
Deficiency of Senescence Marker Protein 30 Exacerbates Cardiac Injury after Ischemia/Reperfusion

 and
and
Abstract
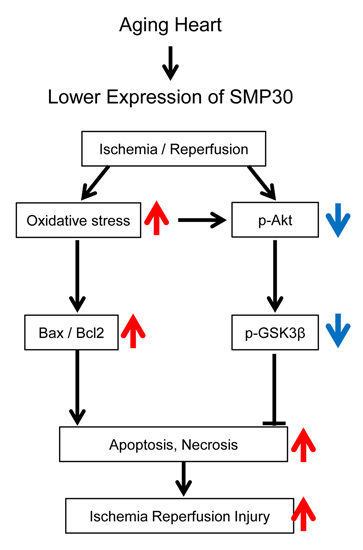
:
1. Introduction
2. Results
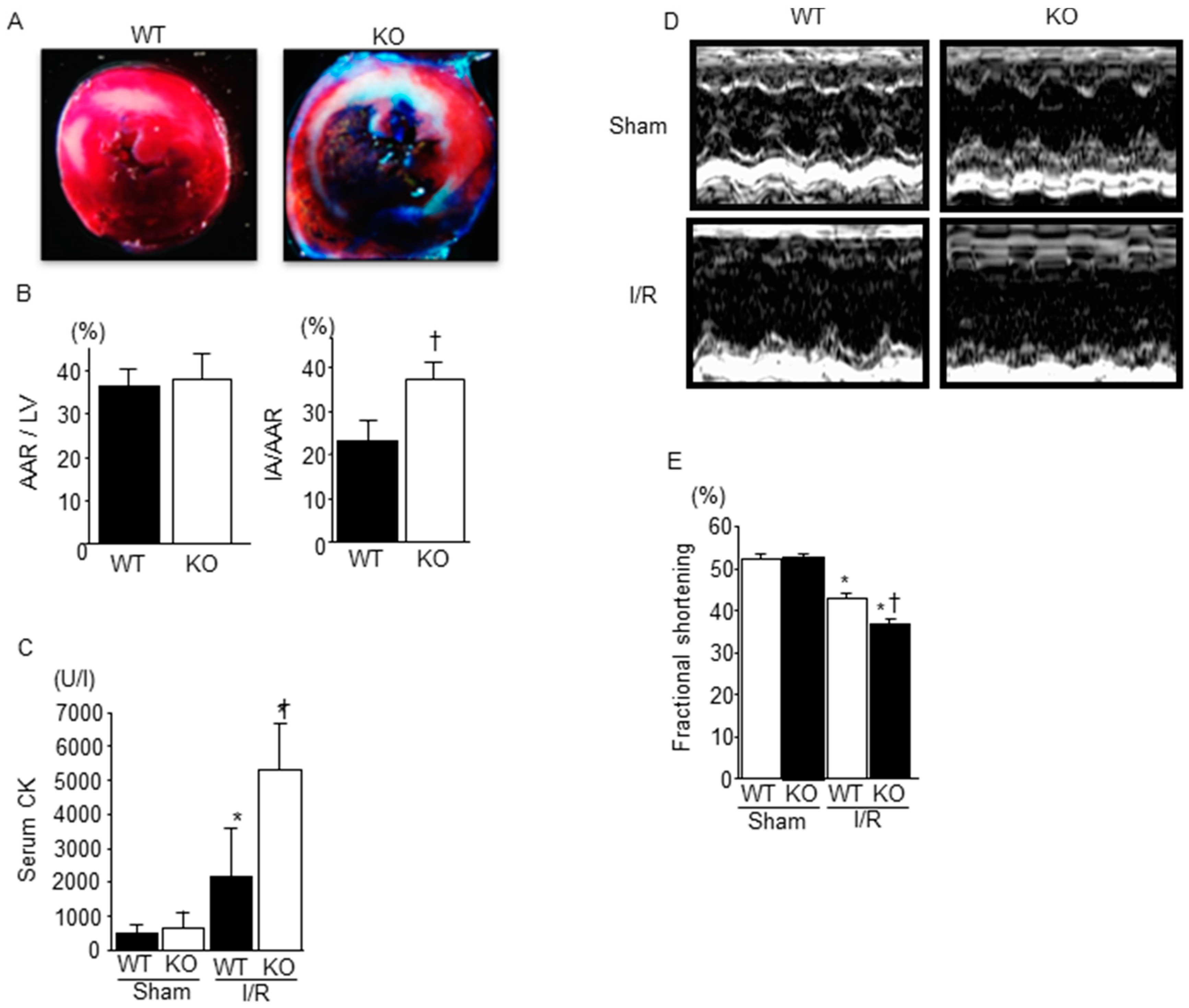
2.1. Deficiency of SMP30 Exacerbates Infarct Size Induced by I/R
2.2. Cardiac Function after I/R Was Exacerbated in SMP30 KO Mice Compared with WT Mice
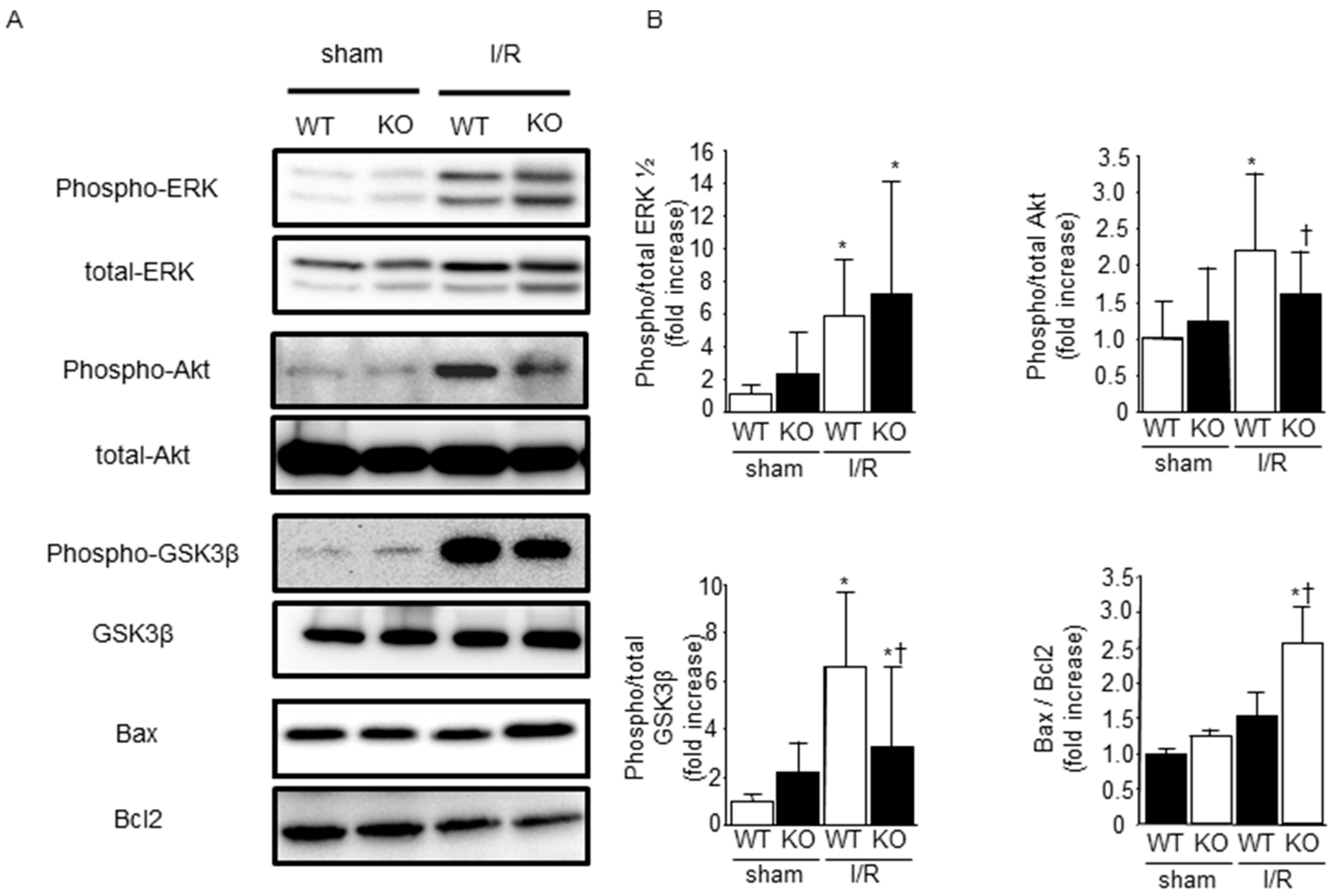
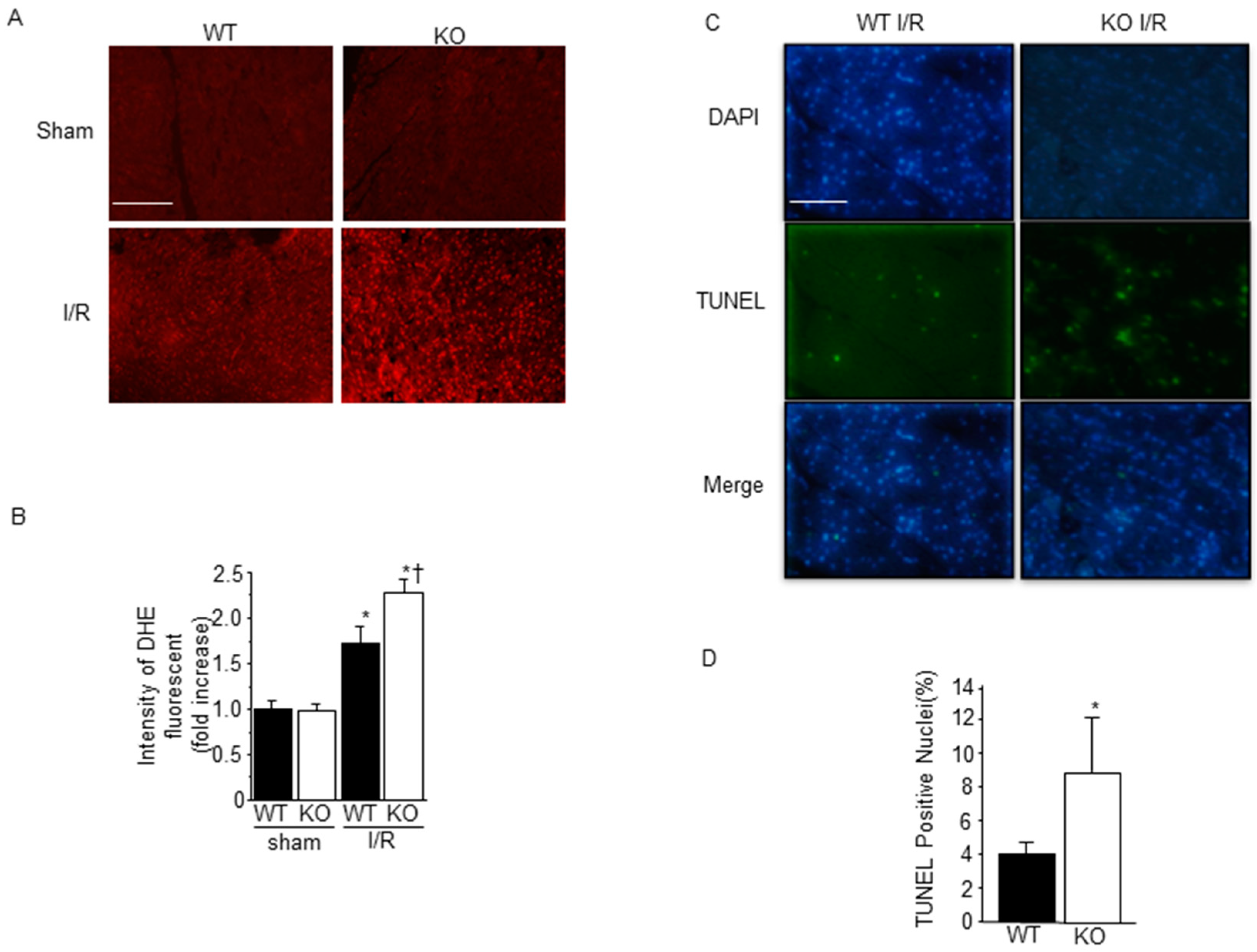
2.3. Effect of SMP30 Deficiency on Oxidative Stress and Myocardial Apoptosis Induced by I/R
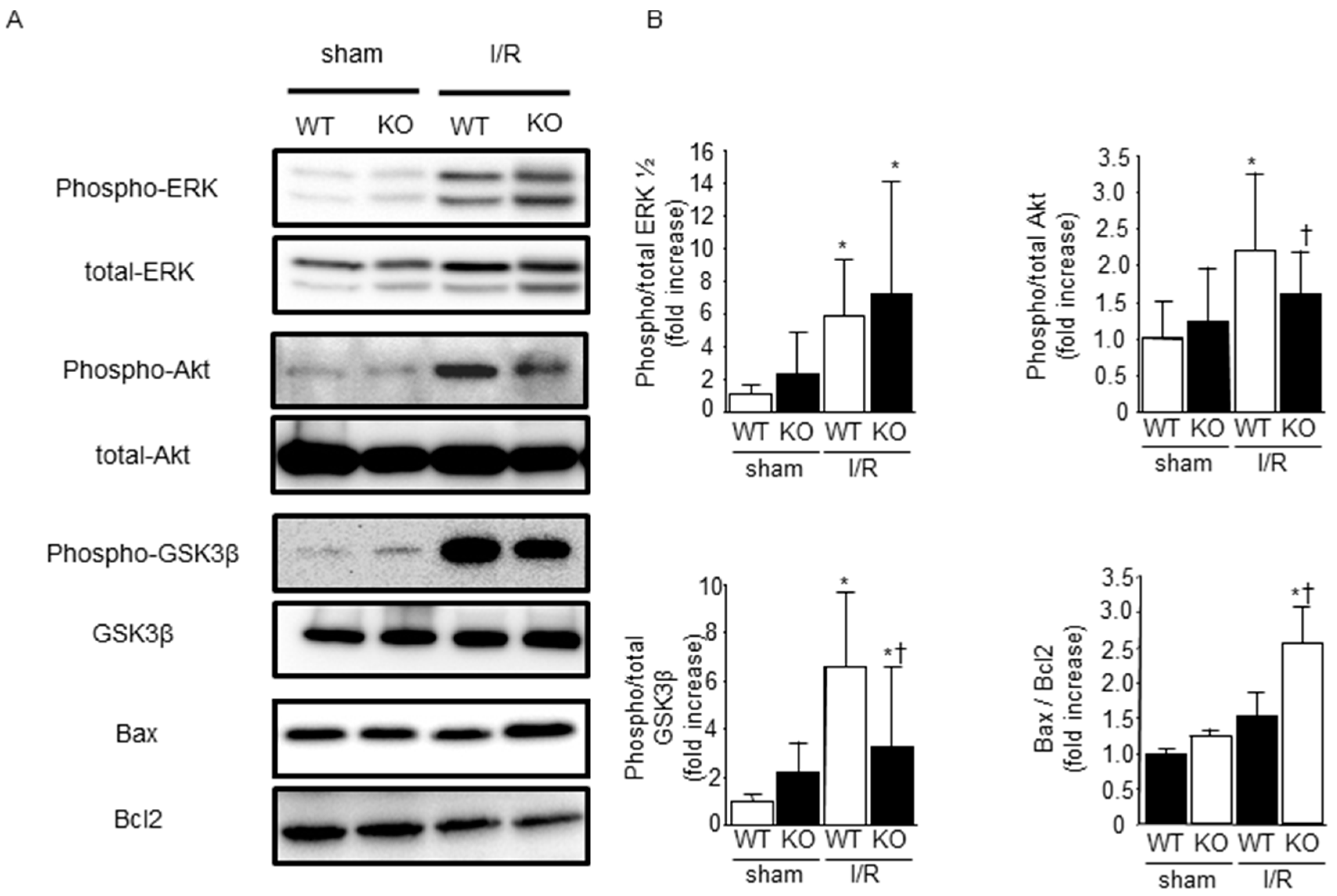
2.4. Phosphorylation of ERK1/2, Akt, and Glycogen Synthase Kinase-3β (GSK-3β) after I/R
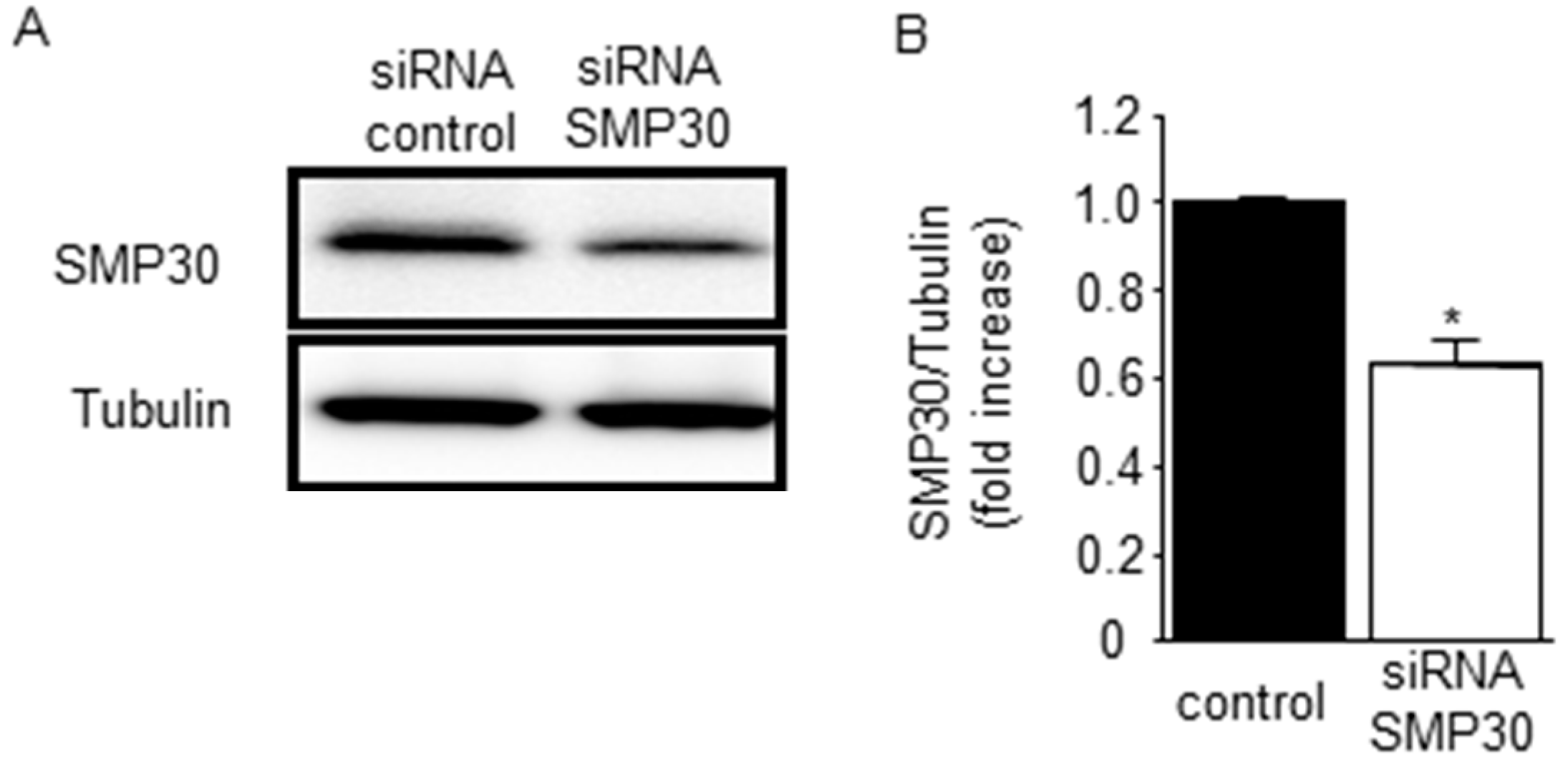
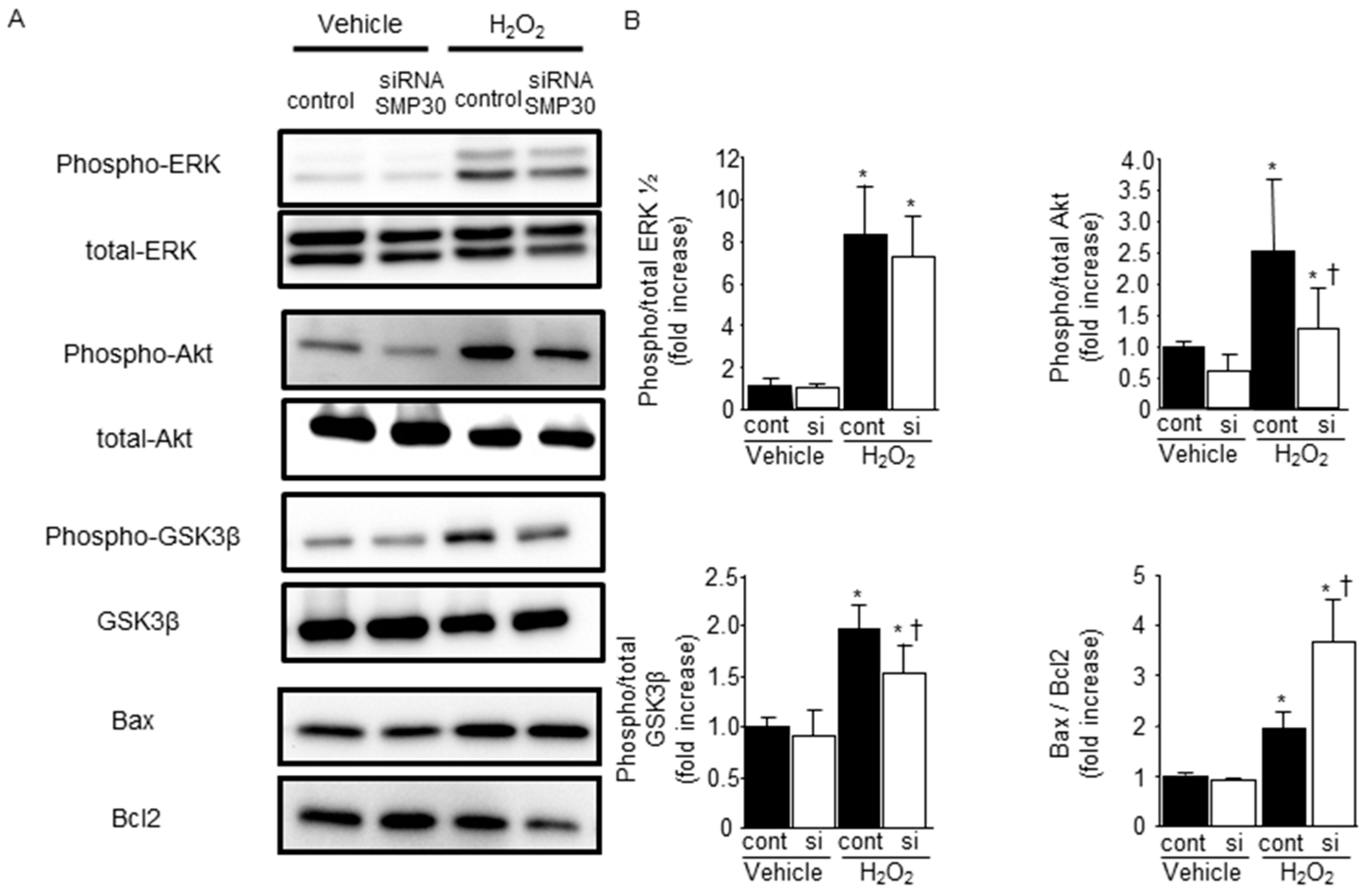
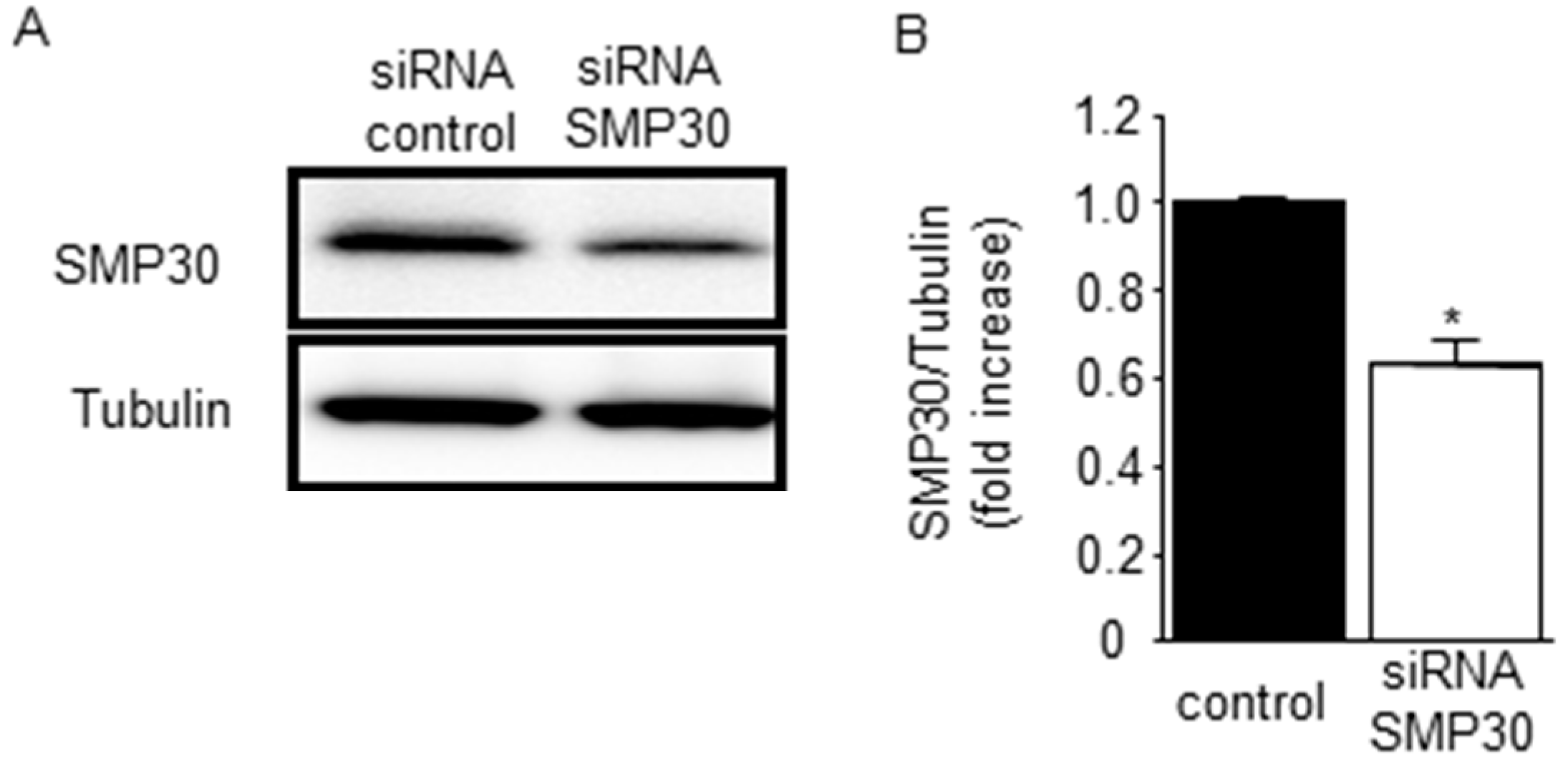
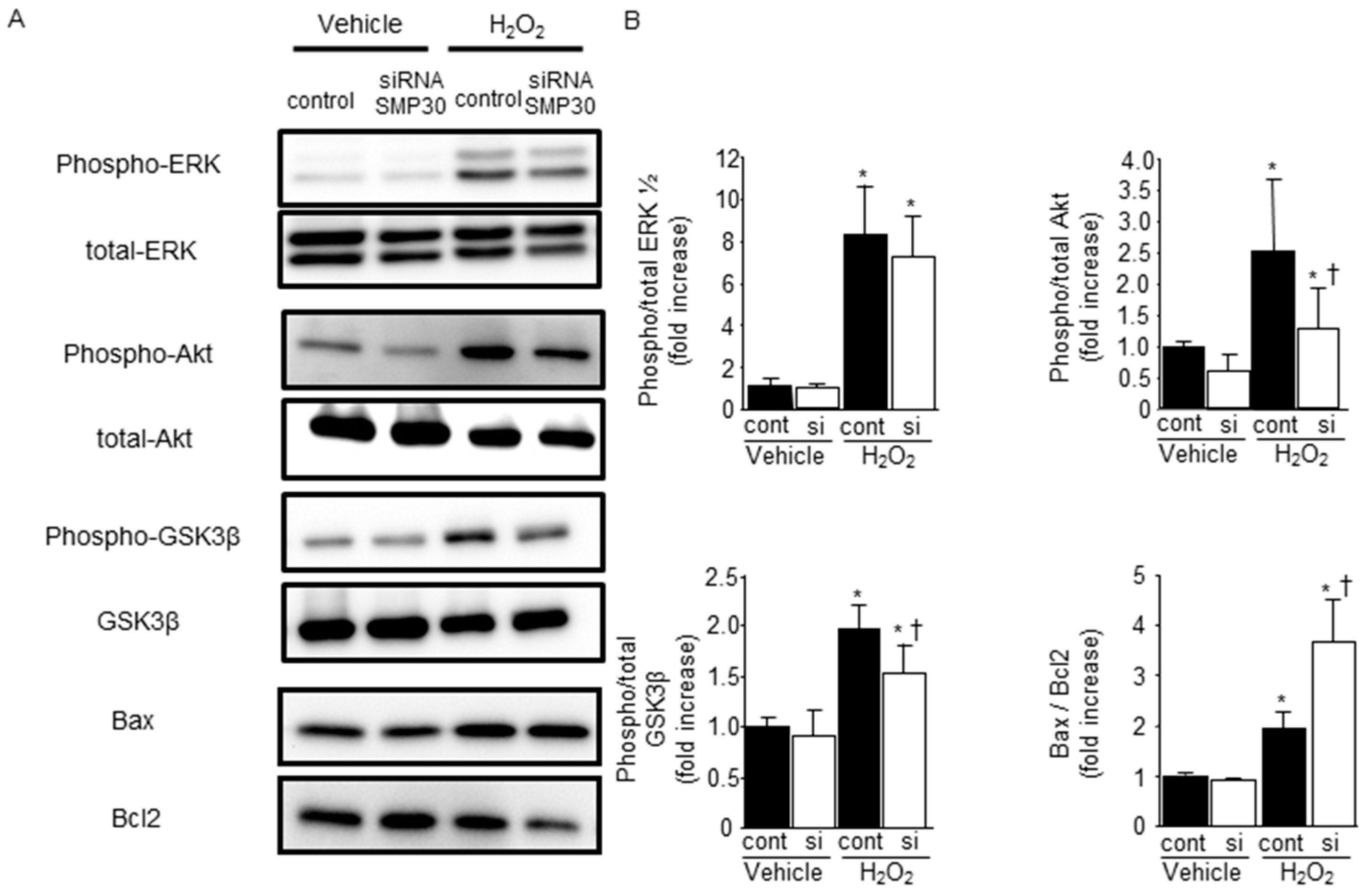
2.5. SMP30 Silencing on ERK1/2, Akt, and GSK-3β Phosphorylation in Cardiomyocytes after H2O2 Stimulation
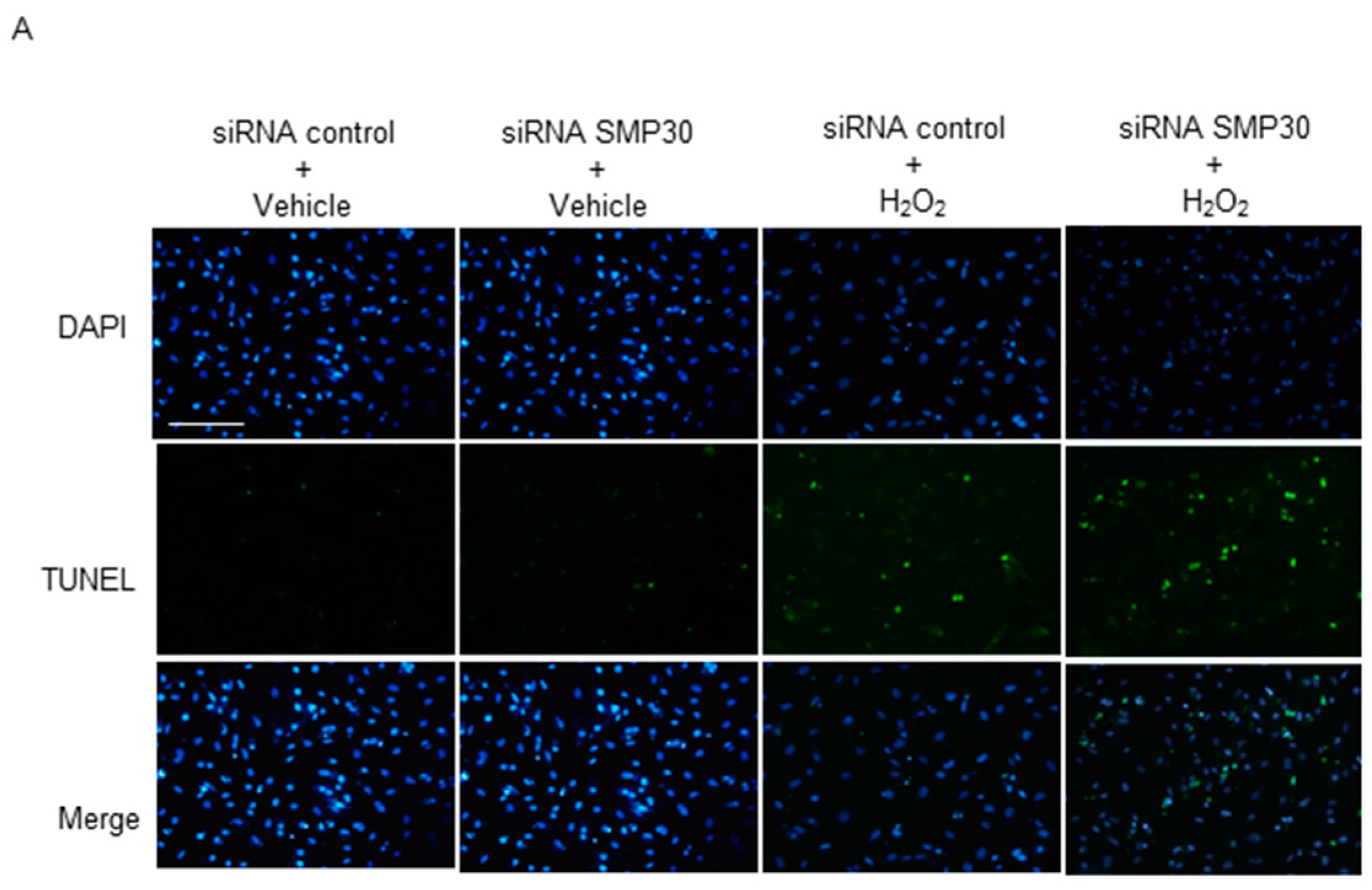
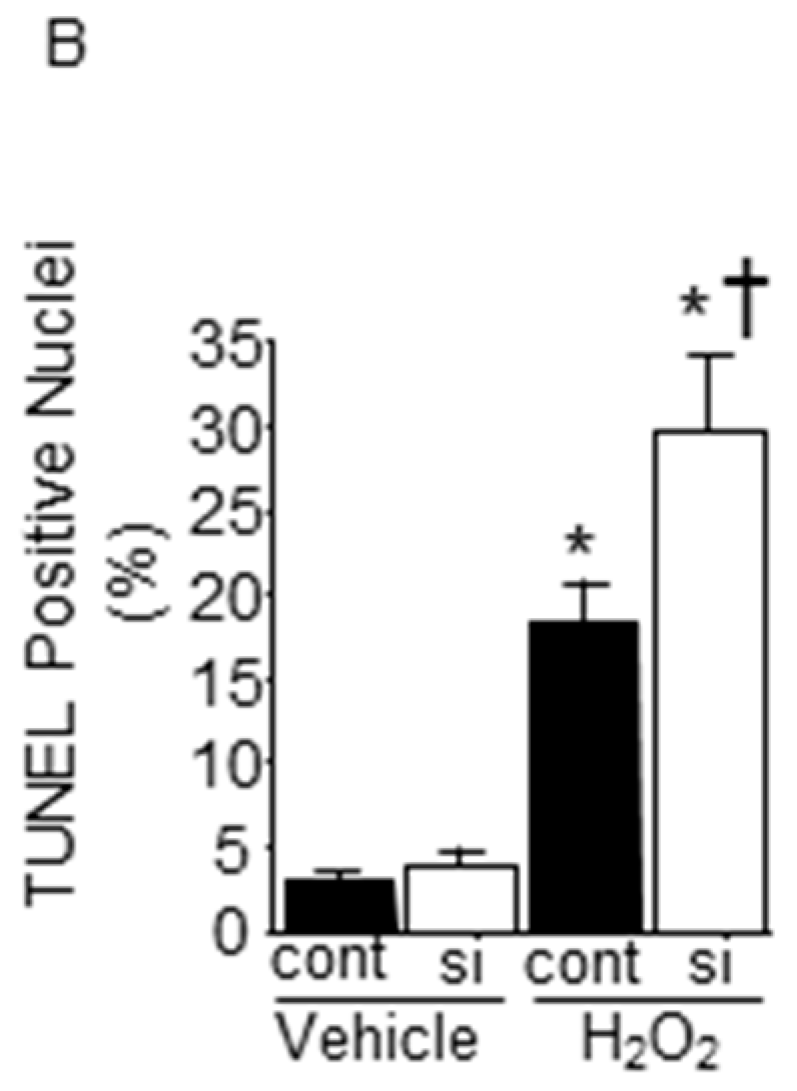
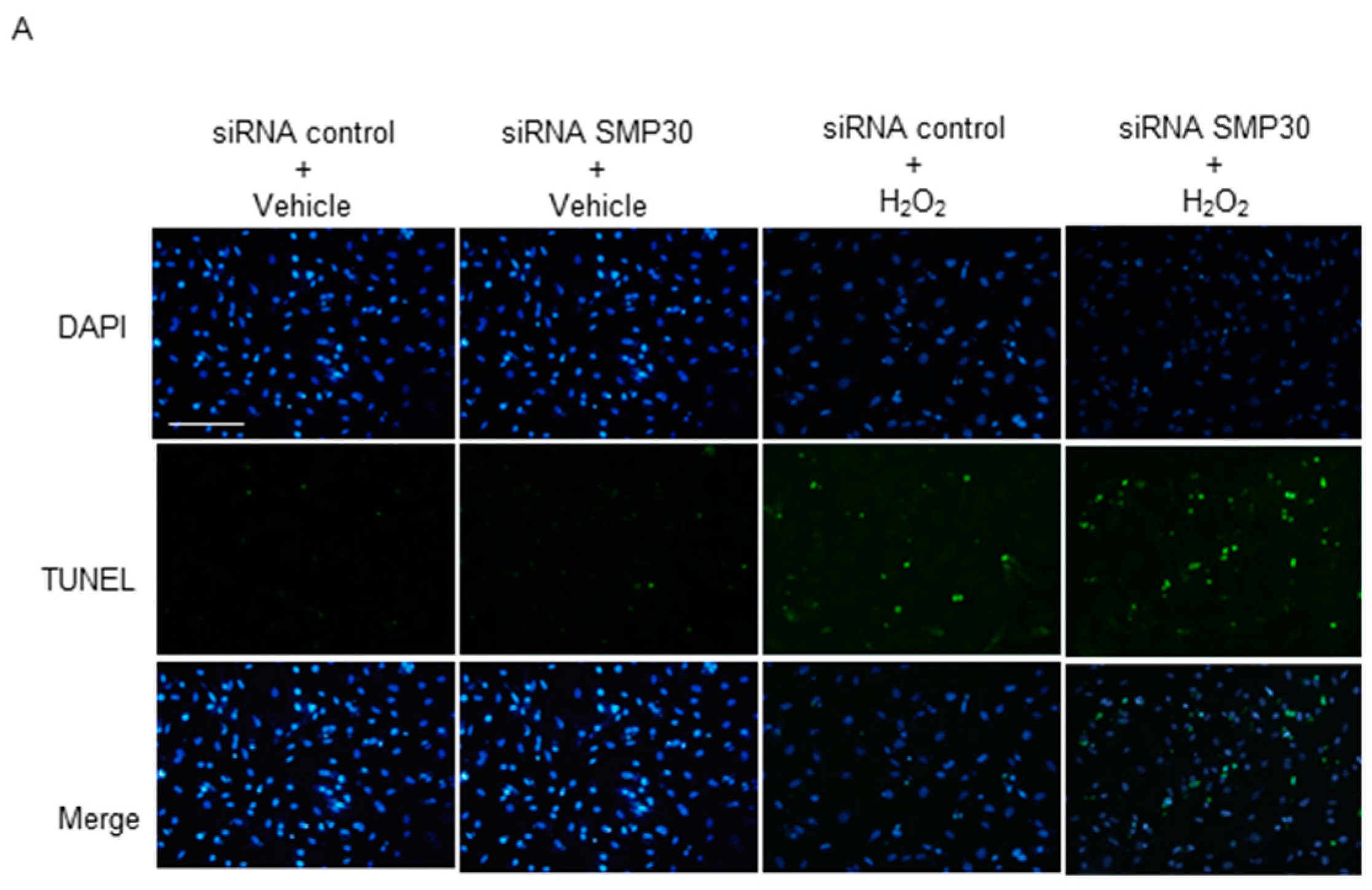
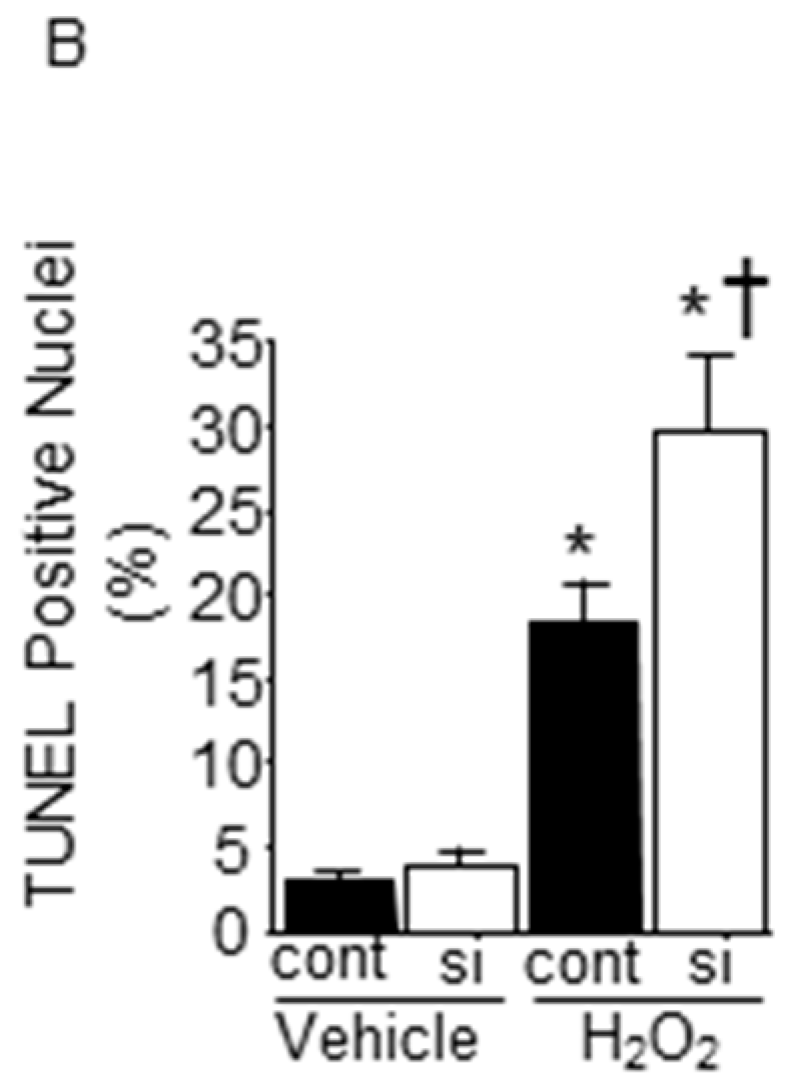
2.6. Impact of Silencing of SMP30 Expression on Cardiomyocyte Apoptosis
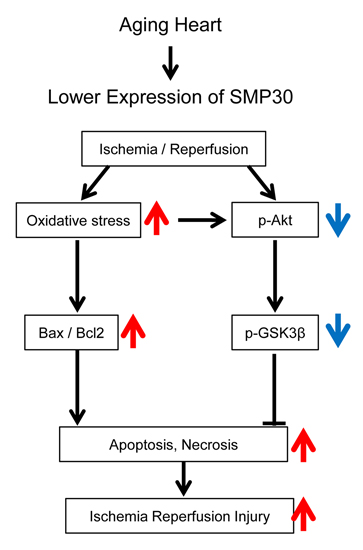
3. Discussion
4. Experimental Section
4.1. Animal Protocol
4.2. Echocardiography
4.3. Heart Surgery of I/R Model
4.4. Western Blot Analysis
4.5. Detection of Apoptosis
4.6. Assessment of Superoxide Generation
4.7. Cultured Neonatal Rat Cardiomyocytes
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Antman, E.M.; Hand, M.; Armstrong, P.W.; Bates, E.R.; Green, L.A.; Halasyamani, L.K.; Hochman, J.S.; Krumholz, H.M.; Lamas, G.A.; Mullany, C.J.; et al. 2007 focused update of the acc/aha 2004 guidelines for the management of patients with st-elevation myocardial infarction: A report of the american college of cardiology/american heart association task force on practice guidelines: Developed in collaboration with the canadian cardiovascular society endorsed by the american academy of family physicians: 2007 writing group to review new evidence and update the acc/aha 2004 guidelines for the management of patients with st-elevation myocardial infarction, writing on behalf of the 2004 writing committee. Circulation 2008, 117, 296–329. [Google Scholar] [PubMed]
- Cannon, C.P.; Gibson, C.M.; Lambrew, C.T.; Shoultz, D.A.; Levy, D.; French, W.J.; Gore, J.M.; Weaver, W.D.; Rogers, W.J.; Tiefenbrunn, A.J. Relationship of symptom-onset-to-balloon time and door-to-balloon time with mortality in patients undergoing angioplasty for acute myocardial infarction. JAMA 2000, 283, 2941–2947. [Google Scholar] [CrossRef] [PubMed]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Elsasser, A.; Suzuki, K.; Lorenz-Meyer, S.; Bode, C.; Schaper, J. The role of apoptosis in myocardial ischemia: A critical appraisal. Basic Res. Cardiol. 2001, 96, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Eefting, F.; Rensing, B.; Wigman, J.; Pannekoek, W.J.; Liu, W.M.; Cramer, M.J.; Lips, D.J.; Doevendans, P.A. Role of apoptosis in reperfusion injury. Cardiovasc. Res. 2004, 61, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.Q.; Morris, C.D.; Budde, J.M.; Wang, N.P.; Muraki, S.; Sun, H.Y.; Guyton, R.A. Inhibition of myocardial apoptosis reduces infarct size and improves regional contractile dysfunction during reperfusion. Cardiovasc. Res. 2003, 59, 132–142. [Google Scholar] [CrossRef]
- Garcia-Dorado, D.; Garcia-del-Blanco, B.; Otaegui, I.; Rodriguez-Palomares, J.; Pineda, V.; Gimeno, F.; Ruiz-Salmeron, R.; Elizaga, J.; Evangelista, A.; Fernandez-Aviles, F.; et al. Intracoronary injection of adenosine before reperfusion in patients with st-segment elevation myocardial infarction: A randomized controlled clinical trial. Int. J. Cardiol. 2014, 177, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Antman, E.M. Early administration of intravenous magnesium to high-risk patients with acute myocardial infarction in the magnesium in coronaries (magic) trial: A randomised controlled trial. Lancet 2002, 360, 1189–1196. [Google Scholar] [CrossRef]
- Boissel, J.-P. Effect of 48-h intravenous trimetazidine on short- and long-term outcomes of patients with acute myocardial infarction, with and without thrombolytic therapy. Eur. Heart J. 2000, 21, 1537–1546. [Google Scholar]
- Shiba, N.; Shimokawa, H. Chronic heart failure in japan: Implications of the chart studies. Vasc. Health Risk Manag. 2008, 4, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, H.; Yajima, J.; Oikawa, Y.; Tanaka, S.; Fukamachi, D.; Suzuki, S.; Sagara, K.; Otsuka, T.; Matsuno, S.; Funada, R.; et al. Impact of aging on the clinical outcomes of japanese patients with coronary artery disease after percutaneous coronary intervention. Heart Vessels 2014, 29, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Pepe, S. Mitochondrial function in ischaemia and reperfusion of the ageing heart. Clin. Exp. Pharmacol. Physiol. 2000, 27, 745–750. [Google Scholar] [CrossRef] [PubMed]
- Lesnefsky, E.J.; Moghaddas, S.; Tandler, B.; Kerner, J.; Hoppel, C.L. Mitochondrial dysfunction in cardiac disease: Ischemia--reperfusion, aging, and heart failure. J. Mol. Cell. Cardiol. 2001, 33, 1065–1089. [Google Scholar] [CrossRef] [PubMed]
- Besse, S.; Delcayre, C.; Chevalier, B.; Hardouin, S.; Heymes, C.; Bourgeois, F.; Moalic, J.M.; Swynghedauw, B. Is the senescent heart overloaded and already failing? Cardiovasc. Drugs Ther. 1994, 8, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Di Lisa, F.; Bernardi, P. Mitochondria and ischemia-reperfusion injury of the heart: Fixing a hole. Cardiovasc. Res. 2006, 70, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Pepe, S. Dysfunctional ischemic preconditioning mechanisms in aging. Cardiovasc. Res. 2001, 49, 11–14. [Google Scholar] [CrossRef]
- Misaka, T.; Suzuki, S.; Miyata, M.; Kobayashi, A.; Shishido, T.; Ishigami, A.; Saitoh, S.; Hirose, M.; Kubota, I.; Takeishi, Y. Deficiency of senescence marker protein 30 exacerbates angiotensin ii-induced cardiac remodelling. Cardiovasc. Res. 2013, 99, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Mandel, J.L.; Shirasawa, T.; Hino, O.; Shirai, T.; Maruyama, N. Isolation of cdna clone encoding human homologue of senescence marker protein-30 (smp30) and its location on the x chromosome. Biochim. Biophys. Acta 1995, 1263, 249–252. [Google Scholar] [CrossRef]
- Fujita, T.; Uchida, K.; Maruyama, N. Purification of senescence marker protein-30 (smp30) and its androgen-independent decrease with age in the rat liver. Biochim. Biophys. Acta 1992, 1116, 122–128. [Google Scholar] [CrossRef]
- Izumi, T.; Yamaguchi, M. Overexpression of regucalcin suppresses cell death and apoptosis in cloned rat hepatoma h4-ii-e cells induced by lipopolysaccharide, pd 98059, dibucaine, or bay k 8644. J. Cell. Biochem. 2004, 93, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Izumi, T.; Yamaguchi, M. Overexpression of regucalcin suppresses cell death in cloned rat hepatoma h4-ii-e cells induced by tumor necrosis factor-alpha or thapsigargin. J. Cell. Biochem. 2004, 92, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Yumura, W.; Imasawa, T.; Suganuma, S.; Ishigami, A.; Handa, S.; Kubo, S.; Joh, K.; Maruyama, N. Accelerated tubular cell senescence in smp30 knockout mice. Histol. Histopathol. 2006, 21, 1151–1156. [Google Scholar] [PubMed]
- Ishigami, A.; Kondo, Y.; Nanba, R.; Ohsawa, T.; Handa, S.; Kubo, S.; Akita, M.; Maruyama, N. Smp30 deficiency in mice causes an accumulation of neutral lipids and phospholipids in the liver and shortens the life span. Biochem. Biophys. Res. Commun. 2004, 315, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Miyata, M.; Suzuki, S.; Misaka, T.; Shishido, T.; Saitoh, S.; Ishigami, A.; Kubota, I.; Takeishi, Y. Senescence marker protein 30 has a cardio-protective role in doxorubicin-induced cardiac dysfunction. PLoS ONE 2013, 8, e79093. [Google Scholar] [CrossRef] [PubMed]
- Misaka, T.; Suzuki, S.; Miyata, M.; Kobayashi, A.; Ishigami, A.; Shishido, T.; Saitoh, S.; Kubota, I.; Takeishi, Y. Senescence marker protein 30 inhibits angiotensin ii-induced cardiac hypertrophy and diastolic dysfunction. Biochem. Biophys. Res. Commun. 2013, 439, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Shishido, T.; Kadowaki, S.; Kitahara, T.; Suzuki, S.; Katoh, S.; Funayama, A.; Netsu, S.; Watanabe, T.; Goto, K.; et al. Diacylglycerol kinase alpha exacerbates cardiac injury after ischemia/reperfusion. Heart Vessels 2014, 29, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. Time to take myocardial reperfusion injury seriously. N. Engl. J. Med. 2008, 359, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, A.; Fujita, T.; Handa, S.; Shirasawa, T.; Koseki, H.; Kitamura, T.; Enomoto, N.; Sato, N.; Shimosawa, T.; Maruyama, N. Senescence marker protein-30 knockout mouse liver is highly susceptible to tumor necrosis factor-alpha- and fas-mediated apoptosis. Am. J. Pathol. 2002, 161, 1273–1281. [Google Scholar] [CrossRef]
- Sato, T.; Seyama, K.; Sato, Y.; Mori, H.; Souma, S.; Akiyoshi, T.; Kodama, Y.; Mori, T.; Goto, S.; Takahashi, K.; et al. Senescence marker protein-30 protects mice lungs from oxidative stress, aging, and smoking. Am. J. Respir. Crit. Care Med. 2006, 174, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial ros-induced ros release: An update and review. Biochim. Biophys. Acta 2006, 1757, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, H.; Saitoh, S.; Machii, H.; Yamada, S.; Hoshino, Y.; Misaka, T.; Ishigami, A.; Takeishi, Y. Senescence marker protein-30 (smp30) deficiency impairs myocardium-induced dilation of coronary arterioles associated with reactive oxygen species. Int. J. Mol. Sci. 2013, 14, 9408–9423. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Kishimoto, Y.; Konishi, T.; Fujita, Y.; Ito, M.; Shimokado, K.; Maruyama, N.; Ishigami, A. Ascorbic acid deficiency affects genes for oxidation-reduction and lipid metabolism in livers from smp30/gnl knockout mice. Biochim. Biophys. Acta 2014, 1840, 2289–2298. [Google Scholar] [CrossRef] [PubMed]
- Son, T.G.; Zou, Y.; Jung, K.J.; Yu, B.P.; Ishigami, A.; Maruyama, N.; Lee, J. Smp30 deficiency causes increased oxidative stress in brain. Mech. Ageing Dev. 2006, 127, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.J.; Ishigami, A.; Maruyama, N.; Takahashi, R.; Goto, S.; Yu, B.P.; Chung, H.Y. Modulation of gene expression of smp-30 by lps and calorie restriction during aging process. Exp. Gerontol. 2004, 39, 1169–1177. [Google Scholar] [CrossRef] [PubMed]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Lai, P.; Yip, N.C.; Michelangeli, F. Regucalcin (RGN/SMP30) alters agonist- and thapsigargin-induced cytosolic [ca2+] transients in cells by increasing serca Ca(2+)atpase levels. FEBS Lett. 2011, 585, 2291–2294. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. Reperfusion injury salvage kinase signalling: Taking a risk for cardioprotection. Heart Fail Rev. 2007, 12, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Inai, Y.; Sato, Y.; Handa, S.; Kubo, S.; Shimokado, K.; Goto, S.; Nishikimi, M.; Maruyama, N.; Ishigami, A. Senescence marker protein 30 functions as gluconolactonase in l-ascorbic acid biosynthesis, and its knockout mice are prone to scurvy. Proc. Natl. Acad. Sci. USA 2006, 103, 5723–5728. [Google Scholar] [CrossRef] [PubMed]
- Funayama, A.; Shishido, T.; Netsu, S.; Narumi, T.; Kadowaki, S.; Takahashi, H.; Miyamoto, T.; Watanabe, T.; Woo, C.H.; Abe, J.I.; et al. Cardiac nuclear high mobility group box 1 prevents the development of cardiac hypertrophy and heart failure. Cardiovasc. Res. 2013, 99, 657–654. [Google Scholar] [CrossRef] [PubMed]
- Netsu, S.; Shishido, T.; Kitahara, T.; Honda, Y.; Funayama, A.; Narumi, T.; Kadowaki, S.; Takahashi, H.; Miyamoto, T.; Arimoto, T.; et al. Midkine exacerbates pressure overload-induced cardiac remodeling. Biochem. Biophys. Res. Commun. 2014, 443, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Narumi, T.; Shishido, T.; Otaki, Y.; Kadowaki, S.; Honda, Y.; Funayama, A.; Honda, S.; Hasegawa, H.; Kinoshita, D.; Yokoyama, M.; et al. High-mobility group box 1-mediated heat shock protein beta 1 expression attenuates mitochondrial dysfunction and apoptosis. J. Mol. Cell. Cardiol. 2015, 82, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shishido, T.; Nozaki, N.; Takahashi, H.; Arimoto, T.; Niizeki, T.; Koyama, Y.; Abe, J.; Takeishi, Y.; Kubota, I. Central role of endogenous toll-like receptor-2 activation in regulating inflammation, reactive oxygen species production, and subsequent neointimal formation after vascular injury. Biochem. Biophys. Res. Commun. 2006, 345, 1446–1453. [Google Scholar] [CrossRef] [PubMed]
- Shishido, T.; Woo, C.H.; Ding, B.; McClain, C.; Molina, C.A.; Yan, C.; Yang, J.; Abe, J. Effects of Mek5/Erk5 association on small ubiquitin-related modification of erk5: Implications for diabetic ventricular dysfunction after myocardial infarction. Circ. Res. 2008, 102, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Honda, Y.; Shishido, T.; Takahashi, T.; Watanabe, T.; Netsu, S.; Kinoshita, D.; Narumi, T.; Kadowaki, S.; Nishiyama, S.; Takahashi, H.; et al. Midkine deteriorates cardiac remodeling via epidermal growth factor receptor signaling in chronic kidney disease. Hypertension 2016. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Echocardiografic Parameter | WT Sham | SMP30 KO Sham | WT I/R | SMP30 KO I/R |
|---|---|---|---|---|
| IVSd (mm) | 0.75 ± 0.05 | 0.73 ± 0.06 | 0.73 ± 0.05 | 0.73 ± 0.04 |
| LVPWd (mm) | 0.71 ± 0.03 | 0.72 ± 0.07 | 0.74 ± 0.05 | 0.71 ± 0.04 |
| LVEDd (mm) | 3.01 ± 0.11 | 3.08 ± 0.08 | 2.92 ± 0.31 | 2.87 ± 0.32 |
| LVESd (mm) | 1.42 ± 0.07 | 1.45 ± 0.05 | 1.65 ± 0.28 ** | 1.81 ± 0.30 **,# |
| LVFS (%) | 52.8 ± 2.1 | 53.0 ± 1.6 | 43.4 ± 5.1 ** | 37.3 ± 4.3 **,# |
| HR (bpm) | 499 ± 37 | 520 ± 44 | 478 ± 46 | 509 ± 25 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kadowaki, S.; Shishido, T.; Sasaki, T.; Sugai, T.; Narumi, T.; Honda, Y.; Otaki, Y.; Kinoshita, D.; Takahashi, T.; Nishiyama, S.; et al. Deficiency of Senescence Marker Protein 30 Exacerbates Cardiac Injury after Ischemia/Reperfusion. Int. J. Mol. Sci. 2016, 17, 542. https://doi.org/10.3390/ijms17040542
Kadowaki S, Shishido T, Sasaki T, Sugai T, Narumi T, Honda Y, Otaki Y, Kinoshita D, Takahashi T, Nishiyama S, et al. Deficiency of Senescence Marker Protein 30 Exacerbates Cardiac Injury after Ischemia/Reperfusion. International Journal of Molecular Sciences. 2016; 17(4):542. https://doi.org/10.3390/ijms17040542
Chicago/Turabian StyleKadowaki, Shinpei, Tetsuro Shishido, Toshiki Sasaki, Takayuki Sugai, Taro Narumi, Yuki Honda, Yoichiro Otaki, Daisuke Kinoshita, Tetsuya Takahashi, Satoshi Nishiyama, and et al. 2016. "Deficiency of Senescence Marker Protein 30 Exacerbates Cardiac Injury after Ischemia/Reperfusion" International Journal of Molecular Sciences 17, no. 4: 542. https://doi.org/10.3390/ijms17040542
APA StyleKadowaki, S., Shishido, T., Sasaki, T., Sugai, T., Narumi, T., Honda, Y., Otaki, Y., Kinoshita, D., Takahashi, T., Nishiyama, S., Takahashi, H., Arimoto, T., Miyamoto, T., Watanabe, T., Ishigami, A., Takeishi, Y., & Kubota, I. (2016). Deficiency of Senescence Marker Protein 30 Exacerbates Cardiac Injury after Ischemia/Reperfusion. International Journal of Molecular Sciences, 17(4), 542. https://doi.org/10.3390/ijms17040542