
Anti-Inflammatory and Cytoprotective Effects of TMC-256C1 from Marine-Derived Fungus Aspergillus sp. SF-6354 via up-Regulation of Heme Oxygenase-1 in Murine Hippocampal and Microglial Cell Lines

Abstract
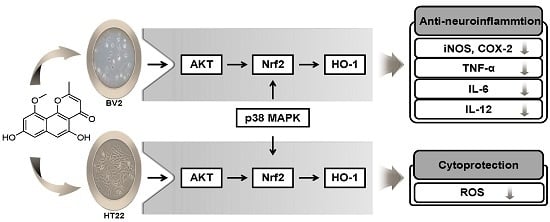
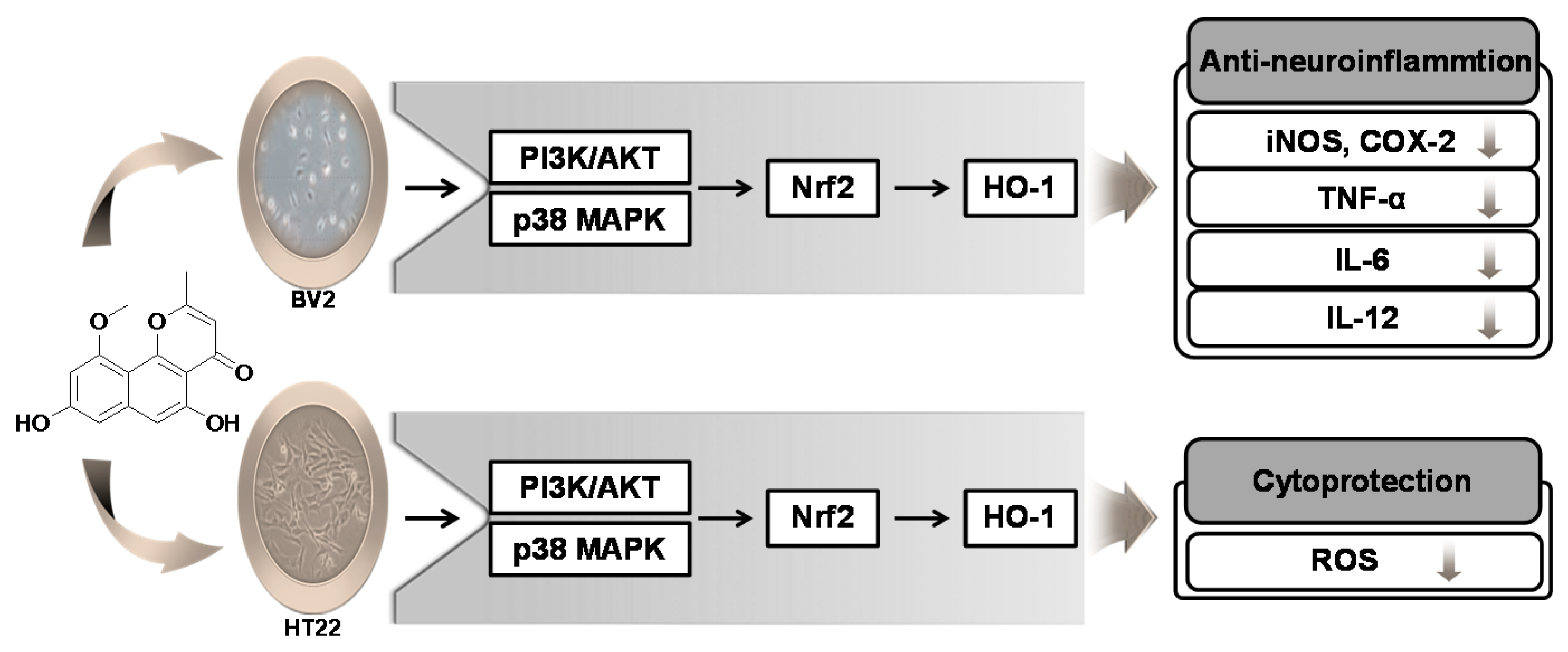
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
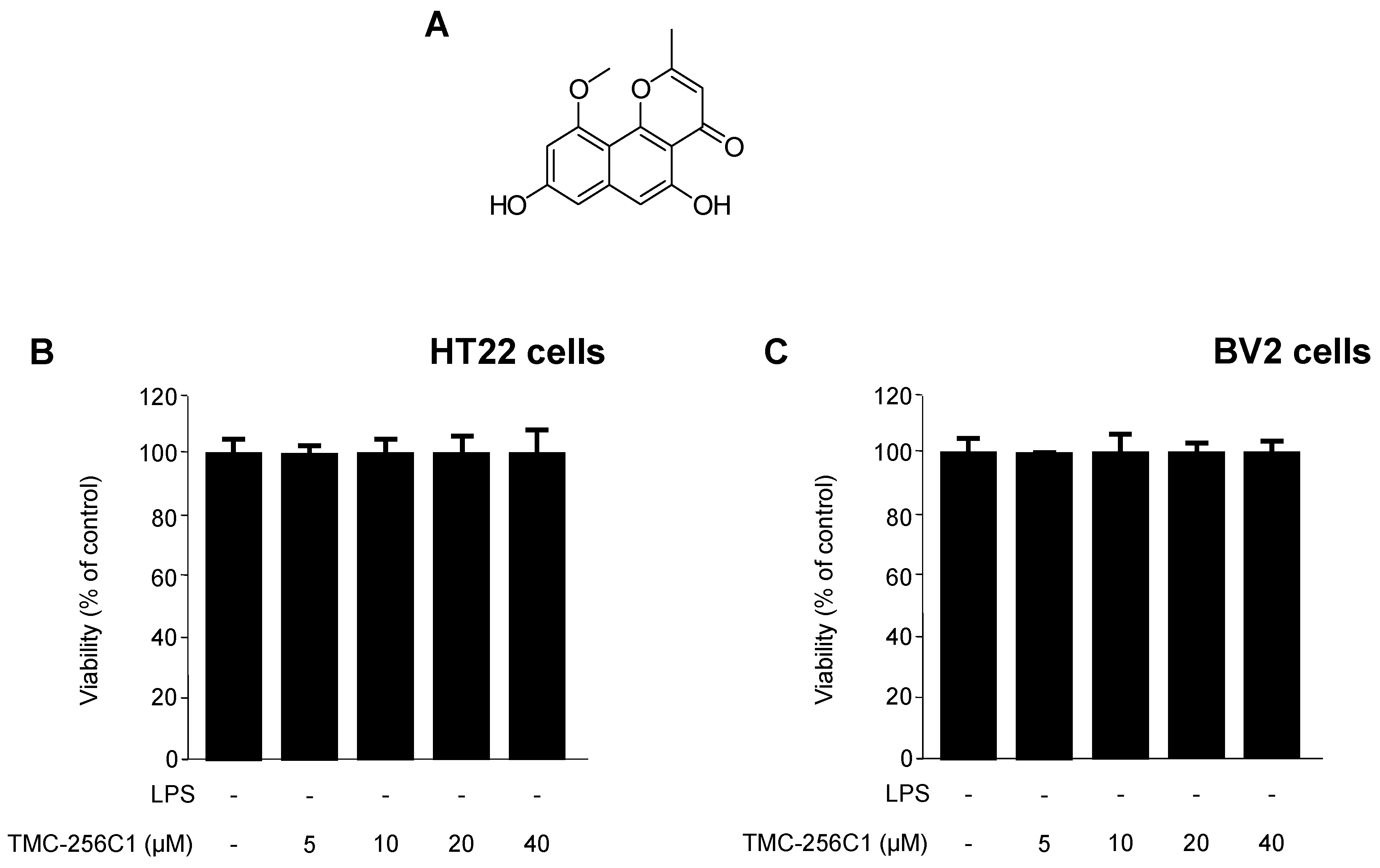
2.1. Structure Determination and Isolation of TMC-256C1, and Effect of TMC-256C1 on the Viability of HT22 and BV2 Cells
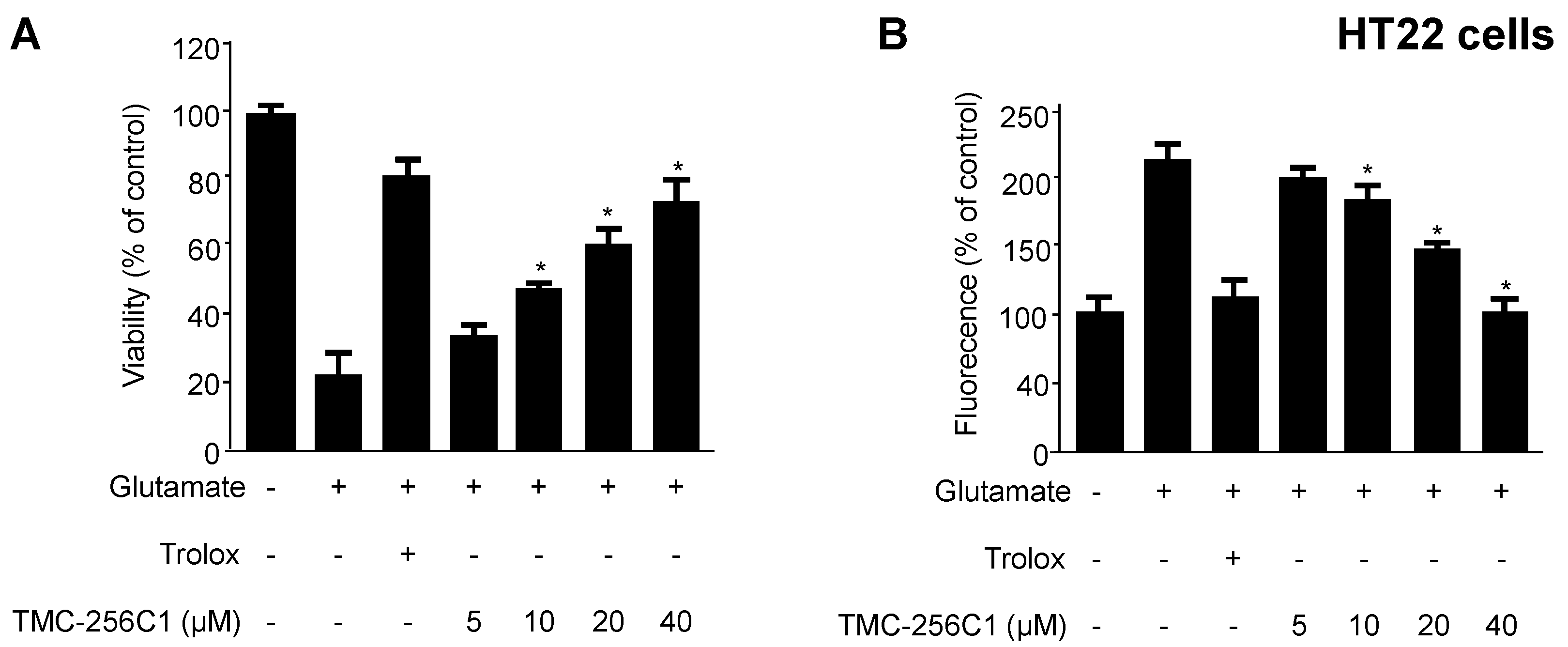
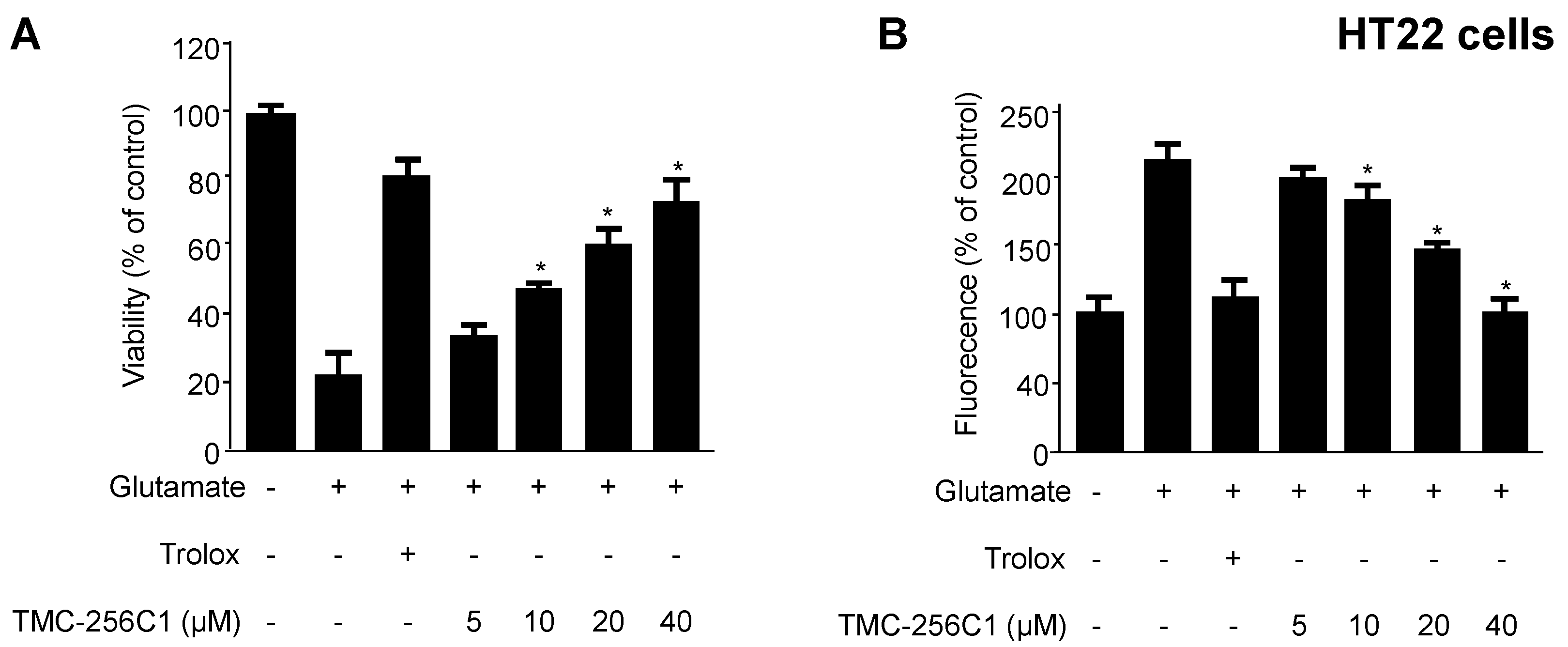
2.2. Effects of TMC-256C1 on Glutamate-Induced Cytotoxicity and Reactive Oxygen Species Generation
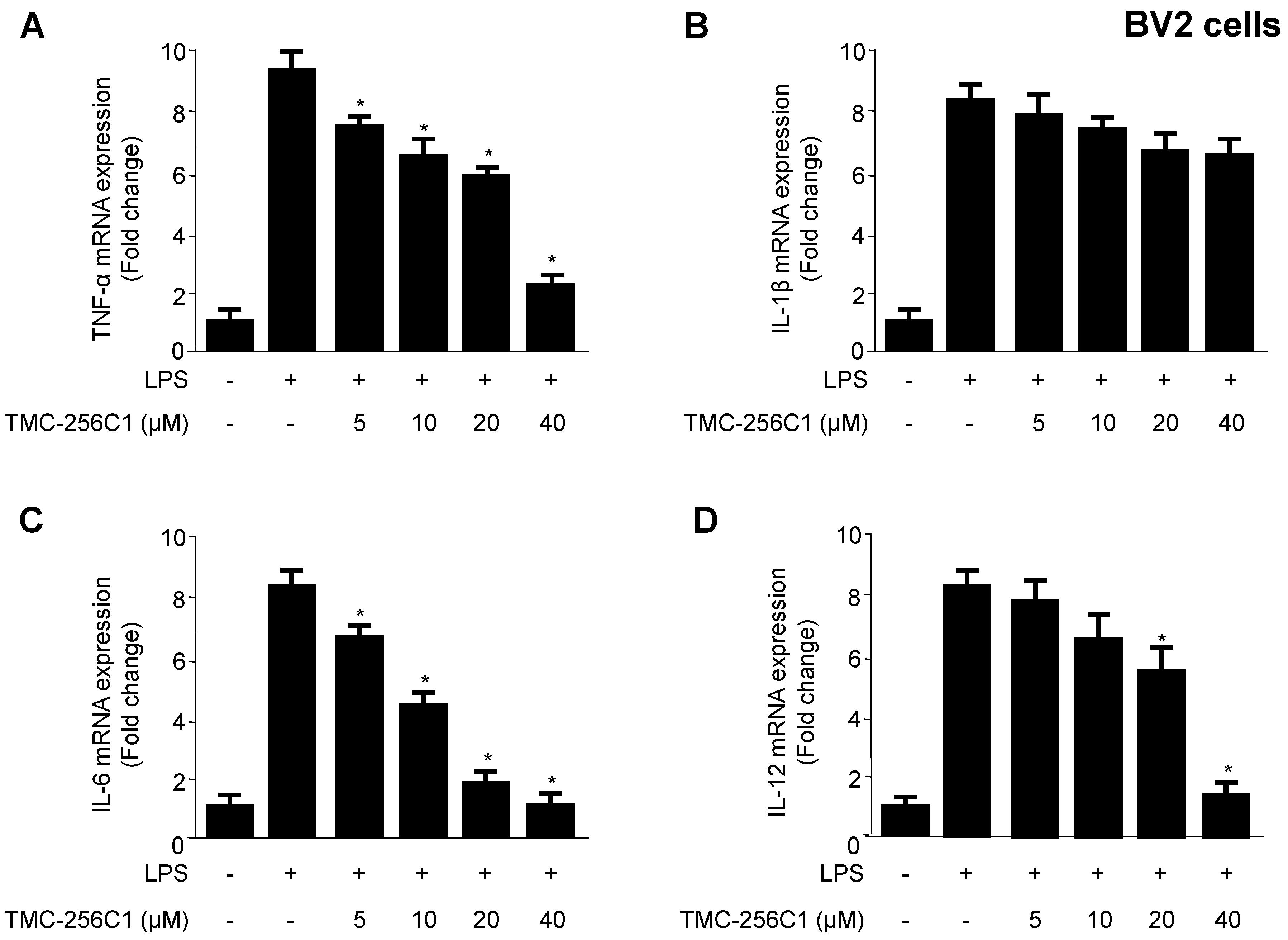
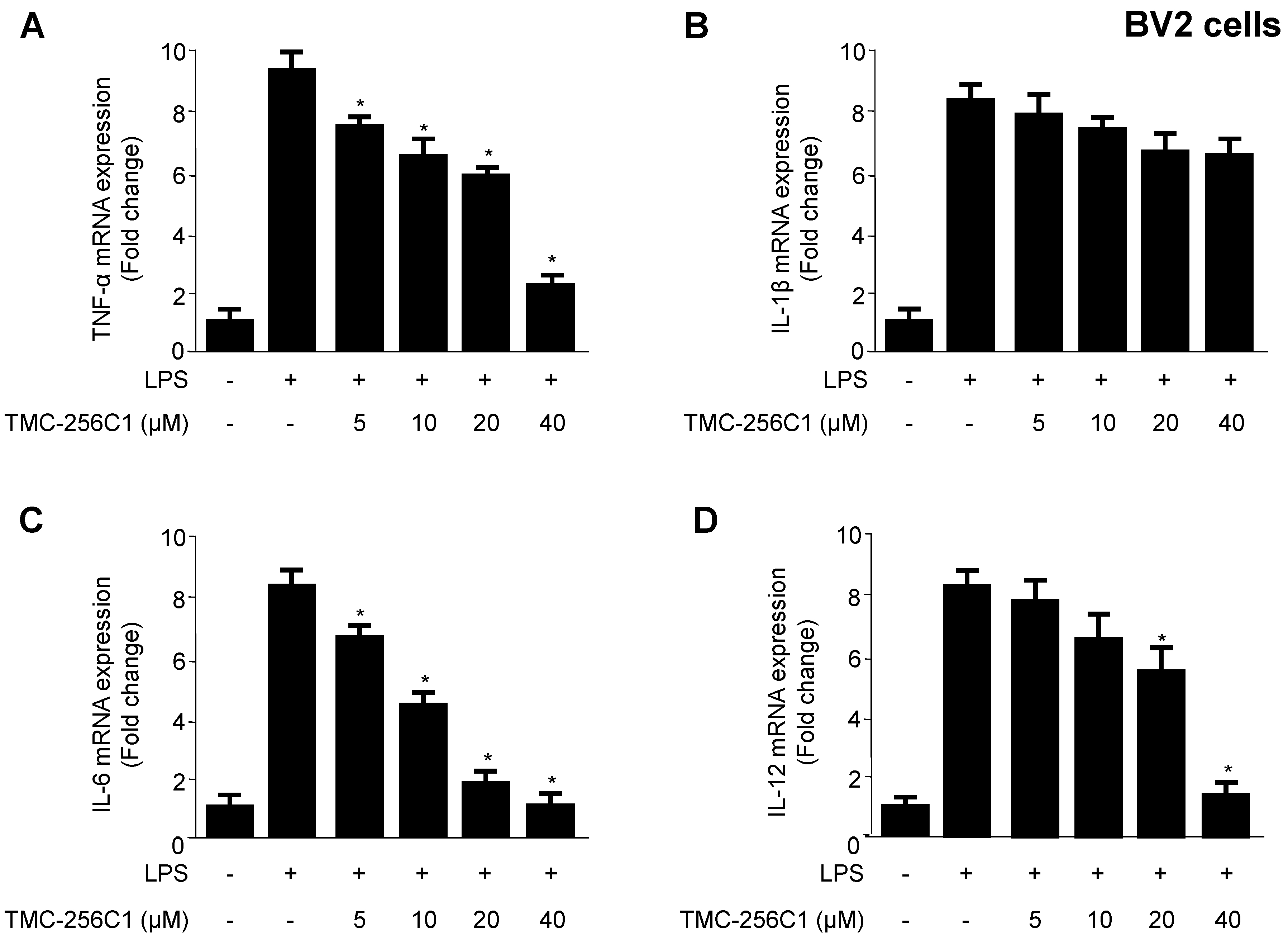
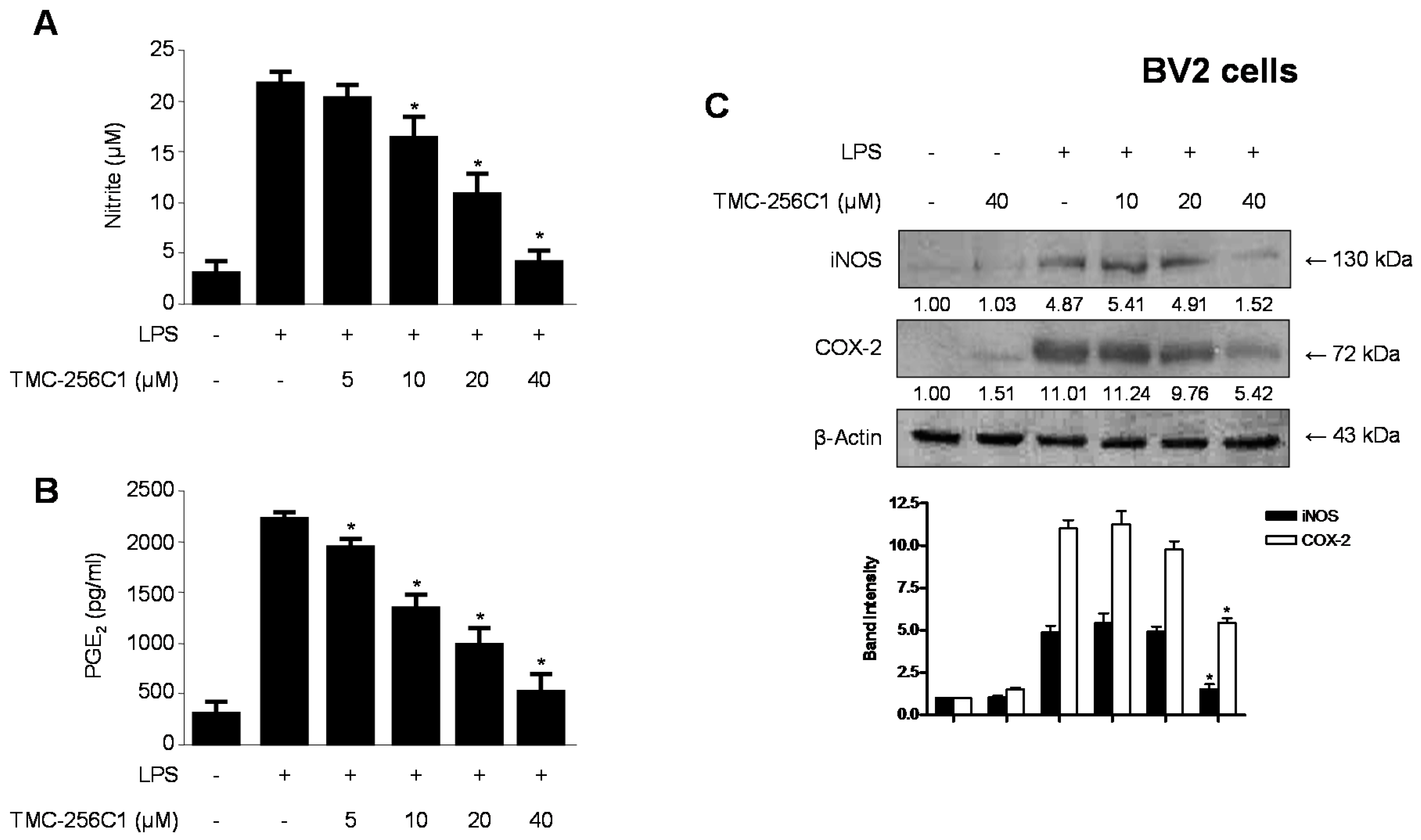
2.3. Effects of TMC-256C1 on Pro-Inflammatory Cytokines in BV2 Cells Stimulated with Lipopolysaccharides (LPS)
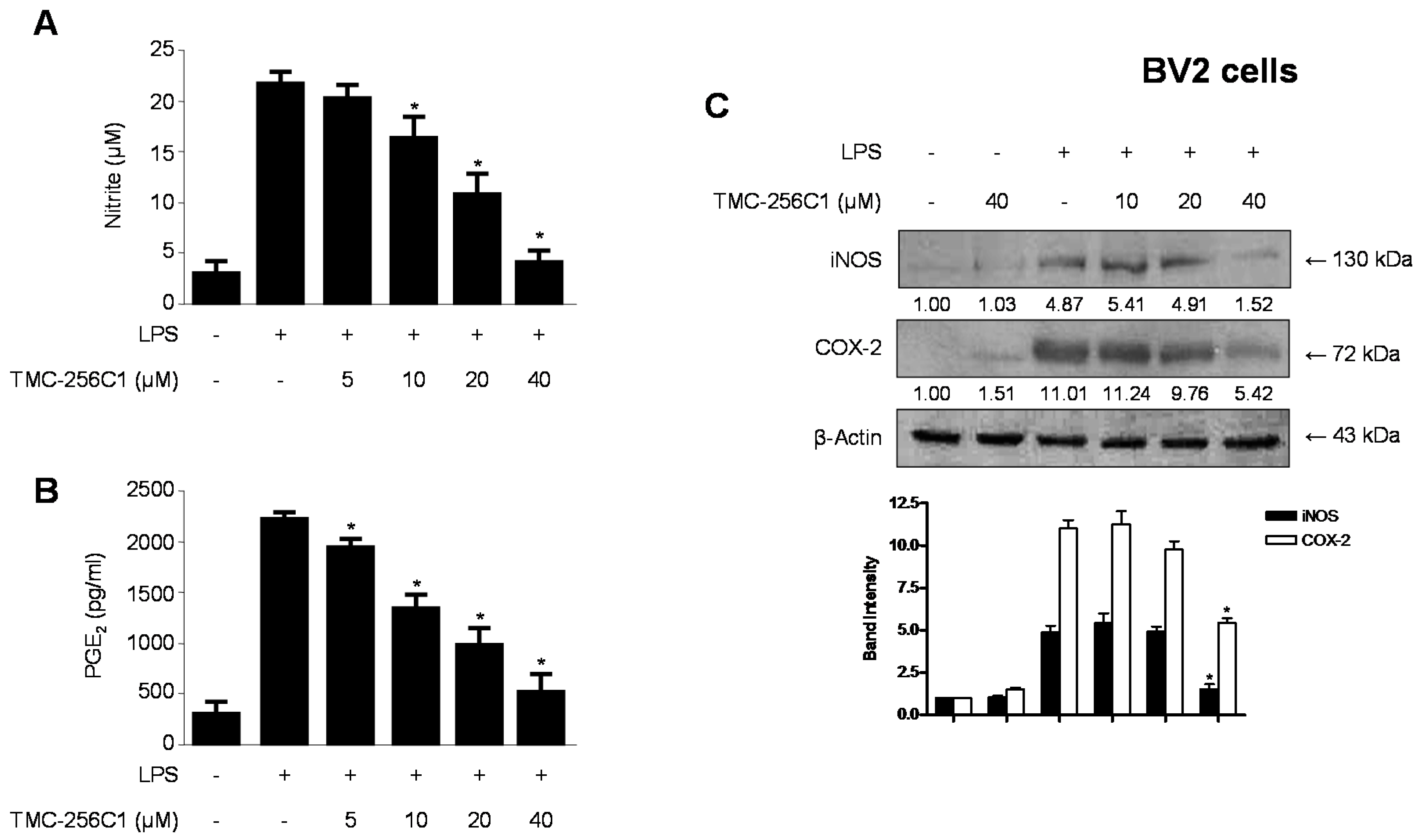
2.4. Effects of TMC-256C1 on the Protein Expression Levels of Inducible Nitric Oxide Synthase (iNOS) and Cyclooxygenase-2 (COX-2) in BV2 Cells Stimulated with LPS
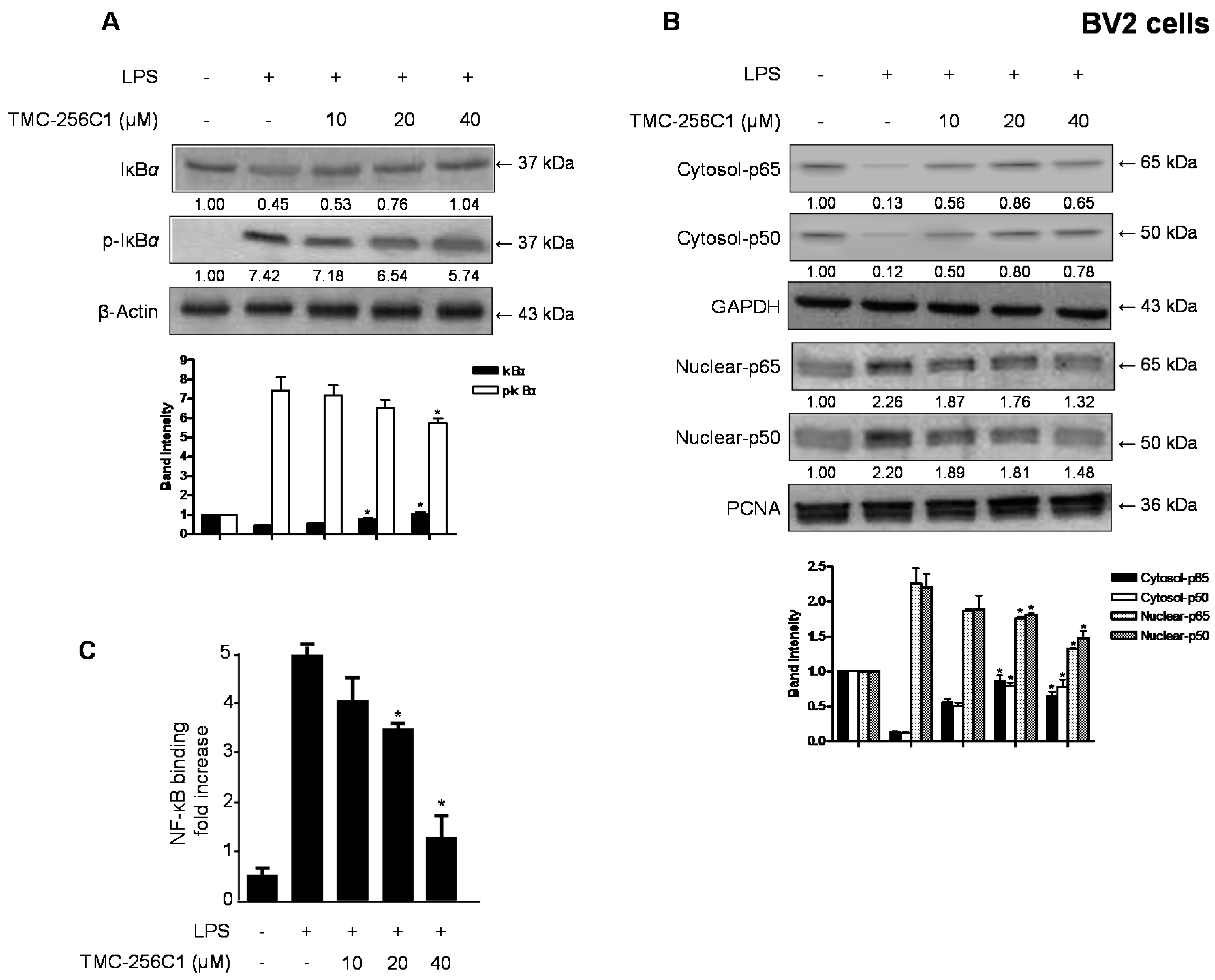
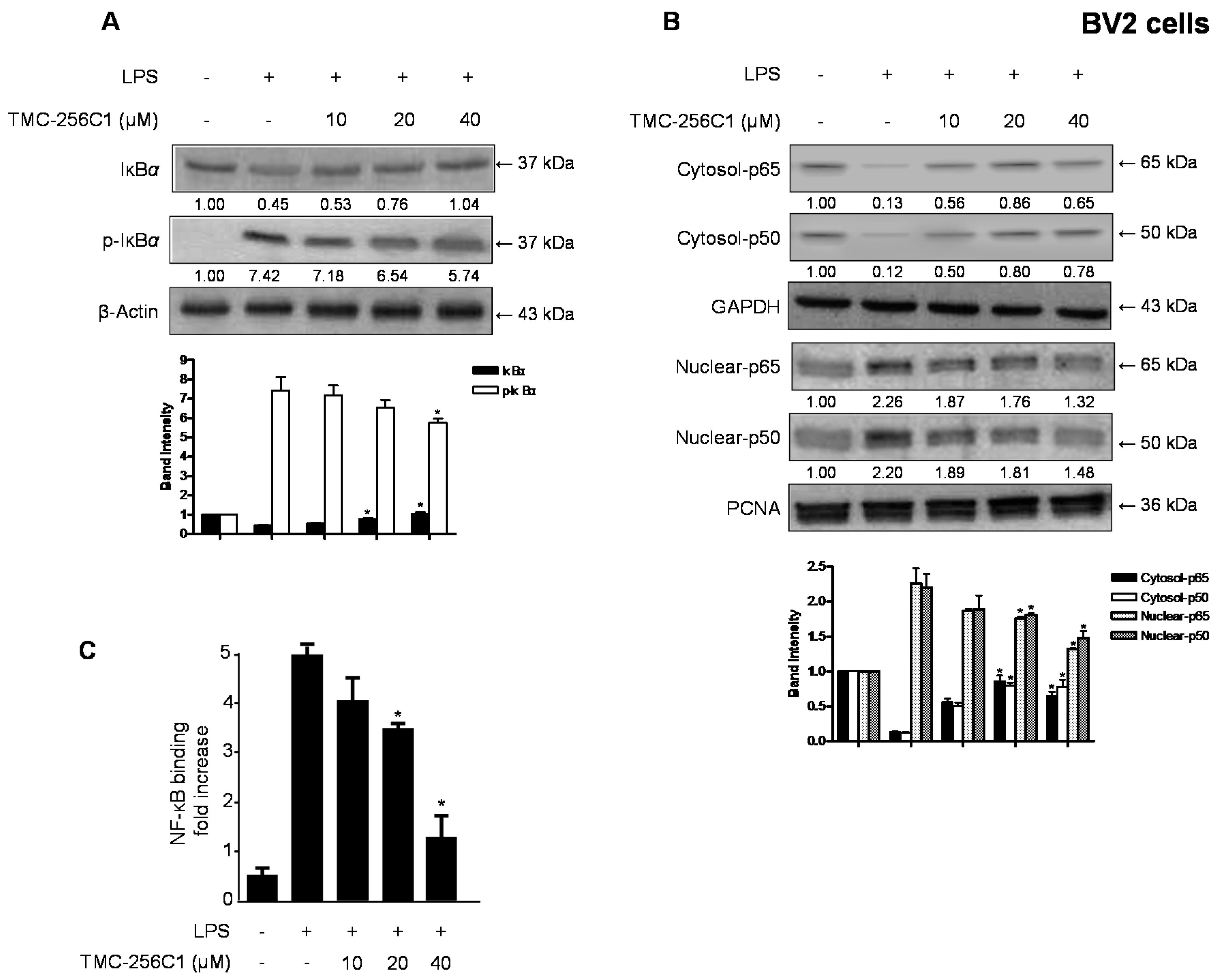
2.5. Effects of TMC-256C1 on NF-κB Activation in BV2 Cells Stimulated with LPS
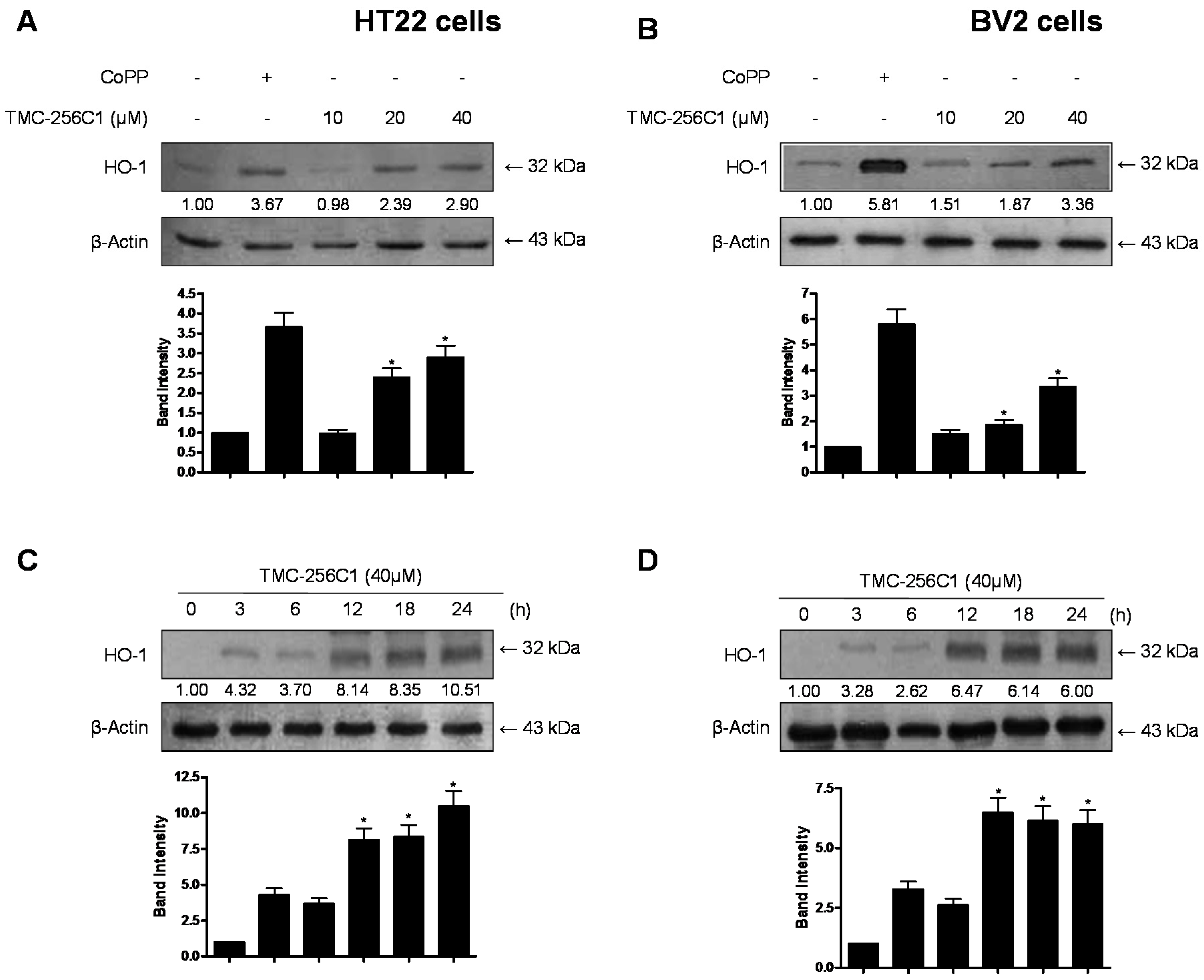
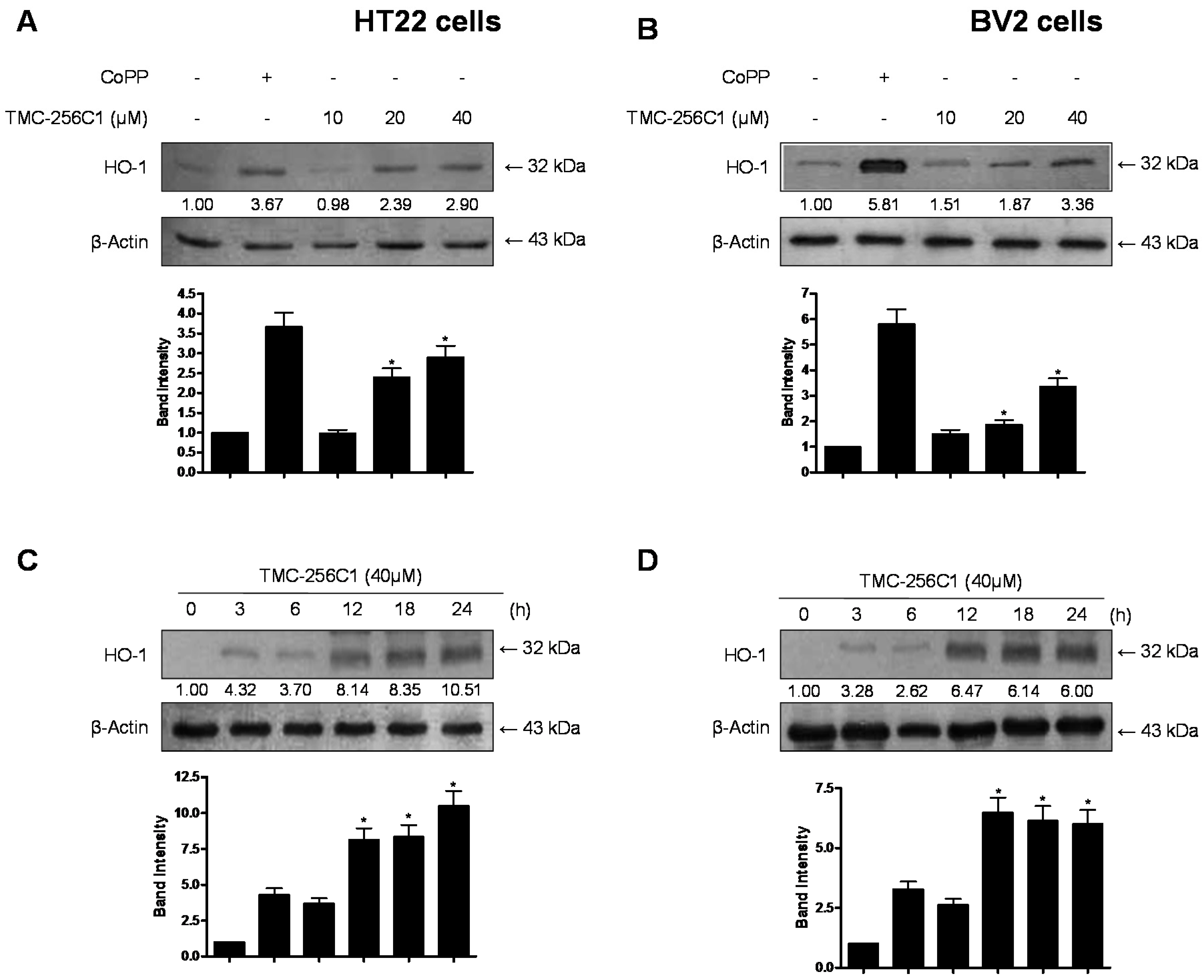
2.6. Effects of TMC-256C1 on HO-1 Expression in HT22 and BV2 Cells
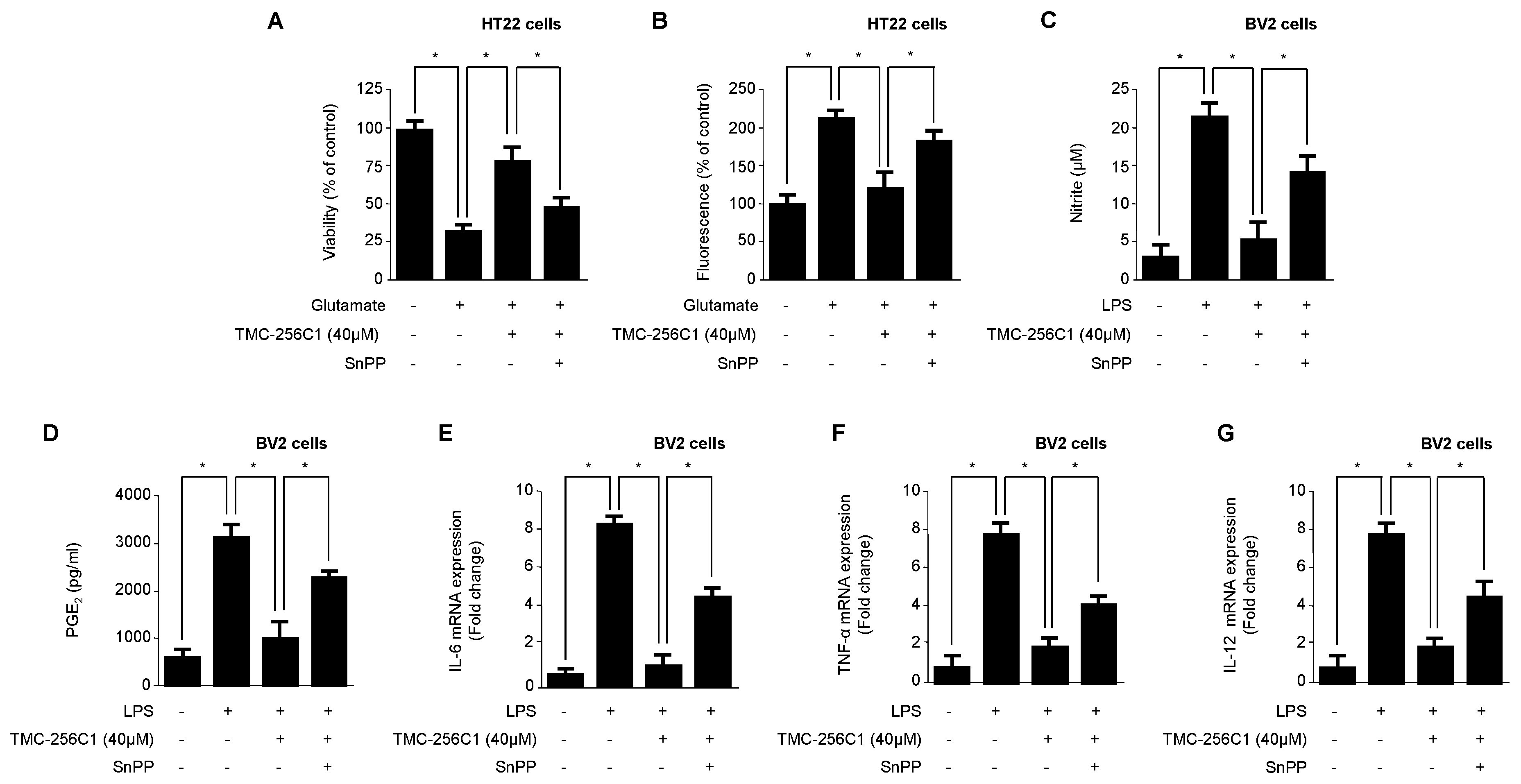
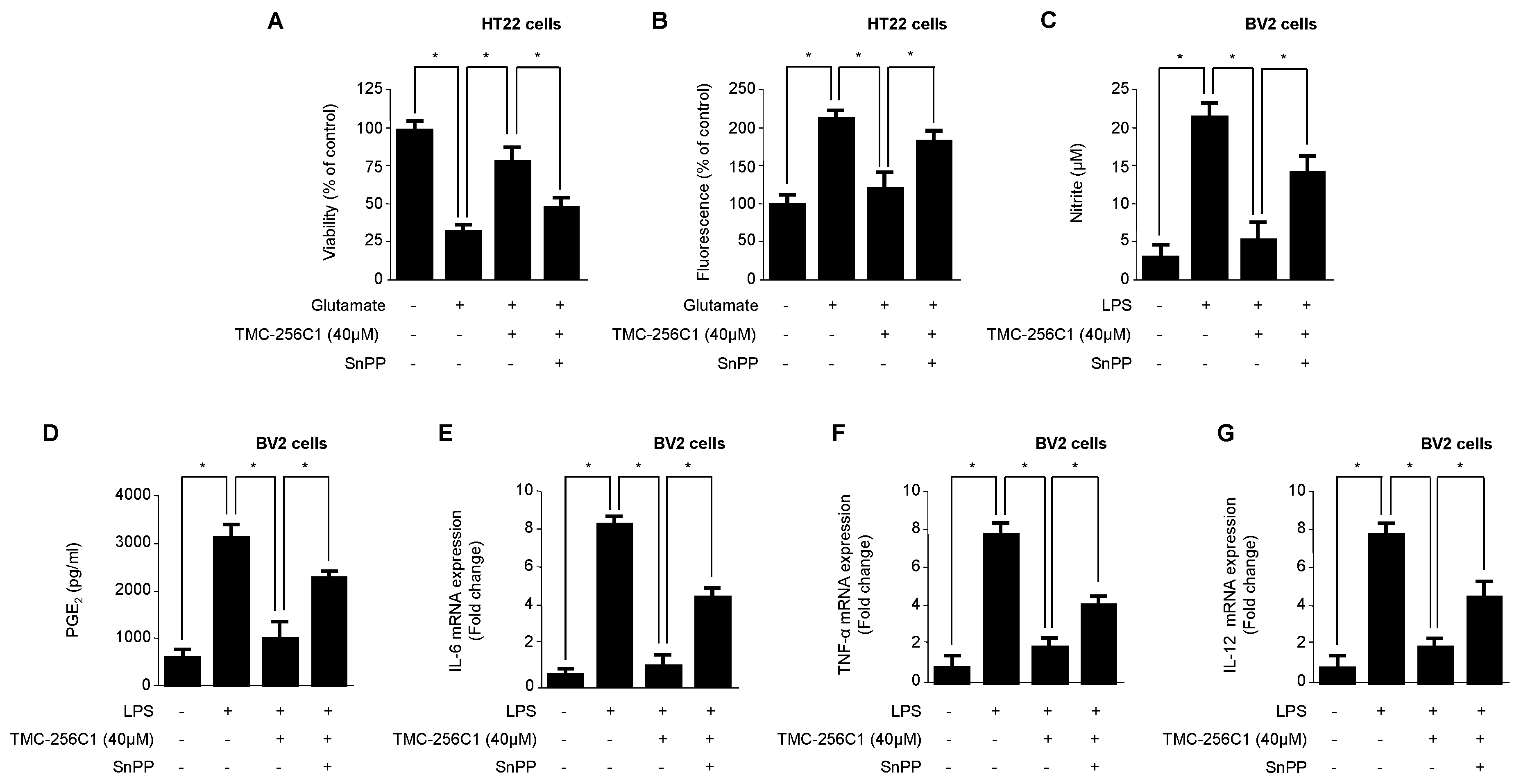
2.7. Effects of HO-1 Expression on Glutamate-Induced Oxidative Neurotoxicity and the Inhibition of Pro-Inflammatory Mediators by TMC-256C1
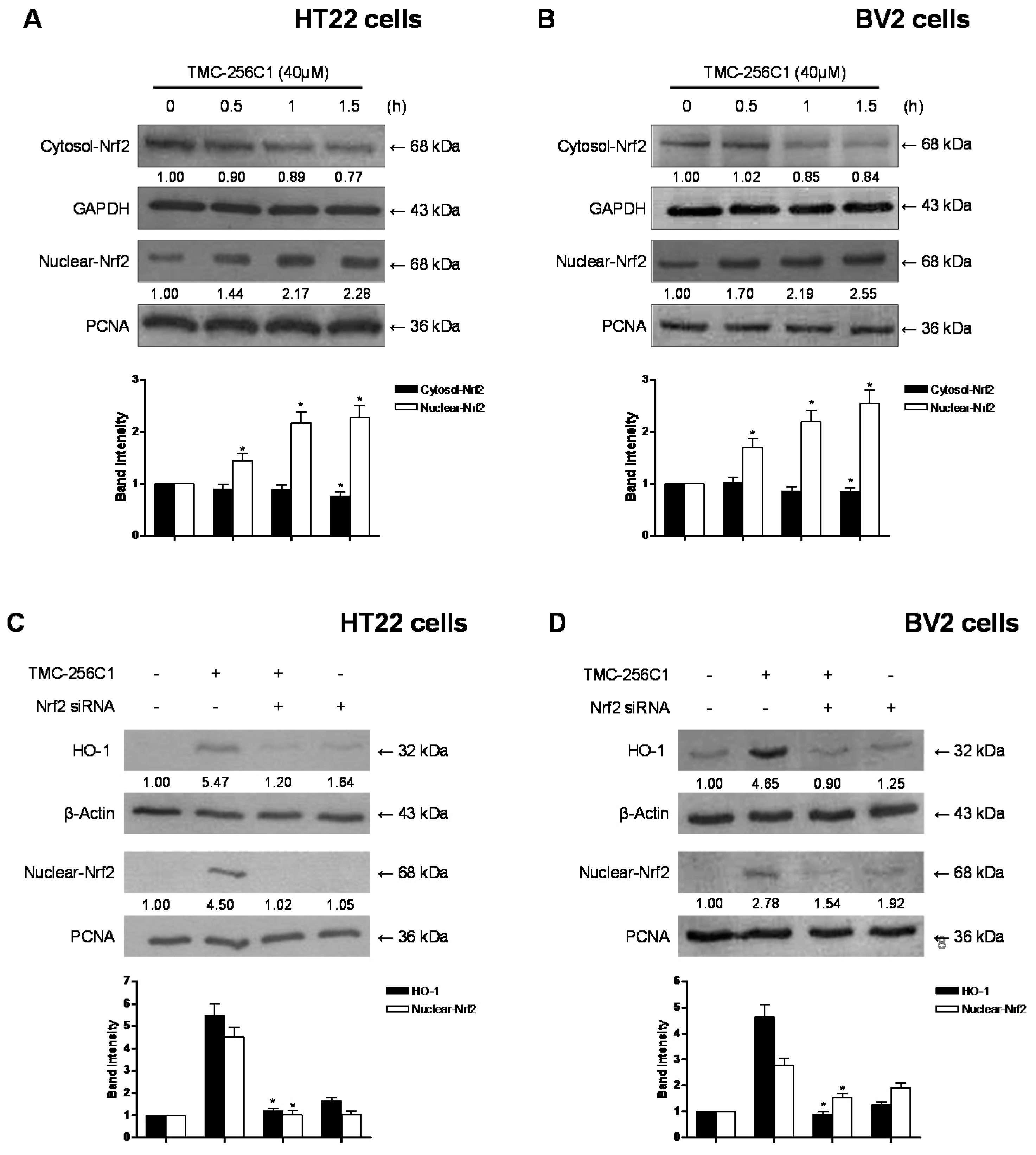
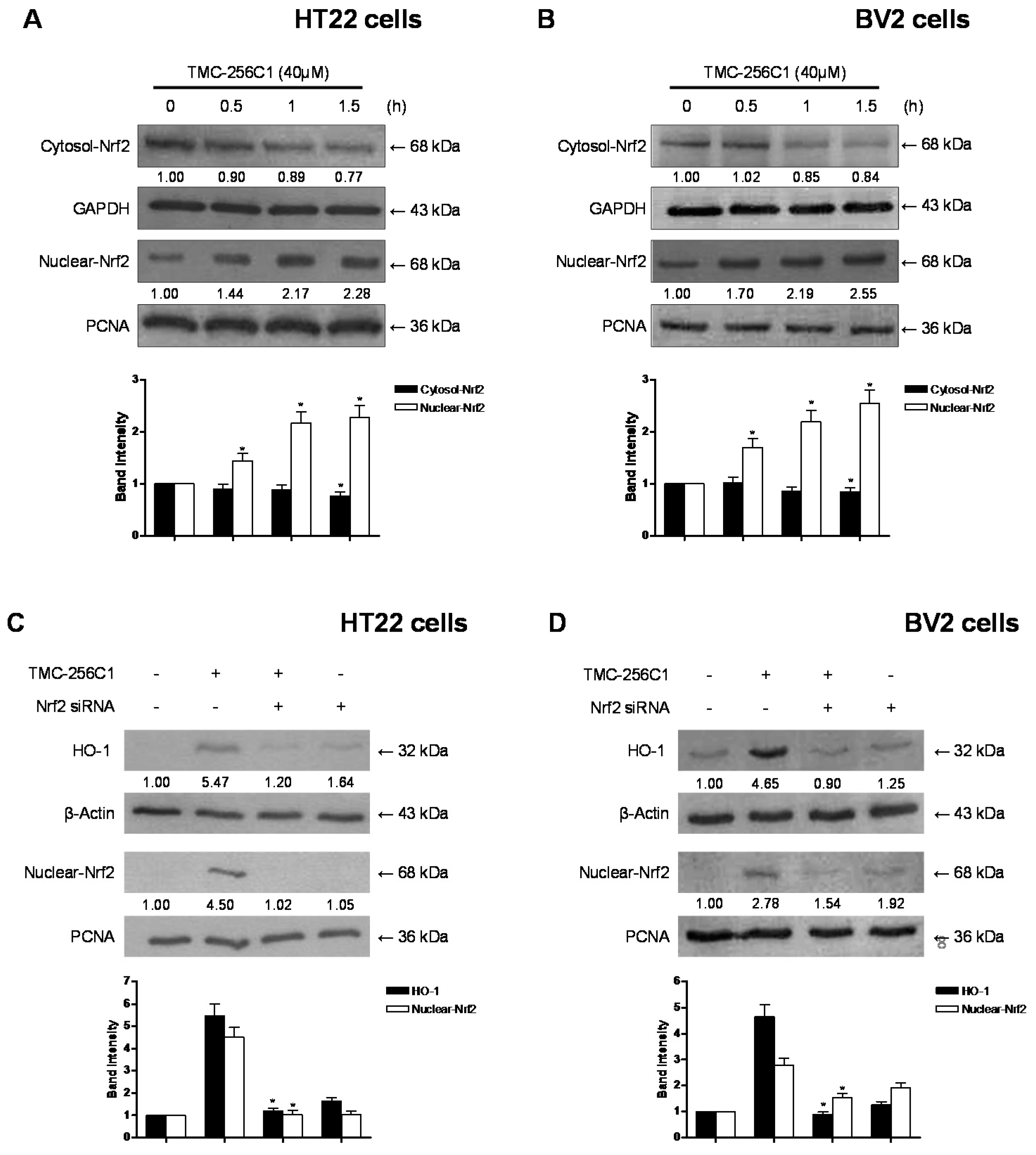
2.8. Effects of TMC-256C1 on the Nuclear Translocation of Nrf2 in HT22 and BV2 Cells
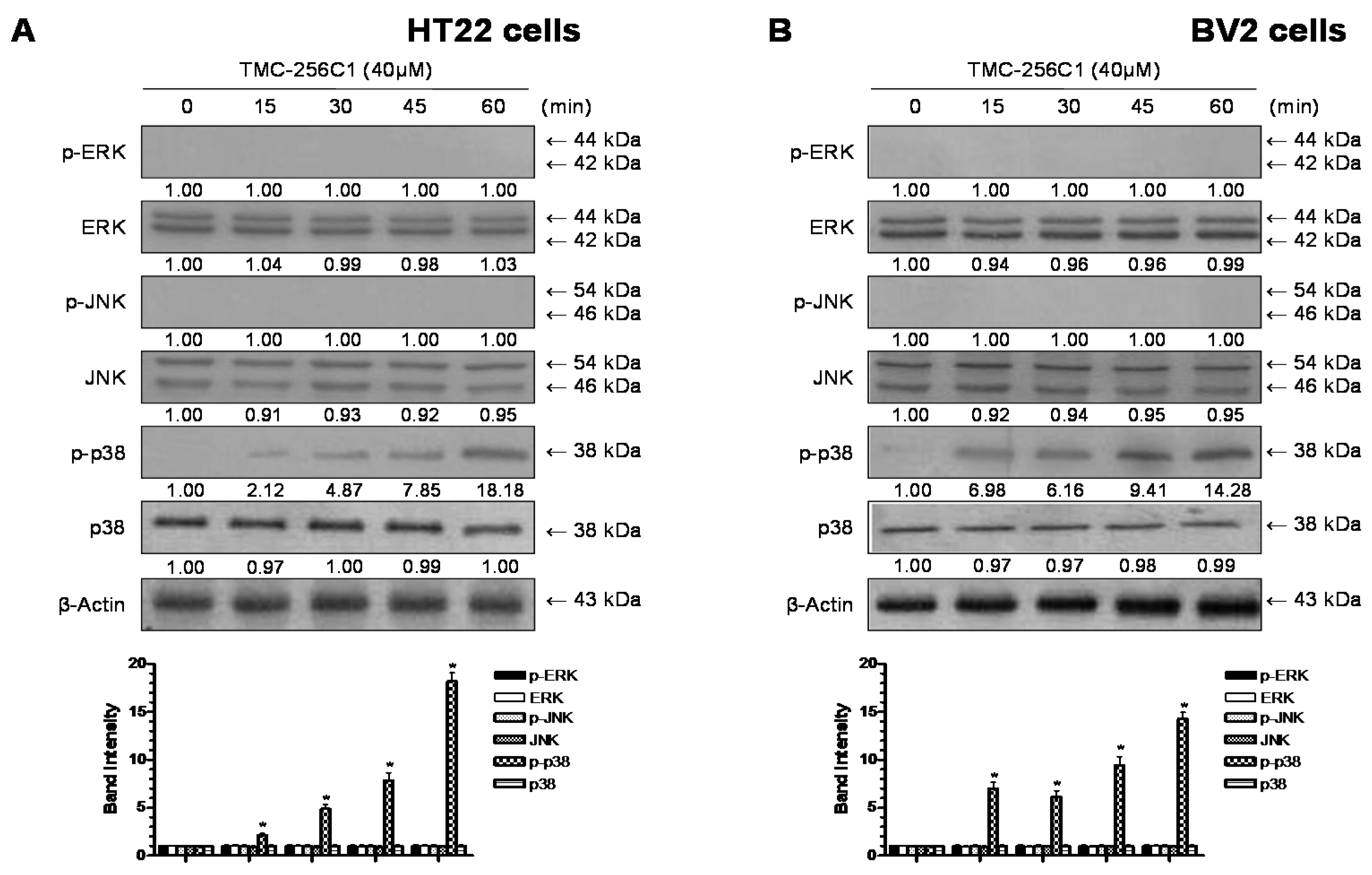
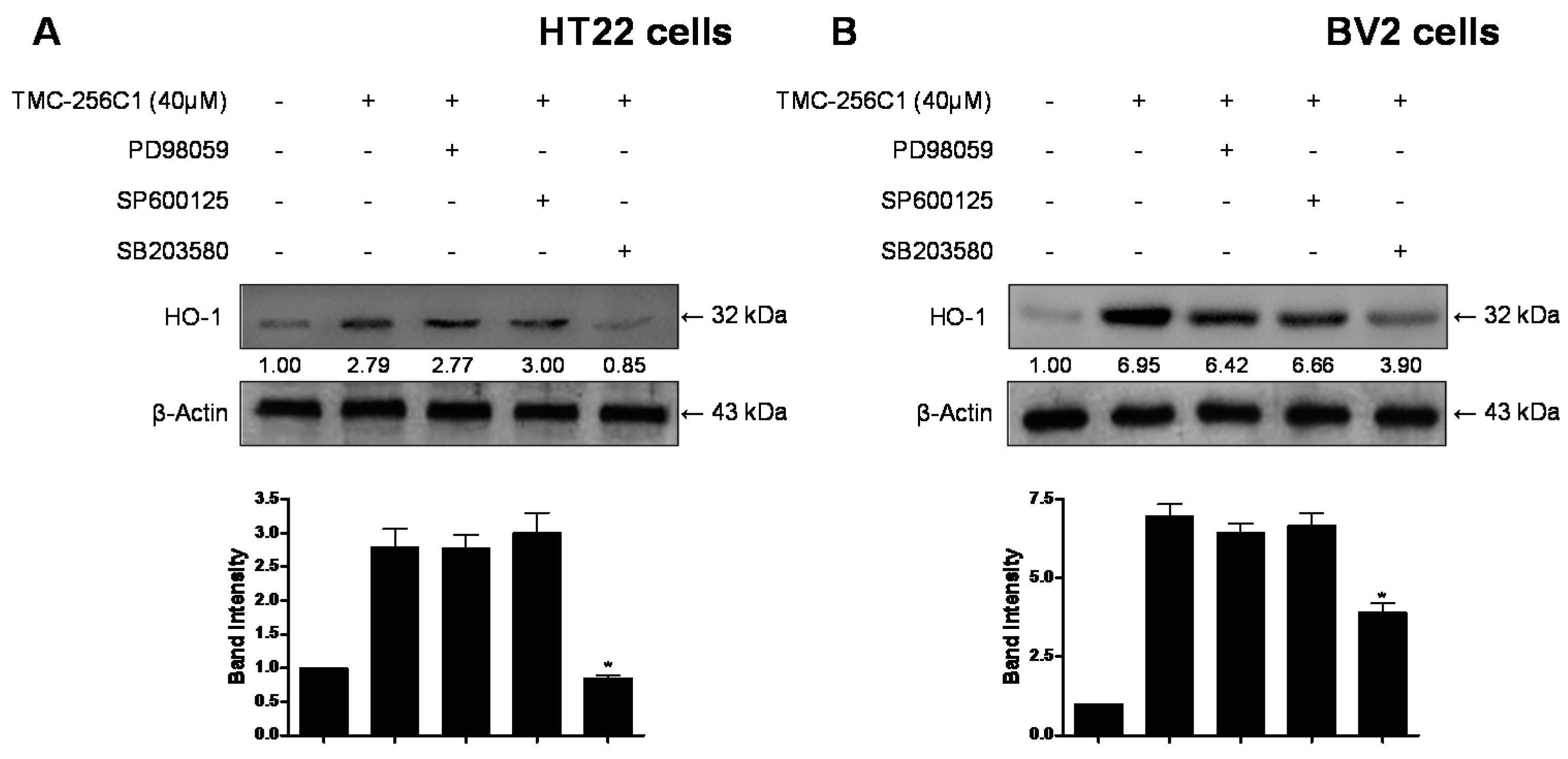
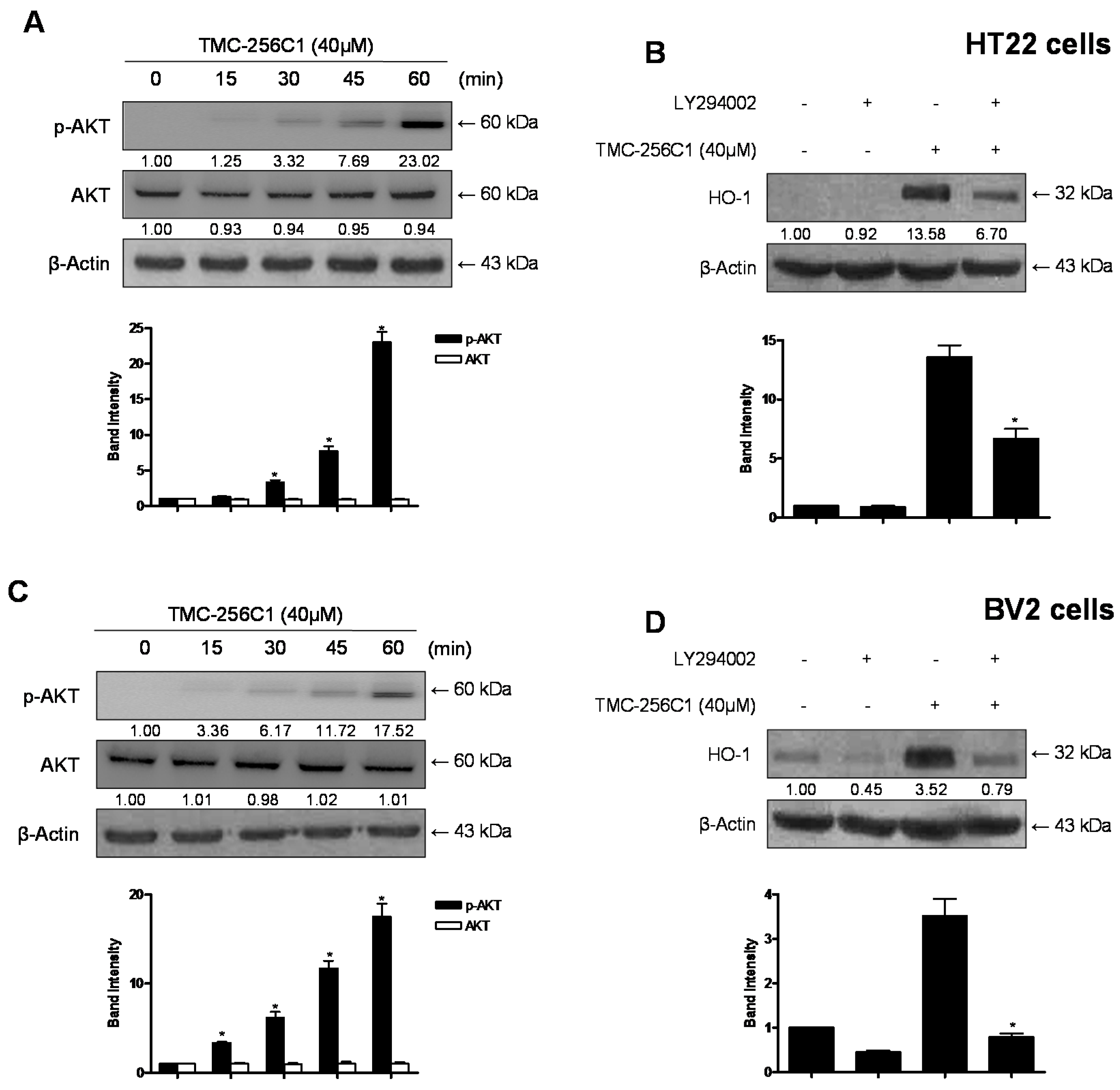
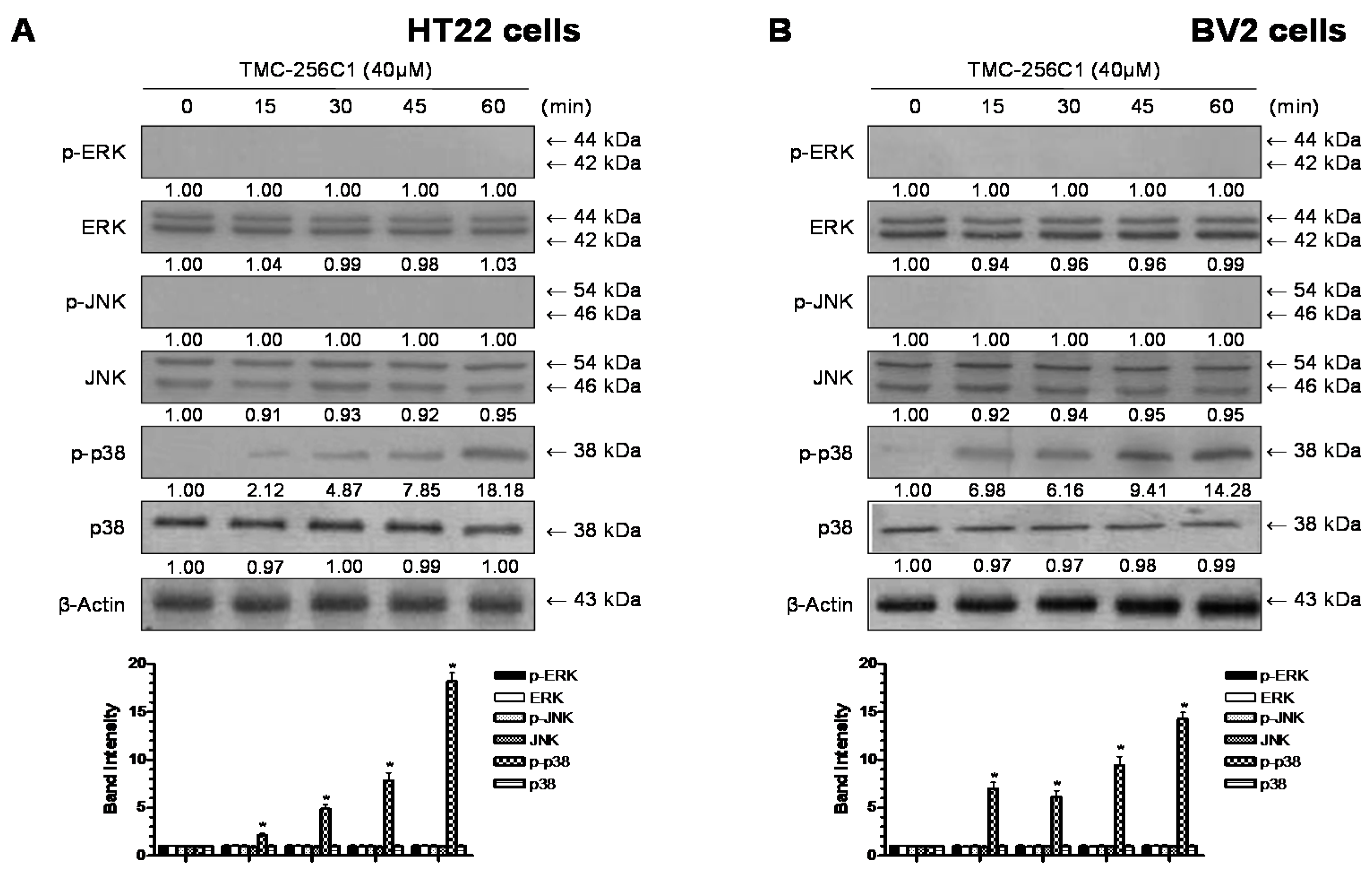
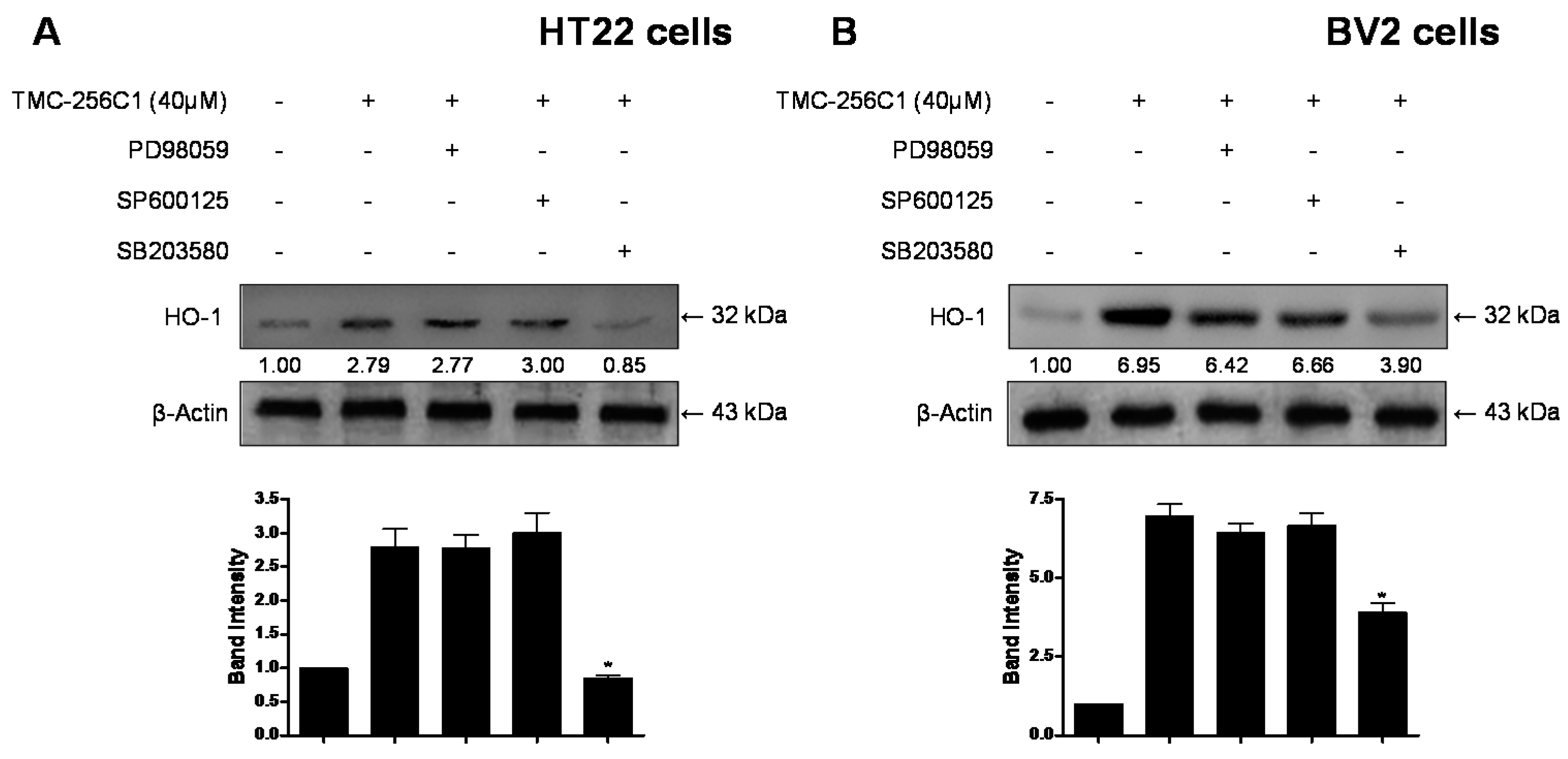
2.9. Involvement of the Mitogen-Activated Protein Kinases (MAPK) Pathways in the Induction of HO-1 Expression by TMC-256C1
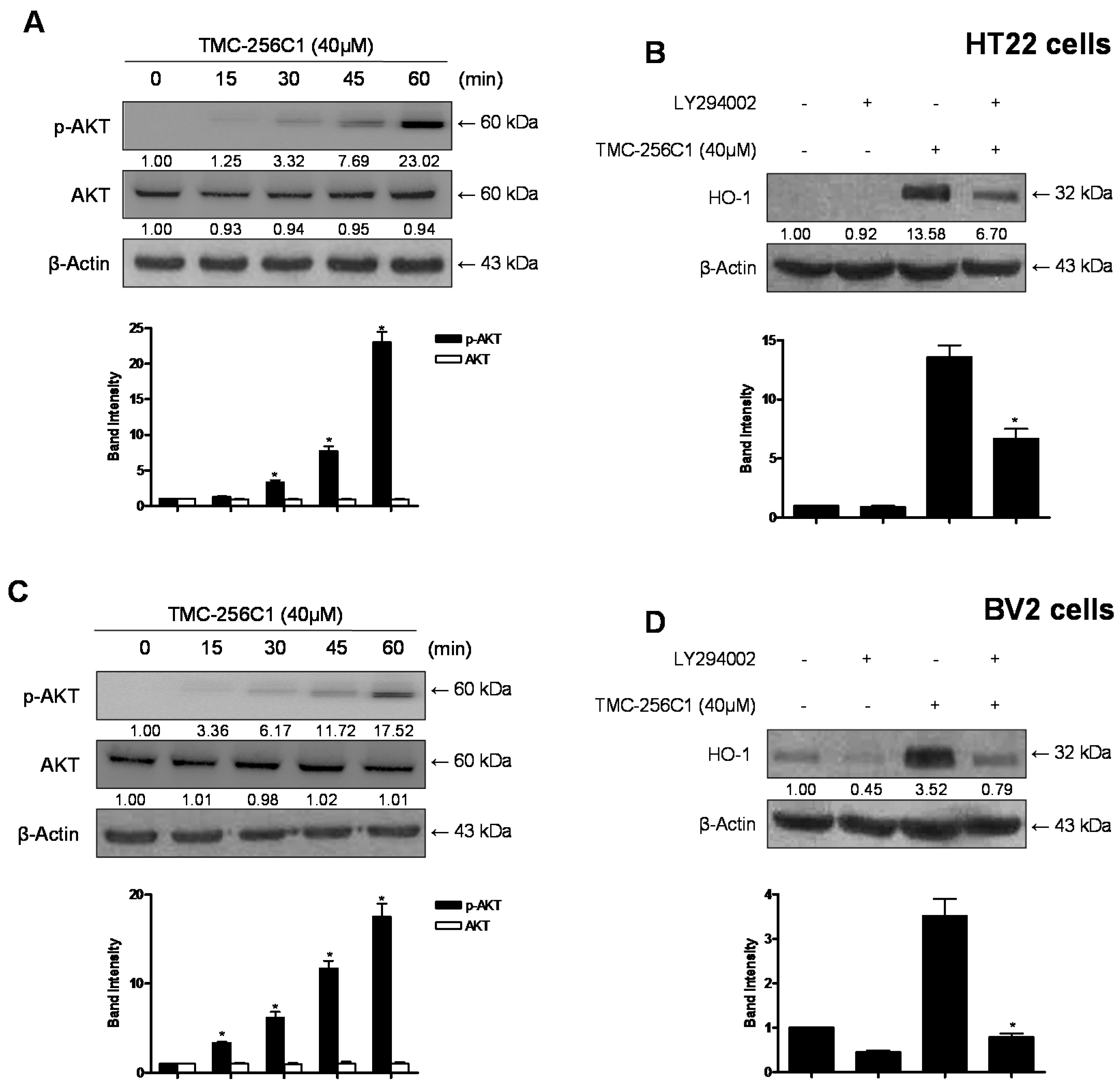
2.10. Involvement of the PI3K/Akt Pathway in the Induction of HO-1 Expression by TMC-256C1
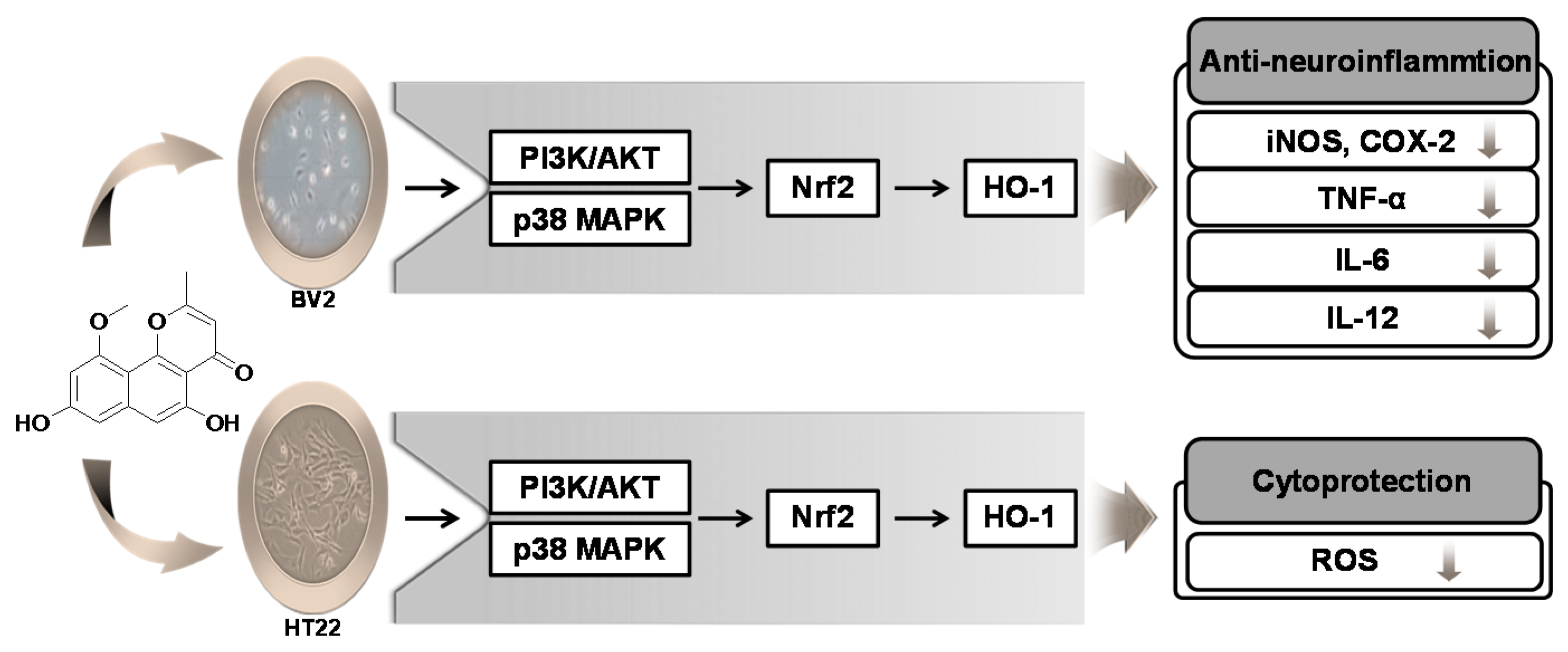
3. Discussion
4. Experimental Section
4.1. Instruments, Fungal Materials and Isolation of TMC-256C1
4.2. Chemicals and Reagents
4.3. Cell Culture and Viability Assay
4.4. Quantitative Reverse-Transcription Polymerase Chain Reaction
4.5. DNA-Binding Activity of NF-κB
4.6. Preparation of Cytosolic and Nuclear Fractions
4.7. Nitrite (NO Production) and PGE2 Determination
4.8. Western Blot Analysis
4.9. Reactive Oxygen Species Measurement
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mosley, R.L.; Benner, E.J.; Kadiu, I.; Thomas, M.; Boska, M.D.; Hasand, K.; Lauriea, C.; Gendelman, H.E. Neuroinflammation, oxidative stress, and the pathogenesis of Parkinson’s disease. Clin. Neurosci. Res. 2006, 6, 261–281. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Rottkamp, C.A.; Nunomura, A.; Raina, A.K.; Perry, G. Oxidative stress in Alzheimer’s disease. Biochim. Biophys. Acta 2000, 1502, 139–144. [Google Scholar] [CrossRef]
- Xu, X.; Chua, C.C.; Kong, J.; Kostrzewa, R.M.; Kumaraguru, U.; Hamdy, R.C.; Chua, B.H. Necrostatin-1 protects against glutamate-induced glutathionedepletion and caspase-independent cell death in HT22 cells. J. Neurochem. 2007, 103, 2004–2014. [Google Scholar] [CrossRef] [PubMed]
- Maher, P.; Davis, J.B. The role of monoamine metabolism in oxidative glutamate toxicity. J. Neurosci. 1996, 16, 6394–6401. [Google Scholar] [PubMed]
- Glezer, I.; Simard, A.R.; Rivest, S. Neuroprotective role of the innate immune system by microglia. Neuroscience 2007, 147, 867–883. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.S.; Knapp, D.J.; Crews, F.T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 55, 55453–55562. [Google Scholar] [CrossRef] [PubMed]
- Cannella, B.; Raine, C.S. The adhesion molecule and cytokine profile of multiple sclerosis lesions. Ann. Neurol. 1995, 37, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Peress, N.S.; Perillo, E.; Seidman, R.J. Glial transforming growth factor (TGF)-β isotypes in multiple sclerosis: Differential glial expression of TGF-β1, 2 and 3 isotypes in multiple sclerosis. J. Neuroimmunol. 1996, 71, 115–123. [Google Scholar] [CrossRef]
- Park, S.E.; Sapkota, K.; Kim, S.; Kim, H.; Kim, S.J. Kaempferol acts through mitogen-activated protein kinases and protein kinase B/AKT to elicit protection in a model of neuroinflammation in BV2 microglial cells. Br. J. Pharmacol. 2011, 164, 1008–1025. [Google Scholar] [CrossRef] [PubMed]
- Chow, Y.L.; Lee, K.H.; Vidyadaran, S.; Lajis, N.H.; Akhtara, M.N.; Israfa, D.A.; Syahida, A. Cardamonin from Alpini arafflesiana inhibits inflammatory responses in IFN-γ/LPS-stimulated BV2 microglia via NF-κB signalling pathway. Int. Immunopharmacol. 2012, 12, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Chen, D.; Hu, Q.; Wang, G. Rotenone directly induces BV2 cell activation via the p38 MAPK pathway. PLoS ONE 2013, 8, e72046. [Google Scholar] [CrossRef] [PubMed]
- Llesuy, S.F.; Tomaro, M.L. Hemeoxygenase and oxidative stress. Evidence of involvement of bilirubin as physiological protector against oxidative damage. Biochim. Biophys. Acta 1994, 1223, 9–14. [Google Scholar] [CrossRef]
- Ryter, S.W.; Otterbein, L.E.; Morse, D.; Choi, A.M. Hemeoxygenase/carbon monoxide signaling pathways: Regulation and functional significance. Mol. Cell. Biochem. 2002, 234–235, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.S.; Tsai, H.L.; Chau, L.Y. Induction of heme oxygenase-1 expression in murine macrophages is essential for the anti-inflammatory effect of low dose 15-deoxy-∆12, 14-prostaglandin J2. J. Biol. Chem. 2003, 278, 19325–19330. [Google Scholar] [CrossRef] [PubMed]
- Gueler, F.; Park, J.K.; Rong, S.; Kirsch, T.; Lindschau, C.; Zheng, W.; Elger, M.; Fiebeler, A.; Fliser, D.; Luft, F.C.; et al. Statins attenuate ischemia-reperfusion injury by inducing heme oxygenase-1 in infiltrating macrophages. Am. J. Pathol. 2007, 170, 1192–1199. [Google Scholar] [CrossRef] [PubMed]
- Oh, G.S.; Pae, H.O.; Lee, B.S.; Kim, B.N.; Kim, J.M.; Kim, H.R.; Jeon, S.B.; Jeone, W.K.; Chae, H.J.; Chung, H.T. Hydrogen sulfide inhibits nitric oxide production and nuclear factor-κB via heme oxygenase-1 expression in RAW264. 7 macrophages stimulated with lipopolysaccharide. Free Radic. Biol. Med. 2006, 41, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Wiesel, P.; Foster, L.C.; Pellacani, A.; Layne, M.D.; Hsieh, C.M.; Huggins, G.S.; Strauss, P.; Yet, S.F.; Perrella, M.A. Thioredoxin facilitates the induction of heme oxygenase-1 in response to inflammatory mediators. J. Biol. Chem. 2000, 275, 24840–24846. [Google Scholar] [CrossRef] [PubMed]
- Surh, Y.J. Cancer chemoprevention with dietary phytochemicals. Nat. Rev. Cancer 2003, 3, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhang, X.; Fan, H.; Liu, Y. Curcumin upregulates transcription factor Nrf2, HO-1 expression and protects rat brains against focal ischemia. Brain Res. 2009, 1282, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Fukui, M.; Choi, H.J.; Zhu, B.T. Mechanism for the protective effect of resveratrol against oxidative stress-induced neuronal death. Free Radic. Biol. Med. 2010, 49, 800–813. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.; Choi, J.H.; Yang, H.; Jeong, E.J.; Lee, K.Y.; Kim, Y.C.; Sung, S.H. Neuroprotective and anti-inflammatory effects of flavonoids isolated from Rhus verniciflua in neuronal HT22 and microglial BV2 cell lines. Food Chem. Toxicol. 2012, 50, 1940–1945. [Google Scholar] [CrossRef] [PubMed]
- Fenical, W.; Jensen, P.R. Developing a new resource for drug discovery: Marine actinomycete bacteria. Nat. Chem. Biol. 2006, 2, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Bugni, T.S.; Ireland, C.M. Marine-derived fungi: A chemically and biologically diverse group of microorganisms. Nat. Prod. Rep. 2004, 21, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H.; Moore, B.S. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol. 2012, 19, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, M.; Kohno, J.; Yamamoto, K.; Okuda, T.; Nishio, M.; Kawano, K.; Ohnuki, T. TMC-256A1 and C1, new inhibitors of IL-4 signal transduction produced by Aspergillus niger var niger TC 1629. J. Antibiot. 2002, 55, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.S.; Shang, Z.; Li, X.M.; Li, C.S.; Cui, C.M.; Wang, B.G. Secondary metabolites produced by solid fermentation of the marine-derived fungus Penicillium commune QSD-17. Biosci. Biotechnol. Biochem. 2012, 76, 358–360. [Google Scholar] [CrossRef] [PubMed]
- Abraham, N.G.; Kappas, A. Pharmacological and clinical aspects of hemeoxygenase. Pharmacol. Rev. 2008, 60, 79–127. [Google Scholar] [CrossRef] [PubMed]
- Paine, A.; Eiz-Vesper, B.; Blasczyk, R.; Immenschuh, S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem. Pharmacol. 2010, 80, 1895–1903. [Google Scholar] [CrossRef] [PubMed]
- Kietzmann, T.; Samoylenko, A.; Immenschuh, S. Transcriptional regulation of heme oxygenase-1 gene expression by MAP kinases of the JNK and p38 pathways in primary cultures of rat hepatocytes. J. Biol. Chem. 2003, 278, 17927–17936. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Rojo, A.I.; Salinas, M.; Diaz, R.; Gallardo, G.; Alam, J.; de Galarreta, C.M.; Cuadrado, A. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3′-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J. Biol. Chem. 2004, 279, 8919–8929. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Cui, W.; Li, G.; Yuan, S.; Xu, D.; Hoi, M.P.; Lin, Z.; Dou, J.; Han, Y.; Lee, S.M. Baicalein Protects against 6-OHDA-Induced Neurotoxicity through Activation of Keap1/Nrf2/HO-1 and Involving PKCα and PI3K/AKT Signaling Pathways. J. Agric. Food Chem. 2012, 60, 8171–8182. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Chen, Y.T.; Huang, Y.W.; Tsai, H.J.; Kuo, C.C. 4-Ketopinoresinol, a novel naturally occurring ARE activator, induces the Nrf2/HO-1 axis and protects against oxidative stress-induced cell injury via activation of PI3K/AKT signaling. Free Radic. Biol. Med. 2012, 52, 1054–1066. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.M.; Main, B.S.; Crack, P.J. Neuroinflammation and oxidative stress: Co-conspirators in the pathology of Parkinson’s disease. Neurochem. Int. 2013, 62, 803–819. [Google Scholar] [CrossRef] [PubMed]
- Mhatre, M.; Floyd, R.A.; Hensley, K. Oxidative stress and neuroinflammation in Alzheimer’s disease and amyotrophic lateral sclerosis: Common links and potential therapeutic targets. J. Alzheimers Dis. 2004, 6, 147–157. [Google Scholar] [PubMed]
- Jin, D.Q.; Lim, C.S.; Hwang, J.K.; Ha, I.; Han, J.S. Anti-oxidant and anti-inflammatory activities of macelignan in murine hippocampal cell line and primary culture of rat microglial cells. Biochem. Biophys. Res. Commun. 2005, 331, 1264–1269. [Google Scholar] [CrossRef] [PubMed]
- Jeong, G.S.; Li, B.; Lee, D.S.; Kim, K.H.; Lee, I.K.; Lee, K.R.; Kim, Y.C. Cytoprotective and anti-inflammatory effects of spinasterol via the induction of heme oxygenase-1 in murine hippocampal and microglial cell lines. Int. Immunopharmacol. 2010, 10, 1587–1594. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.; Yang, H.; Lee, M.; Huh, J.; Kim, H.W.; Kim, H.P.; Sung, S.H. Neuroprotective Benzyl Benzoate Glycosides from Disporum viridescens Roots in HT22 Hippocampal Neuronal Cells. J. Nat. Prod. 2013, 76, 2291–2297. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Jeong, G.S.; Kang, D.G.; Lee, H.S.; Kim, Y.C. Cytoprotective effects of lindenenyl acetate isolated from Lindera strychnifolia on mouse hippocampal HT22 cells. Eur. J. Pharmacol. 2009, 614, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.C.; Hu, S.; Molitor, T.W.; Shaskan, E.G.; Peterson, P.K. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J. Immunol. 1992, 149, 2736–2741. [Google Scholar] [PubMed]
- Bianchi, R.; Giambanco, I.; Donato, R. S100B/RAGE-dependent activation of microglia via NF-κB and AP-1: Co-regulation of COX-2 expression by S100B, IL-1β and TNF-α. Neurobiol. Aging 2010, 31, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Verma, I.M. NF-κB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoha, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayamae, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Alam, J.; Stewart, D.; Touchard, C.; Boinapally, S.; Choi, A.M.; Cook, J.L. Nrf2, a Cap‘n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 1999, 274, 26071–26078. [Google Scholar] [CrossRef] [PubMed]
- Elbirt, K.K.; Whitmarsh, A.J.; Davis, R.J.; Bonkovsky, H.L. Mechanism of sodium arsenite-mediated induction of heme oxygenase-1 in hepatoma cells. Role of mitogen-activated protein kinases. J. Biol. Chem. 1998, 273, 8922–8931. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.S.; Kim, D.C.; Lee, D.S.; Kim, K.S.; Ko, W.; Sohn, J.H.; Yim, J.H.; Kim, Y.C.; Oh, H. Anti-neuroinflammatory effect of aurantiamide acetate from the marine fungus Aspergillus sp. SF-5921: Inhibition of NF-κB and MAPK pathways in lipopolysaccharide-induced mouse BV2 microglial cells. Int. Immunopharmacol. 2014, 23, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.C.; Lee, H.S.; Ko, W.; Lee, D.S.; Sohn, J.H.; Yim, J.H.; Kim, Y.C.; Oh, H. Anti-inflammatory effect of methylpenicinoline from a marine isolate of Penicillium. sp. (SF-5995): Inhibition of NF-κB and MAPK pathways in lipopolysaccharide-induced RAW264.7 macrophages and BV2 microglia. Molecules 2014, 19, 18073–18089. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.S.; Kim, K.S.; Ko, W.; Li, B.; Keo, S.; Jeong, G.S.; Oh, H.; Kim, Y.C. The neoflavonoid latifolin isolated from MeOH extract of Dalbergia odorifera attenuates inflammatory responses by inhibiting NF-κB activation via Nrf2-mediated heme oxygenase-1 expression. Phytother. Res. 2014, 28, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.S.; Cha, B.Y.; Woo, J.T.; Kim, Y.C.; Jang, J.H. Acerogenin A from Acer nikoense Maxim Prevents Oxidative Stress-Induced Neuronal Cell Death through Nrf2-Mediated Heme Oxygenase-1 Expression in Mouse Hippocampal HT22 Cell Line. Molecules 2015, 20, 12545–12557. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.-C.; Cho, K.-H.; Ko, W.; Yoon, C.-S.; Sohn, J.H.; Yim, J.H.; Kim, Y.-C.; Oh, H. Anti-Inflammatory and Cytoprotective Effects of TMC-256C1 from Marine-Derived Fungus Aspergillus sp. SF-6354 via up-Regulation of Heme Oxygenase-1 in Murine Hippocampal and Microglial Cell Lines. Int. J. Mol. Sci. 2016, 17, 529. https://doi.org/10.3390/ijms17040529
Kim D-C, Cho K-H, Ko W, Yoon C-S, Sohn JH, Yim JH, Kim Y-C, Oh H. Anti-Inflammatory and Cytoprotective Effects of TMC-256C1 from Marine-Derived Fungus Aspergillus sp. SF-6354 via up-Regulation of Heme Oxygenase-1 in Murine Hippocampal and Microglial Cell Lines. International Journal of Molecular Sciences. 2016; 17(4):529. https://doi.org/10.3390/ijms17040529
Chicago/Turabian StyleKim, Dong-Cheol, Kwang-Ho Cho, Wonmin Ko, Chi-Su Yoon, Jae Hak Sohn, Joung Han Yim, Youn-Chul Kim, and Hyuncheol Oh. 2016. "Anti-Inflammatory and Cytoprotective Effects of TMC-256C1 from Marine-Derived Fungus Aspergillus sp. SF-6354 via up-Regulation of Heme Oxygenase-1 in Murine Hippocampal and Microglial Cell Lines" International Journal of Molecular Sciences 17, no. 4: 529. https://doi.org/10.3390/ijms17040529
APA StyleKim, D.-C., Cho, K.-H., Ko, W., Yoon, C.-S., Sohn, J. H., Yim, J. H., Kim, Y.-C., & Oh, H. (2016). Anti-Inflammatory and Cytoprotective Effects of TMC-256C1 from Marine-Derived Fungus Aspergillus sp. SF-6354 via up-Regulation of Heme Oxygenase-1 in Murine Hippocampal and Microglial Cell Lines. International Journal of Molecular Sciences, 17(4), 529. https://doi.org/10.3390/ijms17040529