
A New Secondary Structure Assignment Algorithm Using Cα Backbone Fragments

 ,
,
Abstract
:
1. Introduction
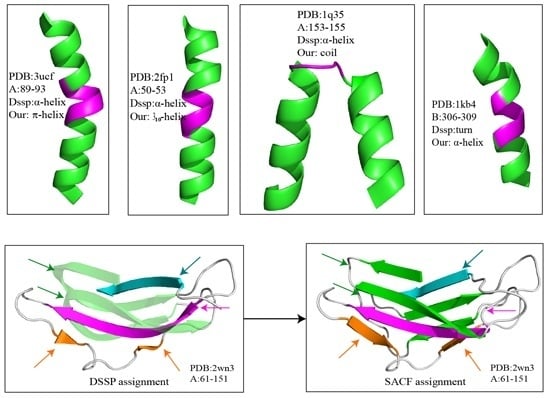
2. Results and Discussion
3. Methods
3.1. The Data Set
3.2. Secondary Structure Assignment by DSSP
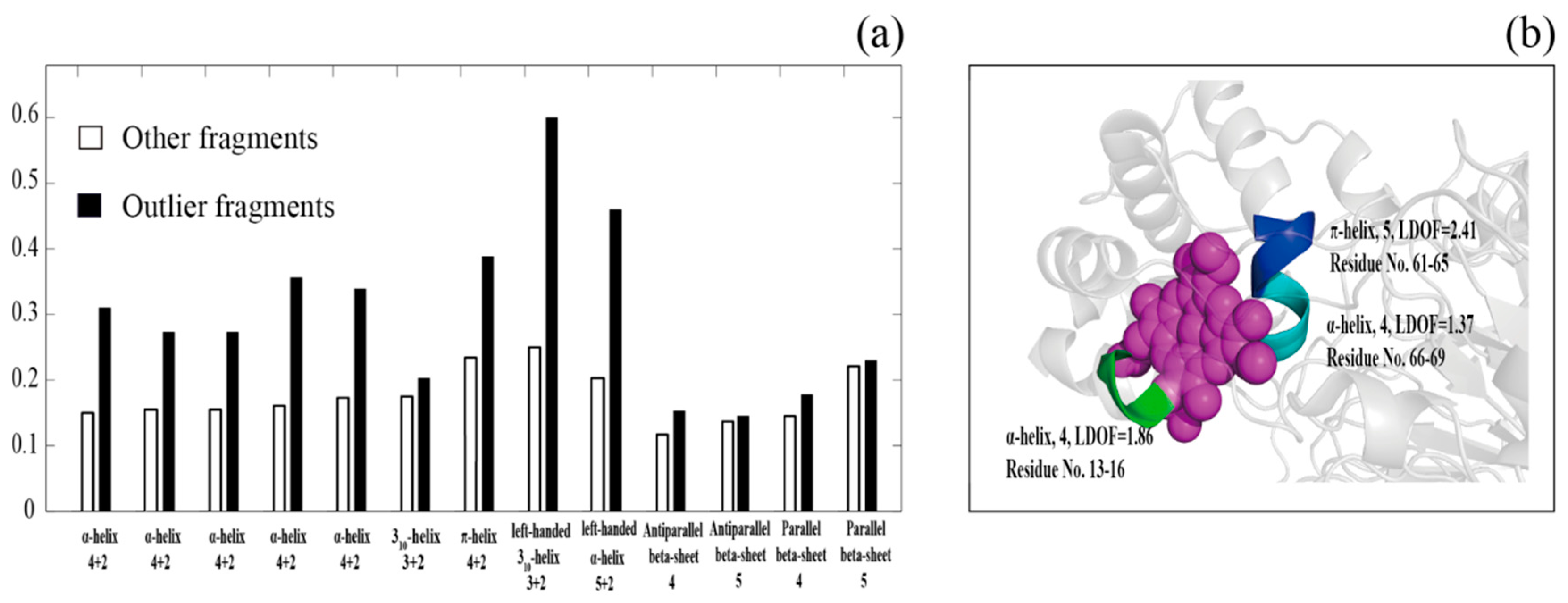
3.3. Outlier Detection
3.4. Clustering and Central Poses Selection
3.5. Our Secondary Structure Assignment Algorithm
3.5.1. Helix Assignment
LET ai = 0, i = 0,…, n // All residues are initialized as coil FOR i < n FOR len (length from 8 to 4) FOR k ∈ α-helix Central Pose Set Index IF dist [Segment (i, i + len), Pose (α-helix, len, k)] < RMSD (α-helix, len, k) THEN a(i + 1, i + len-2) = 1 // Residues from i + 1 to i + len-2 are labeled as α-helix END IF END FOR END FOR END FOR
FOR i < n IF (ai == 0) AND (ai − 1 == 1) AND (ai + 3 == 1) / Merge two adjacent helices less than four residues apart FOR any seven consecutive residues including i, i + 1 and i + 2 FOR k ∈ Helix Kink Pose Set IF dist [The seven residues fragment, Pose (helix kink, 7, k)] < RMSD (helix kink, 7, k) ai = ai + 1 = ai + 2 = 1 // Residues i, i + 1, i + 2 are label as α-helix END IF END FOR END FOR END IF END FOR
3.5.2. Parallel β-sheet Assignment
LET bi = 0, i = 0,…, n // All residues are initialized as coil FOR i < n FOR len (length from 5 to 4) FOR k ∈ Parallel β-strand Pose Set Index IF dist [Segment(i, i + len), Pose(β-strand, len, k)] < RMSD (Parallel β-strand, len, k) THEN b(i, i + len-1) =1 // Residues from i to i + len-1 are label as parallel β-strand END IF END FOR END FOR END FOR
FOR i < n IF (bi == 1) AND (bi + 1 == 1) // Find residues have been assigned as β-strand FOR j = 1 to n (j ≠ i − 1, i, i + 1) // Find the hydrogen bond partner β-strand residues IF (bj > 0) AND (bj + 1 > 0) FOR k ∈ Parallel β-sheet Ladder Pose Index IF dist [Segment(i, i + 1, j + 1, j), Pose(ladder, 4 ,k)] < RMSD (ladder, 4, k) THEN bi++,bi + 1++ END FOR END IF END FOR END IF END FOR
3.6. Comparison Measures
3.7. Secondary Structure Assignment Methods in Comparison
4. The Correlation between Outlier Poses Assigned by DSSP and Protein–Ligand Binding Sites
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pauling, L.; Corey, R.B.; Branson, H.R. The structure of proteins; two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. USA 1951, 37, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Vieira-Pires, R.S.; Morais-Cabral, J.H. 3(10) helices in channels and other membrane proteins. J. Gen. Physiol. 2010, 136, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Wilmot, C.M.; Thornton, J.M. β-turns and their distortions: A proposed new nomenclature. Protein Eng. 1990, 3, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.S.; Getzoff, E.D.; Richardson, D.C. The β bulge: A common small unit of nonrepetitive protein structure. Proc. Natl Acad. Sci. USA 1978, 75, 2574–2578. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, E.G.; Thornton, J.M. Promotif—A program to identify and analyze structural motifs in proteins. Protein Sci. 1996, 5, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Labesse, G.; Colloc’h, N.; Pothier, J.; Mornon, J.P. P-sea: A new efficient assignment of secondary structure from c alpha trace of proteins. Comput. Appl. Biosci. 1997, 13, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.S. Schematic drawings of protein structures. Methods Enzymol. 1985, 115, 359–380. [Google Scholar] [PubMed]
- Sillitoe, I.; Lewis, T.E.; Cuff, A.; Das, S.; Ashford, P.; Dawson, N.L.; Furnham, N.; Laskowski, R.A.; Lee, D.; Lees, J.G.; et al. Cath: Comprehensive structural and functional annotations for genome sequences. Nucleic Acids Res. 2015, 43, D376–D381. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Definition of general topological equivalence in protein structures. A procedure involving comparison of properties and relationships through simulated annealing and dynamic programming. J. Mol. Biol. 1990, 212, 403–428. [Google Scholar] [PubMed]
- Hubbard, T.; Tramontano, A. Update on protein structure prediction: Results of the 1995 irbm workshop. Fold. Des. 1996, 1, R55–R63. [Google Scholar] [CrossRef]
- Levitt, M.; Greer, J. Automatic identification of secondary structure in globular proteins. J. Mol. Biol. 1977, 114, 181–239. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Konagurthu, A.S.; Lesk, A.M.; Allison, L. Minimum message length inference of secondary structure from protein coordinate data. Bioinformatics 2012, 28, i97–105. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Frishman, D.; Argos, P. Knowledge-based protein secondary structure assignment. Proteins 1995, 23, 566–579. [Google Scholar] [CrossRef] [PubMed]
- Fodje, M.N.; Al-Karadaghi, S. Occurrence, conformational features and amino acid propensities for the pi-helix. Protein Eng. 2002, 15, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.; Letellier, G.; Marin, A.; Taly, J.F.; de Brevern, A.G.; Gibrat, J.F. Protein secondary structure assignment revisited: A detailed analysis of different assignment methods. BMC Struct. Biol. 2005, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- King, S.M.; Johnson, W.C. Assigning secondary structure from protein coordinate data. Proteins 1999, 35, 313–320. [Google Scholar] [CrossRef]
- Majumdar, I.; Krishna, S.S.; Grishin, N.V. Palsse: A program to delineate linear secondary structural elements from protein structures. BMC Bioinform. 2005, 6, 202. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Yoo, M.J.; Shin, J.; Cho, K.H. Saba (secondary structure assignment program based on only alpha carbons): A novel pseudo center geometrical criterion for accurate assignment of protein secondary structures. BMB Rep. 2011, 44, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, R.; Rose, G.D. A physical basis for protein secondary structure. Proc. Natl Acad. Sci. USA 1999, 96, 14258–14263. [Google Scholar] [CrossRef] [PubMed]
- Cubellis, M.V.; Cailliez, F.; Lovell, S.C. Secondary structure assignment that accurately reflects physical and evolutionary characteristics. BMC Bioinform. 2005, 6 (Suppl. 4), S8. [Google Scholar] [CrossRef] [PubMed]
- Nagy, G.; Oostenbrink, C. Dihedral-based segment identification and classification of biopolymers i: Proteins. J. Chem. Inform. Model. 2014, 54, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Law, S.M.; Frank, A.T.; Brooks, C.L. Pcasso: A fast and efficient c alpha-based method for accurately assigning protein secondary structure elements. J. Comput. Chem. 2014, 35, 1757–1761. [Google Scholar] [CrossRef] [PubMed]
- Richards, F.M.; Kundrot, C.E. Identification of structural motifs from protein coordinate data: Secondary structure and first-level supersecondary structure. Proteins 1988, 3, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Taylor, W.R. Defining linear segments in protein structure. J. Mol. Biol. 2001, 310, 1135–1150. [Google Scholar] [CrossRef] [PubMed]
- Sklenar, H.; Etchebest, C.; Lavery, R. Describing protein structure: A general algorithm yielding complete helicoidal parameters and a unique overall axis. Proteins 1989, 6, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Dunker, A.K.; Zhou, Y.Q. Assessing secondary structure assignment of protein structures by using pairwise sequence-alignment benchmarks. Proteins 2008, 71, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, J.; Knapp, E.W. Protein secondary structure classification revisited: Processing dssp information with pssc. J. Chem. Inform. Model. 2014, 54, 2166–2179. [Google Scholar] [CrossRef] [PubMed]
- Hodge, V.J.; Austin, J. A survey of outlier detection methodologies. Artif. Intell. Rev. 2004, 22, 85–126. [Google Scholar] [CrossRef]
- Jain, A.K. Data clustering: 50 years beyond k-means. Pattern Recognit. Lett. 2010, 31, 651–666. [Google Scholar] [CrossRef]
- Xu, S.; Zou, S.; Wang, L. A geometric clustering algorithm with applications to structural data. J. Comput. Biol. 2015, 22, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Hutter, M.; Jin, H.D. A new local distance-based outlier detection approach for scattered real-world data. Data Min. Knowl. Discov. 2009, 5476, 813–822. [Google Scholar]
- Colloc'h, N.; Etchebest, C.; Thoreau, E.; Henrissat, B.; Mornon, J.P. Comparison of three algorithms for the assignment of secondary structure in proteins: The advantages of a consensus assignment. Protein Eng. 1993, 6, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.; Henrick, K.; Nakamura, H.; Markley, J.L. The worldwide protein data bank: Ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2007, 35, D301–D303. [Google Scholar] [CrossRef] [PubMed]
- Novotny, M.; Kleywegt, G.J. A survey of left-handed helices in protein structures. J. Mol. Biol. 2005, 347, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. A discussion of the solution for the best rotation to relate two sets of vectors. Acta Crystallogr. Sect. A 1978, 34, 827–828. [Google Scholar] [CrossRef]
- Wilman, H.R.; Shi, J.; Deane, C.M. Helix kinks are equally prevalent in soluble and membrane proteins. Proteins 2014, 82, 1960–1970. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Xu, S.; Wang, L. An algorithm for protein helix assignment using helix geometry. PLoS ONE 2015. [Google Scholar] [CrossRef] [PubMed]
- Zemla, A.; Venclovas, C.; Fidelis, K.; Rost, B. A modified definition of sov, a segment-based measure for protein secondary structure prediction assessment. Proteins 1999, 34, 220–223. [Google Scholar] [CrossRef]
- Rost, B.; Sander, C.; Schneider, R. Redefining the goals of protein secondary structure prediction. J. Mol. Biol. 1994, 235, 13–26. [Google Scholar] [CrossRef]
- Matsuo, K.; Watanabe, H.; Gekko, K. Improved sequence-based prediction of protein secondary structures by combining vacuum-ultraviolet circular dichroism spectroscopy with neural network. Proteins 2008, 73, 104–112. [Google Scholar] [CrossRef] [PubMed]
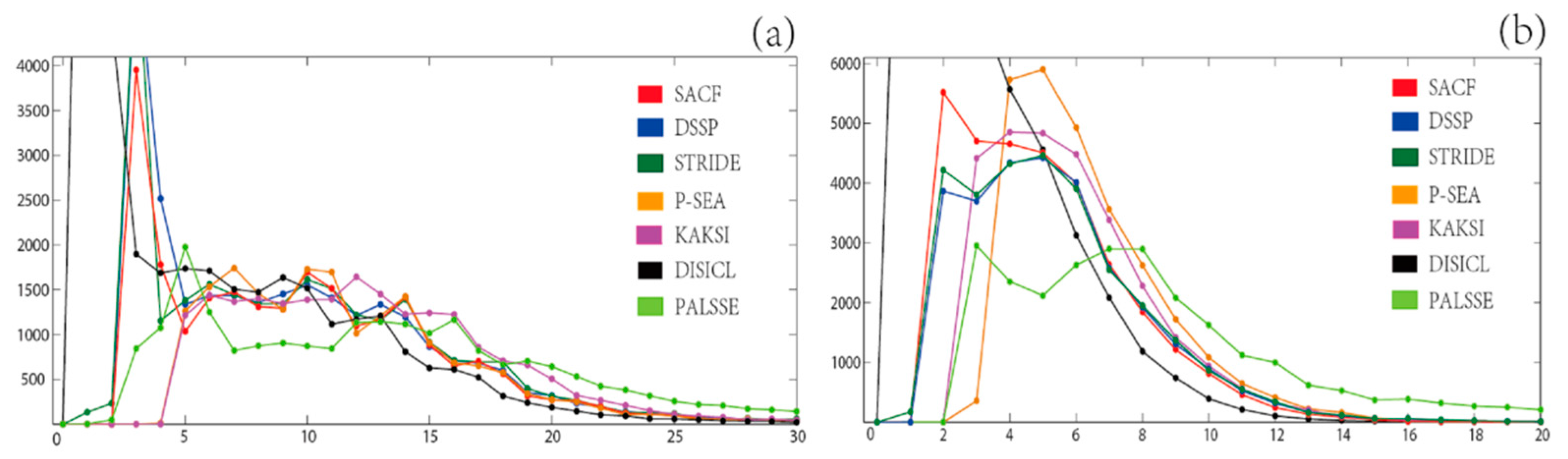

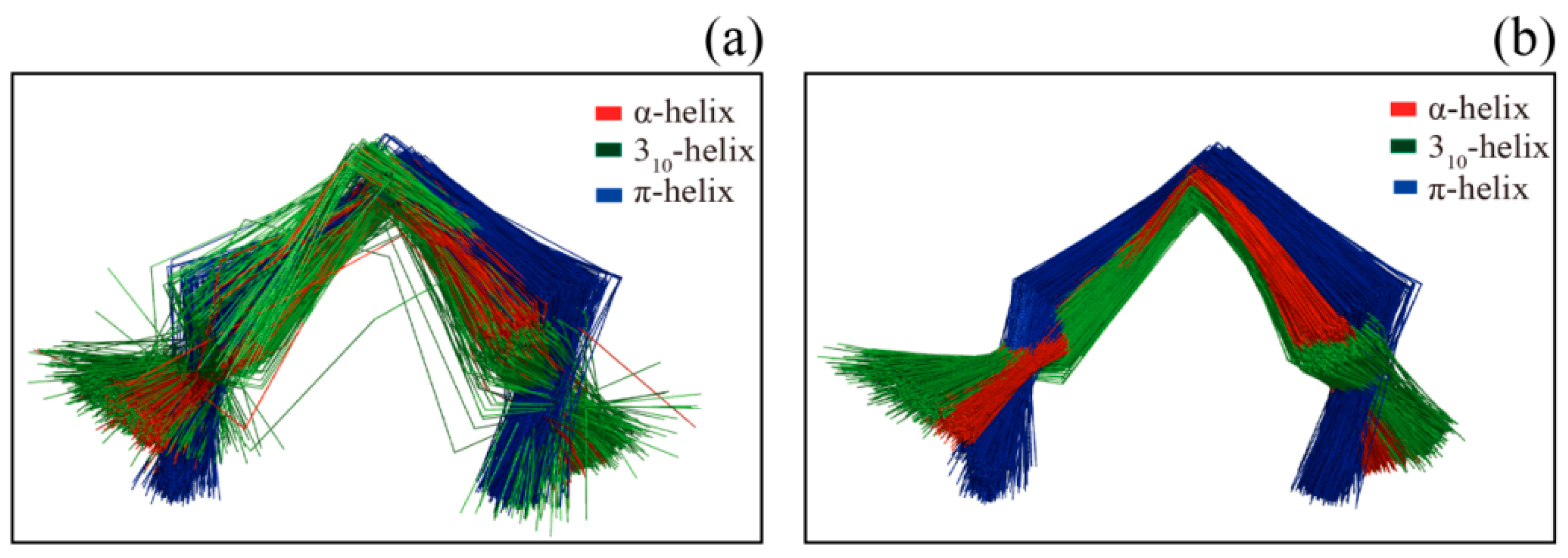
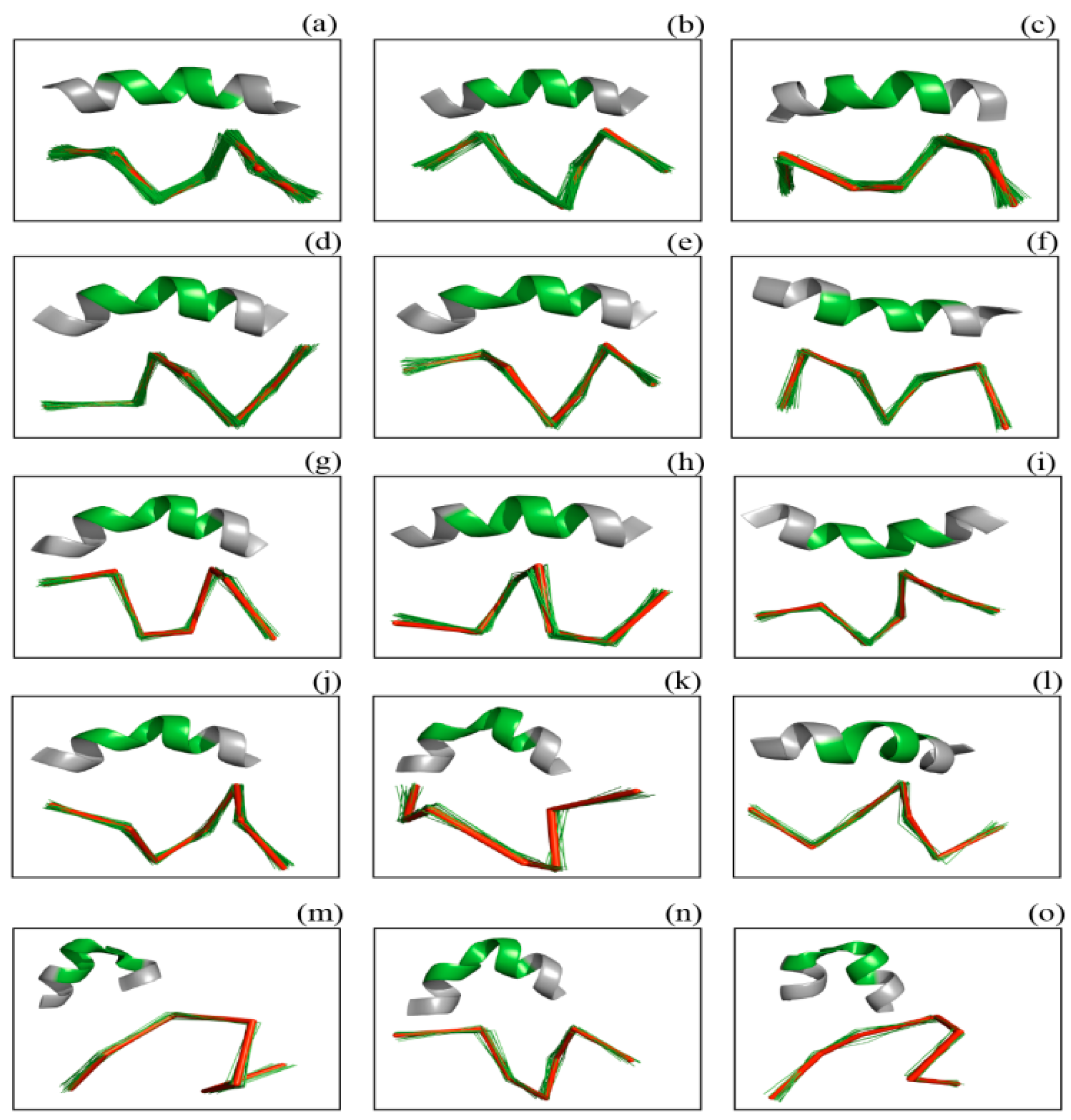

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Dssp | Stride | P-sea | Kaksi | Disicl | Palsse | Segno | Pross | Xtlsstr | Pcasso |
|---|---|---|---|---|---|---|---|---|---|---|
| Sacf | 84.7 | 85.1 | 81.8 | 82.6 | 76.9 | 68.4 | 80.5 | 83.1 | 76.1 | 84.3 |
| Dssp | 95.0 | 80.9 | 83.5 | 78.9 | 72.9 | 83.0 | 84.3 | 77.2 | 93.5 | |
| Stride | 81.1 | 84.1 | 78.4 | 73.6 | 82.5 | 84.8 | 80.2 | 92.0 | ||
| P-sea | 82.3 | 78.3 | 68.8 | 85.9 | 86.2 | 74.4 | 82.1 | |||
| Kaksi | 74.8 | 77.5 | 80.5 | 82.9 | 78.5 | 83.8 | ||||
| Disicl | 63.1 | 80.8 | 81.8 | 74.9 | 79.6 | |||||
| Palsse | 66.3 | 66.1 | 70.6 | 73.6 | ||||||
| Segno | 87.4 | 76.4 | 82.4 | |||||||
| Pross | 79.3 | 84.5 | ||||||||
| Xtlsstr | 79.2 |
| Method | Sacf | Dssp | Stride | P-sea | Kaksi | Disicl | Palsse | Segno | Pross | Xtlsstr | Pcasso |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sacf | 96.6 | 94.1 | 92.6 | 92.6 | 88.3 | 81.7 | 80.1 | 91.2 | 90.3 | 95.2 | |
| Dssp | 91.3 | 93.7 | 86.0 | 88.4 | 82.9 | 81.1 | 75.8 | 86.1 | 89.2 | 94.1 | |
| Stride | 90.1 | 95.2 | 86.2 | 88.0 | 84.4 | 82.5 | 77.1 | 87.4 | 92.6 | 92.7 | |
| P-sea | 96.9 | 96.7 | 94.2 | 95.7 | 91.3 | 84.1 | 83.7 | 95.2 | 91.6 | 96.5 | |
| Kaksi | 93.8 | 96.0 | 93.4 | 92.6 | 84.7 | 86.3 | 79.1 | 92.8 | 91.6 | 95.0 | |
| Disicl | 87.3 | 89.9 | 89.6 | 85.6 | 85.7 | 72.8 | 80.0 | 87.6 | 85.6 | 89.6 | |
| Palsse | 60.3 | 62.4 | 63.1 | 63.7 | 67.1 | 47.8 | 50.5 | 62.2 | 69.0 | 59.7 | |
| Segno | 92.9 | 94.1 | 93.2 | 92.3 | 91.5 | 94.4 | 76.8 | 93.5 | 89.5 | 94.1 | |
| Pross | 95.9 | 97.4 | 97.5 | 95.6 | 96.7 | 93.9 | 83.1 | 86.2 | 93.9 | 97.1 | |
| Xtlsstr | 82.7 | 86.8 | 89.1 | 81.5 | 83.9 | 76.7 | 85.6 | 71.7 | 81.9 | 84.5 | |
| Pcasso | 90.9 | 96.4 | 93.2 | 87.6 | 89.5 | 84.7 | 80.3 | 77.4 | 87.6 | 89.4 |
| Method | Sacf | Dssp | Stride | P-sea | Kaksi | Disicl | Palsse | Segno | Pross | Xtlsstr | Pcasso |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sacf | 86.0 | 85.4 | 78.7 | 86.0 | 78.3 | 68.9 | 80.9 | 78.6 | 71.3 | 87.1 | |
| Dssp | 81.2 | 97.0 | 78.0 | 88.0 | 70.8 | 73.1 | 80.4 | 77.2 | 71.3 | 89.2 | |
| Stride | 79.4 | 96.7 | 87.3 | 70.2 | 73.3 | 80.2 | 75.7 | 70.7 | 87.9 | ||
| P-sea | 78.7 | 78.9 | 78.6 | 83.2 | 77.2 | 70.8 | 87.0 | 79.0 | 68.7 | 80.6 | |
| Kaksi | 84.5 | 92.0 | 91.6 | 83.8 | 77.0 | 76.9 | 86.4 | 80.6 | 73.0 | 91.9 | |
| Disicl | 64.6 | 68.6 | 68.7 | 71.0 | 69.6 | 53.4 | 75.7 | 72.8 | 65.3 | 70.8 | |
| Palsse | 45.7 | 51.3 | 51.6 | 50.4 | 52.1 | 35.8 | 48.9 | 43.4 | 43.0 | 47.5 | |
| Segno | 75.8 | 79.8 | 79.9 | 82.5 | 82.3 | 80.9 | 68.3 | 81.5 | 72.9 | 81.2 | |
| Pross | 81.7 | 83.1 | 83.4 | 83.2 | 84.4 | 88.9 | 64.8 | 91.2 | 76.1 | 84.5 | |
| Xtlsstr | 74.9 | 77.8 | 77.9 | 73.8 | 77.2 | 79.1 | 62.9 | 82.8 | 78.0 | 77.0 | |
| Pcasso | 84.2 | 90.8 | 89.7 | 78.8 | 87.5 | 73.0 | 70.3 | 81.1 | 76.5 | 70.3 |
| Method | Same | N cap | N cap | C cap | C cap | ||||
|---|---|---|---|---|---|---|---|---|---|
| +(1–2) | +(>2) | −(1–2) | −(>2) | +(1-2) | +(>2) | −(1–2) | −(>2) | ||
| Sacf | 5194 | 1407 | 23 | 1919 | 534 | 1865 | 15 | 3142 | 578 |
| Stride | 11,388 | 990 | 34 | 332 | 80 | 801 | 60 | 401 | 62 |
| P-sea | 1639 | 4782 | 678 | 870 | 569 | 4405 | 610 | 1267 | 423 |
| Kaksi | 1761 | 5765 | 153 | 2269 | 217 | 5347 | 131 | 1737 | 92 |
| Disicl | 1310 | 4090 | 252 | 1828 | 369 | 1131 | 96 | 7306 | 587 |
| Palsse | 87 | 7423 | 726 | 121 | 59 | 7153 | 728 | 121 | 26 |
| Segno | 2734 | 5222 | 448 | 913 | 332 | 3344 | 397 | 1182 | 253 |
| Pross | 3037 | 2626 | 117 | 1638 | 796 | 2350 | 107 | 2326 | 592 |
| Xtlsstr | 803 | 5932 | 332 | 1855 | 600 | 1173 | 130 | 4023 | 857 |
| Pcasso | 5950 | 1211 | 50 | 1856 | 347 | 1795 | 35 | 2302 | 272 |
| Method | Same | N cap | N cap | C cap | C cap | ||||
|---|---|---|---|---|---|---|---|---|---|
| +(1–2) | +(>2) | −(1–2) | −(>2) | +(1–2) | +(>2) | −(1–2) | −(>2) | ||
| Sacf | 2375 | 1355 | 16 | 2218 | 535 | 1902 | 11 | 2,897 | 578 |
| Stride | 8352 | 733 | 83 | 285 | 80 | 544 | 69 | 353 | 63 |
| P-sea | 1621 | 3260 | 568 | 853 | 486 | 3267 | 473 | 1,225 | 433 |
| Kaksi | 1473 | 4138 | 71 | 2163 | 317 | 3890 | 73 | 1,638 | 195 |
| Disicl | 815 | 2720 | 182 | 1602 | 371 | 749 | 85 | 5,367 | 591 |
| Palsse | 56 | 5713 | 786 | 116 | 63 | 5513 | 781 | 114 | 28 |
| Segno | 2364 | 3753 | 384 | 851 | 337 | 2322 | 335 | 1085 | 255 |
| Pross | 2481 | 1820 | 83 | 1567 | 802 | 1544 | 84 | 2200 | 594 |
| Xtlsstr | 636 | 4447 | 275 | 1791 | 602 | 829 | 124 | 3507 | 863 |
| Pcasso | 4994 | 867 | 66 | 1267 | 348 | 973 | 48 | 1490 | 273 |
| SSE Name | Len | μ (Å) 1 | Σ 2 | Adj.R-Square | Total Number of SSEs | Number of Outliers | Number of Clusters | Max 3 |
|---|---|---|---|---|---|---|---|---|
| α-helix | 4 + 24 | 0.411 | 0.218 | 0.969 | 4776 | 496 | 18 | 682 |
| α-helix | 5 + 2 | 0.388 | 0.173 | 0.971 | 2842 | 349 | 25 | 276 |
| α-helix | 6 + 2 | 0.393 | 0.150 | 0.979 | 3159 | 357 | 28 | 315 |
| α-helix | 7 + 2 | 0.418 | 0.185 | 0.976 | 3578 | 326 | 33 | 383 |
| α-helix | 8 + 2 | 0.435 | 0.189 | 0.970 | 3521 | 563 | 25 | 273 |
| 310-helix | 3 + 2 | 0.303 | 0.157 | 0.980 | 15,689 | 2334 | 32 | 1830 |
| π-helix | 5 + 2 | 0.516 | 0.437 | 0.955 | 1243 | 224 | 19 | 304 |
| Left-α-helix | 4 + 2 | 1.012 | 8.004 | 0.815 | 72 | 23 | 8 | 16 |
| Left-310-helix | 3 + 2 | 0.596 | 0.239 | 0.898 | 812 | 211 | 21 | 82 |
| Parallel β-ladder | 4 | 0.352 | 0.314 | 0.987 | 62,204 | 6917 | 22 | 7821 |
| Antiparallel β-ladder | 4 | 0.427 | 0.201 | 0.989 | 97,088 | 8562 | 23 | 8787 |
| Parallel β-strand | 4 | 0.383 | 0.189 | 0.999 | 5374 | 689 | 25 | 878 |
| Parallel β-strand | 5 | 0.496 | 0.486 | 0.973 | 5846 | 858 | 28 | 664 |
| Parallel β-strand | 6 | 0.776 | 0.579 | 0.966 | 4678 | 868 | 31 | 670 |
| Parallel β-strand | 7 | 1.400 | 0.623 | 0.898 | 2608 | 419 | 28 | 385 |
| Parallel β-strand | 8 | 1.631 | 1.282 | 0.921 | 1627 | 37 | 32 | 337 |
| Antiparallel β-strand | 4 | 0.543 | 0.571 | 0.984 | 6176 | 821 | 19 | 1048 |
| Antiparallel β-strand | 5 | 0.546 | 0.671 | 0.959 | 6554 | 867 | 28 | 886 |
| Antiparallel β-strand | 6 | 1.367 | 1.825 | 0.926 | 4600 | 672 | 25 | 738 |
| Antiparallel β-strand | 7 | 1.882 | 0.817 | 0.943 | 5217 | 841 | 26 | 909 |
| Antiparallel β-strand | 8 | 1.994 | 0.824 | 0.945 | 4221 | 898 | 31 | 682 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, C.; Wang, G.; Liu, A.; Xu, S.; Wang, L.; Zou, S. A New Secondary Structure Assignment Algorithm Using Cα Backbone Fragments. Int. J. Mol. Sci. 2016, 17, 333. https://doi.org/10.3390/ijms17030333
Cao C, Wang G, Liu A, Xu S, Wang L, Zou S. A New Secondary Structure Assignment Algorithm Using Cα Backbone Fragments. International Journal of Molecular Sciences. 2016; 17(3):333. https://doi.org/10.3390/ijms17030333
Chicago/Turabian StyleCao, Chen, Guishen Wang, An Liu, Shutan Xu, Lincong Wang, and Shuxue Zou. 2016. "A New Secondary Structure Assignment Algorithm Using Cα Backbone Fragments" International Journal of Molecular Sciences 17, no. 3: 333. https://doi.org/10.3390/ijms17030333
APA StyleCao, C., Wang, G., Liu, A., Xu, S., Wang, L., & Zou, S. (2016). A New Secondary Structure Assignment Algorithm Using Cα Backbone Fragments. International Journal of Molecular Sciences, 17(3), 333. https://doi.org/10.3390/ijms17030333