
The Protective Effect of Icariin on Mitochondrial Transport and Distribution in Primary Hippocampal Neurons from 3× Tg-AD Mice

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
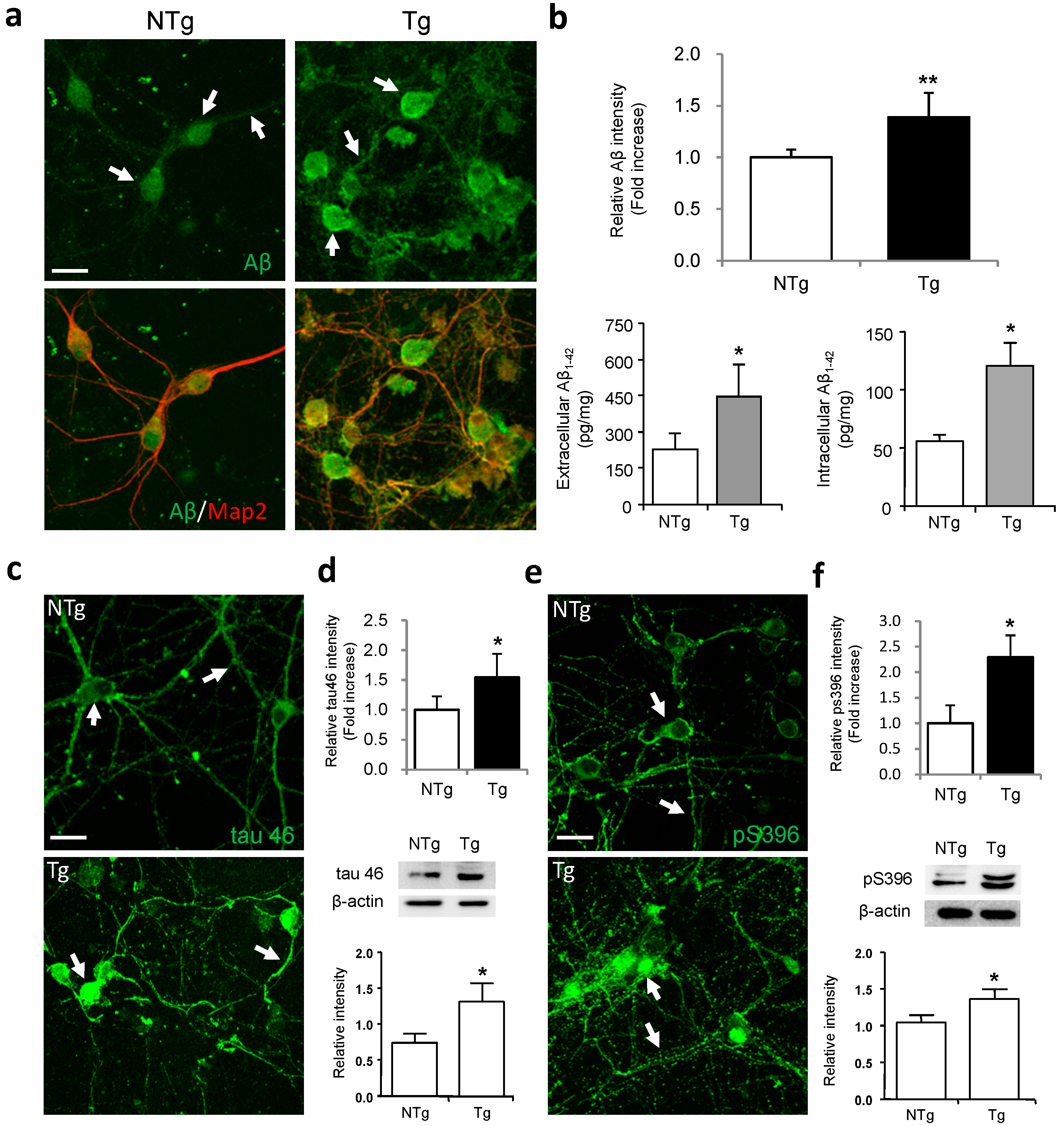
2.1. Characterization of Amyloid-β Peptide (Aβ) and Tau Expression in Primary Hippocampal Neurons from 3× Transgenic (Tg)-Alzheimer’s Disease (AD) Mice
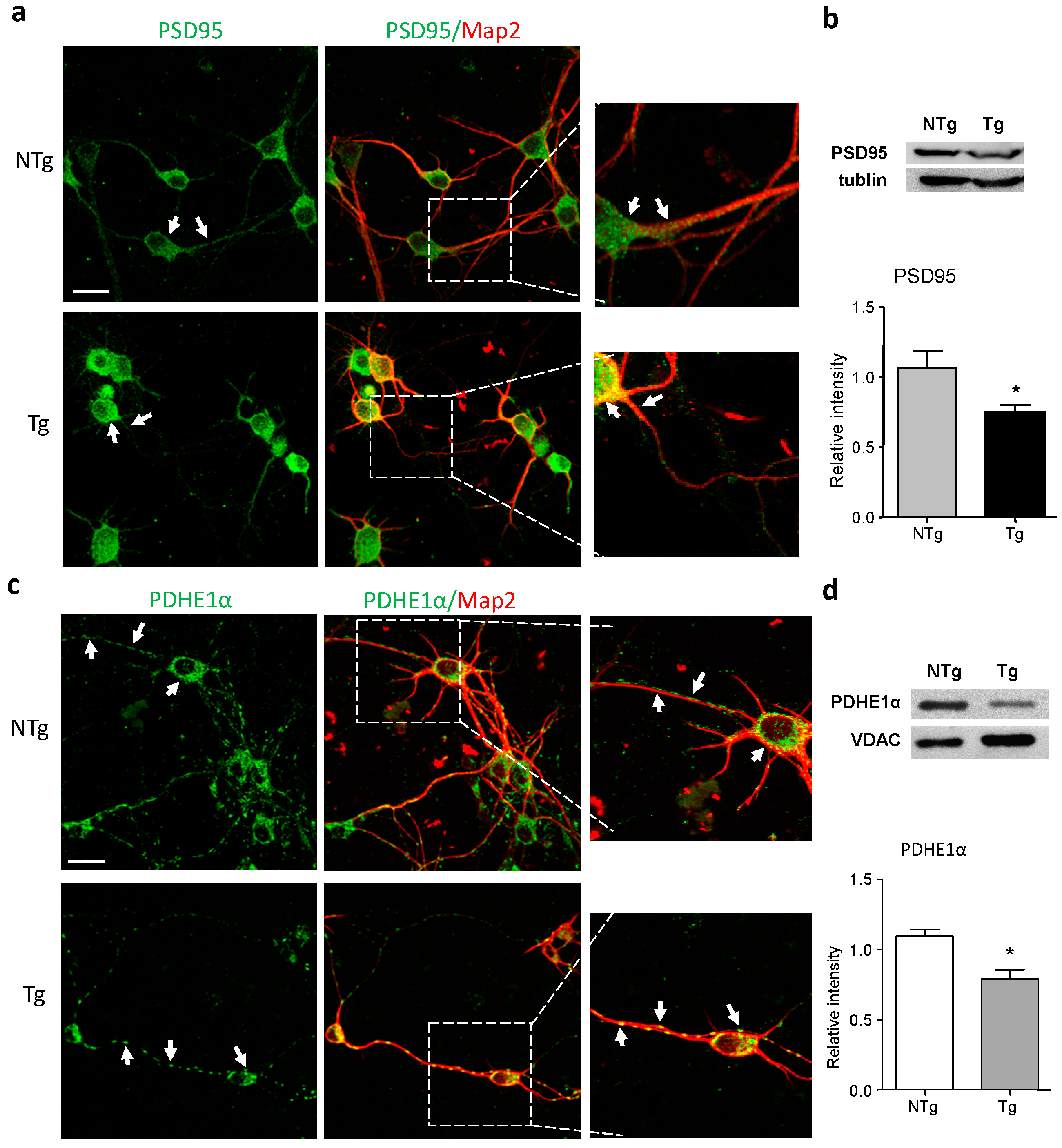
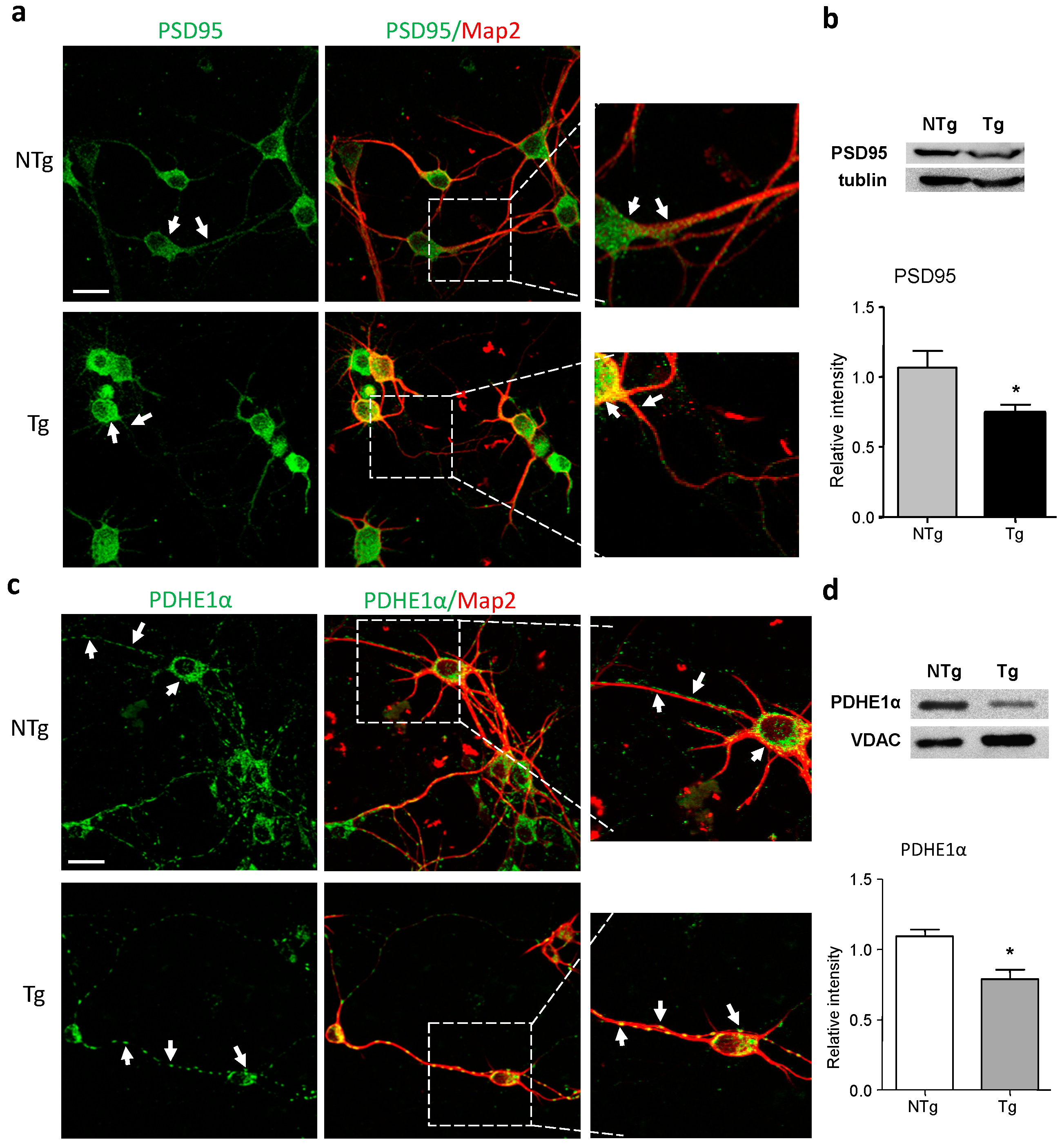
2.2. Alterations in Synaptic and Mitochondrial Protein Expression in AD Neurons
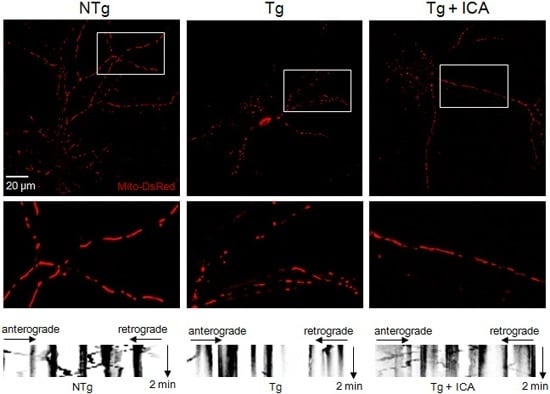
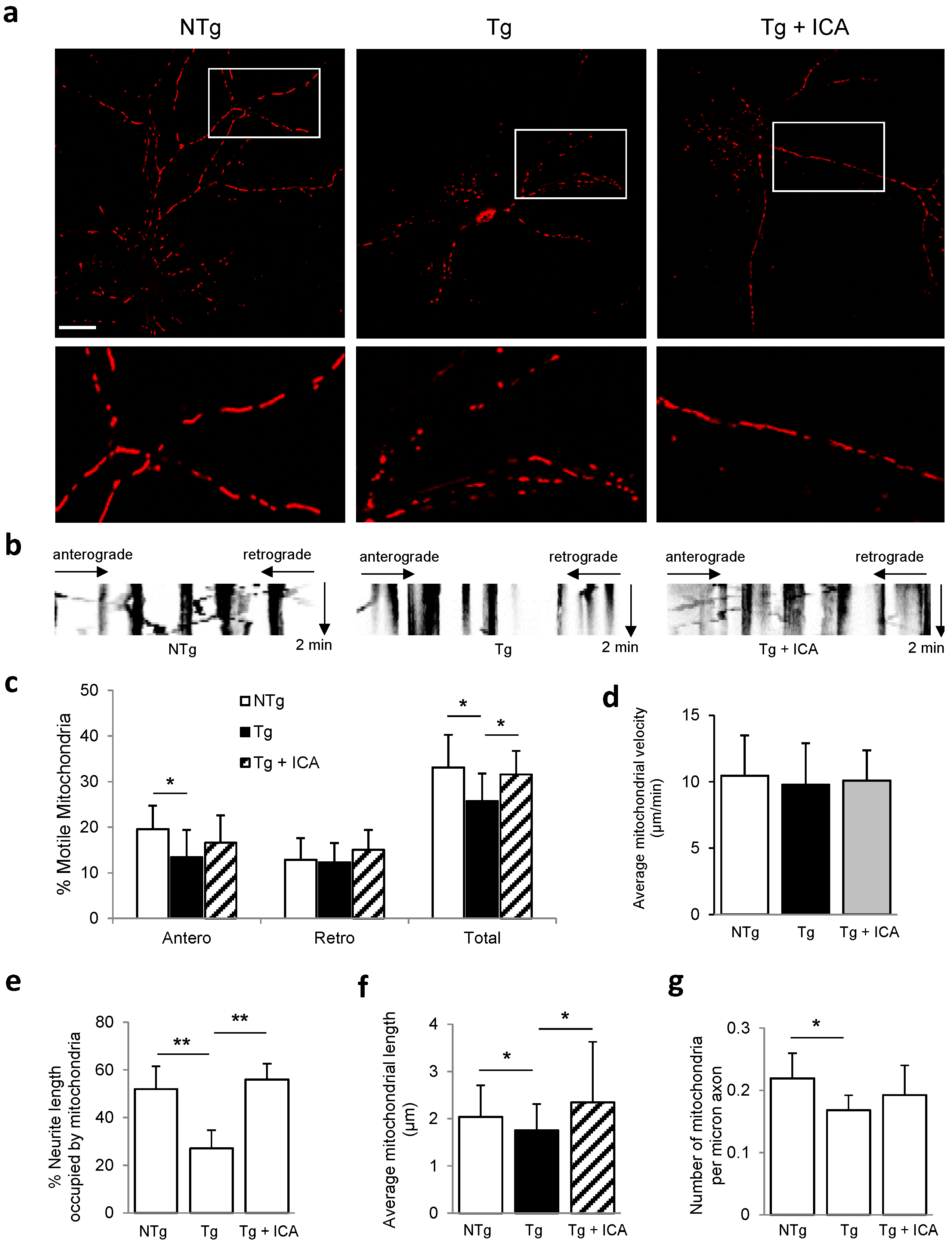
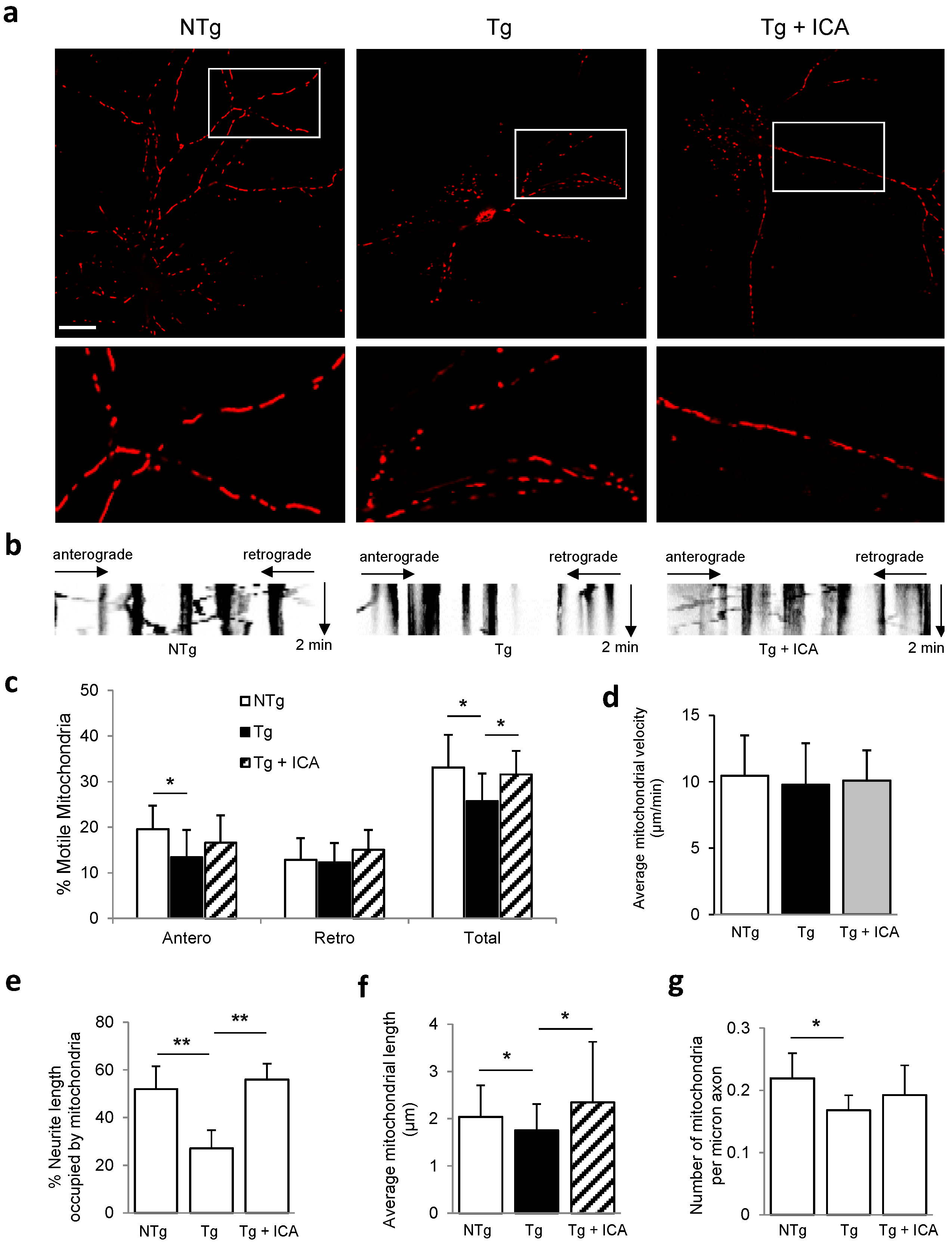
2.3. Icariin Preserved Mitochondrial Mass and Promoted Mitochondrial Transport
2.4. Effects of Icariin on Mitochondrial Dynamics and Synaptic Protein Expression
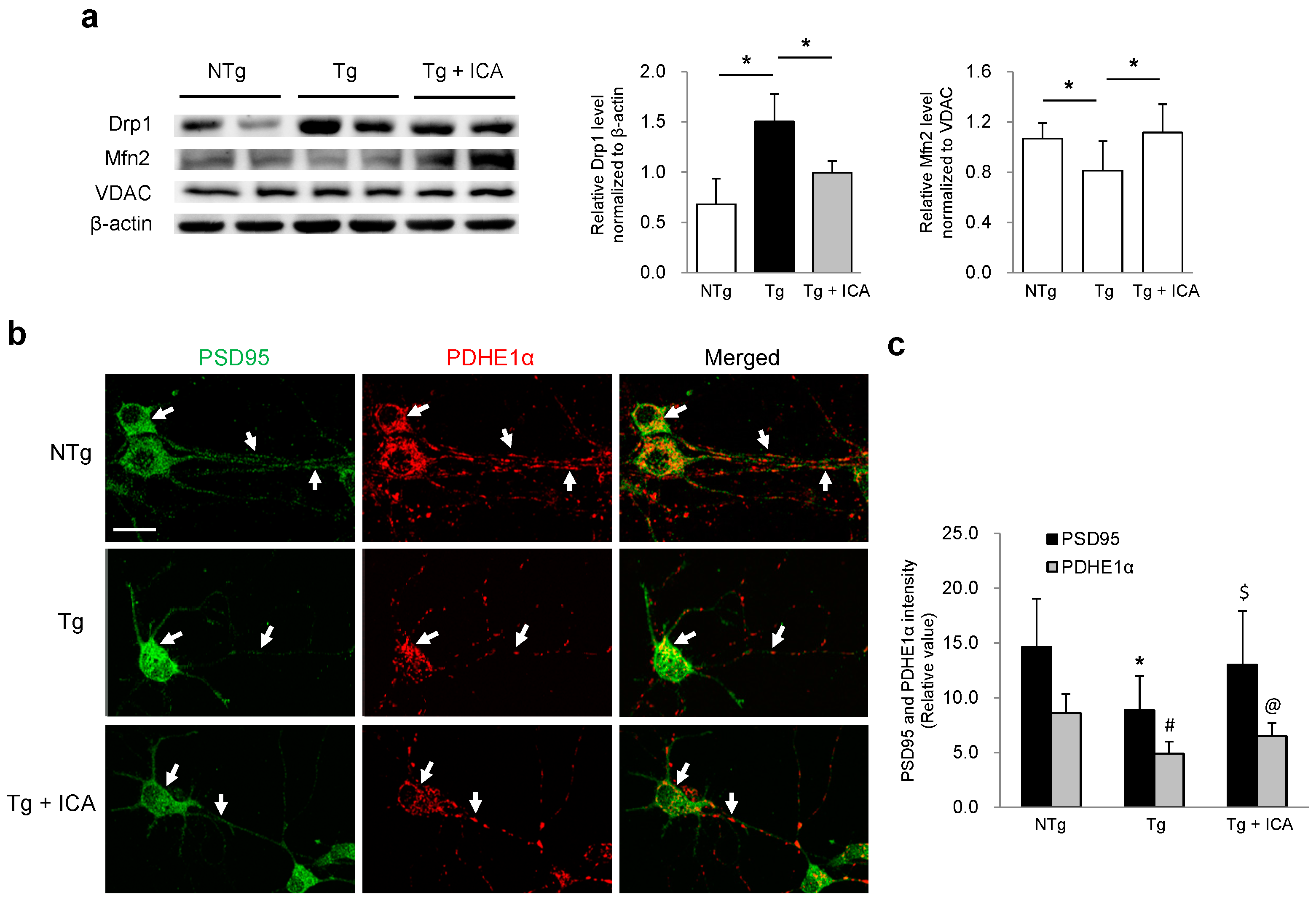
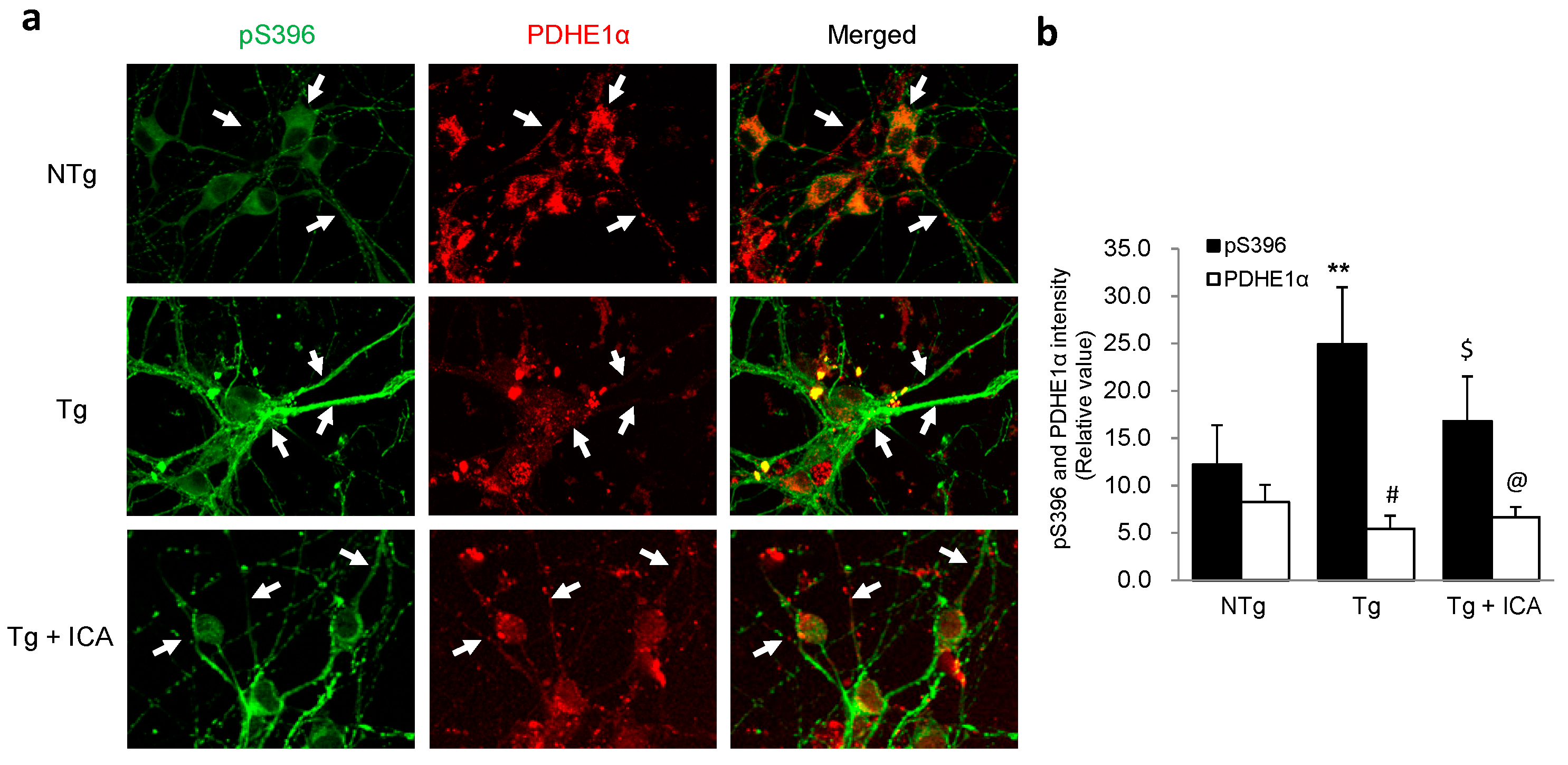
2.5. Aβ Reduction Is Associated with Increased Pyruvate Dehydrogenase-E1α (PDHE1α) Expression in AD Neurons: Effect of Icariin

2.6. Effects of Icariin on Hyperphosphorylated Tau Expression in AD Neurons
3. Discussion
4. Experimental Section
4.1. Ethics Statement
4.2. Materials
4.3. Neuronal Culture and Treatment
4.4. Immunocytochemistry Assay
4.5. Western Blot Assay
4.6. Enzyme Linked Immunosorbent Assay (ELISA)
4.7. Mitochondrial Trafficking Recording and Data Analysis
4.8. Axonal Mitochondrial Content Measurements
4.9. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; Karran, E.; de Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Calkins, M.J.; Reddy, P.H. Amyloid β impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer’s disease neurons. Biochim. Biophys. Acta 2011, 1812, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Rosales-Corral, S.; Acuna-Castroviejo, D.; Tan, D.X.; Lopez-Armas, G.; Cruz-Ramos, J.; Munoz, R.; Melnikov, V.G.; Manchester, L.C.; Reiter, R.J. Accumulation of exogenous amyloid-β peptide in hippocampal mitochondria causes their dysfunction: A protective role for melatonin. Oxid. Med. Cell. Longev. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Guan, J.; Borrelli, L.A.; Xu, J.; Serrano-Pozo, A.; Bacskai, B.J. Mitochondrial alterations near amyloid plaques in an Alzheimer’s disease mouse model. J. Neurosci. 2013, 33, 17042–17051. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Guo, L.; Yan, S.; Sosunov, A.A.; McKhann, G.M.; Yan, S.S. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 18670–18675. [Google Scholar] [CrossRef] [PubMed]
- Perez-Cruz, C.; Nolte, M.W.; van Gaalen, M.M.; Rustay, N.R.; Termont, A.; Tanghe, A.; Kirchhoff, F.; Ebert, U. Reduced spine density in specific regions of CA1 pyramidal neurons in two transgenic mouse models of Alzheimer’s disease. J. Neurosci. 2011, 31, 3926–3934. [Google Scholar] [CrossRef] [PubMed]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. Abad directly links Aβ to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Beal, M.F. Amyloid β, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Rettberg, J.R.; Yao, J.; Brinton, R.D. Estrogen: A master regulator of bioenergetic systems in the brain and body. Front. Neuroendocrinol. 2014, 35, 8–30. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis. J. Alzheimers Dis. 2010, 20 (Suppl. S2), S265–S279. [Google Scholar] [PubMed]
- Takuma, K.; Yao, J.; Huang, J.; Xu, H.; Chen, X.; Luddy, J.; Trillat, A.C.; Stern, D.M.; Arancio, O.; Yan, S.S. Abad enhances Aβ-induced cell stress via mitochondrial dysfunction. FASEB J. 2005, 19, 597–598. [Google Scholar] [PubMed]
- Blalock, E.M.; Geddes, J.W.; Chen, K.C.; Porter, N.M.; Markesbery, W.R.; Landfield, P.W. Incipient Alzheimer’s disease: Microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc. Natl. Acad. Sci. USA 2004, 101, 2173–2178. [Google Scholar] [CrossRef] [PubMed]
- Blass, J.P.; Sheu, R.K.; Gibson, G.E. Inherent abnormalities in energy metabolism in Alzheimer disease. Interaction with cerebrovascular compromise. Ann. N. Y. Acad. Sci. 2000, 903, 204–221. [Google Scholar] [CrossRef] [PubMed]
- Perry, E.K.; Perry, R.H.; Tomlinson, B.E.; Blessed, G.; Gibson, P.H. Coenzyme a-acetylating enzymes in Alzheimer’s disease: Possible cholinergic “compartment” of pyruvate dehydrogenase. Neurosci. Lett. 1980, 18, 105–110. [Google Scholar] [CrossRef]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Ma, M.; Zhao, Q.; Fang, L.; Chang, J.; Wang, Y.; Fei, R.; Song, X. Mitochondrial bioenergetic deficits in the hippocampi of rats with chronic ischemia-induced vascular dementia. Neuroscience 2013, 231, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Aβ and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Kopeikina, K.J.; Carlson, G.A.; Pitstick, R.; Ludvigson, A.E.; Peters, A.; Luebke, J.I.; Koffie, R.M.; Frosch, M.P.; Hyman, B.T.; Spires-Jones, T.L. Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer’s disease brain. Am. J. Pathol. 2011, 179, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Schulz, K.L.; Eckert, A.; Rhein, V.; Mai, S.; Haase, W.; Reichert, A.S.; Jendrach, M.; Muller, W.E.; Leuner, K. A new link to mitochondrial impairment in tauopathies. Mol. Neurobiol. 2012, 46, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Shahpasand, K.; Uemura, I.; Saito, T.; Asano, T.; Hata, K.; Shibata, K.; Toyoshima, Y.; Hasegawa, M.; Hisanaga, S. Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer’s disease. J. Neurosci. 2012, 32, 2430–2441. [Google Scholar] [CrossRef] [PubMed]
- Eckert, A.; Nisbet, R.; Grimm, A.; Gotz, J. March separate, strike together—Role of phosphorylated tau in mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, V.S.; Fenech, M. Mutations that affect mitochondrial functions and their association with neurodegenerative diseases. Mutat. Res. Rev. Mutat. Res. 2014, 759, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Knott, A.B.; Perkins, G.; Schwarzenbacher, R.; Bossy-Wetzel, E. Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kim, K.R.; Ryu, S.Y.; Son, S.; Hong, H.S.; Mook-Jung, I.; Ho, W.K. Impaired short-term plasticity in mossy fiber synapses caused by mitochondrial dysfunction of dentate granule cells is the earliest synaptic deficit in a mouse model of Alzheimer’s disease. J. Neurosci. 2012, 32, 5953–5963. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.J.; Slupe, A.M.; Strack, S. Cell signaling and mitochondrial dynamics: Implications for neuronal function and neurodegenerative disease. Neurobiol. Dis. 2013, 51, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; He, X.; Yang, Y.; Li, M.; Hao, D.; Jia, Z. The genus epimedium: An ethnopharmacological and phytochemical review. J. Ethnopharmacol. 2011, 134, 519–541. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Luo, Y.; Huang, X.N.; Gong, Q.H.; Wu, Q.; Shi, J.S. Icariin inhibits β-amyloid peptide segment 25–35 induced expression of β-secretase in rat hippocampus. Eur. J. Pharmacol. 2010, 626, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Urano, T.; Tohda, C. Icariin improves memory impairment in Alzheimer’s disease model mice (5× FAD) and attenuates amyloid β-induced neurite atrophy. Phytother. Res. 2010, 24, 1658–1663. [Google Scholar] [CrossRef] [PubMed]
- Zeng, K.W.; Ko, H.; Yang, H.O.; Wang, X.M. Icariin attenuates β-amyloid-induced neurotoxicity by inhibition of tau protein hyperphosphorylation in PC12 cells. Neuropharmacology 2010, 59, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Shen, C.; Chu, J.; Zhang, R.; Li, Y.; Li, L. Icariin decreases the expression of APP and BACE-1 and reduces the β-amyloid burden in an APP transgenic mouse model of Alzheimer’s disease. Int. J. Biol. Sci. 2014, 10, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Zheng, H.Y.; Huang, X.X.; Han, S.X.; Zhang, D.S.; Ni, J.Z.; He, X.Y. Neuroprotective effects of icariin on brain metabolism, mitochondrial functions, and cognition in triple-transgenic Alzheimer’s disease mice. CNS Neurosci. Ther. 2015, 22, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Vale, C.; Alonso, E.; Rubiolo, J.A.; Vieytes, M.R.; LaFerla, F.M.; Gimenez-Llort, L.; Botana, L.M. Profile for amyloid-β and tau expression in primary cortical cultures from 3× Tg-AD mice. Cell. Mol. Neurobiol. 2010, 30, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.B. Signal-processing machines at the postsynaptic density. Science 2000, 290, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Perry, G.; Smith, M.A.; Zhu, X. Amyloid-β-derived diffusible ligands cause impaired axonal transport of mitochondria in neurons. Neurodegener. Dis. 2010, 7, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Pinho, C.M.; Teixeira, P.F.; Glaser, E. Mitochondrial import and degradation of amyloid-β peptide. Biochim. Biophys. Acta 2014, 1837, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Mao, P.; Calkins, M.J.; Cornea, A.; Reddy, A.P.; Murphy, M.P.; Szeto, H.H.; Park, B.; Reddy, P.H. Mitochondria-targeted antioxidants protect against amyloid-β toxicity in Alzheimer’s disease neurons. J. Alzheimers Dis. 2010, 20 (Suppl. S2), S609–S631. [Google Scholar] [PubMed]
- Reddy, P.H.; Reddy, T.P.; Manczak, M.; Calkins, M.J.; Shirendeb, U.; Mao, P. Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res. Rev. 2011, 67, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-β overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19318–19323. [Google Scholar] [CrossRef] [PubMed]
- Kanaan, N.M.; Morfini, G.A.; LaPointe, N.E.; Pigino, G.F.; Patterson, K.R.; Song, Y.; Andreadis, A.; Fu, Y.; Brady, S.T.; Binder, L.I. Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J. Neurosci. 2011, 31, 9858–9868. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Ke, Y.D.; Gotz, J. Phosphorylated tau interacts with c-jun N-terminal kinase-interacting protein 1 (JIP1) in Alzheimer disease. J. Biol. Chem. 2009, 284, 20909–20916. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla, R.A.; Matthews-Roberson, T.A.; Dolan, P.J.; Johnson, G.V. Caspase-cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons: Implications for the pathogenesis of Alzheimer disease. J. Biol. Chem. 2009, 284, 18754–18766. [Google Scholar] [CrossRef] [PubMed]
- Moffett, J.R.; Ross, B.; Arun, P.; Madhavarao, C.N.; Namboodiri, A.M. N-acetylaspartate in the CNS: From neurodiagnostics to neurobiology. Prog. Neurobiol. 2007, 81, 89–131. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.K.; Choi, Y.H.; Kwon, H.; Lee, S.B.; Kim, D.H.; Sung, C.K.; Park, Y.I.; Dong, M.S. Estrogenic/antiestrogenic activities of a epimedium koreanum extract and its major components: In vitro and in vivo studies. Food Chem. Toxicol. 2012, 50, 2751–2759. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, D.M.; Tang, M.X.; Stern, Y.; Sano, M.; Marder, K.; Bell, K.L.; Schofield, P.; Dooneief, G.; Gurland, B.; Mayeux, R. Cognitive function in nondemented older women who took estrogen after menopause. Neurology 1998, 50, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Maki, P.M.; Zonderman, A.B.; Resnick, S.M. Enhanced verbal memory in nondemented elderly women receiving hormone-replacement therapy. Am. J. Psychiatry 2001, 158, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Zandi, P.P.; Carlson, M.C.; Plassman, B.L.; Welsh-Bohmer, K.A.; Mayer, L.S.; Steffens, D.C.; Breitner, J.C. Hormone replacement therapy and incidence of Alzheimer disease in older women: The cache county study. JAMA 2002, 288, 2123–2129. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Han, S.; Huang, X.; Ni, J.; He, X. The Protective Effect of Icariin on Mitochondrial Transport and Distribution in Primary Hippocampal Neurons from 3× Tg-AD Mice. Int. J. Mol. Sci. 2016, 17, 163. https://doi.org/10.3390/ijms17020163
Chen Y, Han S, Huang X, Ni J, He X. The Protective Effect of Icariin on Mitochondrial Transport and Distribution in Primary Hippocampal Neurons from 3× Tg-AD Mice. International Journal of Molecular Sciences. 2016; 17(2):163. https://doi.org/10.3390/ijms17020163
Chicago/Turabian StyleChen, Yijing, Shuangxue Han, Xiuxian Huang, Jiazuan Ni, and Xiaoyang He. 2016. "The Protective Effect of Icariin on Mitochondrial Transport and Distribution in Primary Hippocampal Neurons from 3× Tg-AD Mice" International Journal of Molecular Sciences 17, no. 2: 163. https://doi.org/10.3390/ijms17020163
APA StyleChen, Y., Han, S., Huang, X., Ni, J., & He, X. (2016). The Protective Effect of Icariin on Mitochondrial Transport and Distribution in Primary Hippocampal Neurons from 3× Tg-AD Mice. International Journal of Molecular Sciences, 17(2), 163. https://doi.org/10.3390/ijms17020163