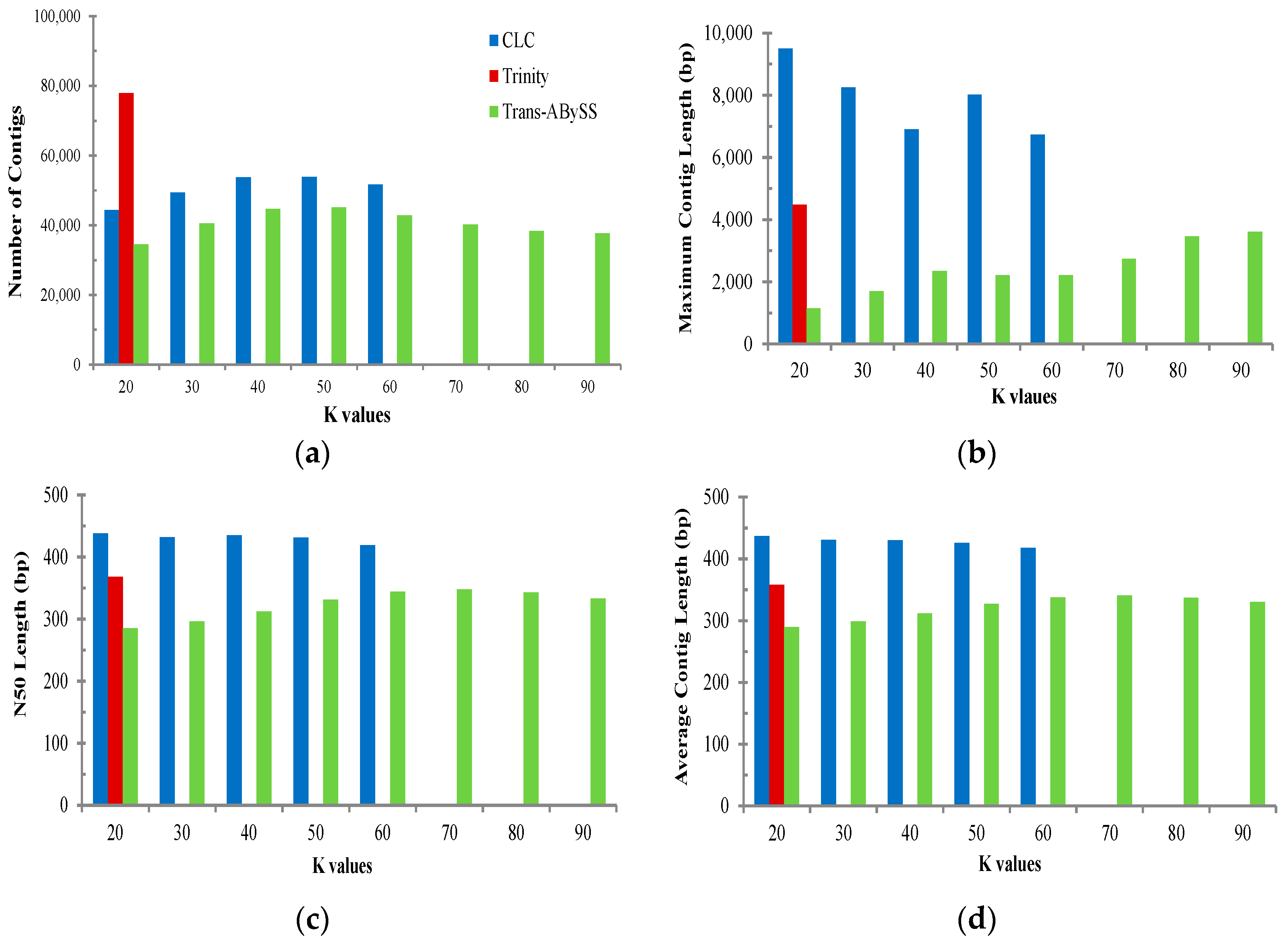
2.2. Transcriptome de Novo Assembly and Comparison of Assemblers
2.3. BLAST Search and Species Distribution
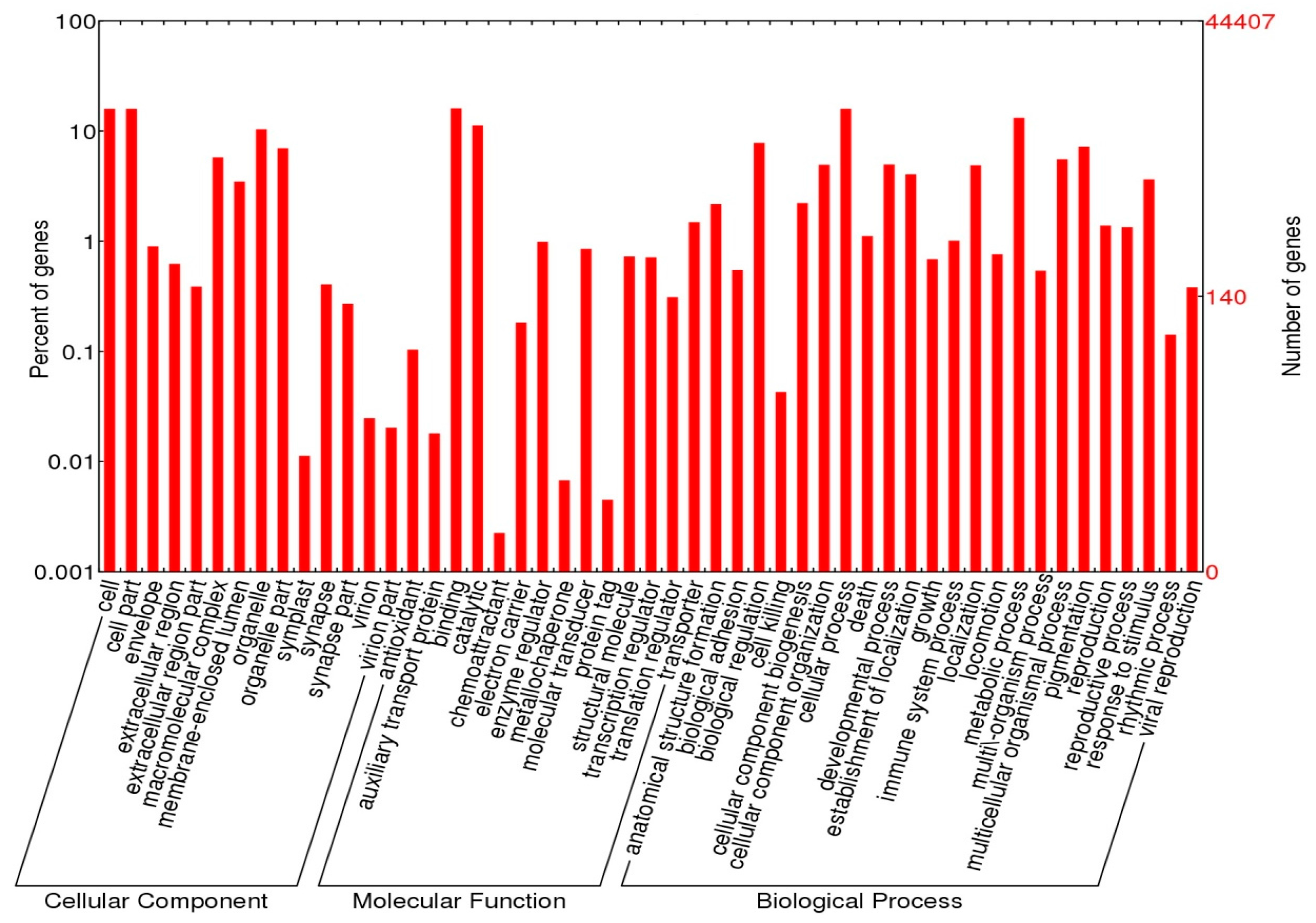
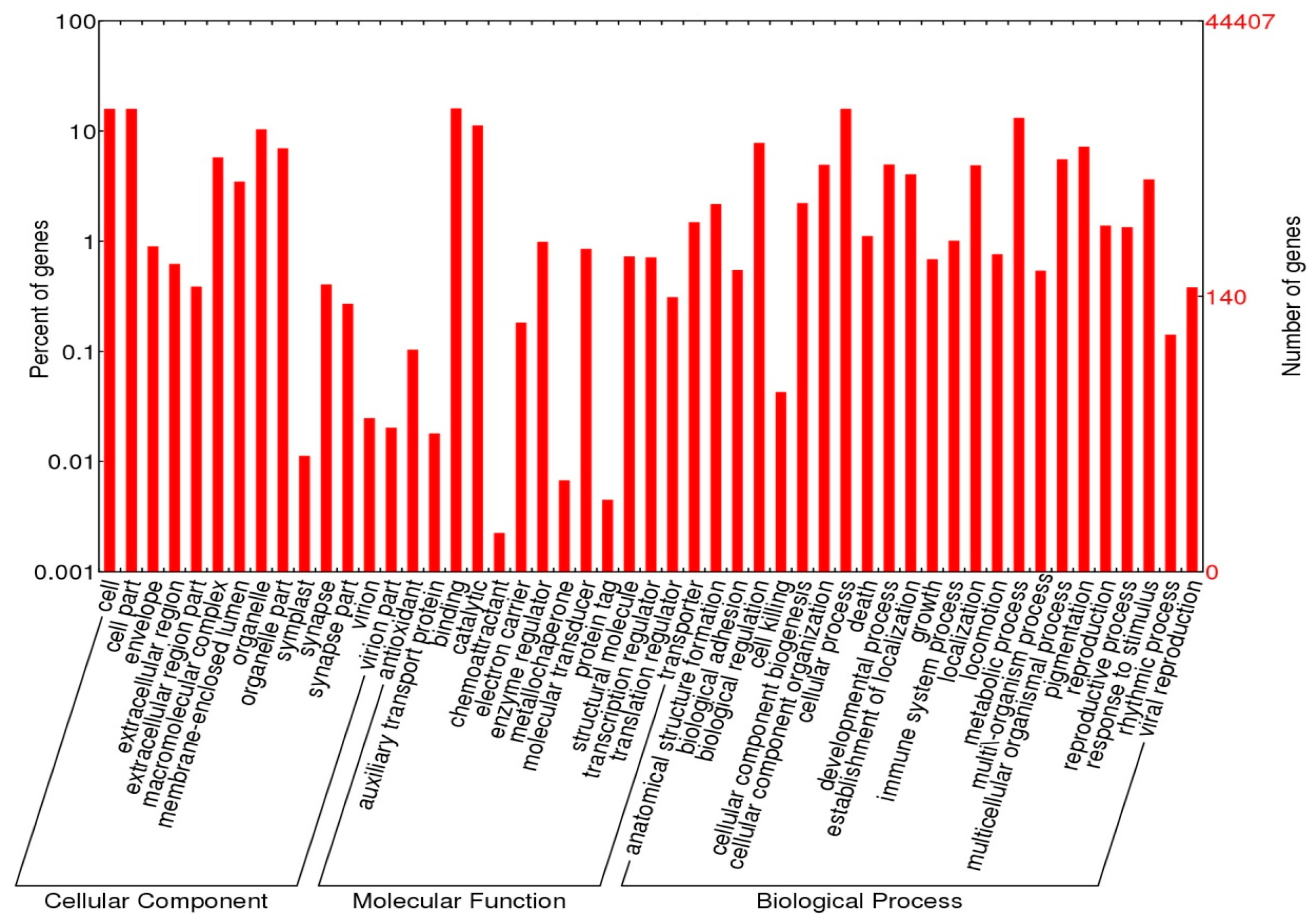
2.4. Functional Annotations
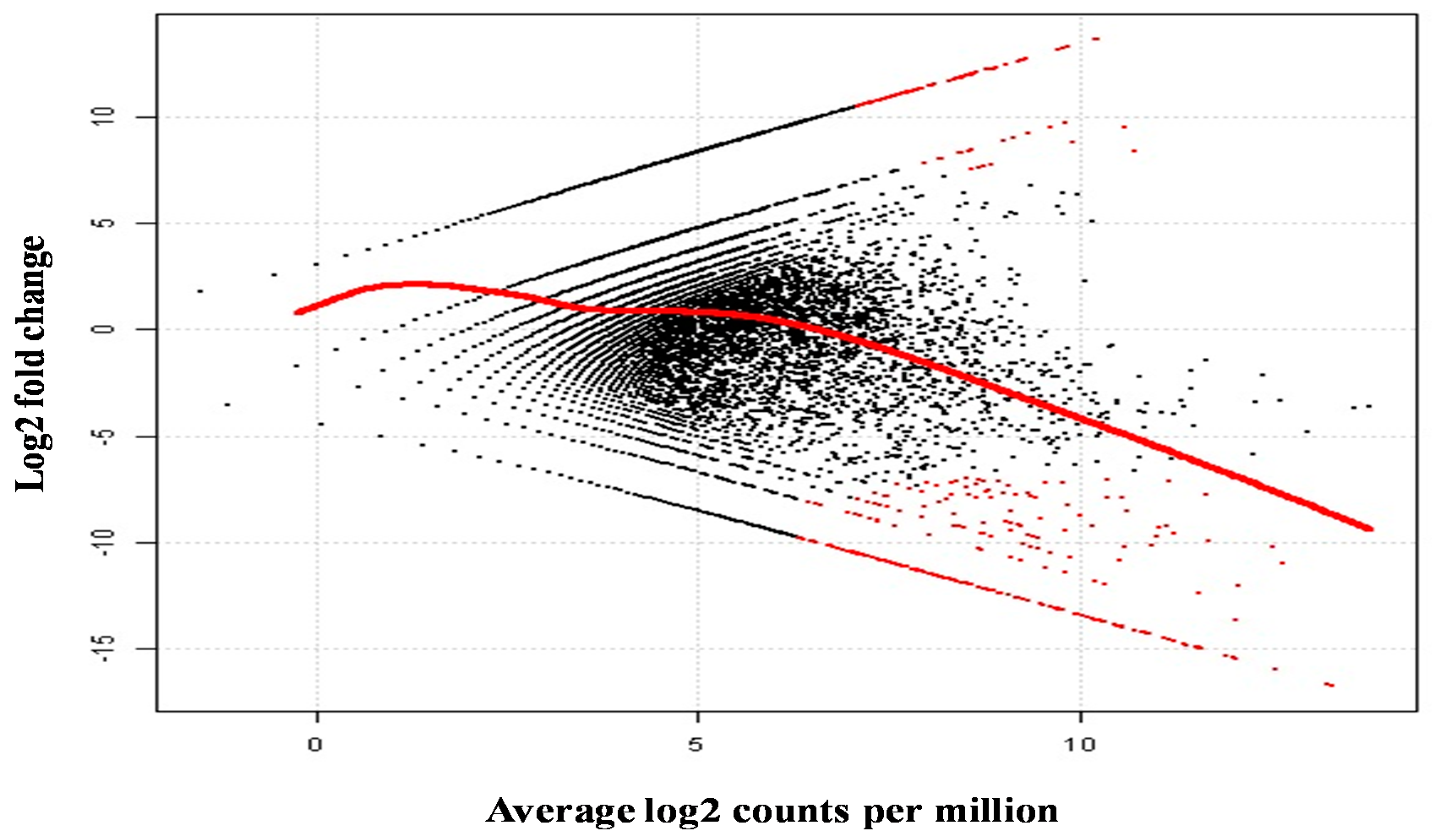
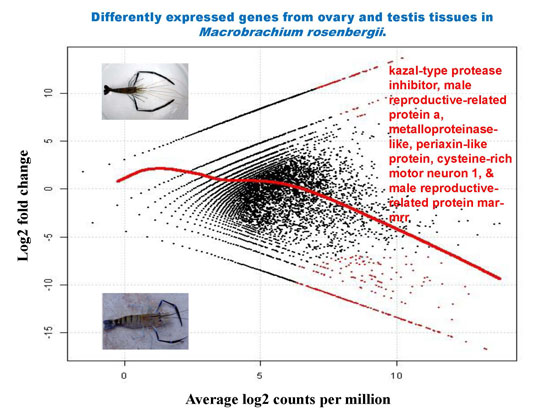
2.5. Differential Analysis of Gene Expression Profiles in M. rosenbergii Ovary and Testis Tissues
2.6. Putative Genes/Proteins Affecting Sex Differentiation and Reproductive Development and/or Function
2.7. Putative Molecular Markers
3. Discussion
3.1. Comparison of Transcriptome Assembly Performance
3.2. Annotation Analysis
3.3. Putative Genes Associated with Reproductive Traits and Development
3.4. Limitations of the Current Study
- (i)
- Limited samples: Prawns (only three tissue sample types) were only collected at final harvest, which resulted in limited information on gonadal development in both sexes without associated histological studies (developmental stages during vitellogenesis and spermatogenesis). Future experiments should be designed to include a wider range of tissues and include multiple stages of gonadal development in both sexes for fine scale gene expression. This approach would likely result in greater differential gene expression between the sexes as well as identify key genes/proteins that are involved in crustacean reproduction.
- (ii)
- A small and single-end read transcriptome dataset: A minimum of 10 Gbp with longer and/or paired-end reads with multiple tissue samples from a range of developmental stages should be used to generate a comprehensive reference transcriptome because different types of reads to assemble a de novo transcriptome (differing in both length and pairing attributes) might have substantial influence on transcriptome downstream analyses/results. More importantly, the effect of paired-end vs. single-end strategy has been reported to have a much greater impact in terms of false positives than sequencing length [62]. Thus, the paired-end approach could be an ideal strategy to significantly improve the tracking of alternate splice junctions, indels, and differential gene expression [63]. In addition, applying multiple assembly strategies (i.e., multiple k-mers) using various assemblers can affect overall efficiency of de novo assembly in terms of contig numbers and average contig length, overcoming pitfalls associated with short reads during de novo assembly. In particular, merging all assemblies from multiple assemblers using CD-HIT-EST and/or EvidentialGene tr2aacds pipelines could provide a more comprehensive and more accurate assembly [25,30].
- (iii)
- Requirement for lab validation: The support from substantial wet lab validation is required to strengthen the quality of data. This approach was not, however, available in the current study, so we recommend the validation of candidate genes (i.e., reproductive-related genes), SSRs, and SNPs in the future. In particular, correlations between developmental stages and gene expression profiles validated using a qRT-PCR approach would allow inferences to be made about specific gene functions related to prawn reproduction.
4. Materials and Methods
4.1. Tissue Material and RNA Extraction
4.2. Sequence Cleaning and Assembly
4.3. Annotation of mRNAs
4.4. Expression Analysis
4.5. Identification of SSR Motifs and SNPs
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jung, H.; Lyons, R.E.; Hurwood, D.A.; Mather, P.B. Genes and growth performance in crustacean species: A review of relevant genomic studies in crustaceans and other taxa. Rev. Aquac. 2013, 5, 77–110. [Google Scholar] [CrossRef]
- Liu, Y.; Hui, M.; Cui, Z.; Luo, D.; Song, C.; Li, Y.; Liu, L. Comparative transcriptome analysis reveals sex-biased gene expression in juvenile Chinese mitten crab Eriocheir sinensis. PLoS ONE 2015, 10, e0133068. [Google Scholar]
- Peng, J.; Wei, P.; Zhang, B.; Zhao, Y.; Zeng, D.; Chen, X.; Li, M.; Chen, X. Gonadal transcriptomic analysis and differentially expressed genes in the testis and ovary of the Pacific white shrimp (Litopenaeus vannamei). BMC Genom. 2015, 16, 1006. [Google Scholar] [CrossRef] [PubMed]
- Sharabi, Q.; Manor, R.; Weil, S.; Aflalo, E.D.; Lezer, Y.; Levy, T.; Aizen, J.; Ventura, T.; Mather, P.B.; Khalaila, I.; et al. Identification and characterization of an insulin-like receptor involved in Crustacean reproduction. Endocrinology 2016, 157, 928–941. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Liu, P.; Jia, F.; Li, J.; Gao, B. De novo transcriptome analysis of Portunus trituberculatus ovary and testis by RNA-Seq: Identification of genes involved in gonadal development. PLoS ONE 2015, 10, e0133659. [Google Scholar] [CrossRef] [PubMed]
- Leelatanawit, R.; Uawisetwathana, U.; Klinbunga, S.; Karoonuthaisiri, N. A cDNA microarray, UniShrimpChip, for identification of genes relevant to testicular development in the black tiger shrimp (Penaeus monodon). BMC Mol. Biol. 2011, 12, 15. [Google Scholar] [CrossRef] [PubMed]
- Qiao, H.; Fu, H.; Jin, S.; Wu, Y.; Jiang, S.; Gong, Y.; Xiong, Y. Constructing and random sequencing analysis of normalized cDNA library of testis tissue from oriental river prawn (Macrobrachium nipponense). Comp. Biochem. Physiol. 2012, 7, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.; Knibb, W.; Remilton, C.; Elizur, A. De-novo transcriptome analysis of the banana shrimp (Fenneropenaeus merguiensis) and identification of genes associated with reproduction and development. Mar. Genom. 2015, 22, 71–78. [Google Scholar] [CrossRef] [PubMed]
- FAO. Fisheries Statistical daTabase, Global Aquaculture Production (Fisheries Global Information System, Online Query). Available online: http://www.fao.org/fishery/statistics/global-commodities-production/en (accessed on 25 November 2015).
- Jung, H.; Lyons, R.E.; Dinh, H.; Hurwood, D.A.; McWilliam, S.; Mather, P.B. Transcriptome of a giant freshwater prawn (Macrobrachium rosenbergii): De novo assembly, annotation and marker discovery. PLoS ONE 2011, 6, e27938. [Google Scholar] [CrossRef] [PubMed]
- Ventura, T.; Manor, R.; Aflalo, E.D.; Chalifa-Caspi, V.; Weil, S.; Sharabi, O.; Sagi, A. Post-embryonic transcriptomes of the prawn Macrobrachium rosenbergii: Multigenic succession through metamorphosis. PLoS ONE 2013, 8, e55322. [Google Scholar] [CrossRef] [PubMed]
- Thanh, N.H.; Zhao, L.; Liu, Q. De novo transcriptome sequencing analysis and comparison of differentially expressed genes (DEGs) in Macrobrachium rosenbergii in China. PLoS ONE 2014, 9, e109656. [Google Scholar]
- Suwansa-ard, S.; Thongbuakaew, T.; Wang, T.; Zhao, M.; Elizur, A.; Hanna, P.J.; Sretarugsa, P.; Cummins, S.F.; Sobhon, P. In silico neuropeptidome of female Macrobrachium rosenbergii based on transcriptome and peptide mining of eyestalk, central nervous system and ovary. PLoS ONE 2015, 10, e123848. [Google Scholar]
- Aflalo, E.D.; Raju, D.V.S.N.; Bommi, N.A.; Verghese, J.T.; Samraj, T.Y.C.; Hulata, G.; Ovadia, O.; Sagi, A. Toward a sustainable production of genetically improved all-male prawn (Macrobrachium rosenbergii): Evaluation of production traits and obtaining neofemales in three Indian strains. Aquaculture 2014, 338–341, 197–207. [Google Scholar]
- Hung, D.; Vu, N.T.; Nguyen, N.H.; Ponzoni, R.W.; Hurwood, D.A.; Mather, P.B. Genetic response to combined family selection for improved mean harvest weight in giant freshwater prawn (Macrobrachium rosenbergii) in Vietnam. Aquaculture 2013, 412–413, 70–73. [Google Scholar]
- Malecha, S. The case for all-female freshwater prawn, Macrobrachium rosenbergii (DeMan), culture. Aquac. Res. 2012, 43, 1038–1048. [Google Scholar]
- Nhan, D.T.; Wille, M.; Hung, L.T.; Sorgeloos, P. Effects of larval stocking density and feeding regime on larval rearing of giant freshwater prawn (Macrobrachium rosenbergii). Aquaculture 2010, 300, 80–86. [Google Scholar] [CrossRef]
- Li, Y.; Qian, Y.Q.; Ma, W.M.; Yang, W.J. Inhibition mechanism and the effects of structure of activity of male reproduction-related peptidase inhibitor Kazal-type (MRPINK) of Macrobrachium rosenbergii. Mar. Biotechnol. 2009, 11, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Ngernsoungnern, P.; Ngernsoungnern, A.; Weerachatyanukul, W.; Meeratana, P.; Hanna, P.J.; Prasert Sobhon, P.; Sretarugs, P. Abalone egg-laying hormone induces rapid ovarian maturation and early spawning of the giant freshwater prawn, Macrobrachium rosenbergii. Aquaculture 2009, 296, 143–149. [Google Scholar] [CrossRef]
- Phoungpectchara, I.; Tinikul, Y.; Poljaroen, J.; Changklungmoa, N.; Siangcham, T.; Sroyraya, M.; Chotwiwatthanakun, C.; Vanichviriyakit, R.; Hanna, P.J.; Sobhon, P. Expression of the male reproduction-related gene (Mar-Mrr) in the spermatic duct of the giant freshwater prawn, Macrobrachium rosenbergii. Cell Tissue Res. 2012, 348, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Ventura, T.; Aflalo, E.D.; Weil, S.; Kashkush, K.; Sagi, A. Isolation and characterization of a female-specific DNA marker in the giant freshwater prawn Macrobrachium rosenbergii. Heredity 2011, 107, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Ventura, T.; Manor, R.; Aflalo, E.D.; Weil, S.; Rosen, O.; Sagi, A. Timing sexual differential: Full functional sex reversal achieved through silencing of a single insulin-like gene in the prawn, Macrobrachium rosenbergii. Biol. Reprod. 2012, 86, 90. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.H.; Qiu, G.F. Female-only sex-linked amplified fragment length polymorphism markers support ZW/ZZ sex determination in the giant freshwater prawn Macrobrachium rosenbergii. Anim. Genet. 2013, 44, 782–785. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhang, Y.; Zhou, Z.; Waldbieser, G.; Sun, F.; Lu, J.; Zhang, J.; Jiang, Y.; Zhang, H.; Wang, X.; et al. Efficient assembly and annotation of the transcriptome of catfish by RNA-Seq analysis of a doubled haploid homozygote. BMC Genom. 2013, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Flicek, P.; Birney, E. Sense from sequence reads: Methods for alignment and assembly. Nat. Methods 2009, 11, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Lyons, R.E.; Li, Y.; Thanh, N.M.; Dinh, H.; Hurwood, D.A.; Salin, K.R.; Mather, P.B. A candidate gene association study for growth performance in an improved giant freshwater prawn (Macribrachium rosenbergii) culture line. Mar. Biotechnol. 2014, 16, 161–180. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Jiang, H.; Cao, D.; Liu, L.; Hu, S.; Wang, Q. Comparative transcriptome analysis of the accessory sex gland and testis from the Chinese mitten crab (Eriocheir sinensis). PLoS ONE 2013, 8, e53915. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.V.; Jung, H.; Nguyen, T.M.; Hurwood, D.; Mather, P. Evaluation of potential candidate genes involved in salinity tolerance in striped catfish (Pangasianodon hypohthalmus) using an RNA-SEQ approach. Mar. Genom. 2016, 25, 75–888. [Google Scholar] [CrossRef] [PubMed]
- Thanh, N.M.; Jung, H.; Lyons, R.E.; Njaci, I.; Yoon, B.H.; Chand, V.; Tuan, N.V.; Thua, V.T.M.; Mather, P. Optimizing de novo transcriptome assembly and extending genomic resources for striped catfish (Pangasianodon hypophthalmus). Mar. Genom. 2015, 23, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Surget-Groba, Y.; Montoya-Burgos, J.I. Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res. 2010, 20, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Sadamoto, H.; Takahashi, H.; Okada, T.; Kenmoku, H.; Toyota, M.; Asakawa, Y. De novo sequencing and transcriptome analysis of the central nervous system of mollusk Lymnaea stagnalis by deep RNA sequencing. PLoS ONE 2012, 7, e42546. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.; Wang, Z.; Li, G. The impacts of read length and transcriptome complexity for de novo assembly: A simulation study. PLoS ONE 2014, 9, e94825. [Google Scholar] [CrossRef] [PubMed]
- Hosomichi, K.; Shiina, T.; Tajima, A.; Inoue, I. The impact of next-generation sequencing technologies of HLA research. J. Hum. Genet. 2015, 60, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Rachanimuk, P.; Rungnapa, L.; Sittikankeaw, K.; Klinbunga, S.; Khamnamtong, B.; Puanglarp, N.; Menasveta, P. Expressed sequence tag analysis for identification and characterization of sex-related genes in the giant tiger shrimp Penaeus monodon. J. Biochem. Mol. Biol. 2007, 40, 501–510. [Google Scholar]
- Zhang, E.F.; Qiu, G.F. A novel Dmrt gene is specifically expressed in the testis of Chinese mitten crab, Eriocheir sinensis. Dev. Genes Evol. 2010, 220, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, F.; Wen, R.; Xiang, J. Identification and characterization of the sex-determiner transformer-2 homologue in Chinese shrimp, Fenneropenaeus chinensis. Sex. Dev. 2012, 6, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Nakkrasae, L.I.; Damrongphol, P. A vasa-like gene in the giant freshwater prawn, Macrobrachium rosenbergii. Mol. Reprod. Dev. 2007, 74, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.; Ren, H.; Gao, S. Insulin-like growth factors (IGFs), IGF receptors, and IGF-binding proteins: Roles in skeletal muscle growth and differentiation. Gen. Comp. Endocrinol. 2010, 167, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Rosen, O.; Manor, R.; Weil, S.; Gafni, O.; Linial, A.; Aflalo, E.D.; Ventura, T.; Sagi, A. A sexual shift induced by silencing of a single insulin like gene in crayfish: Ovarian upregulation and testicular degeneration. PLoS ONE 2010, 5, e15281. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.Y.; Lin, J.Y.; Guo, S.Z.; Chen, Y.; Li, J.L.; Giu, G.F. Molecular characterization and expression analysis of an insulin-like gene from the androgenic gland of the oriental river prawn, Macrobrachium nipponense. Gen. Comp. Endocrinol. 2013, 185, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Buřič, M.; Kouba, A.; Kozak, P. Intra-sex dimorphism in crayfish females. Zoology 2010, 113, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Gopal, C.; Gopikrishna, G.; Krishna, G.; Jahageerdar, S.S.; Rye, M.; Hayes, B.J.; Paulpandia, S.; Kiranb, R.P.; Pillaia, S.M.; Ravichandran, P.; et al. Weight and time of onset of female- superior sexual dimorphism in pond reared Penaeus monodon. Aquaculture 2010, 300, 237–239. [Google Scholar] [CrossRef]
- Qiu, G.F.; Liu, P. On the role of Cdc2 kinase during meiotic maturation of oocyte in the Chinese mitten crab, Eriocheir sinensis. Comp. Biochem. Physiol. 2009, 152, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Visudtiphole, V.; Klinbunga, S.; Kirtikara, K. Molecular characterization and expression profiles of cyclin A and cyclin B during ovarian development of the giant tiger shrimp Penaeus monodon. Comp. Biochem. Physiol. 2009, 152, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Li, W.; Huang, J.; Ye, H. Two transcripts and the expression profiles of cyclin A in ovary of the mud crab, Scylla paramamosain. Mar. Freshw. Behav. Physiol. 2013, 46, 45–58. [Google Scholar] [CrossRef]
- Ma, A.; Wang, Y.; Zou, Z.; Fu, M.; Lin, P.; Zhang, Z. Erk2 in ovarian development of green mud crab Scylla paramamosain. DNA Cell Biol. 2012, 31, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Zhang, Z.; Wang, Y.; Wang, G.; Chen, Y.; Lin, P.; Wang, S.; Zou, Z. Differential expression of ubiquitin-conjugating enzyme E2r in the developing ovary and testis of penaeid shrimp Marsupenaeus japoincus. Mol. Biol. Rep. 2009, 36, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Chen, L.; Wu, P.; Zhao, W.; Li, E.; Qin, L. cDNA cloning and expression of Ubc9 in the developing embryo and ovary of oriental river prawn, Macrobrachium inpponense. Comp. Biochem. Physiol. 2010, 155, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Spees, J.L.; Chang, S.A.; Mykles, D.L.; Snyder, M.J.; Chang, E.S. Molt cycle-dependent molecular chaperone and polyubiquitin gene expression in lobster. Cell Stress Chaperon 2003, 8, 258–264. [Google Scholar] [CrossRef]
- Daniel, F.; Bonnie, B.; Alexander, V. The tails of ubiquitin precursors are ribosomal proteins whose fusion to ubiquitin facilitates ribosome biogenesis. Nature 1989, 338, 394–401. [Google Scholar]
- John, M.; Evelyn, M.; Patrick, J. Novel ubiquitin fusion proteins: Ribosomal protein P1 and actin. J. Mol. Biol. 2003, 328, 771–778. [Google Scholar]
- Wang, Q.; Chen, L.; Wang, Y.; Li, W.; He, L.; Jiang, H. Expression characteristics of two ubiquitin/ribosomal fusion protein genes in the developing testis, accessory gonad and ovary of Chinese mitten crab, Eriocheir sinensis. Mol. Biol. Rep. 2012, 39, 6683–6692. [Google Scholar] [CrossRef] [PubMed]
- Buaklin, A.; Sittikankaew, K.; Khamnamtong, B.; Menasveta, P.; Klinbunga, S. Characterization and expression analysis of the Broad-complex (Br-c) gene of the giant tiger shrimp Penaeus monodon. Comp. Biochem. Physiol. 2013, 164, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.F.; Yamano, K.; Unuma, T. Cathepsin C transcripts are differentially expressed in the final stages of oocyte maturation in kuruma prawn Marsupenaeus japonicas. Comp. Biochem. Physiol. 2005, 140, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.H.; Yang, W.X. Molecular cloning and characterization of KIFC1-like kinesin gene (es-KIFC1) in the testis of the Chinese mitten crab Eriocheir sinensis. Comp. Biochem. Physiol. 2010, 157, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; He, Y.; Hou, L.; Yang, W.X. Myosin Va participates in acrosomal formation and nuclear morphogenesis during spermatogenesis of Chinese mitten crab, Eriocheir sinensis. PLoS ONE 2010, 5, e12738. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Qiu, L.; Jiang, S.; Zhou, F.; Huang, J.; Yang, L.; Su, T.; Zhang, D. Molecular cloning and mRNA expression of M-phase phosphoprotein 6 gene in black tiger shrimp (Penaeus monodon). Mol. Biol. Rep. 2013, 40, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.F.; Qiao, K.; Huang, S.P.; Peng, H.; Huang, W.S.; Chen, B.; Chen, F.Y.; Bo, J.; Wang, K.J. Quantitative gene expression and in situ localization of scygonadin potentially associated with reproductive immunity in tissues of male and female mud crabs, Scylla paramamosain. Fish Shellfish Immunol. 2011, 31, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Ventura, T.; Rosen, O.; Sagi, A. From the discovery of the crustacean androgenic gland to the insulin-like hormone in six decades. Gen. Comp. Endocrinol. 2011, 173, 381–388. [Google Scholar] [CrossRef] [PubMed]
- González, E.; Joly, S. Impact of RNA-seq attributes on false positive rates in differential expression analysis of de novo assembled transcriptomes. BMC Res. Notes 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Chhangawala, S.; Rudy, G.; Mason, C.E.; Rosenfeld, J.A. The impact of read length on quantification of differentially expressed genes and splice junction detection. Genome Biol. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next-generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.; Schein, J.; Chiu, R.; Corbett, R.; Field, M.; Jackman, S.D.; Mungall, K.; Lee, S.; Okada, H.M.; Qian, J.Q.; et al. De novo assembly and analysis of RNA-seq data. Nat. Methods 2010, 7, 909–912. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2008, 36, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [PubMed]
- Götz, S.; Garcia-Gomez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Zheng, H.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Min, X.J.; Butler, G.; Storms, R.; Tsang, A. OrfPredictor: Predicting protein coding regions in EST-derived sequences. Nucleic Acids Res. 2005, 33, W677–W680. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Lu, S.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; DeWeese-Scott, C.; Fong, J.H.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R.; et al. CDD: A Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2011, 39, D225–D229. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Bolger, A.M.; Nagel, A.; Fernie, A.R.; Lunn, J.E.; Stitt, M.; Usadel, B. RobiNA: A user-friendly, integrated software solution for RNA-Seq-based transcriptomics. Nucleic Acids Res. 2012, 40, W622–W627. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Meglécz, E.; Constedoat, C.; Dubut, V.; Gilles, A.; Malausa, T.; Pech, N.; Martin, J.F. QDD: A user-friendly program to select microsatellite markers and design primers from large sequencing projects. Bioinformatics 2010, 26, 403–404. [Google Scholar] [CrossRef] [PubMed]
- Rozen, S.; Skaletsky, H. Primer3 in the WWW for general users and for biologist programmers. Methods Mol. Biol. 2000, 132, 365–386. [Google Scholar] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing Subgroup. The sequence alignment/map (SAM) format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Statistics | 454 FLX | Ion-Torrent | ||||||
|---|---|---|---|---|---|---|---|---|
| Dataset Name | All | Muscle | Ovary | Testis | All | Muscle | Ovary d | Testis |
| TNB before processing (Mbp) a | 244.37 | 114.38 | 86.06 | 43.94 | 1475.49 | 302.71 | 751.09 | 421.69 |
| Total number of reads | 787,731 | 367,379 | 279,393 | 140,959 | 12,945,479 | 3,027,258 | 6,477,802 | 3,440,419 |
| Average read length (bp) | 310 | 311 | 308 | 311 | 113 | 100 | 116 | 123 |
| GC percentage (%) | 48.30 | 50.45 | 44.88 | 49.40 | 42.26 | 48.60 | 47.10 | 46.70 |
| TNB after trimming and processing (Mbp) a | 234.03 | 109.88 | 81.90 | 42.25 | 1269.81 | 252.13 | 644.89 | 374.80 |
| TNR used for assembly b | 754,374 (95.77%) | 352,936 (96.07%) | 265,907 (95.17%) | 135,531 (96.15%) | 11,141,087 (86.06%) | 2,521,259 (83.29%) | 5,561,961 (85.86%) | 3,057,867 (88.88%) |
| ARL after trimming and processing (bp) | 261 | 267 | 258 | 258 | 94 | 87 | 95 | 100 |
| GC percentage (%) c | 48.60 | 49.50 | 44.40 | 49.60 | 42.90 | 47.10 | 38.90 | 46.70 |
| Assembler | Parameter | All | Muscle | Ovary | Testis |
|---|---|---|---|---|---|
| Trans-ABySS (K = 75) | Total base of reads (bp) | 1,294,937,751 | 317,222,362 | 603,806,324 | 343,353,395 |
| No. of total contigs | 40,225 | 4556 | 25,482 | 5335 | |
| Total bases of contigs (bp) | 13,707,795 | 1,629,728 | 8,896,935 | 1,788,051 | |
| No. of contig ≥ 1000 bp | 366 | 83 | 284 | 51 | |
| Contig N50 (bp) | 348 | 357 | 357 | 333 | |
| Mean contig length (bp) | 341 | 358 | 349 | 335 | |
| Largest contig (bp) | 2750 | 4194 | 2387 | 2054 | |
| † CEGMA/Mapping (%) | 50.11/77.71 | ||||
| Trinity (K = 25) | Total base of reads (bp) | 1,294,937,751 | 317,222,362 | 603,806,324 | 343,353,395 |
| No. of total contigs | 78,007 | 11,337 | 70,352 | 17,260 | |
| Total bases of contigs (bp) | 27,946,414 | 4,152,262 | 23,845,796 | 5,580,093 | |
| No. of contig ≥ 1000 bp | 912 | 182 | 439 | 99 | |
| Contig N50 (bp) | 368 | 370 | 344 | 322 | |
| Mean contig length (bp) | 358 | 366 | 339 | 323 | |
| Largest contig (bp) | 4480 | 3223 | 3350 | 2694 | |
| † CEGMA/Mapping (%) | 51.81/77.27 | ||||
| CLC Bio (K = 20) | Total base of reads (bp) | 1,294,937,751 | 317,222,362 | 603,806,324 | 343,353,395 |
| No. of total contigs | 44,407 | 8397 | 35,847 | 10,604 | |
| Total bases of contigs (bp) | 19,415,235 | 3,826,821 | 15,013,223 | 4,260,690 | |
| No. of contig ≥ 1000 bp | 9259 | 1871 | 6860 | 1770 | |
| Contig N50 (bp) | 438 | 448 | 417 | 399 | |
| Mean contig length (bp) | 437 | 456 | 419 | 402 | |
| Largest contig (bp) | 9495 | 8037 | 5139 | 3978 | |
| † CEGMA/Mapping (%) | 53.09/79.26 | ||||
| Contig Number | Putative Function | Length (bp) | E-Value | Log2 Fold Change | Average Log2 Counts Per Million | p-Value | False Discovery Rate |
|---|---|---|---|---|---|---|---|
| 30,784 | kazal-type protease inhibitor | 976 | 4.82 × 10−85 | −16.66 | 13.23 | 6.17 × 10−10 | 1.30 × 10−05 |
| 30,770 | kazal-type proteinase inhibitor | 631 | 2.18 × 10−22 | −14.96 | 11.53 | 1.19 × 10−08 | 4.68 × 10−05 |
| 30,724 | male reproductive-related protein a | 629 | 1.12 × 10−16 | −15.43 | 12.00 | 5.23 × 10−09 | 3.67 × 10−05 |
| 30,822 | 342 | 3.20 × 10−24 | −13.97 | 10.54 | 6.58 × 10−08 | 0.000104 | |
| 30,728 | 432 | 2.54 × 10−08 | −13.97 | 10.5 | 6.60 × 10−08 | 0.000104 | |
| 30,725 | 281 | 1.49 × 10−23 | −13.71 | 10.28 | 1.03 × 10−07 | 0.000129 | |
| 30,740 | 369 | 6.13 × 10−08 | −12.50 | 9.06 | 8.37 × 10−07 | 0.00046 | |
| 30,846 | metalloproteinase-like | 1210 | 3.65 × 10−52 | −15.01 | 11.58 | 1.08 × 10−08 | 4.68 × 10−05 |
| 30,807 | 511 | 2.19 × 10−32 | −13.91 | 10.48 | 7.24 × 10−08 | 0.000109 | |
| 30,868 | periaxin-like protein | 1872 | 4.31 × 10−08 | −14.57 | 11.14 | 2.30 × 10−08 | 6.46 × 10−05 |
| 30,726 | cysteine-rich motor neuron 1 | 742 | 1.09 × 10−07 | −14.52 | 11.09 | 2.54 × 10−08 | 6.66 × 10−05 |
| 30,757 | male reproductive-related protein mar-mrr | 254 | 2.48 × 10−08 | −14.33 | 10.90 | 3.54 × 10−08 | 8.06 × 10−05 |
| 30,716 | 272 | 1.26 × 10−42 | −14.22 | 10.79 | 4.28 × 10−08 | 8.18 × 10−05 | |
| 30,718 | 280 | 9.61 × 10−43 | −12.58 | 9.14 | 7.33 × 10−07 | 0.000428 | |
| 31,029 | blastula protease-10 | 1528 | 1.22 × 10−34 | −13.98 | 10.55 | 6.45 × 10−08 | 0.000104 |
| 19,861 | matrix metalloproteinase-9 | 1103 | 7.60 × 10−20 | −13.88 | 10.45 | 7.65 × 10−08 | 0.000111 |
| 30,737 | keratin associated protein | 771 | 4.77 × 10−19 | −13.76 | 10.33 | 9.47 × 10−08 | 0.000126 |
| 30,785 | 3d domain protein | 510 | 6.23 × 10−06 | −13.67 | 10.24 | 1.10 × 10−07 | 0.000129 |
| 30,768 | lpxtg-motif cell wall anchor domain protein | 1231 | 5.25 × 10−23 | −13.65 | 10.22 | 1.13 × 10−07 | 0.000129 |
| 30,916 | serine proteinase inhibitor | 324 | 1.44 × 10−16 | −13.63 | 10.20 | 1.18 × 10−07 | 0.00013 |
| 30,796 | insulin-like androgenic gland factor | 614 | 1.13 × 10−110 | −13.45 | 10.01 | 1.62 × 10−07 | 0.000159 |
| 30,954 | von willebrand factor d and egf domain-containing | 1632 | 1.12 × 10−27 | −13.36 | 9.93 | 1.87 × 10−07 | 0.000175 |
| 32,194 | apolipoprotein d-like | 513 | 5.53 × 10−08 | −13.12 | 9.69 | 2.85 × 10−07 | 0.000239 |
| 31,036 | hemolectin cg7002-pa | 2221 | 8.78 × 10−25 | −13.05 | 9.62 | 3.21 × 10−07 | 0.000252 |
| 30,739 | epididymal sperm-binding protein 1-like | 1043 | 3.63 × 10−16 | −12.63 | 9.20 | 6.67 × 10−07 | 0.000412 |
| 30,881 | 2 RNA ligase family protein | 364 | 3.61 × 10−06 | −12.50 | 9.06 | 8.41 × 10−07 | 0.00046 |
| 30,750 | keratin associated protein | 668 | 3.21 × 10−11 | −12.49 | 9.05 | 8.52 × 10−07 | 0.00046 |
| 30,749 | fibronectin 1b | 677 | 1.57 × 10−06 | −12.41 | 8.97 | 9.84 × 10−07 | 0.00052 |
| All SNPs | Transitions | Transversions | |||||||||||
| (29,043 (64.69%)) | (15,854 (35.31%)) | ||||||||||||
| CT | GA | AG | TC | AC | GT | GC | AT | TG | CA | CG | TA | ||
| 44,897 | 7290 | 7421 | 7249 | 7083 | 2046 | 2024 | 1304 | 2445 | 2070 | 1905 | 1335 | 2725 | |
| 100 (%) | 16.2 | 16.5 | 16.1 | 15.7 | 4.5 | 4.5 | 2.9 | 5.4 | 4.6 | 4.2 | 2.9 | 6.0 | |
| All Indels | Simple | Complex | |||||||||||
| (8490 (97.15%)) | (249 (2.85%)) | ||||||||||||
| Insertion | Deletion | Insertion | Deletion | ||||||||||
| 8739 | 4369 | 4121 | 207 | 42 | |||||||||
| 100 (%) | 49.99 | 47.16 | 2.37 | 0.48 | |||||||||
| SSR Types | Total | Contigs | Singletons | |||
|---|---|---|---|---|---|---|
| Discovered Motifs | Designed Primers | Discovered Motifs | Designed Primers | Discovered Motifs | Designed Primers | |
| Di-nucleotide | 21,433 | 6643 | 3754 | 977 | 17,679 | 5666 |
| AT/TA | 7598 | 2414 | 1323 | 358 | 6275 | 2056 |
| CA/AC | 5775 | 1752 | 1018 | 259 | 4757 | 1493 |
| CG/GC | 455 | 133 | 50 | 11 | 405 | 122 |
| CT/TC | 0 | 0 | 0 | 0 | 0 | 0 |
| GA/AG | 7605 | 2344 | 1363 | 349 | 6242 | 1995 |
| GT/TG | 0 | 0 | 0 | 0 | 0 | 0 |
| Tri-nucleotide | 7181 | 2218 | 1432 | 375 | 5749 | 1843 |
| AAC/ACA/CAA | 538 | 174 | 80 | 20 | 458 | 154 |
| ACG/CGA/GAC | 199 | 55 | 59 | 13 | 140 | 42 |
| ACT/CTA/TAC | 385 | 120 | 64 | 19 | 321 | 101 |
| AGC/GCA/CAG | 750 | 224 | 225 | 60 | 525 | 164 |
| AGG/GAG/GGA | 631 | 181 | 228 | 52 | 403 | 129 |
| AGT/GTA/TAG | 0 | 0 | 0 | 0 | 0 | 0 |
| CAT/ATC/TCA | 1327 | 402 | 228 | 53 | 1099 | 349 |
| CCA/CAC/ACC | 174 | 49 | 97 | 26 | 77 | 23 |
| CCG/CGC/GCC | 40 | 13 | 27 | 7 | 13 | 6 |
| CCT/CTC/TCC | 0 | 0 | 0 | 0 | 0 | 0 |
| CGG/GGC/GCG | 0 | 0 | 0 | 0 | 0 | 0 |
| CTG/TGC/GCT | 0 | 0 | 0 | 0 | 0 | 0 |
| CTT/TTC/TCT | 0 | 0 | 0 | 0 | 0 | 0 |
| GAA/AAG/AGA | 1616 | 513 | 215 | 61 | 1401 | 452 |
| GAT/ATG/TGA | 0 | 0 | 0 | 0 | 0 | 0 |
| GTT/TGT/TTG | 0 | 0 | 0 | 0 | 0 | 0 |
| TAA/ATA/AAT | 1521 | 487 | 209 | 64 | 1312 | 423 |
| TCG/CGT/GTC | 0 | 0 | 0 | 0 | 0 | 0 |
| TGG/GTG/GGT | 0 | 0 | 0 | 0 | 0 | 0 |
| TTA/TAT/ATT | 0 | 0 | 0 | 0 | 0 | 0 |
| ≥Tetra-nucleotide | 570 | 179 | 52 | 13 | 518 | 166 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, H.; Yoon, B.-H.; Kim, W.-J.; Kim, D.-W.; Hurwood, D.A.; Lyons, R.E.; Salin, K.R.; Kim, H.-S.; Baek, I.; Chand, V.; et al. Optimizing Hybrid de Novo Transcriptome Assembly and Extending Genomic Resources for Giant Freshwater Prawns (Macrobrachium rosenbergii): The Identification of Genes and Markers Associated with Reproduction. Int. J. Mol. Sci. 2016, 17, 690. https://doi.org/10.3390/ijms17050690
Jung H, Yoon B-H, Kim W-J, Kim D-W, Hurwood DA, Lyons RE, Salin KR, Kim H-S, Baek I, Chand V, et al. Optimizing Hybrid de Novo Transcriptome Assembly and Extending Genomic Resources for Giant Freshwater Prawns (Macrobrachium rosenbergii): The Identification of Genes and Markers Associated with Reproduction. International Journal of Molecular Sciences. 2016; 17(5):690. https://doi.org/10.3390/ijms17050690
Chicago/Turabian StyleJung, Hyungtaek, Byung-Ha Yoon, Woo-Jin Kim, Dong-Wook Kim, David A. Hurwood, Russell E. Lyons, Krishna R. Salin, Heui-Soo Kim, Ilseon Baek, Vincent Chand, and et al. 2016. "Optimizing Hybrid de Novo Transcriptome Assembly and Extending Genomic Resources for Giant Freshwater Prawns (Macrobrachium rosenbergii): The Identification of Genes and Markers Associated with Reproduction" International Journal of Molecular Sciences 17, no. 5: 690. https://doi.org/10.3390/ijms17050690
APA StyleJung, H., Yoon, B.-H., Kim, W.-J., Kim, D.-W., Hurwood, D. A., Lyons, R. E., Salin, K. R., Kim, H.-S., Baek, I., Chand, V., & Mather, P. B. (2016). Optimizing Hybrid de Novo Transcriptome Assembly and Extending Genomic Resources for Giant Freshwater Prawns (Macrobrachium rosenbergii): The Identification of Genes and Markers Associated with Reproduction. International Journal of Molecular Sciences, 17(5), 690. https://doi.org/10.3390/ijms17050690