
Induction of Apoptosis by PQ1, a Gap Junction Enhancer that Upregulates Connexin 43 and Activates the MAPK Signaling Pathway in Mammary Carcinoma Cells

Abstract
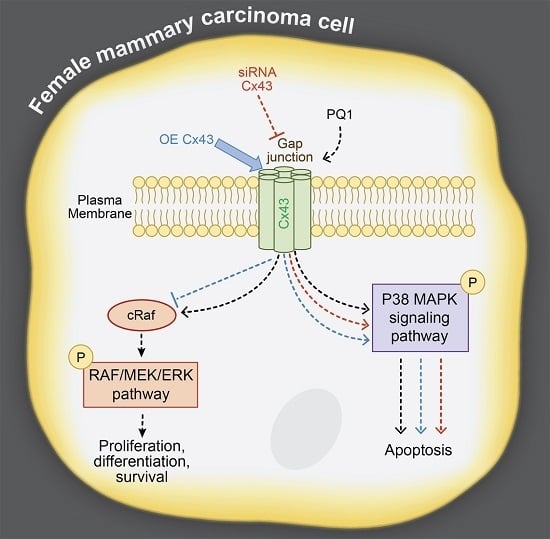
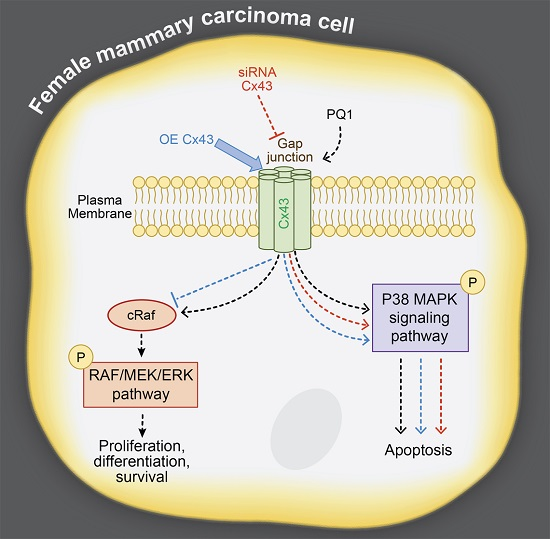
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
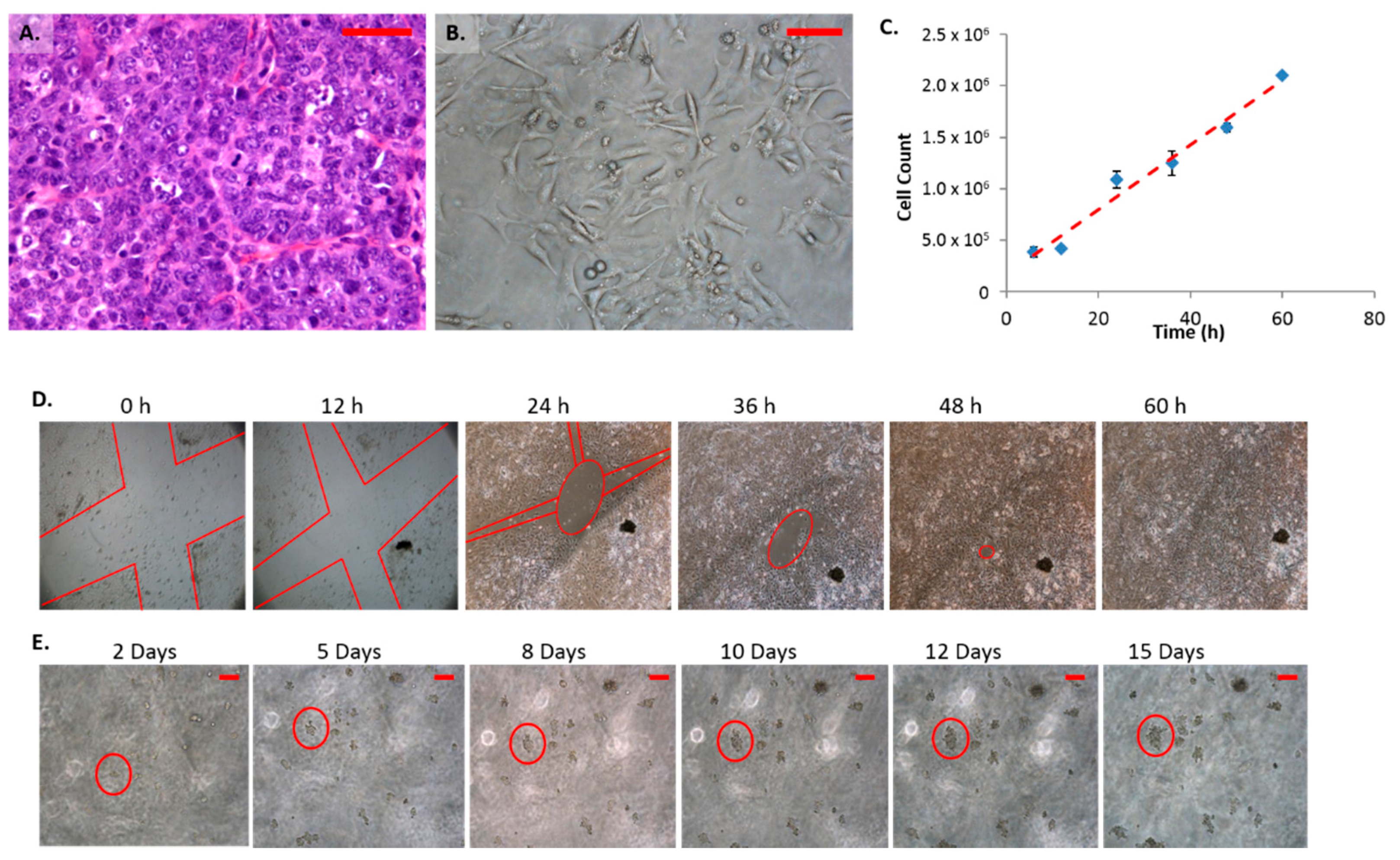
2.1. Morphology and Growth Characteristics of Female Mammary Carcinoma Cells (FMC2u)

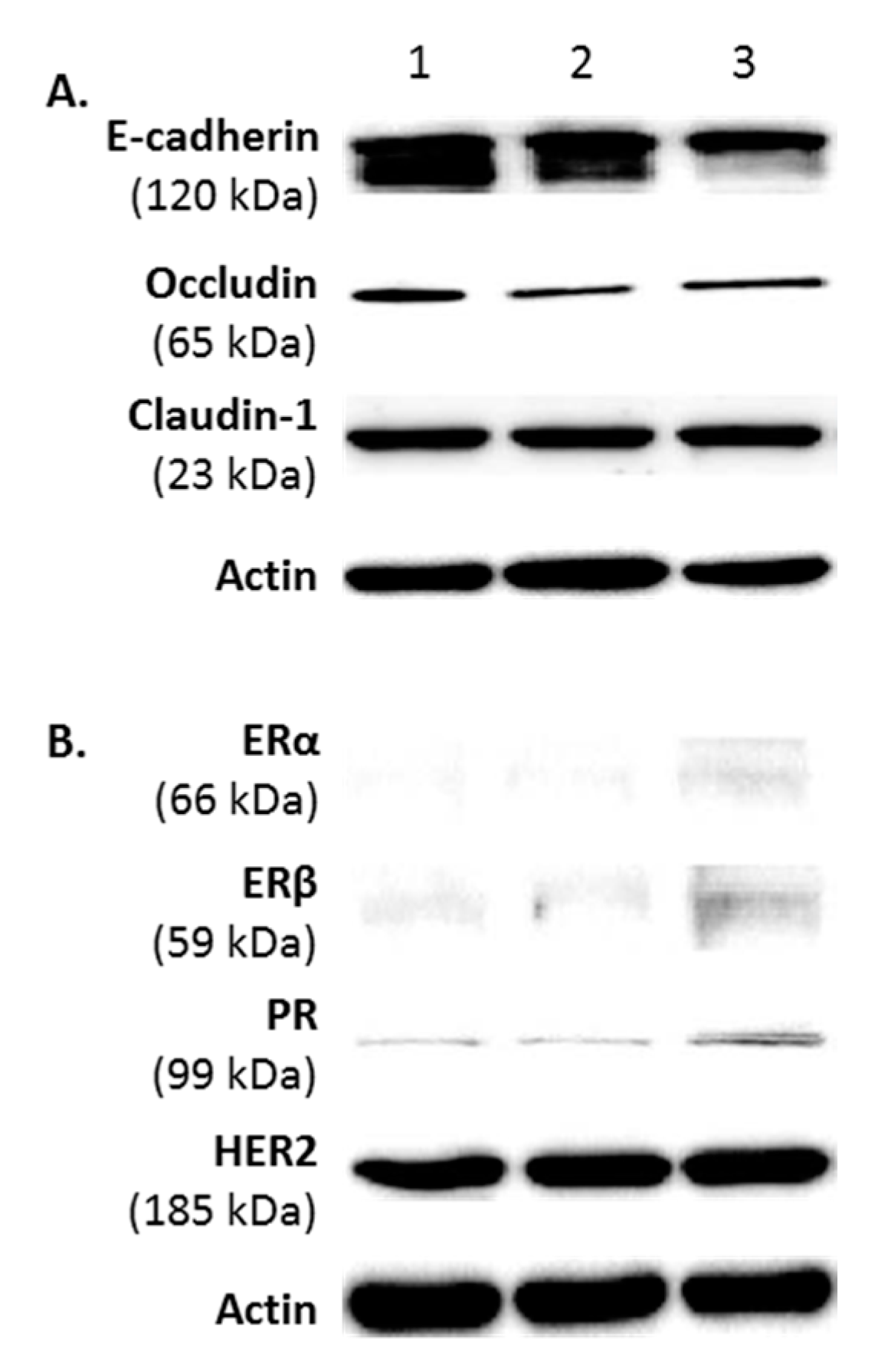
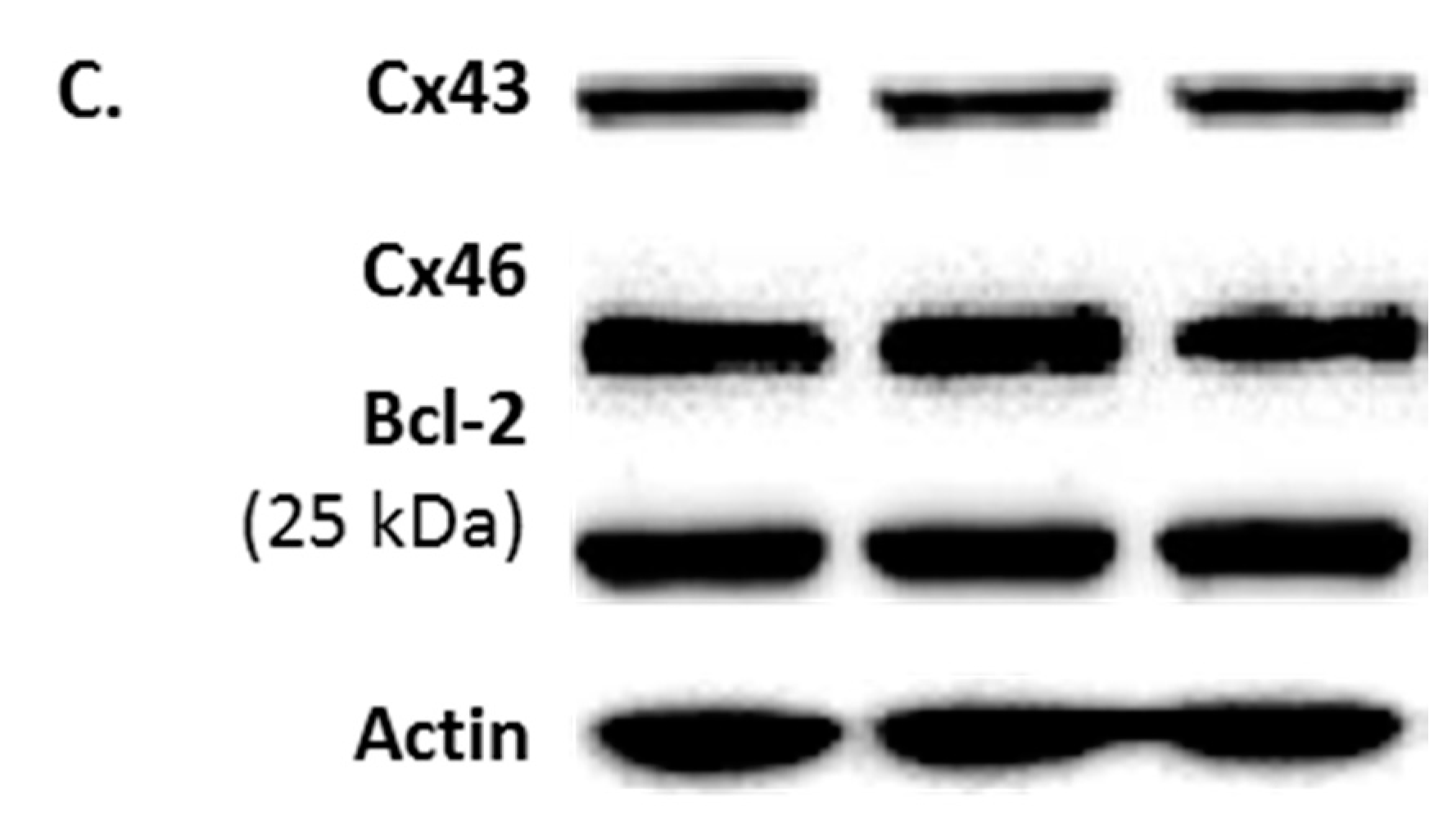
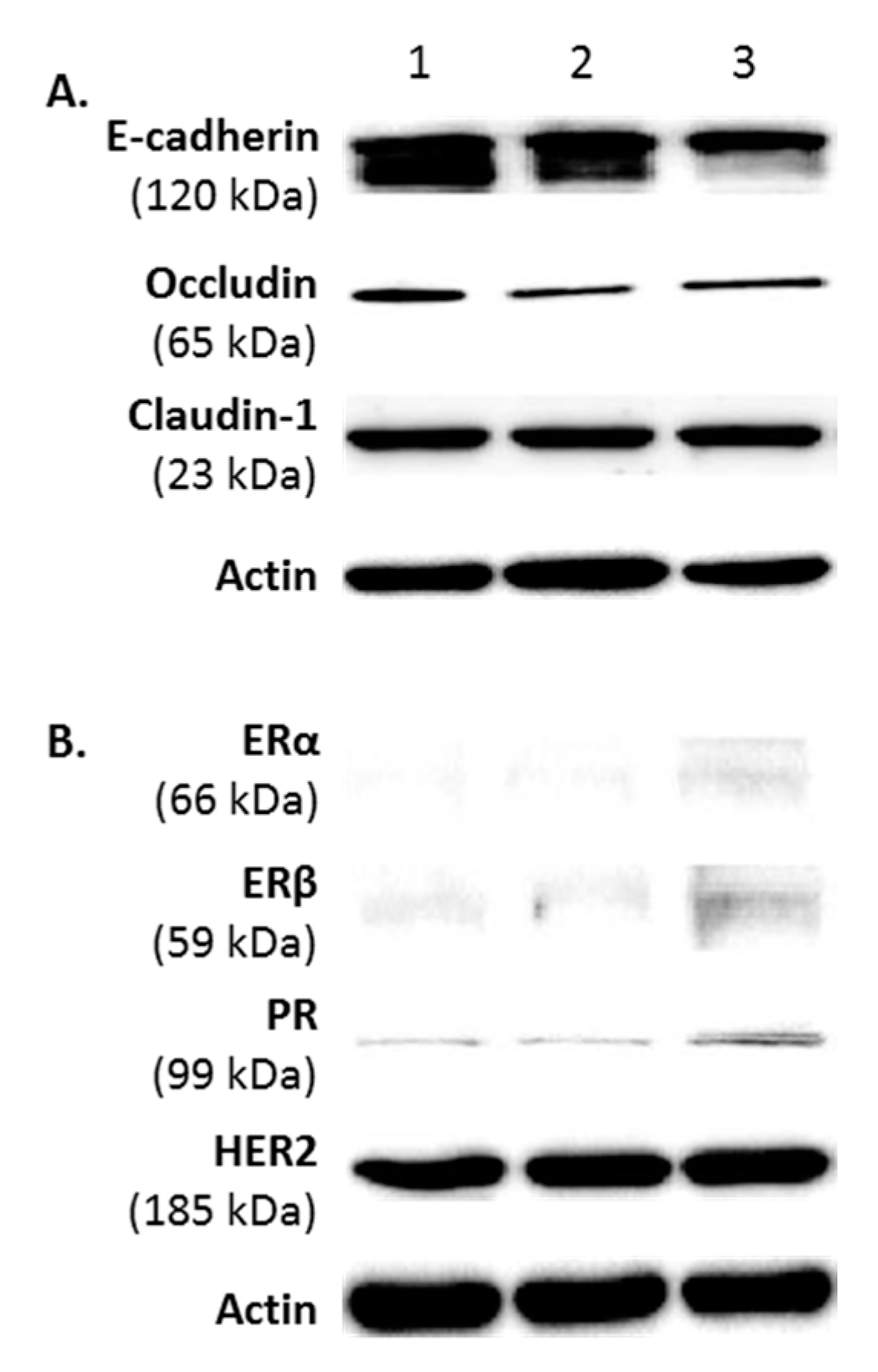
2.2. Protein Phenotype of FMC2u Cells

2.3. Effects of Treatment with Gap Junction Enhancers
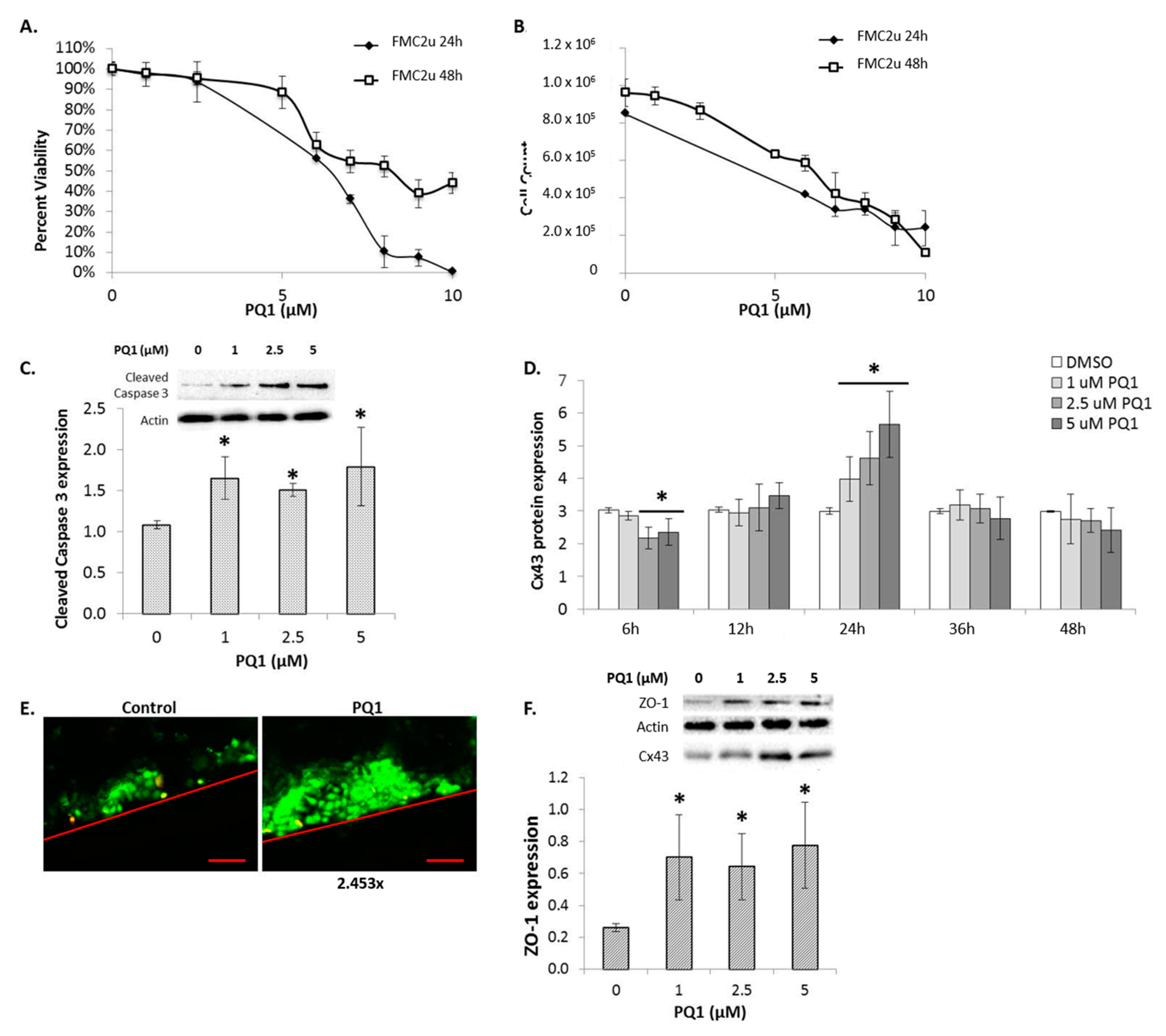
2.4. Cellular Proliferation and Viability
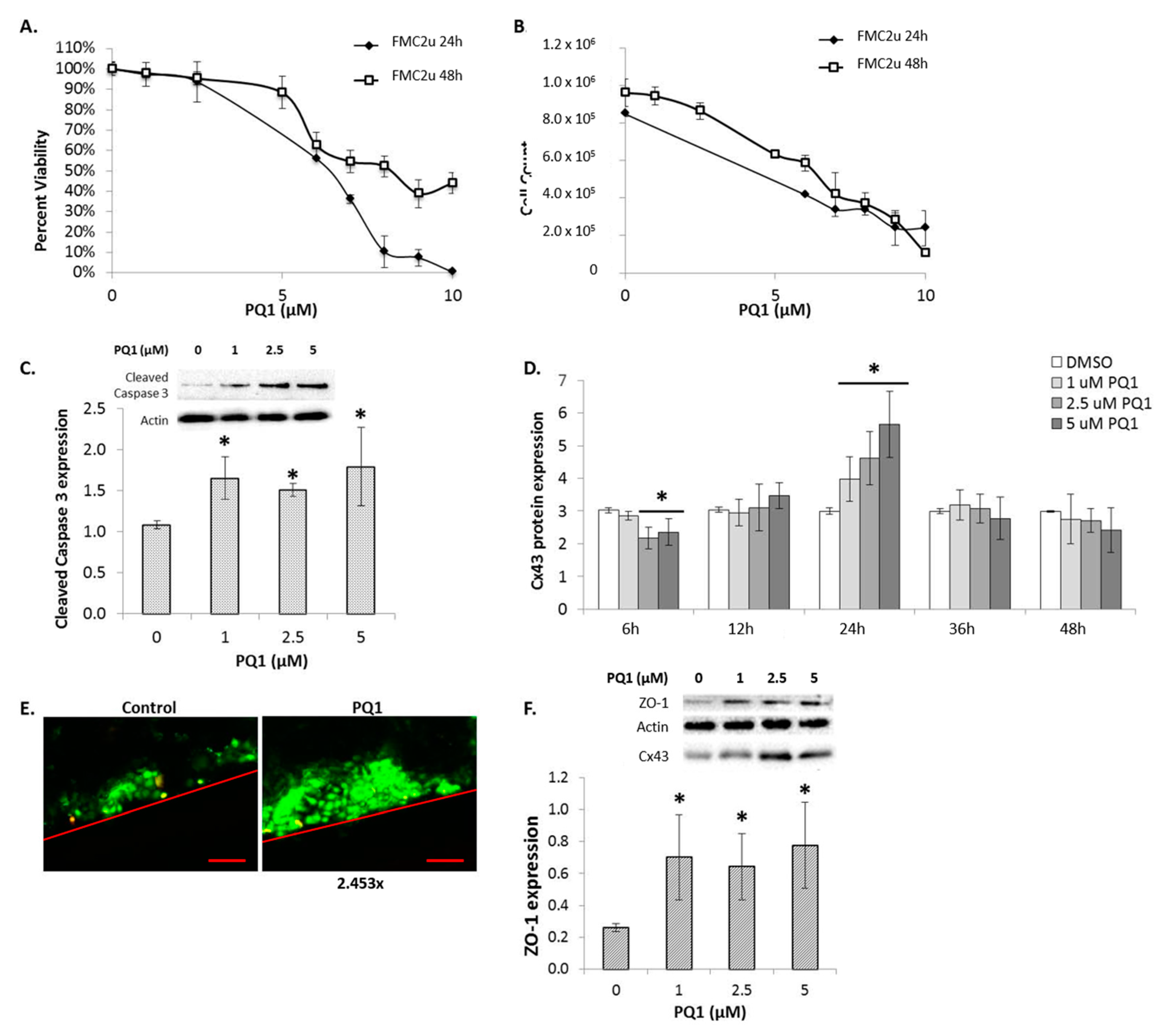
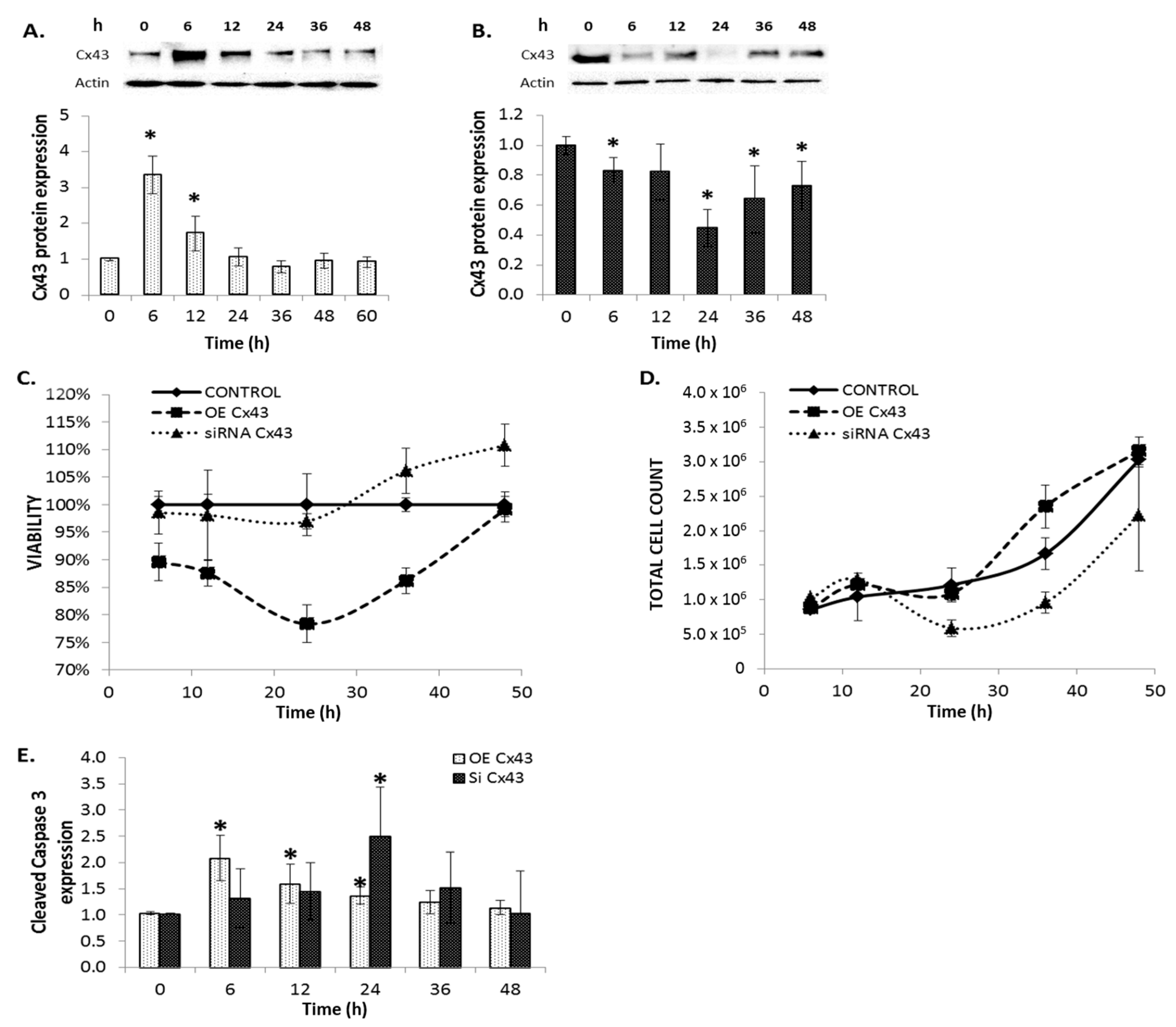
2.5. Connexin Expression
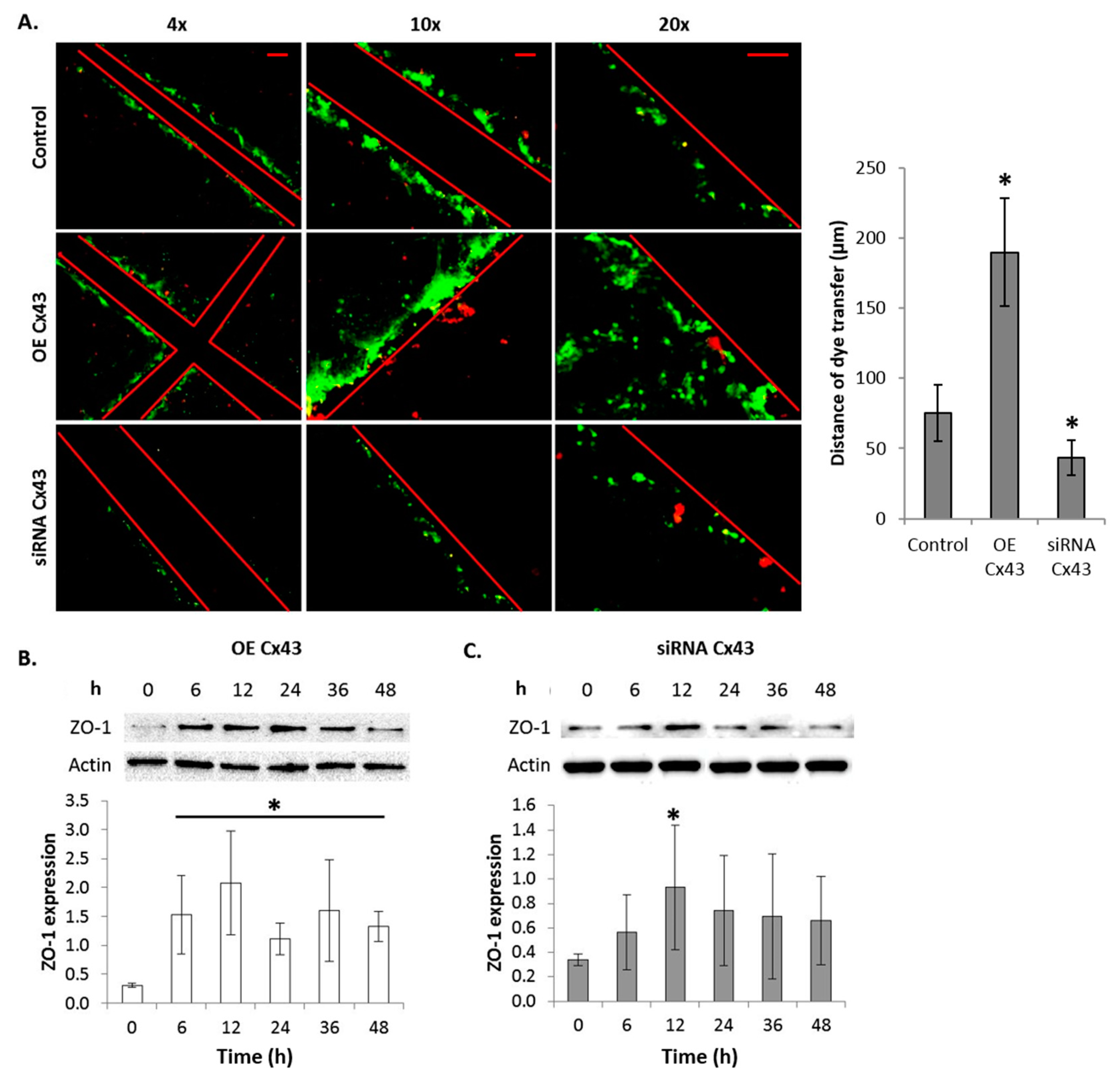
2.6. Gap Junctional Intercellular Communication (GJIC)-Dependent Effect: GJIC and Connexin Protein Binding
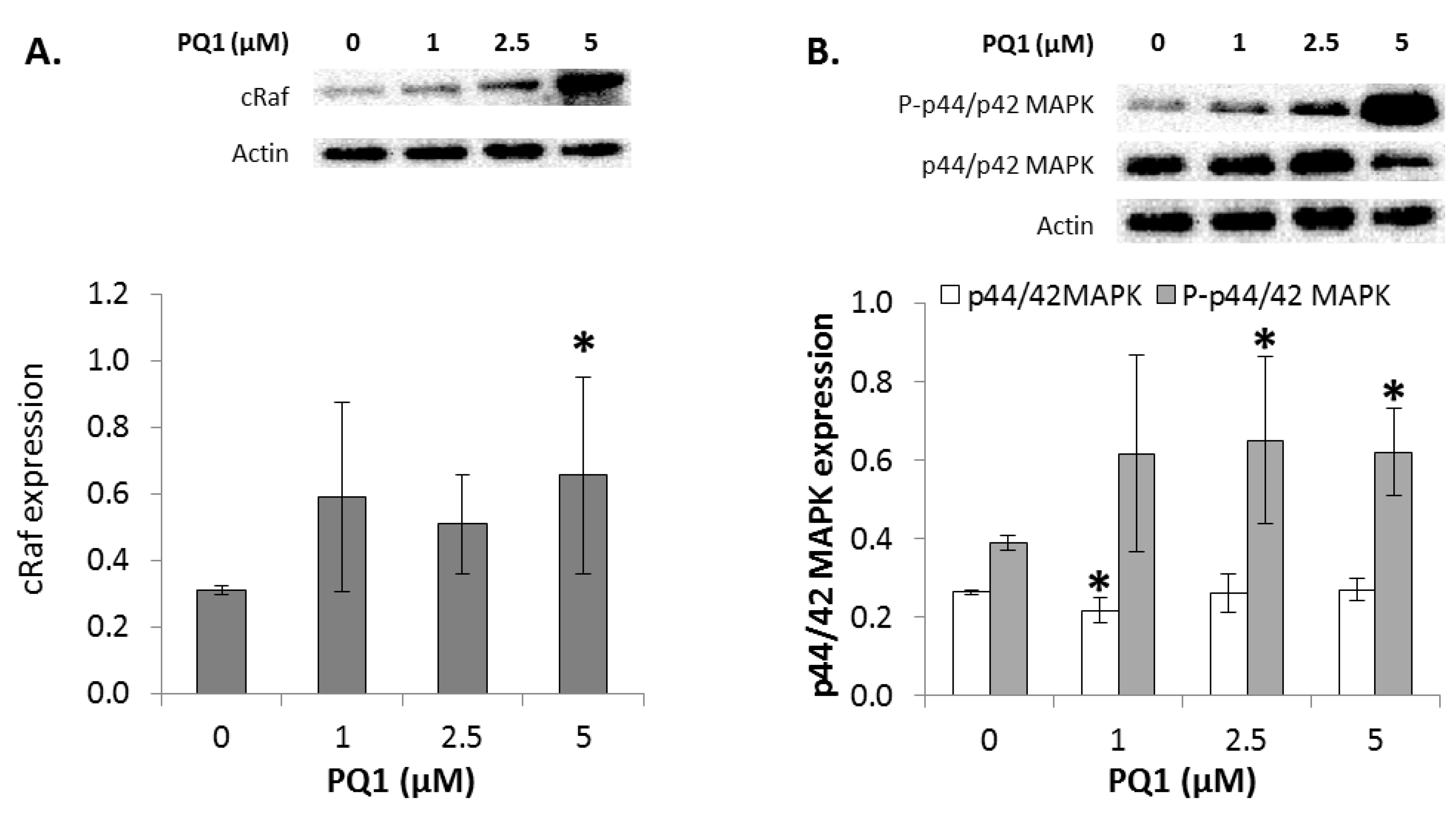
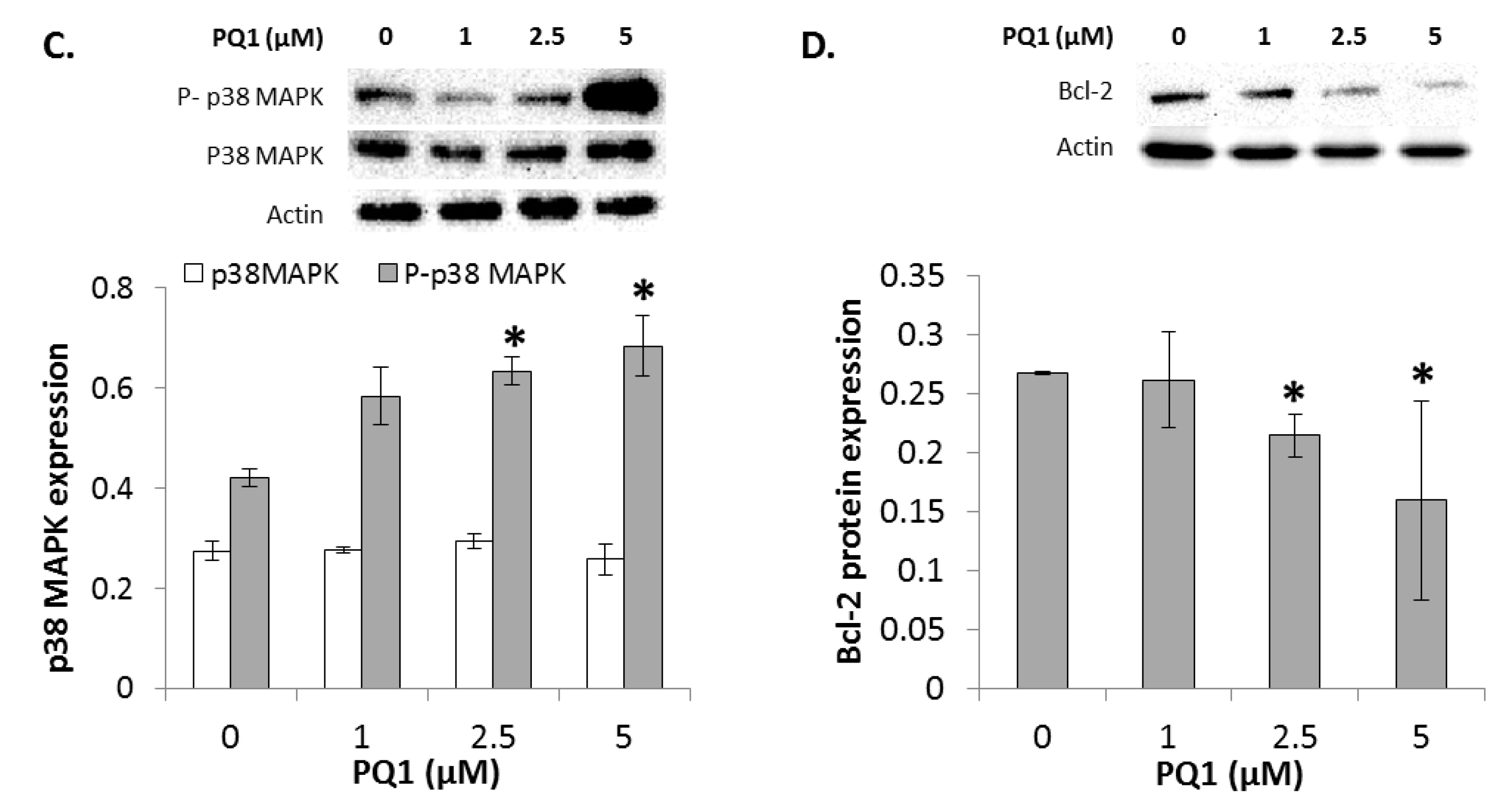
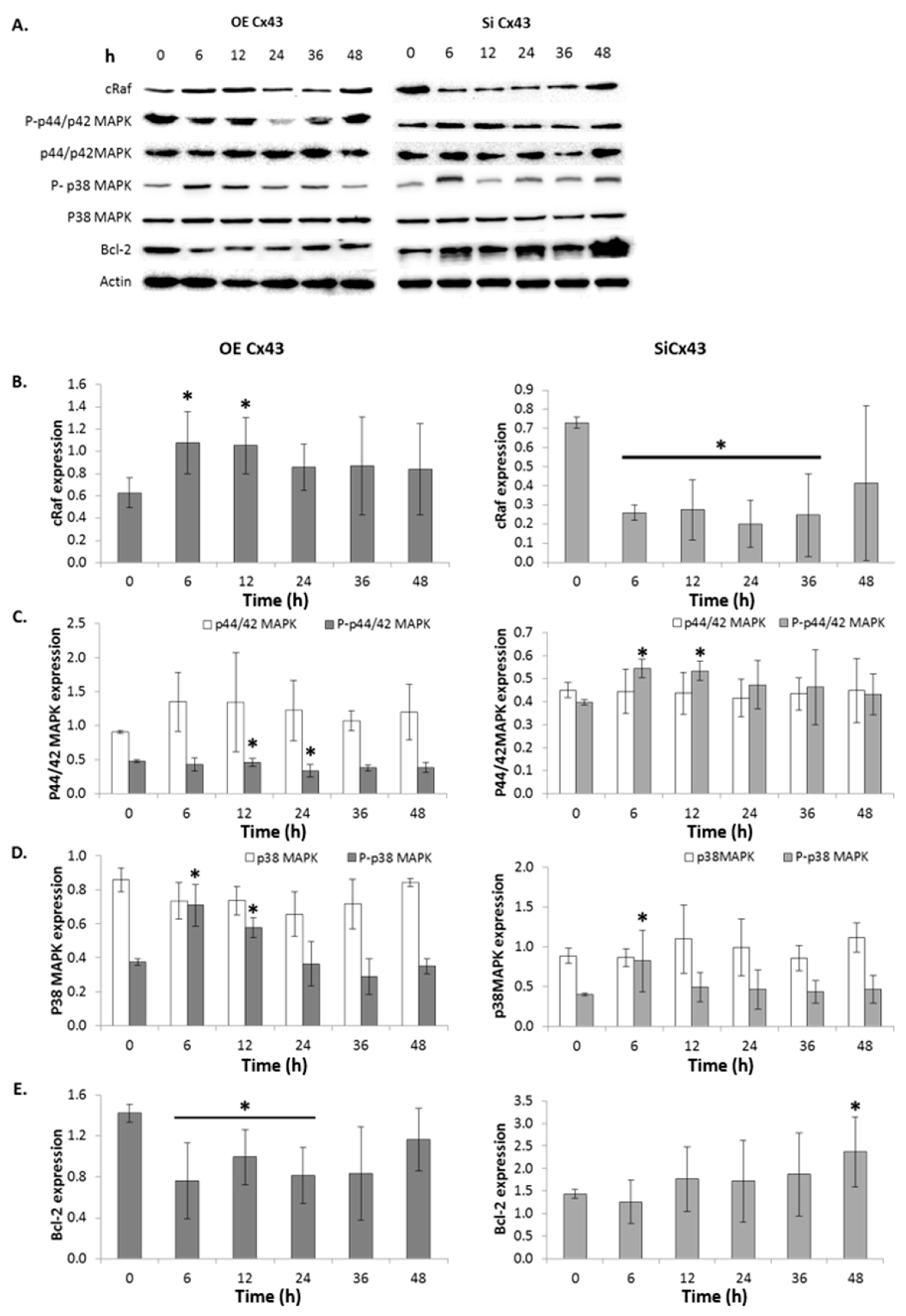
2.7. GJIC-Independent Effect: MAPK Signaling
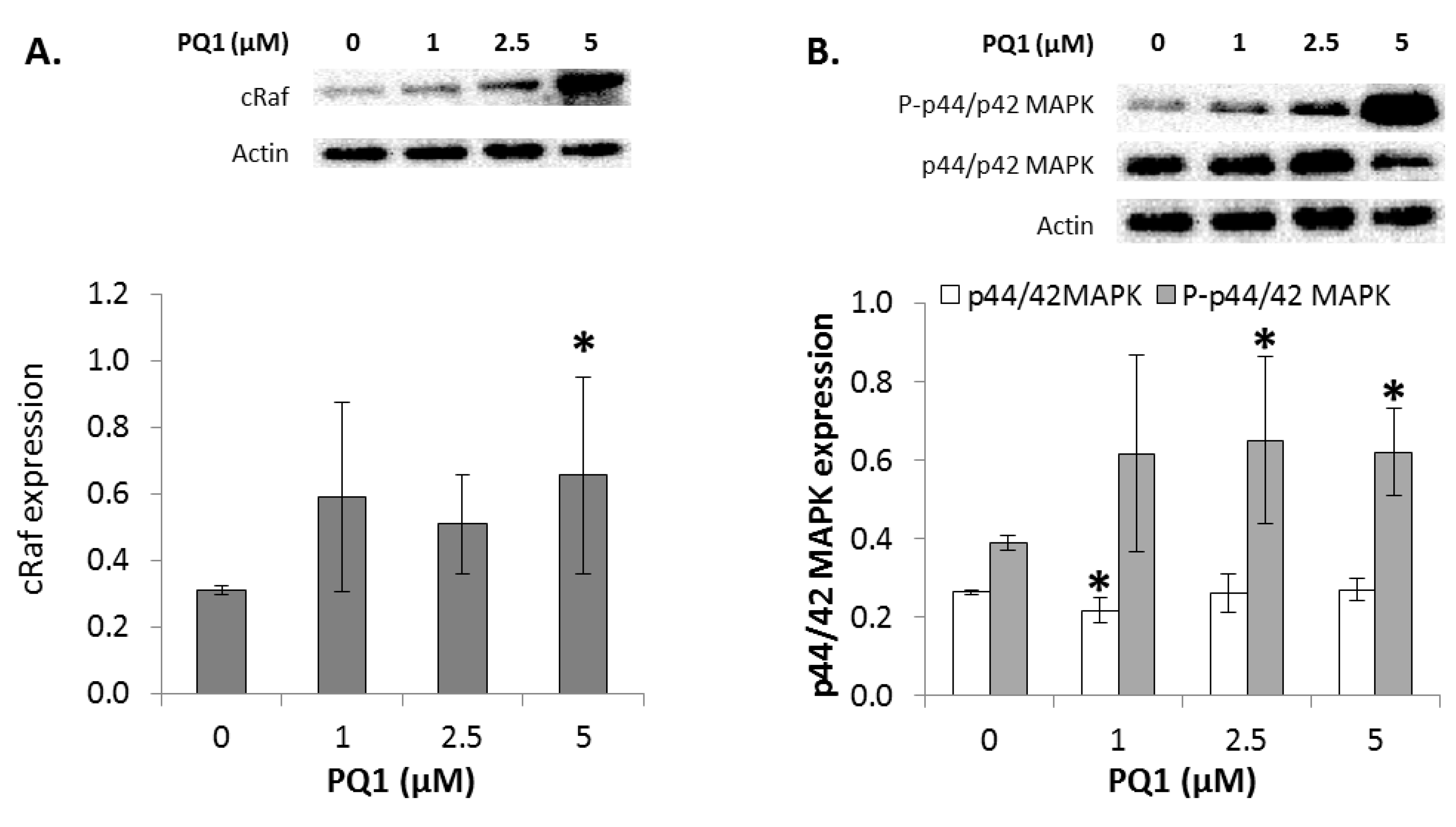
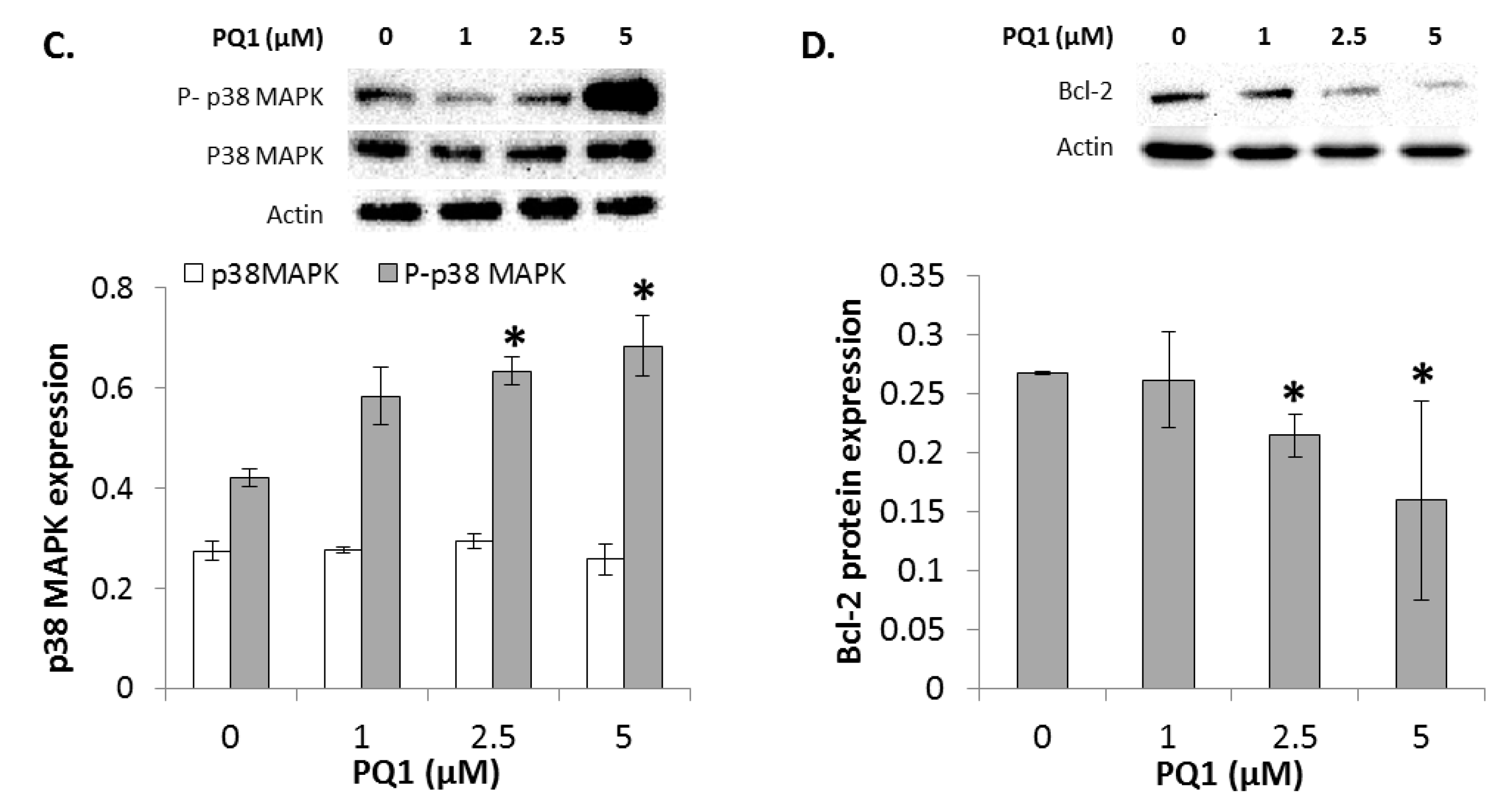
2.8. The Effects of Modulating Connexin 43 Protein Expression
2.9. Cellular Proliferation and Viability
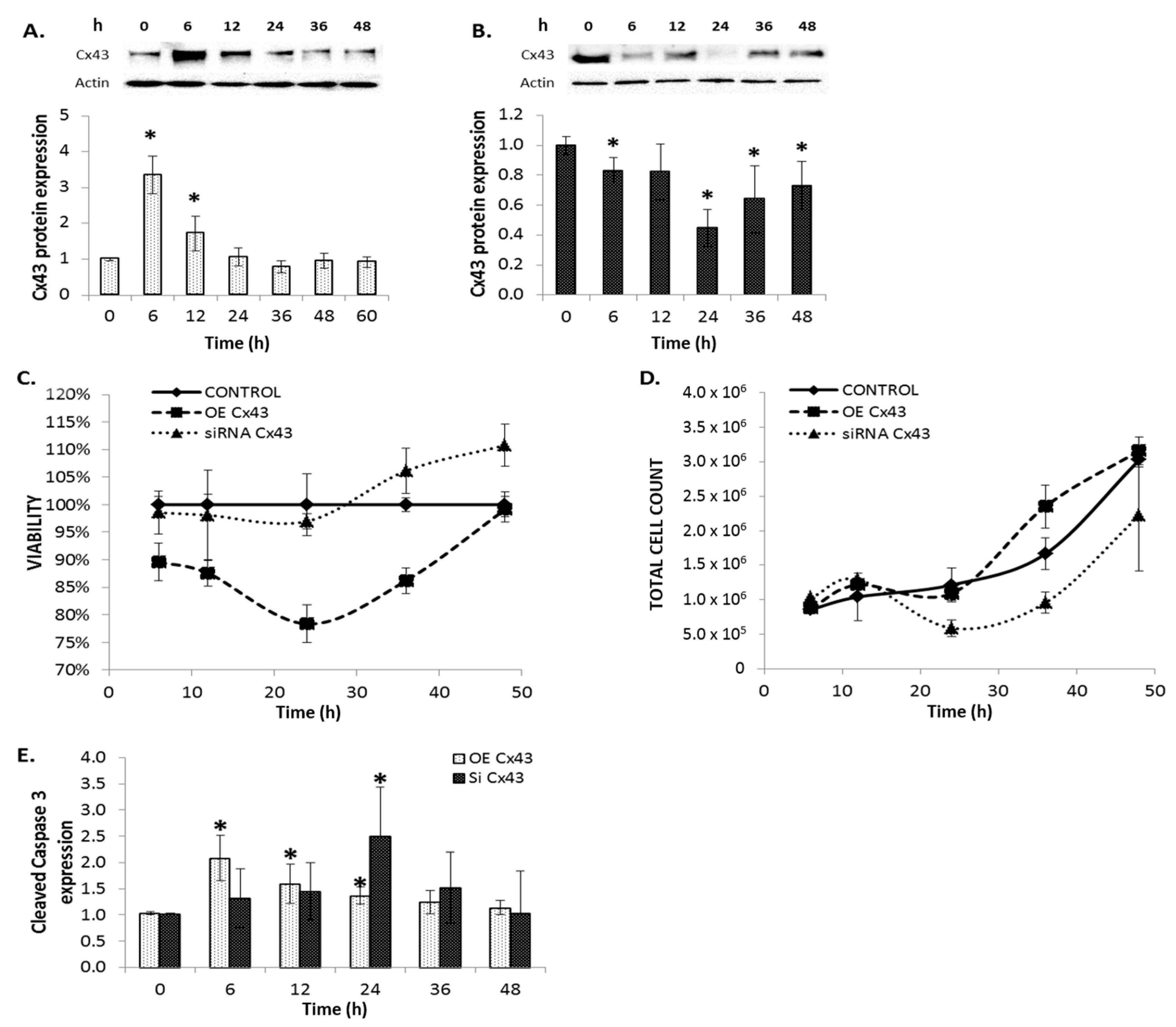
2.10. GJIC-Dependent Effect: GJIC and Connexin Protein Binding
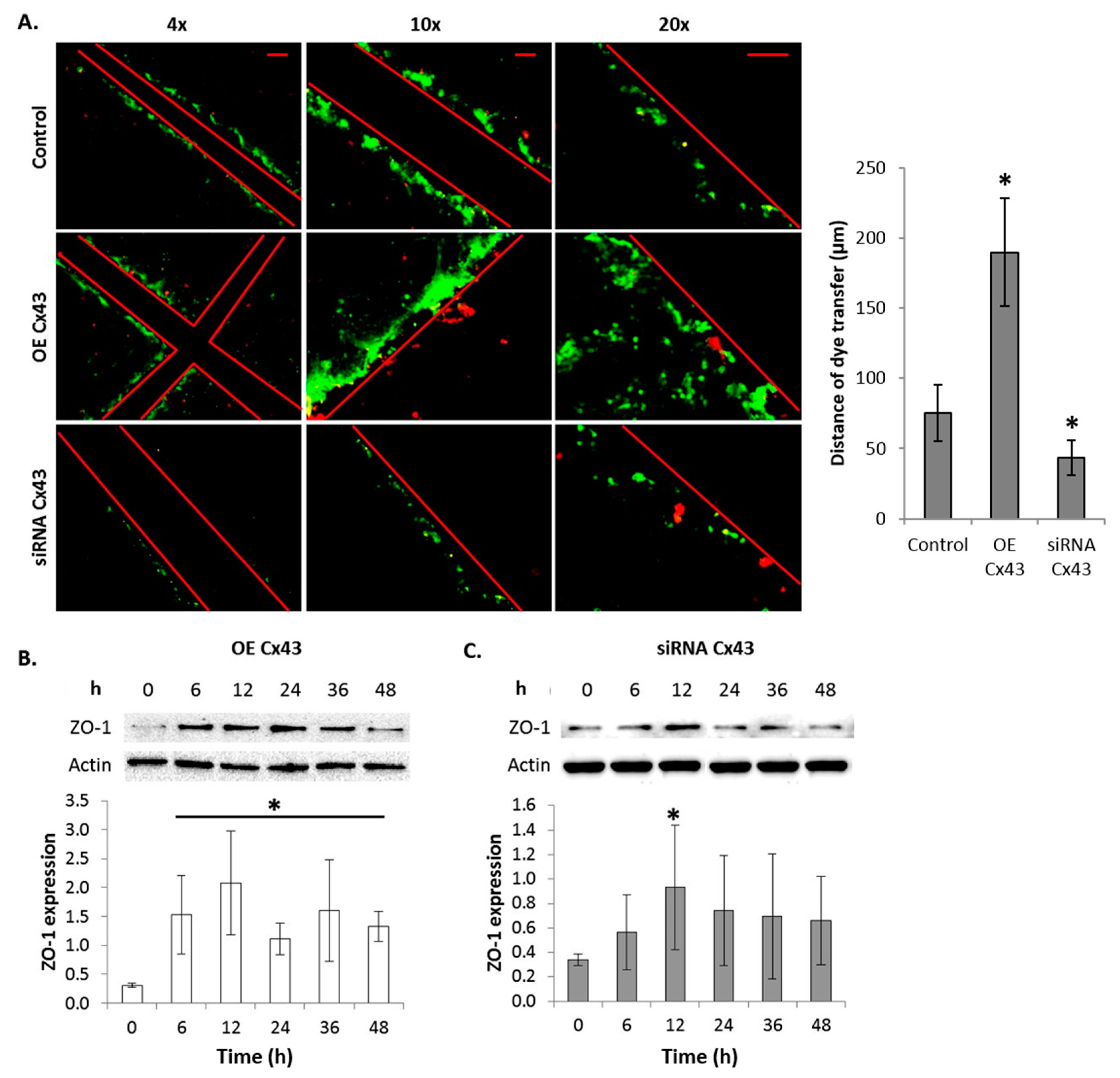
2.11. GJIC-Independent: MAPK Signaling
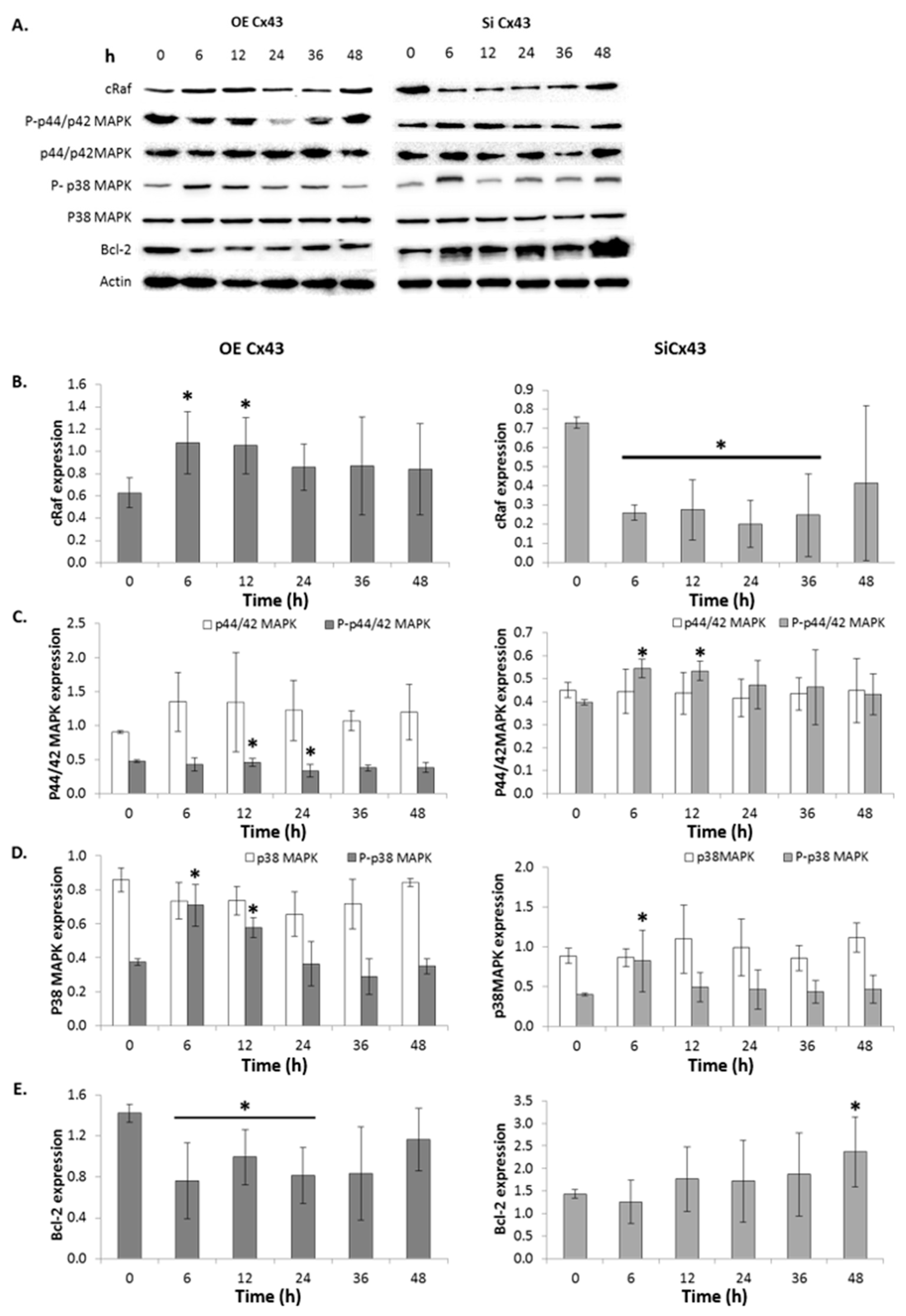
3. Discussion
4. Experimental Section
4.1. Ethics Statement
4.2. Establishment of Cell Cultures
4.3. Media Test
4.4. Cell Doubling Time
4.5. Colony Formation
4.6. Migration Assay
4.7. Invasion Assay
4.8. Cell Recovery
4.9. Antibodies
4.10. Western Blot Assay
4.11. Immunoprecipitation Assay
4.12. Treatment with Gap Junction Enhancers
4.13. Overexpression and Silencing of Connexins
4.14. Proliferation and Viability
4.15. Scrape Load Dye Transfer
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.; Ward, E.; Brawley, O.; Jemal, A. Cancer statistics, 2011: The impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J. Clin. 2011, 61, 212–236. [Google Scholar] [CrossRef] [PubMed]
- Shishido, S.N.; Delahaye, A.; Beck, A.; Nguyen, T.A. The MMTV-PyVT transgenic mouse as a multistage model for mammary carcinoma and the efficacy of antineoplastic treatment. J. Cancer Ther. 2013, 4, 1187–1197. [Google Scholar] [CrossRef]
- Willecke, K.; Eiberger, J.; Degen, J.; Eckardt, D.; Romualdi, A.; Guldenagel, M.; Deutsch, U.; Sohl, G. Structural and functional diversity of connexin genes in the mouse and human genome. Biol. Chem. 2002, 383, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Mesnil, M.; Crespin, S.; Avanzo, J.L.; Zaidan-Dagli, M.L. Defective gap junctional intercellular communication in the carcinogenic process. Biochim. Biophys. Acta 2005, 1719, 125–145. [Google Scholar] [CrossRef] [PubMed]
- Pointis, G.; Fiorini, C.; Gilleron, J.; Carette, D.; Segretain, D. Connexins as precocious markers and molecular targets for chemical and pharmacological agents in carcinogenesis. Curr. Med. Chem. 2007, 14, 2288–2303. [Google Scholar] [CrossRef] [PubMed]
- Naus, C.C.; Laird, D.W. Implications and challenges of connexin connections to cancer. Nat. Rev. 2010, 10, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.X.; Gu, S. Gap junction- and hemichannel-independent actions of connexins. Biochim. Biophys. Acta 2005, 1711, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Gakhar, G.; Hua, D.H.; Nguyen, T.A. Combinational treatment of gap junctional activator and tamoxifen in breast cancer cells. Anticancer Drugs 2010, 21, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Gakhar, G.; Ohira, T.; Shi, A.B.; Hua, D.H.; Nguyen, T.A. Antitumor effect of substituted quinolines in breast cancer cells. Drug Dev. Res. 2008, 69, 526–534. [Google Scholar] [CrossRef]
- Bernzweig, J.; Heiniger, B.; Prasain, K.; Lu, J.; Hua, D.H.; Nguyen, T.A. Anti-breast cancer agents, quinolines, targeting gap junction. Med. Chem. 2011, 7, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.M.; VanBrocklin, M.; McWilliams, M.J.; Leppla, S.H.; Duesbery, N.S.; Vande Woude, G.F. Apoptosis and melanogenesis in human melanoma cells induced by anthrax lethal factor inactivation of mitogen-activated protein kinase kinase. Proc. Natl. Acad. Sci. USA 2002, 99, 3052–3057. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Todokoro, K. Requirement of activation of JNK and p38 for environmental stress-induced erythroid differentiation and apoptosis and of inhibition of ERK for apoptosis. Blood 1999, 94, 853–863. [Google Scholar] [PubMed]
- Cory, S.; Huang, D.C.; Adams, J.M. The Bcl-2 family: Roles in cell survival and oncogenesis. Oncogene 2003, 22, 8590–8607. [Google Scholar] [CrossRef] [PubMed]
- Lipponen, P.; Pietilainen, T.; Kosma, V.M.; Aaltomaa, S.; Eskelinen, M.; Syrjanen, K. Apoptosis suppressing protein Bcl-2 is expressed in well-differentiated breast carcinomas with favourable prognosis. J. Pathol. 1995, 177, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Silvestrini, R.; Veneroni, S.; Daidone, M.G.; Benini, E.; Boracchi, P.; Mezzetti, M.; di Fronzo, G.; Rilke, F.; Veronesi, U. The Bcl-2 protein: A prognostic indicator strongly related to p53 protein in lymph node-negative breast cancer patients. J. Natl. Cancer Inst. 1994, 86, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Paik, S.; Shak, S.; Tang, G.; Kim, C.; Baker, J.; Cronin, M.; Baehner, F.L.; Walker, M.G.; Watson, D.; Park, T.; et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med. 2004, 351, 2817–2826. [Google Scholar] [CrossRef] [PubMed]
- Shi, A.; Nguyen, T.A.; Battina, S.K.; Rana, S.; Takemoto, D.J.; Chiang, P.K.; Hua, D.H. Synthesis and anti-breast cancer activities of substituted quinolines. Bioorg. Med. Chem. Lett. 2008, 18, 3364–3368. [Google Scholar] [CrossRef] [PubMed]
- Heiniger, B.; Gakhar, G.; Prasain, K.; Hua, D.H.; Nguyen, T.A. Second-generation substituted quinolines as anticancer drugs for breast cancer. Anticancer Res. 2010, 30, 3927–3932. [Google Scholar] [PubMed]
- Shishido, S.N.; Delahaye, A.; Beck, A.; Nguyen, T.A. The anticancer effect of PQ1 in the MMTV-PyVT mouse model. Int. J. Cancer 2014, 134, 1474–1483. [Google Scholar] [CrossRef] [PubMed]
- Shishido, S.N.; Nguyen, T.A. Gap junction enhancer increases efficacy of cisplatin to attenuate mammary tumor growth. PLoS ONE 2012, 7, e44963. [Google Scholar] [CrossRef] [PubMed]
- Giepmans, B.N.; Moolenaar, W.H. The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Curr. Biol. 1998, 8, 931–934. [Google Scholar] [CrossRef]
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta 2007, 1773, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Iervolino, A.; Trisciuoglio, D.; Ribatti, D.; Candiloro, A.; Biroccio, A.; Zupi, G.; del Bufalo, D. Bcl-2 overexpression in human melanoma cells increases angiogenesis through VEGF mRNA stabilization and HIF-1-mediated transcriptional activity. FASEB J. 2002, 16, 1453–1455. [Google Scholar] [CrossRef] [PubMed]
- De Chiara, G.; Marcocci, M.E.; Torcia, M.; Lucibello, M.; Rosini, P.; Bonini, P.; Higashimoto, Y.; Damonte, G.; Armirotti, A.; Amodei, S.; et al. Bcl-2 phosphorylation by p38 MAPK–Identification of target sites and biologic consequences. J. Biol. Chem. 2006, 281, 21353–21361. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Nguyen, T.A. PQ1, a quinoline derivative, induces apoptosis in T47D breast cancer cells through activation of caspase-8 and caspase-9. Apoptosis 2013, 18, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.M.; Clark, G.M.; Osborne, C.K.; Allred, D.C. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J. Clin. Oncol. 1999, 17, 1474–1481. [Google Scholar] [PubMed]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Borg, A.; Baldetorp, B.; Ferno, M.; Killander, D.; Olsson, H.; Ryden, S.; Sigurdsson, H. ERBB2 amplification is associated with tamoxifen resistance in steroid-receptor positive breast-cancer. Cancer Lett. 1994, 81, 137–144. [Google Scholar] [CrossRef]
- Wright, C.; Nicholson, S.; Angus, B.; Sainsbury, J.R.C.; Farndon, J.; Cairns, J.; Harris, A.L.; Horne, C.H.W. Relationship between c-erbB-2 protein product expression and response to endocrine therapy in advanced breast-cancer. Br. J. Cancer 1992, 65, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.; Hunter, C.; Bignell, G.; Edkins, S.; Davies, H.; Teague, J.; Stevens, C.; O'Meara, S.; Smith, R.; Parker, A.; et al. Lung cancer: Intragenic ERBB2 kinase mutations in tumours. Nature 2004, 431, 525–526. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; Lane, H.A. Erbb receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Krutovskikh, V.; Mazzoleni, G.; Mironov, N.; Omori, Y.; Aguelon, A.M.; Mesnil, M.; Berger, F.; Partensky, C.; Yamasaki, H. Altered homologous and heterologous gap-junctional intercellular communication in primary human liver tumors associated with aberrant protein localization but not gene mutation of connexin 32. Int. J. Cancer 1994, 56, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Hirschi, K.K.; Xu, C.E.; Tsukamoto, T.; Sager, R. Gap junction genes Cx26 and Cx43 individually suppress the cancer phenotype of human mammary carcinoma cells and restore differentiation potential. Cell Growth Differ. 1996, 7, 861–870. [Google Scholar] [PubMed]
- Jou, Y.S.; Layhe, B.; Matesic, D.F.; Chang, C.C.; de Feijter, A.W.; Lockwood, L.; Welsch, C.W.; Klaunig, J.E.; Trosko, J.E. Inhibition of gap junctional intercellular communication and malignant transformation of rat liver epithelial cells by neu oncogene. Carcinogenesis 1995, 16, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Budunova, I.V.; Carbajal, S.; Slaga, T.J. Effect of diverse tumor promoters on the expression of gap-junctional proteins connexin (Cx)26, Cx31.1, and Cx43 in SENCAR mouse epidermis. Mol. Carcinog. 1996, 15, 202–214. [Google Scholar] [CrossRef]
- Saunders, M.M.; Seraj, M.J.; Li, Z.; Zhou, Z.; Winter, C.R.; Welch, D.R.; Donahue, H.J. Breast cancer metastatic potential correlates with a breakdown in homospecific and heterospecific gap junctional intercellular communication. Cancer Res. 2001, 61, 1765–1767. [Google Scholar] [PubMed]
- Teleki, I.; Krenacs, T.; Szasz, M.A.; Kulka, J.; Wichmann, B.; Leo, C.; Papassotiropoulos, B.; Riemenschnitter, C.; Moch, H.; Varga, Z. The potential prognostic value of connexin 26 and 46 expression in neoadjuvant-treated breast cancer. BMC Cancer 2013, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.P.; Fan, Y.; Hossain, M.Z.; Peng, A.; Zeng, Z.L.; Boynton, A.L. Reversion of the neoplastic phenotype of human glioblastoma cells by connexin 43 (Cx43). Cancer Res. 1998, 58, 5089–5096. [Google Scholar] [PubMed]
- Princen, F.; Robe, P.; Gros, D.; Jarry-Guichard, T.; Gielen, J.; Merville, M.P.; Bours, V. Rat gap junction connexin-30 inhibits proliferation of glioma cell lines. Carcinogenesis 2001, 22, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Herve, J.C.; Bourmeyster, N.; Sarrouilhe, D.; Duffy, H.S. Gap junctional complexes: From partners to functions. Prog. Biophys. Mol. Biol. 2007, 94, 29–65. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.J.; Xu, X.; Lo, C.W. Connexins and cell signaling in development and disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 811–838. [Google Scholar] [CrossRef] [PubMed]
- Barker, R.J.; Price, R.L.; Gourdie, R.G. Increased association of ZO-1 with connexin43 during remodeling of cardiac gap junctions. Circ. Res. 2002, 90, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Duffy, H.S.; Ashton, A.W.; O‘Donnell, P.; Coombs, W.; Taffet, S.M.; Delmar, M.; Spray, D.C. Regulation of connexin43 protein complexes by intracellular acidification. Circ. Res. 2004, 94, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Segretain, D.; Fiorini, C.; Decrouy, X.; Defamie, N.; Prat, J.R.; Pointis, G. A proposed role for ZO-1 in targeting connexin 43 gap junctions to the endocytic pathway. Biochimie 2004, 86, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Laing, J.G.; Chou, B.C.; Steinberg, T.H. ZO-1 alters the plasma membrane localization and function of Cx43 in osteoblastic cells. J. Cell Sci. 2005, 118, 2167–2176. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gilleron, J.; Fiorini, C.; Carette, D.; Avondet, C.; Falk, M.M.; Segretain, D.; Pointis, G. Molecular reorganization of Cx43, ZO-1 and src complexes during the endocytosis of gap junction plaques in response to a non-genomic carcinogen. J. Cell Sci. 2008, 121, 4069–4078. [Google Scholar] [CrossRef] [PubMed]
- Bond, S.L.; Bechberger, J.F.; Khoo, N.K.; Naus, C.C. Transfection of c6 glioma cells with connexin32: The effects of expression of a nonendogenous gap junction protein. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 1994, 5, 179–186. [Google Scholar]
- Musil, L.S.; Cunningham, B.A.; Edelman, G.M.; Goodenough, D.A. Differential phosphorylation of the gap junction protein connexin43 in junctional communication-competent and -deficient cell lines. J. Cell Biol. 1990, 111, 2077–2088. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Shao, Q.; Curtis, H.; Galipeau, J.; Belliveau, D.J.; Wang, T.; Alaoui-Jamali, M.A.; Laird, D.W. Retroviral delivery of connexin genes to human breast tumor cells inhibits in vivo tumor growth by a mechanism that is independent of significant gap junctional intercellular communication. J. Biol. Chem. 2002, 277, 29132–29138. [Google Scholar] [CrossRef] [PubMed]
- Dang, X.T.; Doble, B.W.; Kardami, E. The carboxy-tail of connexin-43 localizes to the nucleus and inhibits cell growth. Mol. Cell. Biochem. 2003, 242, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Moorby, C.; Patel, M. Dual functions for connexins: Cx43 regulates growth independently of gap junction formation. Exp. Cell Res. 2001, 271, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Penninger, J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004, 23, 2838–2849. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Fujii, K.; Zhang, L.; Roberts, T.; Fu, H. Raf-1 promotes cell survival by antagonizing apoptosis signal-regulating kinase 1 through a MEK-ERK independent mechanism. Proc. Natl. Acad. Sci. USA 2001, 98, 7783–7788. [Google Scholar] [CrossRef] [PubMed]
- Trosko, J.E.; Chang, C.C.; Medcalf, A. Mechanisms of tumor promotion: Potential role of intercellular communication. Cancer Invest. 1983, 1, 511–526. [Google Scholar] [CrossRef] [PubMed]
- Rummel, A.M.; Trosko, J.E.; Wilson, M.R.; Upham, B.L. Polycyclic aromatic hydrocarbons with bay-like regions inhibited gap junctional intercellular communication and stimulated MAPK activity. Toxicol. Sci. 1999, 49, 232–240. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shishido, S.N.; Nguyen, T.A. Induction of Apoptosis by PQ1, a Gap Junction Enhancer that Upregulates Connexin 43 and Activates the MAPK Signaling Pathway in Mammary Carcinoma Cells. Int. J. Mol. Sci. 2016, 17, 178. https://doi.org/10.3390/ijms17020178
Shishido SN, Nguyen TA. Induction of Apoptosis by PQ1, a Gap Junction Enhancer that Upregulates Connexin 43 and Activates the MAPK Signaling Pathway in Mammary Carcinoma Cells. International Journal of Molecular Sciences. 2016; 17(2):178. https://doi.org/10.3390/ijms17020178
Chicago/Turabian StyleShishido, Stephanie N., and Thu A. Nguyen. 2016. "Induction of Apoptosis by PQ1, a Gap Junction Enhancer that Upregulates Connexin 43 and Activates the MAPK Signaling Pathway in Mammary Carcinoma Cells" International Journal of Molecular Sciences 17, no. 2: 178. https://doi.org/10.3390/ijms17020178
APA StyleShishido, S. N., & Nguyen, T. A. (2016). Induction of Apoptosis by PQ1, a Gap Junction Enhancer that Upregulates Connexin 43 and Activates the MAPK Signaling Pathway in Mammary Carcinoma Cells. International Journal of Molecular Sciences, 17(2), 178. https://doi.org/10.3390/ijms17020178