
Connecting the Dots: From DNA Damage and Repair to Aging


Abstract
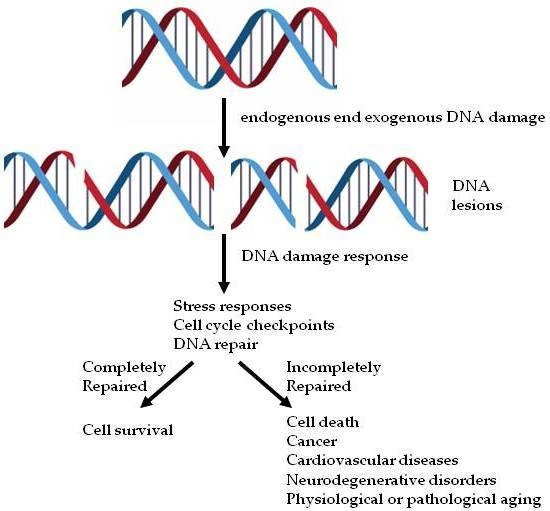
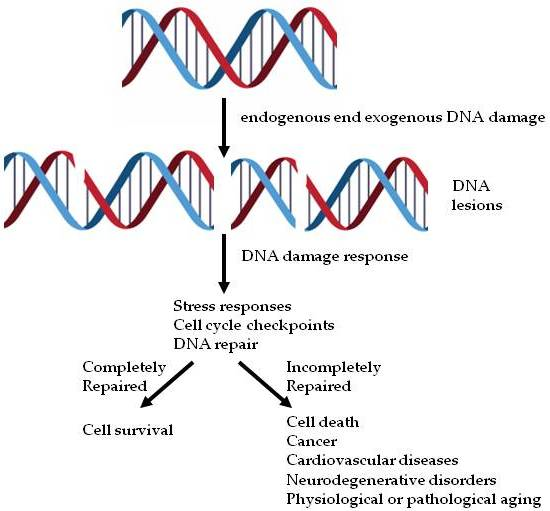
:
1. Introduction
2. The DDR Signaling Cascade
3. The DDR and Human Diseases
3.1. Cancer and DNA Damage
3.2. Cardiovascular Diseases and DNA Damage
3.3. Neurodegenerative Disorders and DNA Damage
3.4. Aging and DNA Damage
4. The Linkage between DNA Damage/Repair and Aging
5. Senescence and Aging
5.1. Evidence from Cellular Study
5.2. Evidence from Animal Models
5.3. Evidence from Human Diseases
6. From Single Cell to Systemic Effect
7. New Dots Connecting DNA Damage and Repair to Aging
7.1. Ubiquitin-Specific Protease 3 (USP3)
7.2. N-Terminal RCC1 Methyltransferase 1 (NRMT1)
7.3. Mitochondria Dysfunction in XP and CS
7.4. Nonenzymatic Post-Translational Modification
7.5. RecQ-Like Helicase Sgs1
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kunkel, T.A. Celebrating DNA’s repair crew. Cell 2015, 163, 1301–1303. [Google Scholar] [CrossRef] [PubMed]
- Haber, J.E. Deciphering the DNA damage response. Cell 2015, 162, 1183–1185. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Pan, L.; Tsai, L.H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Ishida, M.; Tashiro, S.; Yoshizumi, M.; Kihara, Y. Role of DNA damage in cardiovascular disease. Circ. J. 2014, 78, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Chow, H.M.; Herrup, K. Genomic integrity and the ageing brain. Nat. Rev. Neurosci. 2015, 16, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Dobbelstein, M.; Sorensen, C.S. Exploiting replicative stress to treat cancer. Nat. Rev. Drug Discov. 2015, 14, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rocha, H.; Garcia-Garcia, A.; Panayiotidis, M.I.; Franco, R. DNA damage and autophagy. Mutat. Res. 2011, 711, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Seviour, E.G.; Lin, S.Y. The DNA damage response: Balancing the scale between cancer and ageing. Aging 2010, 2, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Ménissier-de Murcia, J.; Molinete, M.; Gradwohl, G.; Simonin, F.; de Murcia, G. Zinc-binding domain of poly(ADP-ribose)polymerase participates in the recognition of singlestrand breaks on DNA. J. Mol. Biol. 1989, 210, 229–233. [Google Scholar] [CrossRef]
- Caldecott, K.W. Protein ADP-ribosylation and the cellular response to DNA strand breaks. DNA Repair 2014, 19, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Elledge, S.J. The DNA damage response: Ten years after. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. ATM Phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar] [CrossRef] [PubMed]
- Podhorecka, M.; Skladanowski, A.; Bozko, P. H2AX phosphorylation: Its role in DNA damage response and cancer therapy. J. Nucleic Acids 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Stiff, T.; O’Driscoll, M.; Rief, N.; Iwabuchi, K.; Lobrich, M.; Jeggo, P.A. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004, 64, 2390–2396. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.; Arbel-Eden, A.; Pilch, D.; Ira, G.; Bonner, W.M.; Petrini, J.H.; Haber, J.E.; Lichten, M. Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr. Biol. 2004, 14, 1703–1711. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, H.; Garcia-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Mortusewicz, O.; Afzal, I.; Lorvellec, M.; Garcia, P.; Helleday, T.; Petermann, E. Increased replication initiation and conflicts with transcription underlie cyclin e-induced replication stress. Oncogene 2013, 32, 3744–3753. [Google Scholar] [CrossRef] [PubMed]
- Shima, N.; Alcaraz, A.; Liachko, I.; Buske, T.R.; Andrews, C.A.; Munroe, R.J.; Hartford, S.A.; Tye, B.K.; Schimenti, J.C. A viable allele of Mcm4 Causes chromosome instability and mammary adenocarcinomas in mice. Nat. Genet. 2007, 39, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Brosh, R.M., Jr. DNA Helicases involved in DNA repair and their roles in cancer. Nat. Rev. Cancer 2013, 13, 542–558. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Raynard, S.; Sehorn, M.G.; Lu, X.; Bussen, W.; Zheng, L.; Stark, J.M.; Barnes, E.L.; Chi, P.; Janscak, P.; et al. RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev. 2007, 21, 3073–3084. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.B.; Hodges, C.A.; Barnes, E.; Vogel, H.; Hassold, T.J.; Luo, G. Defective sister-chromatid cohesion, aneuploidy and cancer predisposition in a mouse model of type II rothmund-thomson syndrome. Hum. Mol. Genet. 2005, 14, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Santoro, I.M.; McDaniel, L.D.; Nishijima, I.; Mills, M.; Youssoufian, H.; Vogel, H.; Schultz, R.A.; Bradley, A. Cancer predisposition caused by elevated mitotic recombination in bloom mice. Nat. Genet. 2000, 26, 424–429. [Google Scholar] [PubMed]
- Gilad, O.; Nabet, B.Y.; Ragland, R.L.; Schoppy, D.W.; Smith, K.D.; Durham, A.C.; Brown, E.J. Combining ATR suppression with oncogenic Ras synergistically increases genomic instability, causing synthetic lethality or tumorigenesis in a dosage-dependent manner. Cancer Res. 2010, 70, 9693–9702. [Google Scholar] [CrossRef] [PubMed]
- Willis, N.A.; Chandramouly, G.; Huang, B.; Kwok, A.; Follonier, C.; Deng, C.; Scully, R. BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature 2014, 510, 556–559. [Google Scholar] [CrossRef] [PubMed]
- Hakem, R.; de la Pompa, J.L.; Sirard, C.; Mo, R.; Woo, M.; Hakem, A.; Wakeham, A.; Potter, J.; Reitmair, A.; Billia, F.; et al. The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell 1996, 85, 1009–1023. [Google Scholar] [CrossRef]
- Xu, X.; Wagner, K.U.; Larson, D.; Weaver, Z.; Li, C.; Ried, T.; Hennighausen, L.; Wynshaw-Boris, A.; Deng, C.X. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat. Genet. 1999, 22, 37–43. [Google Scholar] [PubMed]
- Murga, M.; Bunting, S.; Montana, M.F.; Soria, R.; Mulero, F.; Canamero, M.; Lee, Y.; McKinnon, P.J.; Nussenzweig, A.; Fernandez-Capetillo, O. A mouse model of ATR-seckel shows embryonic replicative stress and accelerated aging. Nat. Genet. 2009, 41, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [PubMed]
- Giordano, F.J. Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Investig. 2005, 115, 500–508. [Google Scholar] [CrossRef] [PubMed]
- De, B.J.; Donker, I.; de, W.J.; Hoeijmakers, J.H.; Weeda, G. Disruption of the mouse xeroderma pigmentosum group D DNA repair/basal transcription gene results in preimplantation lethality. Cancer Res. 1998, 58, 89–94. [Google Scholar]
- De, B.J.; de, W.J.; van, S.H.; Berg, R.J.; Morreau, H.; Visser, P.; Lehmann, A.R.; Duran, M.; Hoeijmakers, J.H.; Weeda, G. A mouse model for the basal transcription/DNA repair syndrome trichothiodystrophy. Mol. Cell 1998, 1, 981–990. [Google Scholar]
- Barlow, C.; Hirotsune, S.; Paylor, R.; Liyanage, M.; Eckhaus, M.; Collins, F.; Shiloh, Y.; Crawley, J.N.; Ried, T.; Tagle, D.; et al. Atm-deficient mice: A paradigm of ataxia telangiectasia. Cell 1996, 86, 159–171. [Google Scholar] [CrossRef]
- Iyama, T.; Wilson, D.M., III. DNA Repair mechanisms in dividing and non-dividing cells. DNA Repair 2013, 12, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Laposa, R.R.; Huang, E.J.; Cleaver, J.E. Increased apoptosis, P53 up-regulation, and cerebellar neuronal degeneration in repair-deficient cockayne syndrome mice. Proc. Natl. Acad. Sci. USA 2007, 104, 1389–1394. [Google Scholar] [CrossRef] [PubMed]
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef] [PubMed]
- Tuppen, H.A.; Blakely, E.L.; Turnbull, D.M.; Taylor, R.W. Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta 2010, 1797, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef] [PubMed]
- Bowman, A.; Birch-Machin, M.A. Age-dependent decrease of mitochondrial complex II activity in human skin fibroblasts. J. Investig. Dermatol. 2016, 136, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Lombard, D.B.; Chua, K.F.; Mostoslavsky, R.; Franco, S.; Gostissa, M.; Alt, F.W. DNA repair, genome stability, and aging. Cell 2005, 120, 497–512. [Google Scholar] [CrossRef] [PubMed]
- De, B.J.; Andressoo, J.O.; de, W.J.; Huijmans, J.; Beems, R.B.; van Steeq, H.; Weeda, G.; van der Horst, G.T.; van Leeuwen, W.; Themmen, A.P.; et al. Premature aging in mice deficient in DNA repair and transcription. Science 2002, 296, 1276–1279. [Google Scholar]
- Papadopoulos, N.; Lindblom, A. Molecular basis of HNPCC: Mutations of MMR genes. Hum. Mutat. 1997, 10, 89–99. [Google Scholar] [CrossRef]
- Boesch, P.; Weber-Lotfi, F.; Ibrahim, N.; Tarasenko, V.; Cosset, A.; Paulus, F.; Lightowlers, R.N.; Dietrich, A. DNA repair in organelles: Pathways, organization, regulation, relevance in disease and aging. Biochim. Biophys. Acta 2011, 1813, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.; Schurman, S.H.; Harboe, C.; de Souza-Pinto, N.C.; Bohr, V.A. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis 2009, 30, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Gorbunova, V.; Seluanov, A.; Mao, Z.; Hine, C. Changes in DNA repair during aging. Nucleic Acids Res. 2007, 35, 7466–7474. [Google Scholar] [CrossRef] [PubMed]
- Vyjayanti, V.N.; Rao, K.S. DNA double strand break repair in brain: Reduced NHEJ activity in aging rat neurons. Neurosci. Lett. 2006, 393, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Vogel, H.; Holcomb, V.B.; Gu, Y.; Hasty, P. Deletion of Ku70, Ku80, or both causes early aging without substantially increased cancer. Mol. Cell. Biol. 2007, 27, 8205–8214. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Campisi, J. Senescent cells, tumor suppression, and organismal aging: Good citizens, and bad neighbors. Cell 2005, 120, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Courtois-Cox, S.; Jones, S.L.; Cichowski, K. Many roads lead to oncogene-induced senescence. Oncogene 2008, 27, 2801–2809. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre’, M.; Nuciforo, P.G.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Tyner, S.D.; Venkatachalam, S.; Choi, J.; Jones, S.; Ghebranious, N.; Igelmann, H.; Lu, X.; Soron, G.; Cooper, B.; Brayton, C.; et al. P53 mutant mice that display early ageing-associated phenotypes. Nature 2002, 415, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of P16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Hyun, M.; Lee, J.; Lee, K.; May, A.; Bohr, V.A.; Ahn, B. Longevity and resistance to stress correlate with DNA repair capacity in Caenorhabditis elegans. Nucleic Acids Res. 2008, 36, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.; Maser, R.S.; Bachoo, R.M.; Menon, J.; Carrasco, D.R.; Gu, Y.; Alt, F.W.; DePinho, R.A. Telomere dysfunction and Atm deficiency compromises organ homeostasis and accelerates ageing. Nature 2003, 421, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Li, W.; Kim, S.; Brodie, S.G.; Deng, C.X. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev. 2003, 17, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Ruzankina, Y.; Pinzon-Guzman, C.; Asare, A.; Ong, T.; Pontano, L.; Cotsarelis, G.; Zediak, V.P.; Velez, M.; Bhandoola, A.; Brown, E.J. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell 2007, 1, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Holcomb, V.B.; Vogel, H.; Hasty, P. Deletion of Ku80 causes early aging independent of chronic inflammation and Rag-1-induced DSBs. Mech. Ageing Dev. 2007, 128, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T.; Karran, P.; Wood, R.D. DNA excision repair pathways. Curr. Opin. Genet. Dev. 1997, 7, 158–169. [Google Scholar] [CrossRef]
- Bertola, D.R.; Cao, H.; Albano, L.M.; Oliveira, D.P.; Kok, F.; Marques-Dias, M.J.; Kim, C.A.; Hegele, R.A. Cockayne syndrome type A: Novel mutations in eight typical patients. J. Hum. Genet. 2006, 51, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Bender, M.M.; Potocki, L.; Metry, D.W. What syndrome is this? Cockayne syndrome. Pediatr. Dermatol. 2003, 20, 538–540. [Google Scholar] [PubMed]
- Robbins, J.H.; Kraemer, K.H.; Lutzner, M.A.; Festoff, B.W.; Coon, H.G. Xeroderma pigmentosum. An inherited diseases with sun sensitivity, multiple cutaneous neoplasms, and abnormal DNA repair. Ann. Intern. Med. 1974, 80, 221–248. [Google Scholar] [CrossRef] [PubMed]
- Robbins, J.H. Xeroderma pigmentosum. Defective DNA repair causes skin cancer and neurodegeneration. Jpn. Automob. Manuf. Assoc. 1988, 260, 384–388. [Google Scholar] [CrossRef]
- Lee, H.W.; Blasco, M.A.; Gottlieb, G.J.; Horner, J.W., 2nd; Greider, C.W.; DePinho, R.A. Essential role of mouse telomerase in highly proliferative organs. Nature 1998, 392, 569–574. [Google Scholar] [PubMed]
- Goodell, M.A.; Rando, T.A. Stem cells and healthy aging. Science 2015, 350, 1199–1204. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Morin, C.L.; Pagliassotti, M.J.; Windmiller, D.; Eckel, R.H. Adipose tissue-derived tumor necrosis factor-α activity is elevated in older rats. J. Gerontol. A Biol. Sci. Med. Sci. 1997, 52, B190–B195. [Google Scholar] [CrossRef] [PubMed]
- Starr, M.E.; Evers, B.M.; Saito, H. Age-associated increase in cytokine production during systemic inflammation: Adipose tissue as a major source of IL-6. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Maggio, M.; Guralnik, J.M.; Longo, D.L.; Ferrucci, L. Interleukin-6 in aging and chronic disease: A magnificent pathway. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Orjalo, A.V.; Desprez, P.Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.E.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 2011, 25, 409–433. [Google Scholar] [CrossRef] [PubMed]
- Nicassio, F.; Corrado, N.; Vissers, J.H.; Areces, L.B.; Bergink, S.; Marteijn, J.A.; Geverts, B.; Houtsmuller, A.B.; Vermeulen, W.; di Fiore, P.P.; et al. Human USP3 is a chromatin modifier required for S phase progression and genome stability. Curr. Biol. 2007, 17, 1972–1977. [Google Scholar] [CrossRef] [PubMed]
- Lancini, C.; van den Berk, P.C.; Vissers, J.H.; Gargiulo, G.; Song, J.Y.; Hulsman, D.; Serresi, M.; Tanger, E.; Blom, M.; Vens, C.; et al. Tight regulation of ubiquitin-mediated DNA damage response by USP3 preserves the functional integrity of hematopoietic stem cells. J. Exp. Med. 2014, 211, 1759–1777. [Google Scholar] [CrossRef] [PubMed]
- Tooley, C.E.; Petkowski, J.J.; Muratore-Schroeder, T.L.; Balsbaugh, J.L.; Shabanowitz, J.; Sabat, M.; Minor, W.; Hunt, D.F.; Macara, I.G. NRMT is an α-N-methyltransferase that methylates RCC1 and retinoblastoma protein. Nature 2010, 466, 1125–1128. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Fu, L.; Wang, Z.; Gan, N.; Dai, X.; Wang, Y. α-N-methylation of damaged DNA-binding protein 2 (DDB2) and its function in nucleotide excision repair. J. Biol. Chem. 2014, 289, 16046–16056. [Google Scholar] [CrossRef] [PubMed]
- Bonsignore, L.A.; Tooley, J.G.; Van Hoose, P.M.; Wang, E.; Cheng, A.; Cole, M.P.; Schaner Tooley, C.E. NRMT1 knockout mice exhibit phenotypes associated with impaired DNA repair and premature aging. Mech. Ageing Dev. 2015, 146–148, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD+/SIRT1 reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef] [PubMed]
- Scheibye-Knudsen, M.; Mitchell, S.J.; Fang, E.F.; Iyama, T.; Ward, T.; Wang, J.; Dunn, C.A.; Singh, N.; Veith, S.; Hasan-Olive, M.M.; et al. A high-fat diet and NAD+ activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 2014, 20, 840–855. [Google Scholar] [CrossRef] [PubMed]
- Soskic, V.; Groebe, K.; Schrattenholz, A. Nonenzymatic posttranslational protein modifications in ageing. Exp. Gerontol. 2008, 43, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.E.; Brunette, S.; Puente, L.G.; Megeney, L.A. Metacaspase Yca1 is required for clearance of insoluble protein aggregates. Proc. Natl. Acad. Sci. USA 2010, 107, 13348–13353. [Google Scholar] [CrossRef] [PubMed]
- Witko-Sarsat, V.; Nguyen-Khoa, T.; Jungers, P.; Drueke, T.B.; Descamps-Latscha, B. Advanced oxidation protein products as a novel molecular basis of oxidative stress in uraemia. Nephrol. Dial. Transplant. 1999, 14, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Chung, W.H.; Shim, E.Y.; Lee, S.E.; Ira, G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 2008, 34, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Böhm, S.; Mihalevic, M.J.; Casal, M.A.; Bernstein, K.A. Disruption of SUMO-targeted ubiquitin ligases Slx5-Slx8/RNF4 alters RecQ-like helicase Sgs1/BLM localization in yeast and human cells. DNA Repair 2015, 26, 1–14. [Google Scholar] [CrossRef] [PubMed]
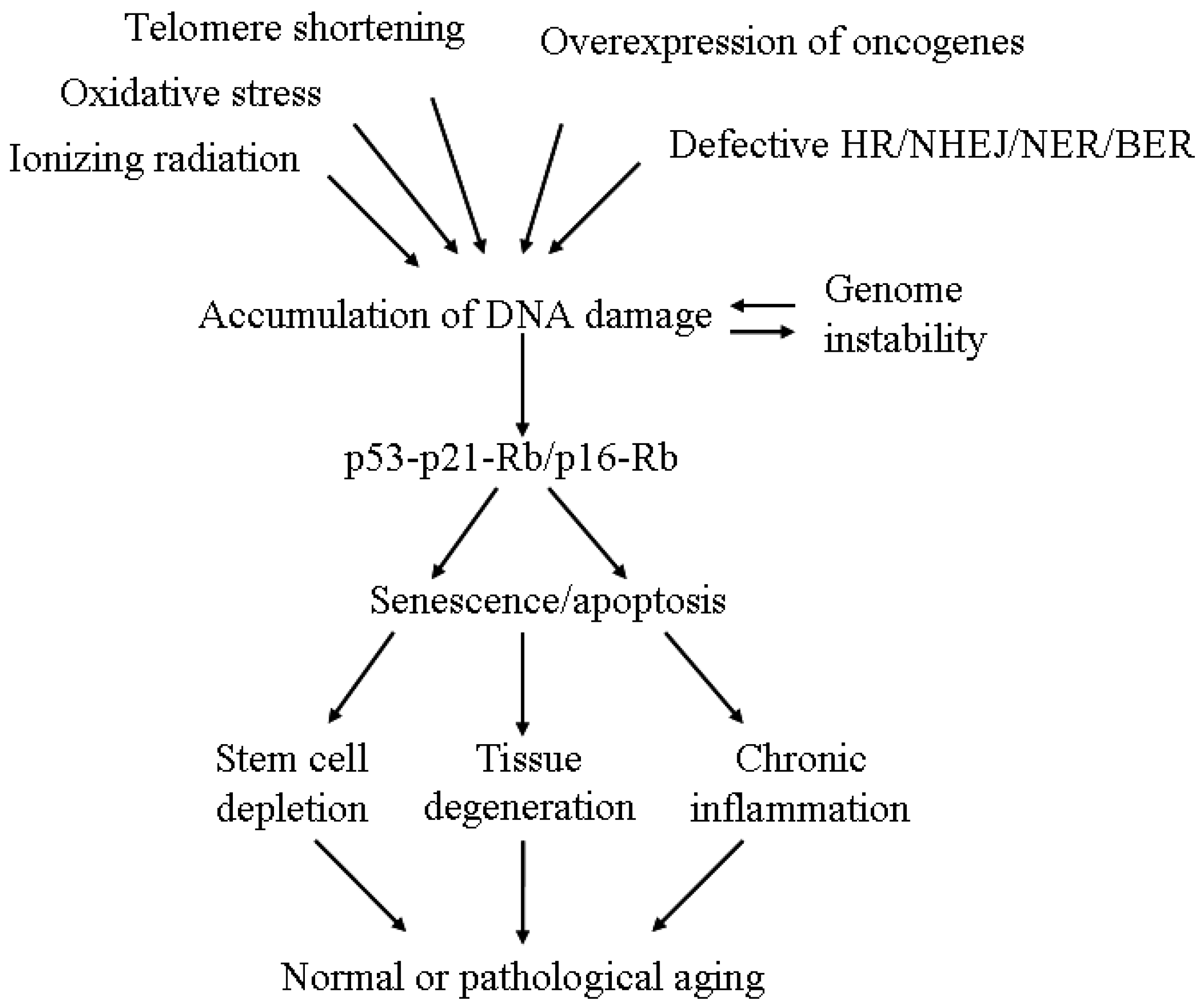

{kind=link}
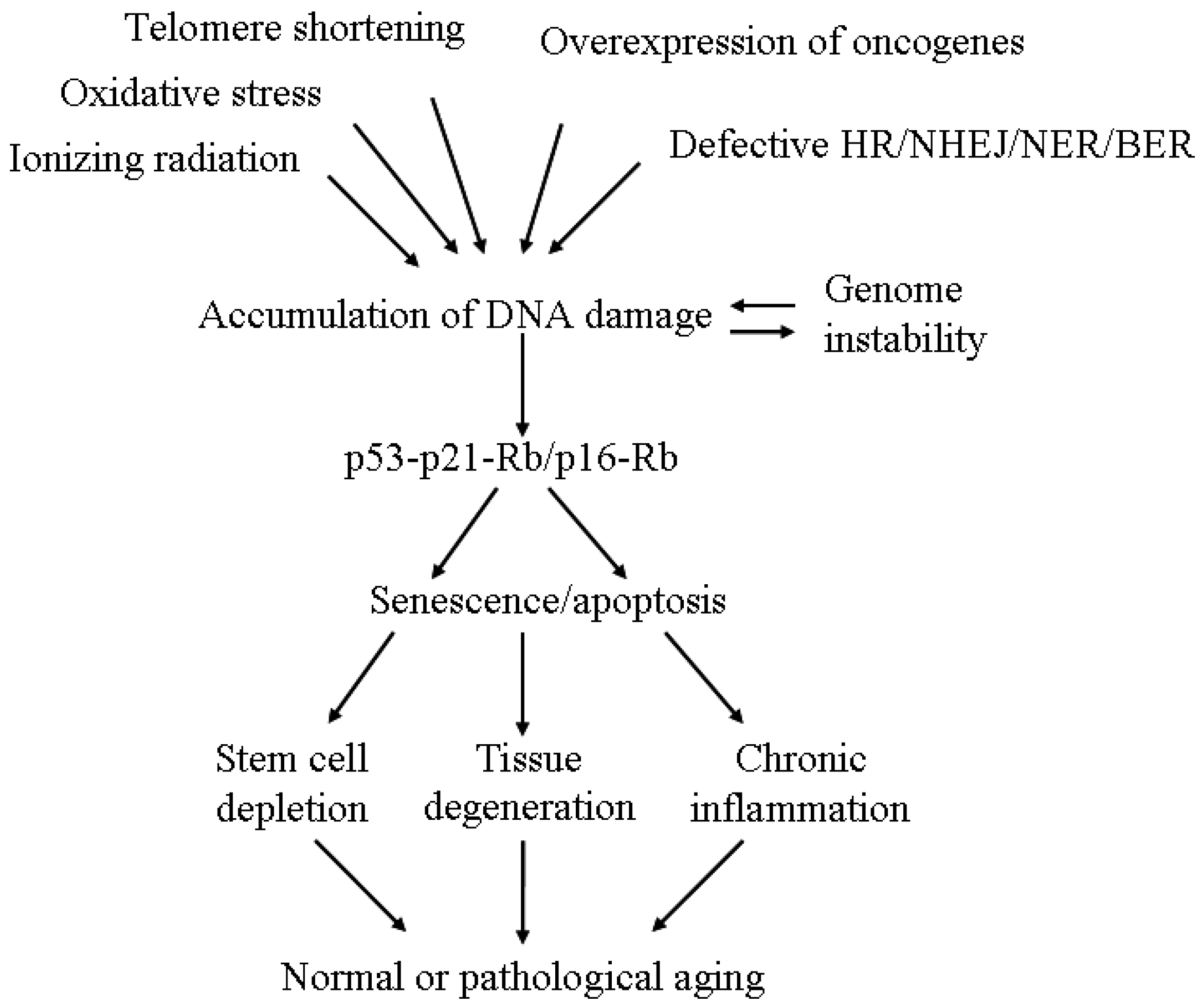{kind=link}
| Disease | Mutated Genes | Repair Pathways Affected |
|---|---|---|
| Breast cancer; ovarian cancer | BRCA1; BRCA2; MRE11 | HR |
| Ataxia telangiectasia | ATM | DSB repair |
| Nijmegen breakage syndrome | NBS1 | DSB repair; telomere maintenance |
| Bloom syndrome | BLM | Mitotic recombination |
| Fanconi anemia | FANC; BRCA2 | DNA crosslink repair |
| Breast cancer; sarcoma; brain cancer; adenocotical carcinoma | P53 | HR; BER; NER; NHEJ |
| Cockayne syndrome | CSA; CSB | TC-NER; GG-NER |
| Trichothiodystrophy | XPB; XPD; XPG | TC-NER; GG-NER |
| Hutchison-Gilford progeria syndrome | LMNA | Nuclear lamina function |
| Xeroderma pigmentosum | XPC | GG-NER |
| Werner syndrome | WRN | telomere maintenance; DNA recombination repair |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, M.-R.; Li, K.; Lin, S.-Y.; Hung, W.-C. Connecting the Dots: From DNA Damage and Repair to Aging. Int. J. Mol. Sci. 2016, 17, 685. https://doi.org/10.3390/ijms17050685
Pan M-R, Li K, Lin S-Y, Hung W-C. Connecting the Dots: From DNA Damage and Repair to Aging. International Journal of Molecular Sciences. 2016; 17(5):685. https://doi.org/10.3390/ijms17050685
Chicago/Turabian StylePan, Mei-Ren, Kaiyi Li, Shiaw-Yih Lin, and Wen-Chun Hung. 2016. "Connecting the Dots: From DNA Damage and Repair to Aging" International Journal of Molecular Sciences 17, no. 5: 685. https://doi.org/10.3390/ijms17050685
APA StylePan, M.-R., Li, K., Lin, S.-Y., & Hung, W.-C. (2016). Connecting the Dots: From DNA Damage and Repair to Aging. International Journal of Molecular Sciences, 17(5), 685. https://doi.org/10.3390/ijms17050685