
Implications of MicroRNAs in the Treatment of Gefitinib-Resistant Non-Small Cell Lung Cancer


 ,
,  and
and
Abstract
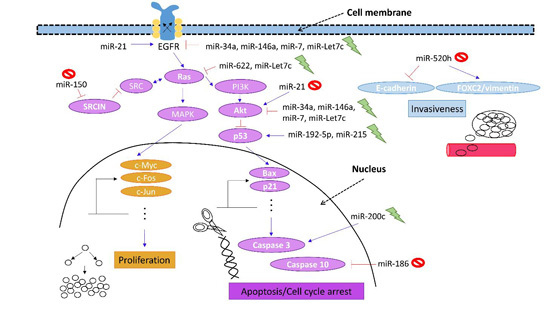
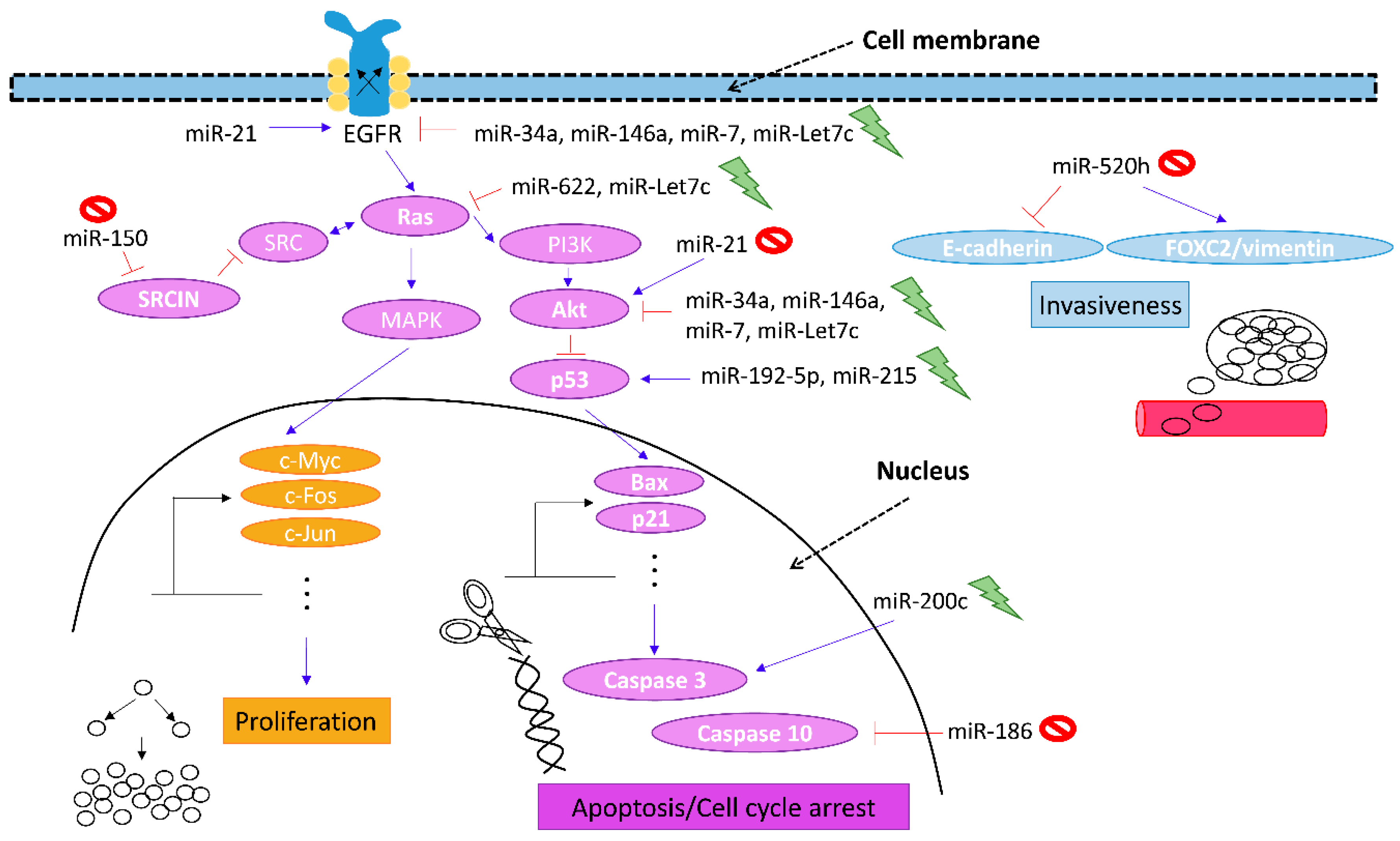
:
1. Introduction
2. Overcoming Gefitinib Resistance: Not Only an Issue of EGFR
3. Does Combination Treatment Enhance the Therapeutic Capacity of EGFR-TKIs?
4. Therapeutic Potential of MicroRNAs to Revert Gefitinib Competence

{kind=link}
{kind=link}
| miRNAs | Models of EGFR-TKI Resistance | Molecular Targets | References |
|---|---|---|---|
| miR-34a | 1. HGF/gefitinib-treated HCC827 2. xenografts | ↑ PARP cleavage ↓ p-MET/MET ↓ p-EGFR | [20] |
| miR-7 | 1. A549 | ↓ EGFR | [21] |
| miR-146a | 1. H1975 2. Human NSCLC | ↑ Caspase 3/7 activity ↓ p-EGFR/EGFR ↓ p-Akt | [22] |
| miR-9-3 | 1. H1975 | ↑ Caspase 3 activity ↑ DNA fragmentation | [23] |
| miR-210 | 1. A549 | ↓ OXPHOS ↑ HIF-1α | [24] |
| miR-150 | 1. Human lung cancer samples 2. A549/H1975 | ↓ SRCIN1 | [25] |
| miR-21 | 1. PC9 xenografts 2. Human NSCLC samples | ↑ p-Akt/Akt | [26] |
| miR-101 | 1. H157 | ? ATM | [27] |
| miR-Let-7c | 1. H441 | ↓ p-STAT3 | [28] |
| 2. H1975 | ↓ Ras/p-Akt | [29] |
5. The Need of Clear, Functional Dissection of miRNAs
6. Synergy with Natural Phytochemicals
| Compounds of Interest | Changes in miRNA | Signalling Markers Involved | References |
|---|---|---|---|
| Antrocin | ↑ miR-Let-7c | ↓ Akt, JAK1/2, STAT3 ↑ Bax, cleavage of caspase 3 | [28] |
| Resveratrol | ↑ miR-335 ↑ miR-582-3p ↑ miR-338-3p ↑ miR194 and more | A number of genes related to apoptosis, cell cycle arrest and proliferation (Predicted) | [44] |
| ↑ miR-622 | ↓ k-Ras | [45] | |
| Augments the effects of miR-200c | ↑ caspase 3/9, CHOP, p-JNK | [32] | |
| ? miR-21 | ↑ Bcl2 | [46] | |
| ↓ miR-520h | ↑ PP2A, E-cadherin ↓ p-Akt, FOXC2, vimentin | [39] | |
| Curcumin | ↑ miR-192-5p, miR-215 | ↑ p53, p21 | [47] |
| ↓ miR-186 | ↑ caspase 10 | [48] |
7. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| Akt | Protein kinase B |
| AMPK | AMP-activated protein kinase |
| ATM | Ataxia telangiectasia mutated |
| ATP | Adenosine triphosphate |
| CD31 | Cluster of differentiation 31 |
| EGF(R) | Epidermal growth factor (receptor) |
| EGFR-TKI | Epidermal growth factor receptor tyrosine kinase inhibitor |
| HIF-1α | Hypoxia-inducing factor 1 alpha |
| HGF | Hepatocyte growth factor |
| IC50 | Half maximal inhibitory concentration |
| IGFR | Insulin growth factor receptor |
| JAK1/2 | Janus kinase 1/2 |
| MET | Hepatocyte growth factor receptor |
| miRNA | MicroRNA |
| mTOR | Mammalian target of rapamycin (mTOR) |
| NSCLC | Non-small cell lung cancer |
| PARP | poly ADP-ribose polymerase (PARP) |
| PCNA | Proliferating cell nuclear antigen |
| PDH | Pyruvate dehydrogenase |
| PI3K | Phosphatidylinositol-3-kinase |
| PP2A | Protein phosphatase 2 |
| RISC | RNA-induced silencing complex |
| ROS | Reactive oxygen species |
| siRNA | Small-interfering RNA |
| SRCIN1 | SRC kinase signalling inhibitor 1 |
| STAT3 | Signal transducer and activator of transcription 3 |
| VEGF | Vascular endothelial growth factor |
| UTR | Untranslated region |
References
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Q.; Liu, J.; Gao, L.; Wang, R.; Chu, W.; Zhang, Y.; Tan, G.; Zhao, X.; Lv, B. MicroRNA-200a targets EGFR and c-Met to inhibit migration, invasion, and gefitinib resistance in non-small cell lung cancer. Cytogenet. Genome Res. 2015, 146, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.; Rizvi, S.M.; Belani, C.P. Afatinib for the treatment of metastatic non-small cell lung cancer. Cancer Manag. Res. 2015, 7, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, C.H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; Park, E.; Yun, C.H.; Sng, N.J.; Lucena-Araujo, A.R.; Yeo, W.L.; Huberman, M.S.; Cohen, D.W.; Nakayama, S.; Ishioka, K.; et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Arcila, M.E.; Oxnard, G.R.; Nafa, K.; Riely, G.J.; Solomon, S.B.; Zakowski, M.F.; Kris, M.G.; Pao, W.; Miller, V.A.; Ladanyi, M. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin. Cancer Res. 2011, 17, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Yang, J.C.; Shih, J.Y.; Su, W.C.; Hsia, T.C.; Tsai, C.M.; Ou, S.H.; Yu, C.J.; Chang, G.C.; Ho, C.L.; Sequist, L.V.; et al. Afatinib for patients with lung adenocarcinoma and epidermal growth factor receptor mutations (LUX-Lung 2): A phase 2 trial. Lancet Oncol. 2012, 13, 539–548. [Google Scholar] [CrossRef]
- Yang, J.C.; Sequist, L.V.; Geater, S.L.; Tsai, C.M.; Mok, T.S.; Schuler, M.; Yamamoto, N.; Yu, C.J.; Ou, S.H.; Zhou, C.; et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: A combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol. 2015, 16, 830–838. [Google Scholar] [CrossRef]
- Li, H.; Takayama, K.; Wang, S.; Shiraishi, Y.; Gotanda, K.; Harada, T.; Furuyama, K.; Iwama, E.; Ieiri, I.; Okamoto, I.; et al. Addition of bevacizumab enhances antitumor activity of erlotinib against non-small cell lung cancer xenografts depending on VEGF expression. Cancer Chemother. Pharmacol. 2014, 74, 1297–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Li, Q.; Zhang, S.; Chen, J.; Huang, L.; Ren, J.; Chang, Y.; Liang, Y.; Wu, G. NVP-BEZ235 overcomes gefitinib-acquired resistance by down-regulating PI3K/AKT/mTOR phosphorylation. Oncol. Targets Ther. 2015, 8, 269–277. [Google Scholar]
- Okon, I.S.; Coughlan, K.A.; Zhang, M.; Wang, Q.; Zou, M.H. Gefitinib-mediated ROS instigates mitochondrial dysfunction and drug resistance in lung cancer cells. J. Biol. Chem. 2015, 290, 9101–9110. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Armstrong, E.A.; Benavente, S.; Chinnaiyan, P.; Harari, P.M. Dual-agent molecular targeting of the epidermal growth factor receptor (EGFR): Combining anti-EGFR antibody with tyrosine kinase inhibitor. Cancer Res. 2004, 64, 5355–5362. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Rho, J.K.; Jeon, B.S.; Choi, S.J.; Park, S.C.; Lee, S.S.; Kim, H.R.; Kim, C.H.; Lee, J.C. Combined inhibition of IGFR enhances the effects of gefitinib in H1650: A lung cancer cell line with EGFR mutation and primary resistance to EGFR-TK inhibitors. Cancer Chemother. Pharmacol. 2010, 66, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Janjigian, Y.Y.; Azzoli, C.G.; Krug, L.M.; Pereira, L.K.; Rizvi, N.A.; Pietanza, M.C.; Kris, M.G.; Ginsberg, M.S.; Pao, W.; Miller, V.A.; et al. Phase I/II trial of cetuximab and erlotinib in patients with lung adenocarcinoma and acquired resistance to erlotinib. Clin. Cancer Res. 2011, 17, 2521–2527. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.; Nag, S.A.; Su, Y.; Voruganti, S.; Qin, J.J.; Zhang, R.; Cho, W.C. miRNAs in cancer prevention and treatment and as molecular targets for natural product anticancer agents. Curr. Cancer Drug Targets 2013, 13, 519–541. [Google Scholar] [CrossRef] [PubMed]
- MacDonagh, L.; Gray, S.G.; Finn, S.P.; Cuffe, S.; O’Byrne, K.J.; Barr, M.P. The emerging role of microRNAs in resistance to lung cancer treatments. Cancer Treat. Rev. 2015, 41, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.Y.; Chen, X.; Zhao, J.; Bao, Z.; Zhang, P.; Liu, Z.F. MicroRNA-34a overcomes HGF-mediated gefitinib resistance in EGFR mutant lung cancer cells partly by targeting MET. Cancer Lett. 2014, 351, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Zheng, L.; Huang, M.; Wang, Y.; Bi, F. MicroRNA expression profiles associated with acquired gefitinib-resistance in human lung adenocarcinoma cells. Mol. Med. Rep. 2015, 11, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Umelo, I.A.; Lv, S.; Teugels, E.; Fostier, K.; Kronenberger, P.; Dewaele, A.; Sadones, J.; Geers, C.; de Greve, J. miR-146a inhibits cell growth, cell migration and induces apoptosis in non-small cell lung cancer cells. PLoS ONE 2013, 8, e60317. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, B.; Han, L.; Li, X.; Tao, H.; Zhang, S.; Hu, Y. Demethylation of miR-9–3 and miR-193a genes suppresses proliferation and promotes apoptosis in non-small cell lung cancer cell lines. Cell. Physiol. Biochem. 2013, 32, 1707–1719. [Google Scholar] [CrossRef] [PubMed]
- Grosso, S.; Doyen, J.; Parks, S.K.; Bertero, T.; Paye, A.; Cardinaud, B.; Gounon, P.; Lacas-Gervais, S.; Noel, A.; Pouyssegur, J. MiR-210 promotes a hypoxic phenotype and increases radioresistance in human lung cancer cell lines. Cell Death Dis. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Hou, D.; Liang, H.; Gong, F.; Wang, Y.; Yan, X.; Jiang, X.; Wang, C.; Zhang, J.; Zen, K.; et al. miR-150 promotes the proliferation and migration of lung cancer cells by targeting SRC kinase signalling inhibitor 1. Eur. J. Cancer 2014, 50, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Zhu, F.; Liu, J.; Xu, T.; Pei, D.; Wang, R.; Qian, Y.; Li, Q.; Wang, L.; Shi, Z.; et al. Alteration in Mir-21/PTEN expression modulates gefitinib resistance in non-small cell lung cancer. PLoS ONE 2014, 9, e0103305. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, H.; Ng, W.L.; Curran, W.J.; Wang, Y. Radiosensitizing effects of ectopic miR-101 on non-small-cell lung cancer cells depend on the endogenous miR-101 level. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.T.; Huang, W.C.; Rao, Y.K.; Ye, M.; Lee, W.H.; Wang, L.S.; Tzeng, D.T.; Wu, C.H.; Shieh, Y.S.; Huang, C.Y.; et al. A sesquiterpene lactone antrocin from Antrodia camphorata negatively modulates JAK2/STAT3 signaling via microRNA let-7c and induces apoptosis in lung cancer cells. Carcinogenesis 2013, 34, 2918–2928. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Liu, J.; Wang, R.; Qian, X.; Xu, R.; Xu, T.; Li, Q.; Wang, L.; Shi, Z.; Zheng, J.; et al. Fulvestrant increases gefitinib sensitivity in non-small cell lung cancer cells by upregulating let-7c expression. Biomed. Pharmacother. 2014, 68, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Fan, X.; Li, W.; Ping, W.; Deng, Y.; Fu, X. miR-138–5p reverses gefitinib resistance in non-small cell lung cancer cells via negatively regulating G protein-coupled receptor 124. Biochem. Biophys. Res. Commun. 2014, 446, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Park, D.H.; Jeon, H.S.; Lee, S.Y.; Choi, Y.Y.; Lee, H.W.; Yoon, S.; Lee, J.C.; Yoon, Y.S.; Kim, D.S.; Na, M.J.; et al. MicroRNA-146a inhibits epithelial mesenchymal transition in non-small cell lung cancer by targeting insulin receptor substrate 2. Int. J. Oncol. 2015, 47, 1545–1553. [Google Scholar] [CrossRef] [PubMed]
- Bai, T.; Dong, D.S.; Pei, L. Synergistic antitumor activity of resveratrol and miR-200c in human lung cancer. Oncol. Rep. 2014, 31, 2293–2297. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Lin, H.; Fan, J.; Lan, J.; Zhong, Y.; Yang, Y.; Li, H.; Wang, Z. CD59 is overexpressed in human lung cancer and regulates apoptosis of human lung cancer cells. Int. J. Oncol. 2013, 43, 850–858. [Google Scholar] [PubMed]
- Li, B.; Ren, S.; Li, X.; Wang, Y.; Garfield, D.; Zhou, S.; Chen, X.; Su, C.; Chen, M.; Kuang, P.; et al. MiR-21 overexpression is associated with acquired resistance of EGFR-TKI in non-small cell lung cancer. Lung Cancer 2014, 83, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Liu, Y.; Bai, Y.; Sun, Y.; Xiao, F.; Guo, Y. Gene expression profiling of drug-resistant small cell lung cancer cells by combining microRNA and cDNA expression analysis. Eur. J. Cancer 2010, 46, 1692–1702. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, M.; Romano, G.; Di Leva, G.; Nuovo, G.; Jeon, Y.J.; Ngankeu, A.; Sun, J.; Lovat, F.; Alder, H.; Condorelli, G.; et al. EGFR and MET receptor tyrosine kinase-altered microRNA expression induces tumorigenesis and gefitinib resistance in lung cancers. Nat. Med. 2012, 18, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhao, J.J.; Zhang, L.; Xu, Q.F.; Zhao, Y.M.; Shi, X.Y.; Xu, A.G. Serum miR-21 level: A potential diagnostic and prognostic biomarker for non-small cell lung cancer. Int. J. Clin. Exp. Med. 2015, 8, 14759–14763. [Google Scholar] [PubMed]
- Xu, L.; Li, L.; Li, J.; Li, H.; Shen, Q.; Ping, J.; Ma, Z.; Zhong, J.; Dai, L. Overexpression of miR-1260b in non-small cell lung cancer is associated with lymph node metastasis. Aging Dis. 2015, 6, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.H.; Chen, H.A.; Chen, P.S.; Cheng, Y.J.; Hsu, W.H.; Chang, Y.W.; Chen, Y.H.; Jan, Y.; Hsiao, M.; Chang, T.Y.; et al. MiR-520h-mediated FOXC2 regulation is critical for inhibition of lung cancer progression by resveratrol. Oncogene 2013, 32, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Hirata, H.; Hinoda, Y.; Shahryari, V.; Deng, G.; Tanaka, Y.; Tabatabai, Z.L.; Dahiya, R. Genistein downregulates onco-miR-1260b and upregulates sFRP1 and Smad4 via demethylation and histone modification in prostate cancer cells. Br. J. Cancer 2014, 110, 1645–1654. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ding, M.; Xia, M.; Chen, S.; Van Le, A.; Soto-Gil, R.; Shen, Y.; Wang, N.; Wang, J.; Gu, W.; et al. A five-miRNA panel identified from a multicentric case-control study serves as a novel diagnostic tool for ethnically diverse non-small-cell lung cancer patients. EBioMedicine 2015, 2, 1377–1385. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, X.; Ren, S.; Chen, X.; Zhang, Y.; Zhou, F.; Zhao, M.; Zhao, C.; Cheng, N.; Zhao, Y.; et al. miR-200c overexpression is associated with better efficacy of EGFR-TKIs in non-small cell lung, cancer patients with EGFR wild-type. Oncotarget 2014, 5, 7902–7916. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Liu, X.; Cho, W.C. Genetic and epigenetic studies for determining molecular targets of natural product anticancer agents. Curr. Cancer Drug Targets 2013, 13, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Lee, E.M.; Cha, H.J.; Kim, K.; Yoon, Y.; Lee, H.; Kim, J.; Kim, Y.J.; Lee, H.G.; Jeung, H.K.; et al. Resveratrol alters microRNA expression profiles in A549 human non-small cell lung cancer cells. Mol. Cells 2011, 32, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Yang, Q.; Liu, B.; Wu, J.; Li, Y.; Yang, C.; Jiang, Y. MicroRNA-622 functions as a tumor suppressor by targeting K-Ras and enhancing the anticarcinogenic effect of resveratrol. Carcinogenesis 2012, 33, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Liang, H.; Xia, Q.; Li, P.; Kong, H.; Lei, P.; Wang, S.; Tu, Z. Resveratrol induces apoptosis of pancreatic cancers cells by inhibiting miR-21 regulation of BCL-2 expression. Clin. Transl. Oncol. 2013, 15, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Zhang, J.; Miao, Q.; Yao, L. Curcumin promotes apoptosis by activating the p53-miR-192–5p/215-XIAP pathway in non-small cell lung cancer. Cancer Lett. 2015, 357, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Du, Y.; Wu, C.; Ren, X.; Ti, X.; Shi, J.; Zhao, F.; Yin, H. Curcumin promotes apoptosis in human lung adenocarcinoma cells through miR-186 * signaling pathway. Oncol. Rep. 2010, 24, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Stahlhut, C.; Slack, F.J. Combinatorial action of microRNAs let-7 and miR-34 effectively synergizes with erlotinib to suppress non-small cell lung cancer cell proliferation. Cell Cycle 2015, 14, 2171–2180. [Google Scholar] [CrossRef] [PubMed]
- Takamizawa, J.; Konishi, H.; Yanagisawa, K.; Tomida, S.; Osada, H.; Endoh, H.; Harano, T.; Yatabe, Y.; Nagino, M.; Nimura, Y.; et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004, 64, 3753–3756. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, T.; Ti, X.; Shi, J.; Wu, C.; Ren, X.; Yin, H. Curcumin promotes apoptosis in A549/DDP multidrug-resistant human lung adenocarcinoma cells through an miRNA signaling pathway. Biochem. Biophys. Res. Commun. 2010, 399, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Bai, W. MiR-21 suppresses the anticancer activities of curcumin by targeting PTEN gene in human non-small cell lung cancer A549 cells. Clin. Transl. Oncol. 2014, 16, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Sheth, S.; Jajoo, S.; Kaur, T.; Mukherjea, D.; Sheehan, K.; Rybak, L.P.; Ramkumar, V. Resveratrol reduces prostate cancer growth and metastasis by inhibiting the Akt/MicroRNA-21 pathway. PLoS ONE 2012, 7, e51655. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Schetter, A.J.; Mollerup, S.; Kohno, T.; Skaug, V.; Bowman, E.D.; Mathe, E.A.; Takenoshita, S.; Yokota, J.; Haugen, A.; et al. The association of microRNA expression with prognosis and progression in early-stage, non-small cell lung adenocarcinoma: A retrospective analysis of three cohorts. Clin. Cancer Res. 2011, 17, 1875–1882. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sin, T.K.; Wang, F.; Meng, F.; Wong, S.C.C.; Cho, W.C.S.; Siu, P.M.; Chan, L.W.C.; Yung, B.Y.M. Implications of MicroRNAs in the Treatment of Gefitinib-Resistant Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2016, 17, 237. https://doi.org/10.3390/ijms17020237
Sin TK, Wang F, Meng F, Wong SCC, Cho WCS, Siu PM, Chan LWC, Yung BYM. Implications of MicroRNAs in the Treatment of Gefitinib-Resistant Non-Small Cell Lung Cancer. International Journal of Molecular Sciences. 2016; 17(2):237. https://doi.org/10.3390/ijms17020237
Chicago/Turabian StyleSin, Thomas K., Fengfeng Wang, Fei Meng, S. C. Cesar Wong, William C. S. Cho, Parco M. Siu, Lawrence W. C. Chan, and Benjamin Y. M. Yung. 2016. "Implications of MicroRNAs in the Treatment of Gefitinib-Resistant Non-Small Cell Lung Cancer" International Journal of Molecular Sciences 17, no. 2: 237. https://doi.org/10.3390/ijms17020237
APA StyleSin, T. K., Wang, F., Meng, F., Wong, S. C. C., Cho, W. C. S., Siu, P. M., Chan, L. W. C., & Yung, B. Y. M. (2016). Implications of MicroRNAs in the Treatment of Gefitinib-Resistant Non-Small Cell Lung Cancer. International Journal of Molecular Sciences, 17(2), 237. https://doi.org/10.3390/ijms17020237