
Molecular and Cellular Mechanisms of Myelodysplastic Syndrome: Implications on Targeted Therapy

Abstract
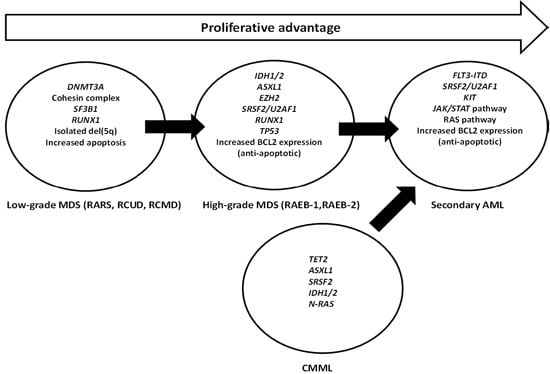
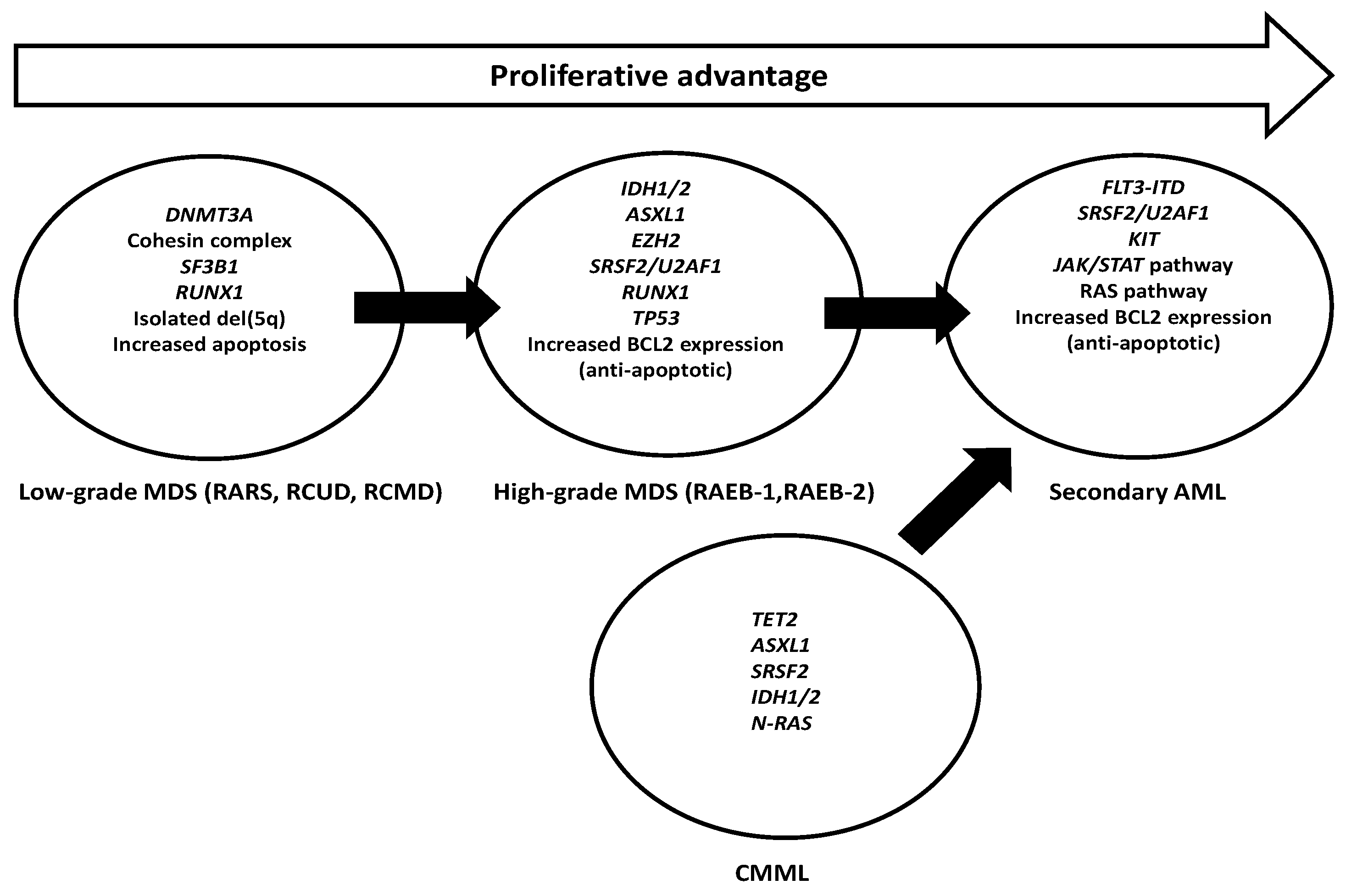
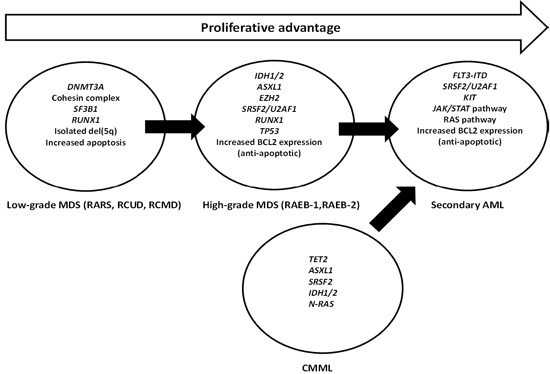
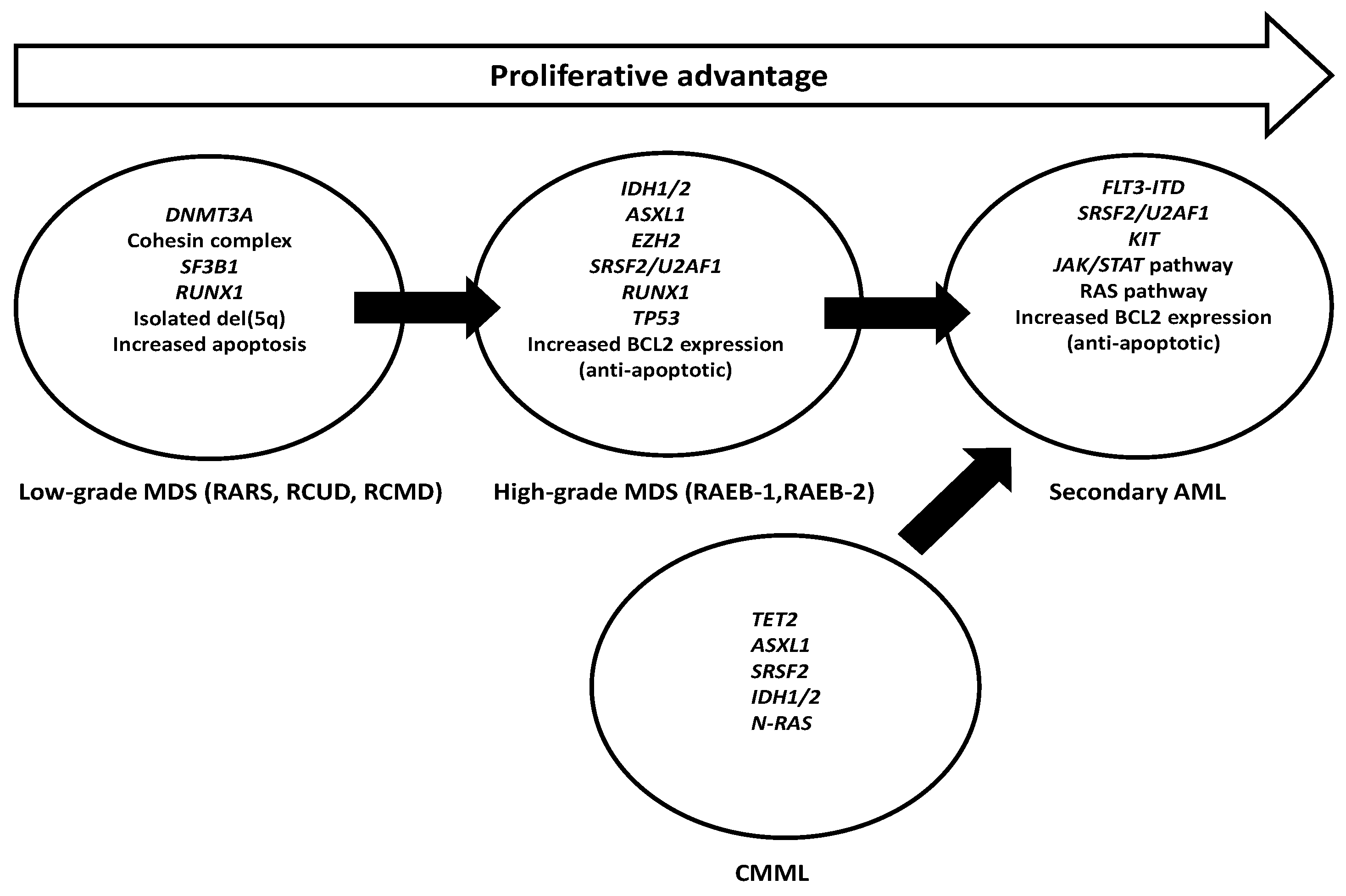
:
1. Introduction
2. Targeting Genes Involved in DNA Methylation
2.1. TET2 Mutations
2.2. DNMT3A Mutations
2.3. Isocitrate Dehydrogenases 1 and 2 (IDH1 and IDH2) Mutations
3. Targeting Mutation in Histone Modification Genes
3.1. EZH2 Mutations
3.2. ASXL1 Mutations
3.3. UTX Mutations
4. Targeting Mutations in the RNA Splicing Genes
4.1. SF3B1 Mutations
4.2. SRSF2 Mutations
4.3. ZRSR2 Mutations
4.4. U2AF1 Mutations
5. Targeting Mutations in Transcription Factor Genes
5.1. RUNX1 Mutations
5.2. BCOR/BCORL1 Mutations
5.3. ETV6 Mutations
5.4. SETBP1 Mutations
5.5. GATA2 Mutations
6. Targeting DNA Repair/Tumor Suppressor Genes
TP53 Mutations
7. Targeting Mutations in Signal Transduction
7.1. JAK2 Mutations
7.2. FLT3 Mutations
7.3. KIT Mutations
7.4. CBL Mutations
7.5. Mutations in the RAS Pathway
7.6. Mutations in the Cohesin Gene Family
8. Other Novel Targeted Approaches
8.1. BCL-2 Inhibition
8.2. Polo-Like Kinases
8.3. Mitogen-Activated Protein Kinase (MAPK) and Mammalian Target of Rapamycin (mTOR)
8.4. Arsenic Trioxide
8.5. Monoclonal Antibodies and Novel Immunotherapy
9. Conclusions and Future Directions
Author Contributions
Conflicts of Interest
References
- Ades, L.; Itzykson, R.; Fenaux, P. Myelodysplastic syndromes. Lancet 2014, 383, 2239–2252. [Google Scholar] [CrossRef]
- Greenberg, P.; Cox, C.; LeBeau, M.M.; Fenaux, P.; Morel, P.; Sanz, G.; Sanz, M.; Vallespi, T.; Hamblin, T.; Oscier, D.; et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997, 89, 2079–2088. [Google Scholar] [PubMed]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Sole, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Ades, L. Review of azacitidine trials in intermediate-2-and high-risk myelodysplastic syndromes. Leuk. Res. 2009, 33, S7–S11. [Google Scholar] [CrossRef]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A.; et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. Lancet Oncol. 2009, 10, 223–232. [Google Scholar] [CrossRef]
- Kantarjian, H.; Oki, Y.; Garcia-Manero, G.; Huang, X.; O’Brien, S.; Cortes, J.; Faderl, S.; Bueso-Ramos, C.; Ravandi, F.; Estrov, Z.; et al. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood 2007, 109, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; O’Brien, S.; Huang, X.; Garcia-Manero, G.; Ravandi, F.; Cortes, J.; Shan, J.; Davisson, J.; Bueso-Ramos, C.E.; Issa, J.P. Survival advantage with decitabine versus intensive chemotherapy in patients with higher risk myelodysplastic syndrome: Comparison with historical experience. Cancer 2007, 109, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Baer, M.R.; Slack, J.L.; Buckstein, R.; Godley, L.A.; Garcia-Manero, G.; Albitar, M.; Larsen, J.S.; Arora, S.; Cullen, M.T.; et al. Multicenter study of decitabine administered daily for 5 days every 4 weeks to adults with myelodysplastic syndromes: The alternative dosing for outpatient treatment (ADOPT) trial. J. Clin. Oncol. 2009, 27, 3842–3848. [Google Scholar] [CrossRef] [PubMed]
- Prebet, T.; Gore, S.D.; Esterni, B.; Gardin, C.; Itzykson, R.; Thepot, S.; Dreyfus, F.; Rauzy, O.B.; Recher, C.; Ades, L.; et al. Outcome of high-risk myelodysplastic syndrome after azacitidine treatment failure. J. Clin. Oncol. 2011, 29, 3322–3327. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Garcia-Manero, G.; Batty, N.; Shan, J.; O’Brien, S.; Cortes, J.; Ravandi, F.; Issa, J.P.; Kantarjian, H. Outcome of patients with myelodysplastic syndrome after failure of decitabine therapy. Cancer 2010, 116, 3830–3834. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Padron, E.; Lancet, J. The molecular basis and clinical significance of genetic mutations identified in myelodysplastic syndromes. Leuk. Res. 2015, 39, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Harada, Y. Recent advances in myelodysplastic syndromes: Molecular pathogenesis and its implications for targeted therapies. Cancer Sci. 2015, 106, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R.; Steensma, D.P. Recent developments in myelodysplastic syndromes. Blood 2014, 124, 2793–2803. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Podoltsev, N.; Gore, S.D.; Zeidan, A.M. The evolving field of prognostication and risk stratification in MDS: Recent developments and future directions. Blood Rev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Meggendorfer, M.; de Albuquerque, A.; Nadarajah, N.; Alpermann, T.; Kern, W.; Steuer, K.; Perglerova, K.; Haferlach, C.; Schnittger, S.; Haferlach, T. Karyotype evolution and acquisition of FLT3 or RAS pathway alterations drive progression of myelodysplastic syndrome to acute myeloid leukemia. Haematologica 2015, 100, e487–e490. [Google Scholar] [CrossRef] [PubMed]
- Gill, H.; Man, C.H.; Ip, A.H.; Choi, W.W.; Chow, H.C.; Kwong, Y.L.; Leung, A.Y. Azacitidine as post-remission consolidation for sorafenib-induced remission of FMS-like tyrosine kinase-3 internal tandem duplication positive acute myeloid leukemia. Haematologica 2015, 100, e250–e253. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Delhommeau, F.; Dupont, S.; della Valle, V.; James, C.; Trannoy, S.; Masse, A.; Kosmider, O.; Le Couedic, J.P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Bravo, G.M.; Lee, E.; Merchan, B.; Kantarjian, H.M.; Garcia-Manero, G. Integrating genetics and epigenetics in myelodysplastic syndromes: Advances in pathogenesis and disease evolution. Br. J. Haematol. 2014, 166, 646–659. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R.; Levine, R.; Ebert, B.L. Unraveling the molecular pathophysiology of myelodysplastic syndromes. J. Clin. Oncol. 2011, 29, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, A.M.; Szpurka, H.; Tiu, R.V.; Makishima, H.; Afable, M.; Huh, J.; O’Keefe, C.L.; Ganetzky, R.; McDevitt, M.A.; Maciejewski, J.P. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood 2009, 113, 6403–6410. [Google Scholar] [CrossRef] [PubMed]
- Itzykson, R.; Kosmider, O.; Renneville, A.; Gelsi-Boyer, V.; Meggendorfer, M.; Morabito, M.; Berthon, C.; Ades, L.; Fenaux, P.; Beyne-Rauzy, O.; et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J. Clin. Oncol. 2013, 31, 2428–2436. [Google Scholar] [CrossRef] [PubMed]
- Kosmider, O.; Gelsi-Boyer, V.; Ciudad, M.; Racoeur, C.; Jooste, V.; Vey, N.; Quesnel, B.; Fenaux, P.; Bastie, J.N.; Beyne-Rauzy, O.; et al. TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica 2009, 94, 1676–1681. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R.; Stevenson, K.E.; Caughey, B.A.; Abdel-Wahab, O.; Steensma, D.P.; Galili, N.; Raza, A.; Kantarjian, H.; Levine, R.L.; Neuberg, D.; et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J. Clin. Oncol. 2012, 30, 3376–3382. [Google Scholar] [CrossRef] [PubMed]
- Jurkowska, R.Z.; Jurkowski, T.P.; Jeltsch, A. Structure and function of mammalian DNA methyltransferases. Chembiochemistry 2011, 12, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Sportoletti, P.; Brunetti, L.; Martelli, M.P. Perspectives for therapeutic targeting of gene mutations in acute myeloid leukemia with normal cytogenetics. Br. J. Haematol. 2015, 170, 305–322. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, A.; Benedetti, R.; Conte, M.; Iside, C.; Altucci, L. Genetic mutations in epigenetic modifiers as therapeutic targets in acute myeloid leukemia. Expert Opin. Ther. Targets 2015, 19, 1187–1202. [Google Scholar] [CrossRef] [PubMed]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef] [PubMed]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukemic hematopoietic stem cells in acute leukemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.J.; Ding, L.; Shen, D.; Shao, J.; Grillot, M.; McLellan, M.; Fulton, R.; Schmidt, H.; Kalicki-Veizer, J.; O’Laughlin, M.; et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia 2011, 25, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Kolquist, K.A.; Schultz, R.A.; Furrow, A.; Brown, T.C.; Han, J.Y.; Campbell, L.J.; Wall, M.; Slovak, M.L.; Shaffer, L.G.; Ballif, B.C. Microarray-based comparative genomic hybridization of cancer targets reveals novel, recurrent genetic aberrations in the myelodysplastic syndromes. Cancer Genet. 2011, 204, 603–628. [Google Scholar] [CrossRef] [PubMed]
- Traina, F.; Visconte, V.; Elson, P.; Tabarroki, A.; Jankowska, A.M.; Hasrouni, E.; Sugimoto, Y.; Szpurka, H.; Makishima, H.; O’Keefe, C.L.; et al. Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms. Leukemia 2014, 28, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, G.; Maharry, K.; Wu, Y.Z.; Radmacher, M.D.; Mrozek, K.; Margeson, D.; Holland, K.B.; Whitman, S.P.; Becker, H.; Schwind, S.; et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: A Cancer and Leukemia Group B study. J. Clin. Oncol. 2010, 28, 2348–2355. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, M.M.; Hanson, C.A.; Hodnefield, J.M.; Lasho, T.L.; Finke, C.M.; Knudson, R.A.; Ketterling, R.P.; Pardanani, A.; Tefferi, A. Differential prognostic effect of IDH1 versus IDH2 mutations in myelodysplastic syndromes: a Mayo Clinic study of 277 patients. Leukemia 2012, 26, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Hou, H.A.; Chou, W.C.; Kuo, Y.Y.; Liu, C.Y.; Chen, C.Y.; Lai, Y.J.; Tseng, M.H.; Huang, C.F.; Chiang, Y.C.; et al. IDH mutations are closely associated with mutations of DNMT3A, ASXL1 and SRSF2 in patients with myelodysplastic syndromes and are stable during disease evolution. Am. J. Hematol. 2014, 89, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, M.M.; Lasho, T.L.; Finke, C.M.; Gangat, N.; Caramazza, D.; Holtan, S.G.; Pardanani, A.; Knudson, R.A.; Ketterling, R.P.; Chen, D.; et al. WHO-defined “myelodysplastic syndrome with isolated del(5q)” in 88 consecutive patients: survival data, leukemic transformation rates and prevalence of JAK2, MPL and IDH mutations. Leukemia 2010, 24, 1283–1289. [Google Scholar] [CrossRef] [PubMed]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, S.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E.; et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Gross, S.; Shen, M.; Straley, K.S.; Pragani, R.; Lea, W.A.; Popovici-Muller, J.; DeLaBarre, B.; Artin, E.; Thorne, N.; et al. Biochemical, cellular, and biophysical characterization of a potent inhibitor of mutant isocitrate dehydrogenase IDH1. J. Biol. Chem. 2014, 289, 13717–13725. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Travins, J.; DeLaBarre, B.; Penard-Lacronique, V.; Schalm, S.; Hansen, E.; Straley, K.; Kernytsky, A.; Liu, W.; Gliser, C.; et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 2013, 340, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, O.; Figueroa, M.E. Interpreting new molecular genetics in myelodysplastic syndromes. Hematol. Am. Soc. Hematol. Educ. Program. 2012, 2012, 56–64. [Google Scholar]
- Lee, T.I.; Jenner, R.G.; Boyer, L.A.; Guenther, M.G.; Levine, S.S.; Kumar, R.M.; Chevalier, B.; Johnstone, S.E.; Cole, M.F.; Isono, K.; et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 2006, 125, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Gelsi-Boyer, V.; Trouplin, V.; Adelaide, J.; Bonansea, J.; Cervera, N.; Carbuccia, N.; Lagarde, A.; Prebet, T.; Nezri, M.; Sainty, D.; et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukemia. Br. J. Haematol. 2009, 145, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Gelsi-Boyer, V.; Trouplin, V.; Roquain, J.; Adélaïde, J.; Carbuccia, N.; Esterni, B.; Finetti, P.; Murati, A.; Arnoulet, C.; Zerazhi, H.; et al. ASXL1 mutation is associated with poor prognosis and acute transformation in chronic myelomonocytic leukemia. Br. J. Haematol. 2010, 151, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Agger, K.; Cloos, P.A.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Thieme, S.; Gyarfas, T.; Richter, C.; Ozhan, G.; Fu, J.; Alexopoulou, D.; Muders, M.H.; Michalk, I.; Jakob, C.; Dahl, A.; et al. The histone demethylase UTX regulates stem cell migration and hematopoiesis. Blood 2013, 121, 2462–2473. [Google Scholar] [CrossRef] [PubMed]
- Padgett, R.A. New connections between splicing and human disease. Trends Genet. 2012, 28, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.S.; Hellstrom-Lindberg, E.; Gambacorti-Passerini, C.; et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N. Engl. J. Med. 2011, 365, 1384–1395. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, M.M.; Lasho, T.L.; Hodnefield, J.M.; Knudson, R.A.; Ketterling, R.P.; Garcia-Manero, G.; Steensma, D.P.; Pardanani, A.; Hanson, C.A.; Tefferi, A. SF3B1 mutations are prevalent in myelodysplastic syndromes with ring sideroblasts but do not hold independent prognostic value. Blood 2012, 119, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Makishima, H.; Visconte, V.; Sakaguchi, H.; Jankowska, A.M.; Abu Kar, S.; Jerez, A.; Przychodzen, B.; Bupathi, M.; Guinta, K.; Afable, M.G.; et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 2012, 119, 3203–3210. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.J.; Kuo, Y.Y.; Hou, H.A.; Li, L.Y.; Tseng, M.H.; Huang, C.F.; Lee, F.Y.; Liu, M.C.; Liu, C.W.; Lin, C.T.; et al. The clinical implication of SRSF2 mutation in patients with myelodysplastic syndrome and its stability during disease evolution. Blood 2012, 120, 3106–3111. [Google Scholar] [CrossRef] [PubMed]
- Thol, F.; Kade, S.; Schlarmann, C.; Loffeld, P.; Morgan, M.; Krauter, J.; Wlodarski, M.W.; Kolking, B.; Wichmann, M.; Gorlich, K.; et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood 2012, 119, 3578–3584. [Google Scholar] [CrossRef] [PubMed]
- Graubert, T.A.; Shen, D.; Ding, L.; Okeyo-Owuor, T.; Lunn, C.L.; Shao, J.; Krysiak, K.; Harris, C.C.; Koboldt, D.C.; Larson, D.E.; et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat. Genet. 2012, 44, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Song, W.J.; Sullivan, M.G.; Legare, R.D.; Hutchings, S.; Tan, X.; Kufrin, D.; Ratajczak, J.; Resende, I.C.; Haworth, C.; Hock, R.; et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukemia. Nat. Genet. 1999, 23, 166–175. [Google Scholar] [PubMed]
- Harada, H.; Harada, Y.; Niimi, H.; Kyo, T.; Kimura, A.; Inaba, T. High incidence of somatic mutations in the AML1/RUNX1 gene in myelodysplastic syndrome and low blast percentage myeloid leukemia with myelodysplasia. Blood 2004, 103, 2316–2324. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Gibbons, R.J.; Mesa, R.A.; Tefferi, A.; Higgs, D.R. Somatic point mutations in RUNX1/CBFA2/AML1 are common in high-risk myelodysplastic syndrome, but not in myelofibrosis with myeloid metaplasia. Eur. J. Haematol. 2005, 74, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K.D.; Fischle, W.; Verdin, E.; Bardwell, V.J. BCoR, a novel corepressor involved in BCL-6 repression. Genes Dev. 2000, 14, 1810–1823. [Google Scholar] [PubMed]
- Li, M.; Collins, R.; Jiao, Y.; Ouillette, P.; Bixby, D.; Erba, H.; Vogelstein, B.; Kinzler, K.W.; Papadopoulos, N.; Malek, S.N. Somatic mutations in the transcriptional corepressor gene BCORL1 in adult acute myelogenous leukemia. Blood 2011, 118, 5914–5917. [Google Scholar] [CrossRef] [PubMed]
- Pagan, J.K.; Arnold, J.; Hanchard, K.J.; Kumar, R.; Bruno, T.; Jones, M.J.; Richard, D.J.; Forrest, A.; Spurdle, A.; Verdin, E.; et al. A novel corepressor, BCOR-L1, represses transcription through an interaction with CtBP. J. Biol. Chem. 2007, 282, 15248–15257. [Google Scholar] [CrossRef] [PubMed]
- Damm, F.; Chesnais, V.; Nagata, Y.; Yoshida, K.; Scourzic, L.; Okuno, Y.; Itzykson, R.; Sanada, M.; Shiraishi, Y.; Gelsi-Boyer, V.; et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood 2013, 122, 3169–3177. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, C.; Bacher, U.; Schnittger, S.; Alpermann, T.; Zenger, M.; Kern, W.; Haferlach, T. ETV6 rearrangements are recurrent in myeloid malignancies and are frequently associated with other genetic events. Genes Chromosomes Cancer 2012, 51, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Inoue, D.; Kitaura, J.; Matsui, H.; Hou, H.A.; Chou, W.C.; Nagamachi, A.; Kawabata, K.C.; Togami, K.; Nagase, R.; Horikawa, S.; et al. SETBP1 mutations drive leukemic transformation in ASXL1-mutated MDS. Leukemia 2015, 29, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.A.; Kuo, Y.Y.; Tang, J.L.; Chou, W.C.; Yao, M.; Lai, Y.J.; Lin, C.C.; Chen, C.Y.; Liu, C.Y.; Tseng, M.H.; et al. Clinical implications of the SETBP1 mutation in patients with primary myelodysplastic syndrome and its stability during disease progression. Am. J. Hematol. 2014, 89, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Petitjean, A.; Achatz, M.I.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. TP53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Petitjean, A.; Mathe, E.; Kato, S.; Ishioka, C.; Tavtigian, S.V.; Hainaut, P.; Olivier, M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007, 28, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Hirano, N.; Toyoshima, H.; Chiba, S.; Mano, H.; Takaku, F.; Yazaki, Y.; Hirai, H. Mutations of the p53 gene in myelodysplastic syndrome (MDS) and MDS-derived leukemia. Blood 1993, 81, 3022–3026. [Google Scholar] [PubMed]
- Caceres, G.; McGraw, K.; Yip, B.H.; Pellagatti, A.; Johnson, J.; Zhang, L.; Liu, K.; Zhang, L.M.; Fulp, W.J.; Lee, J.H.; et al. TP53 suppression promotes erythropoiesis in del(5q) MDS, suggesting a targeted therapeutic strategy in lenalidomide-resistant patients. Proc. Natl. Acad. Sci. USA 2013, 110, 16127–16132. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Cortes, J.E. New treatment for acute myelogenous leukemia. Expert Opin. Pharm. 2015, 16, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Eghtedar, A.; Verstovsek, S.; Estrov, Z.; Burger, J.; Cortes, J.; Bivins, C.; Faderl, S.; Ferrajoli, A.; Borthakur, G.; George, S.; et al. Phase 2 study of the JAK kinase inhibitor ruxolitinib in patients with refractory leukemias, including postmyeloproliferative neoplasm acute myeloid leukemia. Blood 2012, 119, 4614–4618. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.Y.; Man, C.H.; Kwong, Y.L. FLT3 inhibition: A moving and evolving target in acute myeloid leukaemia. Leukemia 2013, 27, 260–268. [Google Scholar] [CrossRef] [PubMed]
- He, B.L.; Man, C.H.; Shi, X.G.; Ma, A.C.H.; Leung, A.Y.H. A Zebrafish Model of FMS-Like Tyrosine Kinase 3 (FLT3) in definitive hematopoiesis and human acute myeloid leukemia. Exp. Hematol. 2013, 41, S48–S48. [Google Scholar] [CrossRef]
- Man, C.H.; Fung, T.K.; Ho, C.; Han, H.H.; Chow, H.C.; Ma, A.C.; Choi, W.W.; Lok, S.; Cheung, A.M.; Eaves, C.; et al. Sorafenib treatment of FLT3-ITD+ acute myeloid leukemia: favorable initial outcome and mechanisms of subsequent nonresponsiveness associated with the emergence of a D835 mutation. Blood 2012, 119, 5133–5143. [Google Scholar] [CrossRef] [PubMed]
- Frohling, S.; Schlenk, R.F.; Breitruck, J.; Benner, A.; Kreitmeier, S.; Tobis, K.; Dohner, H.; Dohner, K. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: A study of the AML Study Group Ulm. Blood 2002, 100, 4372–4380. [Google Scholar] [CrossRef] [PubMed]
- Schnittger, S.; Schoch, C.; Dugas, M.; Kern, W.; Staib, P.; Wuchter, C.; Loffler, H.; Sauerland, C.M.; Serve, H.; Buchner, T.; et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: Correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood 2002, 100, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Kiyoi, H.; Towatari, M.; Yokota, S.; Hamaguchi, M.; Ohno, R.; Saito, H.; Naoe, T. Internal tandem duplication of the FLT3 gene is a novel modality of elongation mutation which causes constitutive activation of the product. Leukemia 1998, 12, 1333–1337. [Google Scholar] [CrossRef] [PubMed]
- Leischner, H.; Albers, C.; Grundler, R.; Razumovskaya, E.; Spiekermann, K.; Bohlander, S.; Ronnstrand, L.; Gotze, K.; Peschel, C.; Duyster, J. SRC is a signaling mediator in FLT3-ITD-but not in FLT3-TKD-positive AML. Blood 2012, 119, 4026–4033. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.P.; Jones, D.; Qiao, W.; Cortes, J.E.; Ravandi, F.; Estey, E.E.; Verma, D.; Kantarjian, H.; Borthakur, G. Prognostic value of FLT3 mutations among different cytogenetic subgroups in acute myeloid leukemia. Cancer 2011, 117, 2145–2155. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Strati, P.; Jabbour, E.; Kadia, T.; Luthra, R.; Wang, S.; Patel, K.; Ravandi, F.; Cortes, J.; Dong, X.Q.; et al. FLT3 mutations in myelodysplastic syndrome and chronic myelomonocytic leukemia. Am. J. Hematol. 2013, 88, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Horiike, S.; Yokota, S.; Nakao, M.; Iwai, T.; Sasai, Y.; Kaneko, H.; Taniwaki, M.; Kashima, K.; Fujii, H.; Abe, T.; et al. Tandem duplications of the FLT3 receptor gene are associated with leukemic transformation of myelodysplasia. Leukemia 1997, 11, 1442–1446. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, R.F.; de Sa Moreira, E.; Silva, M.R.; Alberto, F.L.; Chauffaille Mde, L. FLT3 internal tandem duplication during myelodysplastic syndrome follow-up: A marker of transformation to acute myeloid leukemia. Cancer Genet. Cytogenet. 2008, 183, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Bacher, U.; Haferlach, T.; Kern, W.; Haferlach, C.; Schnittger, S. A comparative study of molecular mutations in 381 patients with myelodysplastic syndrome and in 4130 patients with acute myeloid leukemia. Haematologica 2007, 92, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Bains, A.; Luthra, R.; Medeiros, L.J.; Zuo, Z. FLT3 and NPM1 mutations in myelodysplastic syndromes: Frequency and potential value for predicting progression to acute myeloid leukemia. Am. J. Clin. Pathol. 2011, 135, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Dicker, F.; Haferlach, C.; Sundermann, J.; Wendland, N.; Weiss, T.; Kern, W.; Haferlach, T.; Schnittger, S. Mutation analysis for RUNX1, MLL-PTD, FLT3-ITD, NPM1 and NRAS in 269 patients with MDS or secondary AML. Leukemia 2010, 24, 1528–1532. [Google Scholar] [CrossRef] [PubMed]
- Badar, T.; Patel, K.P.; Thompson, P.A.; DiNardo, C.; Takahashi, K.; Cabrero, M.; Borthakur, G.; Cortes, J.; Konopleva, M.; Kadia, T.; et al. Detectable FLT3-ITD or RAS mutation at the time of transformation from MDS to AML predicts for very poor outcomes. Leuk. Res. 2015, 39, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Shih, L.Y.; Huang, C.F.; Wang, P.N.; Wu, J.H.; Lin, T.L.; Dunn, P.; Kuo, M.C. Acquisition of FLT3 or N-ras mutations is frequently associated with progression of myelodysplastic syndrome to acute myeloid leukemia. Leukemia 2004, 18, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, G.; Karali, V.; Zouvelou, C.; Kyriakou, E.; Dimou, M.; Chrisochoou, S.; Greka, P.; Dufexis, D.; Vervesou, E.; Dimitriadou, E.; et al. Serial determination of FLT3 mutations in myelodysplastic syndrome patients at diagnosis, follow up or acute myeloid leukaemia transformation: Incidence and their prognostic significance. Br. J. Haematol. 2006, 134, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Jabbour, E.; Wang, X.; Luthra, R.; Bueso-Ramos, C.; Patel, K.; Pierce, S.; Yang, H.; Wei, Y.; Daver, N.; et al. Dynamic acquisition of FLT3 or RAS alterations drive a subset of patients with lower risk MDS to secondary AML. Leukemia 2013, 27, 2081–2083. [Google Scholar] [CrossRef] [PubMed]
- Konig, H.; Levis, M. Targeting FLT3 to treat leukemia. Expert Opin. Ther. Targets 2015, 19, 37–54. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.; Stone, R.M.; Deangelo, D.J.; Galinsky, I.; Estey, E.; Lanza, C.; Fox, E.; Ehninger, G.; Feldman, E.J.; Schiller, G.J.; et al. Phase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J. Clin. Oncol. 2010, 28, 4339–4345. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; DeAngelo, D.J.; Klimek, V.; Galinsky, I.; Estey, E.; Nimer, S.D.; Grandin, W.; Lebwohl, D.; Wang, Y.; Cohen, P.; et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood 2005, 105, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.D.; Levis, M.; Beran, M.; Giles, F.; Kantarjian, H.; Berg, K.; Murphy, K.M.; Dauses, T.; Allebach, J.; Small, D. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood 2004, 103, 3669–3676. [Google Scholar] [CrossRef] [PubMed]
- Levis, M.; Ravandi, F.; Wang, E.S.; Baer, M.R.; Perl, A.; Coutre, S.; Erba, H.; Stuart, R.K.; Baccarani, M.; Cripe, L.D.; et al. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood 2011, 117, 3294–3301. [Google Scholar] [CrossRef] [PubMed]
- Knapper, S.; Burnett, A.K.; Littlewood, T.; Kell, W.J.; Agrawal, S.; Chopra, R.; Clark, R.; Levis, M.J.; Small, D. A phase 2 trial of the FLT3 inhibitor lestaurtinib (CEP701) as first-line treatment for older patients with acute myeloid leukemia not considered fit for intensive chemotherapy. Blood 2006, 108, 3262–3270. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, W.; Serve, H.; Dohner, H.; Schwittay, M.; Ottmann, O.G.; O’Farrell, A.M.; Bello, C.L.; Allred, R.; Manning, W.C.; Cherrington, J.M.; et al. A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood 2005, 105, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Pratz, K.W.; Cortes, J.; Roboz, G.J.; Rao, N.; Arowojolu, O.; Stine, A.; Shiotsu, Y.; Shudo, A.; Akinaga, S.; Small, D.; et al. A pharmacodynamic study of the FLT3 inhibitor KW-2449 yields insight into the basis for clinical response. Blood 2009, 113, 3938–3946. [Google Scholar] [CrossRef] [PubMed]
- Zarrinkar, P.P.; Gunawardane, R.N.; Cramer, M.D.; Gardner, M.F.; Brigham, D.; Belli, B.; Karaman, M.W.; Pratz, K.W.; Pallares, G.; Chao, Q.; et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood 2009, 114, 2984–2992. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Konopleva, M.; Shi, Y.X.; McQueen, T.; Harris, D.; Ling, X.; Estrov, Z.; Quintas-Cardama, A.; Small, D.; Cortes, J.; et al. Mutant FLT3: A direct target of sorafenib in acute myelogenous leukemia. J. Natl. Cancer Inst. 2008, 100, 184–198. [Google Scholar] [CrossRef] [PubMed]
- Ravenscroft, G.; Jackaman, C.; Bringans, S.; Papadimitriou, J.M.; Griffiths, L.M.; McNamara, E.; Bakker, A.J.; Davies, K.E.; Laing, N.G.; Nowak, K.J. Mouse models of dominant ACTA1 disease recapitulate human disease and provide insight into therapies. Brain 2011, 134, 1101–1115. [Google Scholar] [CrossRef] [PubMed]
- Borthakur, G.; Kantarjian, H.; Ravandi, F.; Zhang, W.; Konopleva, M.; Wright, J.J.; Faderl, S.; Verstovsek, S.; Mathews, S.; Andreeff, M.; et al. Phase I study of sorafenib in patients with refractory or relapsed acute leukemias. Haematologica 2011, 96, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Metzelder, S.; Wang, Y.; Wollmer, E.; Wanzel, M.; Teichler, S.; Chaturvedi, A.; Eilers, M.; Enghofer, E.; Neubauer, A.; Burchert, A. Compassionate use of sorafenib in FLT3-ITD-positive acute myeloid leukemia: Sustained regression before and after allogeneic stem cell transplantation. Blood 2009, 113, 6567–6571. [Google Scholar] [CrossRef] [PubMed]
- Pratz, K.W.; Cho, E.; Levis, M.J.; Karp, J.E.; Gore, S.D.; McDevitt, M.; Stine, A.; Zhao, M.; Baker, S.D.; Carducci, M.A.; et al. A pharmacodynamic study of sorafenib in patients with relapsed and refractory acute leukemias. Leukemia 2010, 24, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; Fischer, T.; Paquette, R.; Schiller, G.; Schiffer, C.A.; Ehninger, G.; Cortes, J.; Kantarjian, H.M.; DeAngelo, D.J.; Huntsman-Labed, A.; et al. Phase IB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemia. Leukemia 2012, 26, 2061–2068. [Google Scholar] [CrossRef] [PubMed]
- Metzelder, S.K.; Schroeder, T.; Finck, A.; Scholl, S.; Fey, M.; Gotze, K.; Linn, Y.C.; Kroger, M.; Reiter, A.; Salih, H.R.; et al. High activity of sorafenib in FLT3-ITD-positive acute myeloid leukemia synergizes with allo-immune effects to induce sustained responses. Leukemia 2012, 26, 2353–2359. [Google Scholar] [CrossRef] [PubMed]
- Hatzimichael, E.; Tsolas, E.; Briasoulis, E. Profile of pacritinib and its potential in the treatment of hematologic disorders. J. Blood Med. 2014, 5, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Silverman, L.R.; Demakos, E.P.; Peterson, B.L.; Kornblith, A.B.; Holland, J.C.; Odchimar-Reissig, R.; Stone, R.M.; Nelson, D.; Powell, B.L.; DeCastro, C.M.; et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: A study of the cancer and leukemia group B. J. Clin. Oncol. 2002, 20, 2429–2440. [Google Scholar] [CrossRef] [PubMed]
- Cashen, A.F.; Schiller, G.J.; O’Donnell, M.R.; DiPersio, J.F. Multicenter, phase II study of decitabine for the first-line treatment of older patients with acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Gattermann, N.; Seymour, J. F.; Hellstrom-Lindberg, E.; Mufti, G.J.; Duehrsen, U.; Gore, S.D.; Ramos, F.; Beyne-Rauzy, O.; List, A.; et al. Prolonged survival with improved tolerability in higher-risk myelodysplastic syndromes: Azacitidine compared with low dose ara-C. Br. J. Haematol. 2010, 149, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Bowen, D.; Gattermann, N.; Hellstrom-Lindberg, E.; Hofmann, W.K.; Pfeilstocker, M.; Sanz, G.; Santini, V. Practical use of azacitidine in higher-risk myelodysplastic syndromes: An expert panel opinion. Leuk. Res. 2010, 34, 1410–1416. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Gattermann, N.; Germing, U.; Sanz, G.; List, A.F.; Gore, S.; Seymour, J.F.; et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Itzykson, R.; Thepot, S.; Quesnel, B.; Dreyfus, F.; Beyne-Rauzy, O.; Turlure, P.; Vey, N.; Recher, C.; Dartigeas, C.; Legros, L.; et al. Prognostic factors for response and overall survival in 282 patients with higher-risk myelodysplastic syndromes treated with azacitidine. Blood 2011, 117, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Seymour, J.F.; Fenaux, P.; Silverman, L.R.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; List, A.F.; Gore, S.D.; Backstrom, J.; McKenzie, D.; et al. Effects of azacitidine compared with conventional care regimens in elderly (≥75 years) patients with higher-risk myelodysplastic syndromes. Crit. Rev. Oncol. Hematol. 2010, 76, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Maurillo, L.; Venditti, A.; Spagnoli, A.; Gaidano, G.; Ferrero, D.; Oliva, E.; Lunghi, M.; D’Arco, A.M.; Levis, A.; Pastore, D.; et al. Azacitidine for the treatment of patients with acute myeloid leukemia: Report of 82 patients enrolled in an Italian Compassionate Program. Cancer 2012, 118, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Soncini, M.; Santoro, F.; Gutierrez, A.; Frige, G.; Romanenghi, M.; Botrugno, O.A.; Pallavicini, I.; Pelicci, P.; di Croce, L.; Minucci, S. The DNA demethylating agent decitabine activates the TRAIL pathway and induces apoptosis in acute myeloid leukemia. Biochim. Biophys. Acta 2013, 1832, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Hollenbach, P.W.; Nguyen, A.N.; Brady, H.; Williams, M.; Ning, Y.; Richard, N.; Krushel, L.; Aukerman, S.L.; Heise, C.; MacBeth, K.J. A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS ONE 2010, 5, e9001. [Google Scholar] [CrossRef] [PubMed]
- Negrotto, S.; Ng, K.P.; Jankowska, A.M.; Bodo, J.; Gopalan, B.; Guinta, K.; Mulloy, J.C.; Hsi, E.; Maciejewski, J.; Saunthararajah, Y. CpG methylation patterns and decitabine treatment response in acute myeloid leukemia cells and normal hematopoietic precursors. Leukemia 2012, 26, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Ravandi, F.; Alattar, M.L.; Grunwald, M.R.; Rudek, M.A.; Rajkhowa, T.; Richie, M.A.; Pierce, S.; Daver, N.; Garcia-Manero, G.; Faderl, S.; et al. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood 2013, 121, 4655–4662. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Yang, X.; Knapper, S.; White, P.; Smith, B.D.; Galkin, S.; Small, D.; Burnett, A.; Levis, M. FLT3 ligand impedes the efficacy of FLT3 inhibitors in vitro and in vivo. Blood 2011, 117, 3286–3293. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Bailey, E.; Nguyen, B.; Yang, X.; Piloto, O.; Levis, M.; Small, D. Further activation of FLT3 mutants by FLT3 ligand. Oncogene 2011, 30, 4004–4014. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.B.; Kambhampati, S.; Fiskus, W.; Wick, J.; Dutreix, C.; Ganguly, S.; Aljitawi, O.; Reyes, R.; Fleming, A.; Abhyankar, S.; et al. Preclinical and phase I results of decitabine in combination with midostaurin (PKC412) for newly diagnosed elderly or relapsed/refractory adult patients with acute myeloid leukemia. Pharmacotherapy 2013, 33, 1341–1352. [Google Scholar] [CrossRef] [PubMed]
- Solanilla, A.; Grosset, C.; Lemercier, C.; Dupouy, M.; Mahon, F.X.; Schweitzer, K.; Reiffers, J.; Weksler, B.; Ripoche, J. Expression of FLT3-ligand by the endothelial cell. Leukemia 2000, 14, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Chklovskaia, E.; Nissen, C.; Landmann, L.; Rahner, C.; Pfister, O.; Wodnar-Filipowicz, A. Cell-surface trafficking and release of FLT3 ligand from T lymphocytes is induced by common cytokine receptor gamma-chain signaling and inhibited by cyclosporin A. Blood 2001, 97, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Levis, M.; Piloto, O.; Brown, P.; Baldwin, B.R.; Gorin, N.C.; Beran, M.; Zhu, Z.; Ludwig, D.; Hicklin, D.; et al. FLT3 ligand causes autocrine signaling in acute myeloid leukemia cells. Blood 2004, 103, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Drexler, H.G. Expression of FLT3 receptor and response to FLT3 ligand by leukemic cells. Leukemia 1996, 10, 588–599. [Google Scholar] [PubMed]
- Daver, N.; Cortes, J.; Ravandi, F.; Patel, K.P.; Burger, J.A.; Konopleva, M.; Kantarjian, H. Secondary mutations as mediators of resistance to targeted therapy in leukemia. Blood 2015, 125, 3236–3245. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Kim, D.W.; Pinilla-Ibarz, J.; le Coutre, P.; Paquette, R.; Chuah, C.; Nicolini, F.E.; Apperley, J.F.; Khoury, H.J.; Talpaz, M.; et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 2013, 369, 1783–1796. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.P.; Talpaz, M.; Deininger, M.W.; Mauro, M.J.; Flinn, I.W.; Bixby, D.; Lustgarten, S.; Gozgit, J.M.; Clackson, T.; Turner, C.D.; et al. Ponatinib in patients with refractory acute myeloid leukaemia: Findings from a phase 1 study. Br. J. Haematol. 2013, 162, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Gozgit, J.M.; Wong, M.J.; Wardwell, S.; Tyner, J.W.; Loriaux, M.M.; Mohemmad, Q.K.; Narasimhan, N.I.; Shakespeare, W.C.; Wang, F.; Druker, B.J.; et al. Potent activity of ponatinib (AP24534) in models of FLT3-driven acute myeloid leukemia and other hematologic malignancies. Mol. Cancer Ther. 2011, 10, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Fathi, A.T. Emergence of crenolanib for FLT3-mutant AML. Blood 2013, 122, 3547–3548. [Google Scholar] [CrossRef] [PubMed]
- Galanis, A.; Ma, H.; Rajkhowa, T.; Ramachandran, A.; Small, D.; Cortes, J.; Levis, M. Crenolanib is a potent inhibitor of FLT3 with activity against resistance-conferring point mutants. Blood 2014, 123, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Pemmaraju, N.; Kantarjian, H.; Andreeff, M.; Cortes, J.; Ravandi, F. Investigational FMS-like tyrosine kinase 3 inhibitors in treatment of acute myeloid leukemia. Expert Opin. Investig. Drugs 2014, 23, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.C.; Levine, R.L. Translational implications of somatic genomics in acute myeloid leukaemia. Lancet Oncol. 2014, 15, e382–e394. [Google Scholar] [CrossRef]
- Paschka, P.; Marcucci, G.; Ruppert, A.S.; Mrozek, K.; Chen, H.; Kittles, R.A.; Vukosavljevic, T.; Perrotti, D.; Vardiman, J.W.; Carroll, A.J.; et al. Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8;21): A Cancer and Leukemia Group B Study. J. Clin. Oncol. 2006, 24, 3904–3911. [Google Scholar] [CrossRef] [PubMed]
- Kohlmann, A.; Grossmann, V.; Klein, H.U.; Schindela, S.; Weiss, T.; Kazak, B.; Dicker, F.; Schnittger, S.; Dugas, M.; Kern, W.; et al. Next-generation sequencing technology reveals a characteristic pattern of molecular mutations in 72.8% of chronic myelomonocytic leukemia by detecting frequent alterations in TET2, CBL, RAS, and RUNX1. J. Clin. Oncol. 2010, 28, 3858–3865. [Google Scholar] [CrossRef] [PubMed]
- Schwaab, J.; Ernst, T.; Erben, P.; Rinke, J.; Schnittger, S.; Strobel, P.; Metzgeroth, G.; Mossner, M.; Haferlach, T.; Cross, N.C.; et al. Activating CBL mutations are associated with a distinct MDS/MPN phenotype. Ann. Hematol. 2012, 91, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Kao, H.W.; Sanada, M.; Liang, D.C.; Lai, C.L.; Lee, E.H.; Kuo, M.C.; Lin, T.L.; Shih, Y.S.; Wu, J.H.; Huang, C.F.; et al. A high occurrence of acquisition and/or expansion of C-CBL mutant clones in the progression of high-risk myelodysplastic syndrome to acute myeloid leukemia. Neoplasia 2011, 13, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Bogenberger, J.M.; Kornblau, S.M.; Pierceall, W.E.; Lena, R.; Chow, D.; Shi, C.X.; Mantei, J.; Ahmann, G.; Gonzales, I.M.; Choudhary, A.; et al. BCL-2 family proteins as 5-Azacytidine-sensitizing targets and determinants of response in myeloid malignancies. Leukemia 2014, 28, 1657–1665. [Google Scholar] [CrossRef] [PubMed]
- Jilg, S.; Reidel, V.; Muller-Thomas, C.; Konig, J.; Schauwecker, J.; Hockendorf, U.; Huberle, C.; Gorka, O.; Schmidt, B.; Burgkart, R.; et al. Blockade of BCL-2 proteins efficiently induces apoptosis in progenitor cells of high-risk myelodysplastic syndromes patients. Leukemia 2016, 30, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Bogenberger, J.M.; Delman, D.; Hansen, N.; Valdez, R.; Fauble, V.; Mesa, R.A.; Tibes, R. Ex vivo activity of BCL-2 family inhibitors ABT-199 and ABT-737 combined with 5-azacytidine in myeloid malignancies. Leuk. Lymphoma. 2015, 56, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Slape, C.I.; Saw, J.; Jowett, J.B.; Aplan, P.D.; Strasser, A.; Jane, S.M.; Curtis, D.J. Inhibition of apoptosis by BCL2 prevents leukemic transformation of a murine myelodysplastic syndrome. Blood 2012, 120, 2475–2483. [Google Scholar] [CrossRef] [PubMed]
- Cluzeau, T.; Robert, G.; Mounier, N.; Karsenti, J.M.; Dufies, M.; Puissant, A.; Jacquel, A.; Renneville, A.; Preudhomme, C.; Cassuto, J.P.; et al. BCL2L10 is a predictive factor for resistance to azacitidine in MDS and AML patients. Oncotarget 2012, 3, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Cluzeau, T.; Robert, G.; Puissant, A.; Jean-Michel, K.; Cassuto, J.P.; Raynaud, S.; Auberger, P. Azacitidine-resistant SKM1 myeloid cells are defective for AZA-induced mitochondrial apoptosis and autophagy. Cell Cycle 2011, 10, 2339–2343. [Google Scholar] [CrossRef] [PubMed]
- Gjertsen, B.T.; Schoffski, P. Discovery and development of the Polo-like kinase inhibitor volasertib in cancer therapy. Leukemia 2015, 29, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Sekeres, M.A. Arsenic trioxide as a treatment for myelodysplastic syndrome. Curr. Hematol. Malig. Rep. 2006, 1, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Wang, Y.; Tong, H.; Qian, W.; Jin, J. Downregulation of hTERT: An important As2O3 induced mechanism of apoptosis in myelodysplastic syndrome. PLoS ONE 2014, 9, e113199. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, S.; Guerrini, F.; Salvi, F.; Petrini, I.; Gioia, D.; Messa, E.; Palumbo, G.A.; Cilloni, D.; Petrini, M.; Levis, A. Arsenic trioxide and ascorbic acid interfere with the BCL2 family genes in patients with myelodysplastic syndromes: An ex vivo study. J. Hematol. Oncol. 2012, 5. [Google Scholar] [CrossRef] [PubMed]
- Schiller, G.J.; Slack, J.; Hainsworth, J.D.; Mason, J.; Saleh, M.; Rizzieri, D.; Douer, D.; List, A.F. Phase II multicenter study of arsenic trioxide in patients with myelodysplastic syndromes. J. Clin. Oncol. 2006, 24, 2456–2464. [Google Scholar] [CrossRef] [PubMed]
- Vey, N.; Bosly, A.; Guerci, A.; Feremans, W.; Dombret, H.; Dreyfus, F.; Bowen, D.; Burnett, A.; Dennis, M.; Ribrag, V.; et al. Arsenic trioxide in patients with myelodysplastic syndromes: A phase II multicenter study. J. Clin. Oncol. 2006, 24, 2465–2471. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Zhou, F.; Zhang, Y.; Guo, L.; Shi, H.; Hou, J. A combination of thalidomide and arsenic trioxide is effective and well tolerated in patients with myelodysplastic syndromes. Leuk. Res. 2012, 36, 715–719. [Google Scholar] [CrossRef] [PubMed]
- Sekeres, M.A.; Maciejewski, J.P.; Erba, H.P.; Afable, M.; Englehaupt, R.; Sobecks, R.; Advani, A.; Seel, S.; Chan, J.; Kalaycio, M.E. A Phase 2 study of combination therapy with arsenic trioxide and gemtuzumab ozogamicin in patients with myelodysplastic syndromes or secondary acute myeloid leukemia. Cancer 2011, 117, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.S.; Klco, J.M.; Gao, F.; Procknow, E.; Uy, G.L.; Stockerl-Goldstein, K.E.; Abboud, C.N.; Westervelt, P.; DiPersio, J.F.; Hassan, A.; et al. Combination decitabine, arsenic trioxide, and ascorbic acid for the treatment of myelodysplastic syndrome and acute myeloid leukemia: A phase I study. Am. J. Hematol. 2011, 86, 796–800. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Gionfriddo, I.; Cecchetti, F.; Ballanti, S.; Pettirossi, V.; Martelli, M.P. Acute myeloid leukemia with mutated nucleophosmin (NPM1): Any hope for a targeted therapy? Blood Rev. 2011, 25, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Bueso-Ramos, C.; DiNardo, C.; Estecio, M. R.; Davanlou, M.; Geng, Q.R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2014, 28, 1280–1288. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.J.; Shen, D.; Ding, L.; Shao, J.; Koboldt, D.C.; Chen, K.; Larson, D.E.; McLellan, M.D.; Dooling, D.; Abbott, R.; et al. Clonal architecture of secondary acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Kwok, B.; Hall, J.M.; Witte, J.S.; Xu, Y.; Reddy, P.; Lin, K.; Flamholz, R.; Dabbas, B.; Yung, A.; Al-Hafidh, J.; et al. MDS associated somatic mutations and clonal hematopoiesis are common in idiopathic cytopenia of undetermined significance. Blood 2015, 126, 2355–2361. [Google Scholar] [CrossRef] [PubMed]
- Bejar, R.; Stevenson, K.E.; Caughey, B.; Lindsley, R.C.; Mar, B.G.; Stojanov, P.; Getz, G.; Steensma, D.P.; Ritz, J.; Soiffer, R.; et al. Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem-cell transplantation. J. Clin. Oncol. 2014, 32, 2691–2698. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Study | Patient No. | Treatment | FLT3 Mutations at Dx | sAML No. | FLT3 Mutations at sAML | HMA Use in Acquired FLT3 Mutation |
|---|---|---|---|---|---|---|
| Shih et al. [86] | 70 * | Supportive; Ara-C; oral chemotherapy | ITD: 3 (4.3%) | 70 * | ITD: 10 (14.2%) | No |
| TKD: 1 (1.4%) | TKD: 4 (5.7%) | |||||
| Georgiou et al. [87] | 97 | Not reported | ITD: 1 (1%) | 42 | ITD: 5 /42 (12%) | Not reported |
| TKD 1 (1%) | TKD: 1/42 (2.4%) | |||||
| ITD + FLT-TKD: 1 (1%) | ITD + TKD: 1/42 (2.4%) | |||||
| Georgiou et al. [81] | 50 | Not reported | ITD: 0 | 2 | ITD: 2 # | Not reported |
| Dicker et al. [84] | 20 ** | Supportive care | ITD: 0 | 20 ** | ITD: 4 (20%) | No |
| Takahashi et al. [88] | 278 | Supportive/HMA/induction chemotherapy/HSCT | ITD: 2 | 74 | ITD: 11 (15%) | ITD: 6/11 (54.5%) |
| TKD: 2 | TKD: 4 (5.4%) | TKD: 2/4 (50%) | ||||
| Badar et al. [85] | 102 | HMA: 75 (73%) | ITD: 0 | 27 | ITD: 5 (19%) | Not reported |
| Immunomodulators: 7 (7%) | TKD: 0 | |||||
| Growth factors: 4 (4%) | ||||||
| HSCT: 10 (10%) | ||||||
| Others: 16 (16%) | ||||||
| Meggendorfer et al. [15] | 38 | Not reported | ITD: 0 | 38 | ITD: 6 (16%) | Not reported |
| TKD: 0 | TKD: 3 (8%) |
| Mutated Genes | Function | Frequency | Prognostic Impact | Potential Targetted Therapy | References |
|---|---|---|---|---|---|
| DNA methylation | |||||
| TET2 | Conversion of 5mC to 5-hmC | 20%–30% in MDS 50%–60% in CMML | None | DNA methyltransferase inhibitor (DNMTI) | [1,11,17,18,19,20,21,22,23,24,25] |
| DNMT3A | DNA methyltransferase, histone methylation and transcription repression | 10% in MDS | Unfavorable | DNMT1 | [26,27,28,29,32,33,34] |
| IDH1/IDH2 | Convert isocitrate to α-KG, regulates TET2 | 5% | Unfavorable | DNMT1, IDH1/2 inhibitors | [27,28,35,36,37,38,39,40,41] |
| Histone modification | |||||
| EZH2 | Histone methyltransferase, gene repression | 5% | Unfavorable | HDAC inhibitors, EZH2 inhibitors | [28] |
| ASXL1 | H3 methylation | 15%–20% | Unfavorable | HDAC inhibitors | [11,28,42,43,44,45] |
| UTX | H3K27 demethylase | 1% | None | HDAC inhibitors | [11,28,46,47] |
| RNA splicing | |||||
| SF3B1 | Pre-mRNA splicing | 15%–30% | Favorable | None | [11,19,49,50] |
| SRSF2 | Spliceosome assembly | 10%–20% | Unfavorable | None | [11,51,52,53] |
| ZRSR2 | Spliceosome assembly | <5% | None | None | [54] |
| U2AF1 | Spliceosome assembly | 5%–10% | None | None | [11,54] |
| Transcription | |||||
| RUNX1 | Regulates myeloid differentiation | 10% | Unfavorable | None | [11,55,56,57] |
| BCOR/BCORL1 | BCL6 co-repressor | 5% | Unfavorable | HDAC inhibitors | [58,59,60,61] |
| ETV6 | ETS transcription factor | <5% | Unfavorable | None | [11,62] |
| SETBP1 | Cell proliferation | 2%–5% | Unfavorable | None | [11,63,64] |
| GATA2 | Transcriptional activator | Rare | Unfavorable | None | [11] |
| DNA repair | |||||
| TP53 | Cell cycle regulation, tumor suppressor gene | 10% | Unfavorable | Antisense TP53 oligonucleotide | [11,65,66,67,68] |
| Signal transduction | |||||
| JAK2 | Tyrosine kinase activation | 5% | None | JAK1/2 inhibitors | [11,69,70] |
| FLT3 | Tyrosine kinase activation | <5% | Unfavorable | FLT3 inhibitors | [15,16,71,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105] |
| KIT | Tyrosine kinase activation | Rare | None | TKI (imatinib, dasatinib) | [132,133] |
| CBL | Proto-oncogene | 5% | Unfavorable | None | [11,12,134,135,136] |
| RAS pathway | GTPase signal transduction | 10% | Unfavorable | None | |
| Cohesin complex | |||||
| STAG2 | Control of cell division | 5%–10% | Unfavorable | None | [11] |
| RAD21 | Component of cohesin complex | <3% | None | None | [11] |
| SMC3 | Structure and function role in cohesin complex | <3% | None | None | [11] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gill, H.; Leung, A.Y.H.; Kwong, Y.-L. Molecular and Cellular Mechanisms of Myelodysplastic Syndrome: Implications on Targeted Therapy. Int. J. Mol. Sci. 2016, 17, 440. https://doi.org/10.3390/ijms17040440
Gill H, Leung AYH, Kwong Y-L. Molecular and Cellular Mechanisms of Myelodysplastic Syndrome: Implications on Targeted Therapy. International Journal of Molecular Sciences. 2016; 17(4):440. https://doi.org/10.3390/ijms17040440
Chicago/Turabian StyleGill, Harinder, Anskar Y. H. Leung, and Yok-Lam Kwong. 2016. "Molecular and Cellular Mechanisms of Myelodysplastic Syndrome: Implications on Targeted Therapy" International Journal of Molecular Sciences 17, no. 4: 440. https://doi.org/10.3390/ijms17040440
APA StyleGill, H., Leung, A. Y. H., & Kwong, Y.-L. (2016). Molecular and Cellular Mechanisms of Myelodysplastic Syndrome: Implications on Targeted Therapy. International Journal of Molecular Sciences, 17(4), 440. https://doi.org/10.3390/ijms17040440