
Effect of Estradiol on Neurotrophin Receptors in Basal Forebrain Cholinergic Neurons: Relevance for Alzheimer’s Disease



{kind=link}
Abstract
:1. Introduction
2. Basal Forebrain Cholinergic Neurons
2.1. Cholinergic Neurons in Specific Basal Forebrain Cholinergic Neuron Subregions
2.1.1. Nucleus Basalis Magnocellularis
2.1.2. Medial Septum and Diagonal Band of Broca
3. Estrogens
4. Estrogen Receptor Signaling Pathways
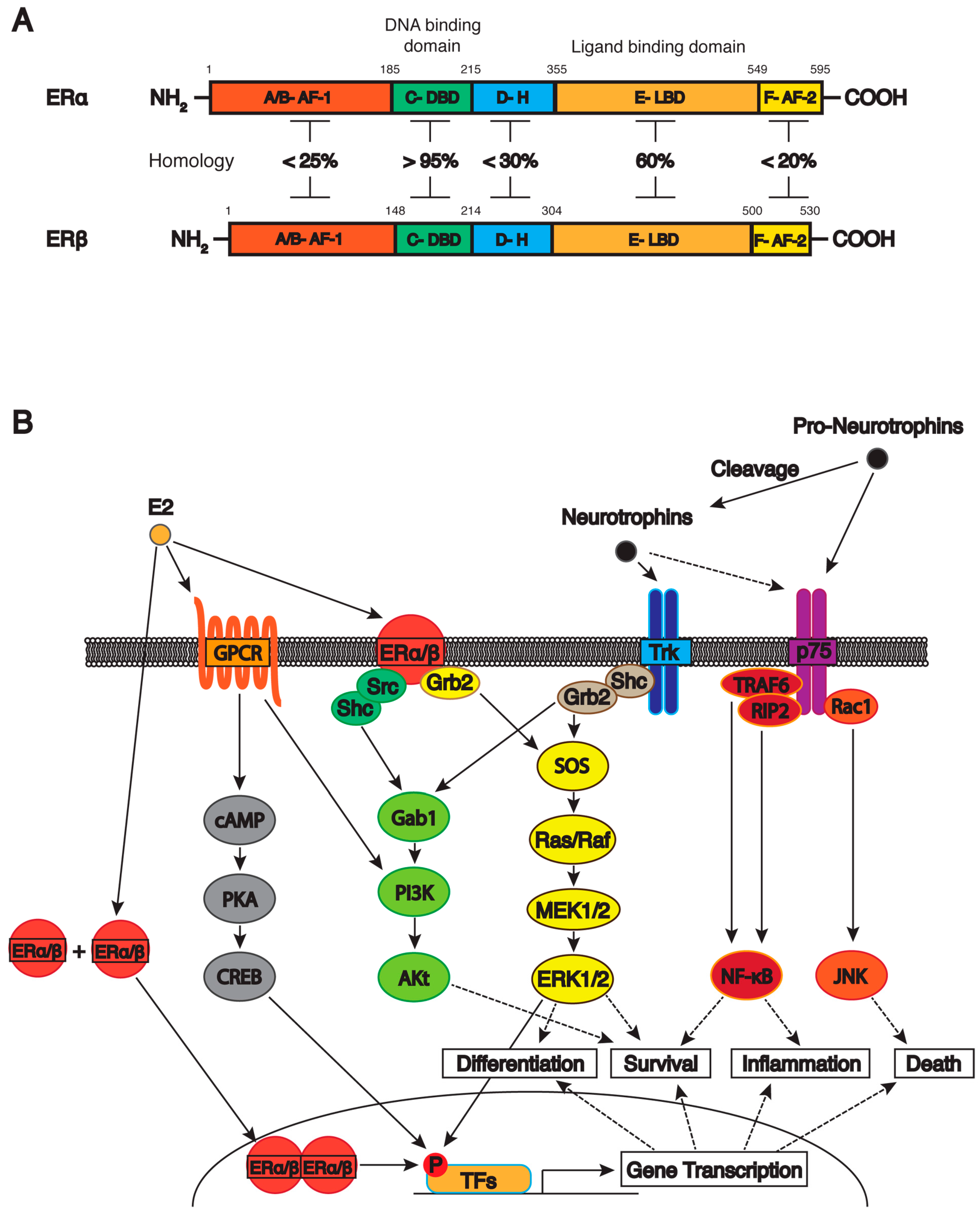
4.1. Estrogen Receptors
4.2. Classical Estrogen Signaling Pathway
4.3. Non-Classical Estrogen Signaling Pathway
4.4. Neuroprotective Effects of Estrogens in the Central Nervous System
5. Neurotrophins
5.1. Neurotrophin Receptors
5.2. Neurotrophin Peptides
5.3. Role of the Neurotrophin System in Neuroprotection
5.4. Neurotrophins in the Basal Forebrain
6. Relationship between Neurotrophins, Estrogens and AD in the Basal Forebrain
7. Conclusions: Therapeutic Potential of Estrogens for AD Treatment and Prevention
Acknowledgments
Conflicts of Interest
References
- Robertson, H.A.; King, G.J. Plasma concentrations of progesterone, oestrone, oestradiol-17β and of oestrone sulphate in the pig at implantation, during pregnancy and at parturition. J. Reprod. Fertil. 1974, 40, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Merrill, R.C. Estriol: A review. Physiol. Rev. 1958, 38, 463–480. [Google Scholar] [PubMed]
- Brown, J.B. Urinary excretion of oestrogens during the menstrual cycle. Lancet 1955, 268, 320–323. [Google Scholar] [CrossRef]
- McEwen, B.S. Invited review: Estrogens effects on the brain: Multiple sites and molecular mechanisms. J. Appl. Physiol. 2001, 91, 2785–2801. [Google Scholar] [PubMed]
- Rapp, P.R.; Morrison, J.H.; Roberts, J.A. Cyclic estrogen replacement improves cognitive function in aged ovariectomized rhesus monkeys. J. Neurosci. 2003, 23, 5708–5714. [Google Scholar] [PubMed]
- Ohm, D.T.; Bloss, E.B.; Janssen, W.G.; Dietz, K.C.; Wadsworth, S.; Lou, W.; Gee, N.A.; Lasley, B.L.; Rapp, P.R.; Morrison, J.H. Clinically relevant hormone treatments fail to induce spinogenesis in prefrontal cortex of aged female rhesus monkeys. J. Neurosci. 2012, 32, 11700–11705. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Rapp, P.R.; Leffler, A.E.; Leffler, S.R.; Janssen, W.G.M.; Lou, W.; McKay, H.; Roberts, J.A.; Wearne, S.L.; Hof, P.R.; et al. Estrogen alters spine number and morphology in prefrontal cortex of aged female rhesus monkeys. J. Neurosci. 2006, 26, 2571–2578. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, Z. Effects of estradiol benzoate on learning-memory behavior and synaptic structure in ovariectomized mice. Life Sci. 2006, 79, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Day, M.; Muniz, L.C.; Bitran, D.; Arias, R.; Revilla-Sanchez, R.; Grauer, S.; Zhang, G.; Kelley, C.; Pulito, V.; et al. Activation of estrogen receptor-β regulates hippocampal synaptic plasticity and improves memory. Nat. Neurosci. 2008, 11, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Wintermantel, T.M.; Campbell, R.E.; Porteous, R.; Bock, D.; Gröne, H.-J.; Todman, M.G.; Korach, K.S.; Greiner, E.; Pérez, C.A.; Schütz, G.; et al. Definition of estrogen receptor pathway critical for estrogen positive feedback to gonadotropin-releasing hormone neurons and fertility. Neuron 2006, 52, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Koszegi, Z.; Szego, É.; Cheong, R.Y.; Tolod-Kemp, E.; Ábrahám, I.M. Postlesion estradiol treatment increases cortical cholinergic innervations via estrogen receptor-α dependent nonclassical estrogen signaling in vivo. Endocrinology 2011, 152, 3471–3482. [Google Scholar] [CrossRef] [PubMed]
- Horvath, K.M.; Hårtig, W.; van der Veen, R.; Keijser, J.N.; Mulder, J.; Ziegert, M.; van der Zee, E.A.; Harkany, T.; Luiten, P.G.M. 17β-estradiol enhances cortical cholinergic innervation and preserves synaptic density following excitotoxic lesions to the rat nucleus basalis magnocellularis. Neuroscience 2002, 110, 489–504. [Google Scholar] [CrossRef]
- Bora, S.H.; Liu, Z.; Kecojevic, A.; Merchenthaler, I.; Koliatsos, V.E. Direct, complex effects of estrogens on basal forebrain cholinergic neurons. Exp. Neurol. 2005, 194, 506–522. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Meyer, E.M.; Millard, W.J.; Simpkins, J.W. Ovarian steroid deprivation results in a reversible learning impairment and compromised cholinergic function in female sprague-dawley rats. Brain Res. 1994, 644, 305–312. [Google Scholar] [CrossRef]
- Paganini-Hill, A.; Henderson, V.W. Estrogen deficiency and risk of Alzheimer’s disease in women. Am. J. Epidemiol. 1994, 140, 256–261. [Google Scholar] [PubMed]
- Rocca, W.A.; Grossardt, B.R.; Shuster, L.T. Oophorectomy, menopause, estrogen treatment, and cognitive aging: Clinical evidence for a window of opportunity. Brain Res. 2011, 1379, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Kawas, C.; Resnick, S.; Morrison, A.; Brookmeyer, R.; Corrada, M.; Zonderman, A.; Bacal, C.; Lingle, D.D.; Metter, E. A prospective study of estrogen replacement therapy and the risk of developing Alzheimer’s disease: The baltimore longitudinal study of aging. Neurology 1997, 48, 1517–1521. [Google Scholar] [CrossRef] [PubMed]
- Baldereschi, M.; di Carlo, A.; Lepore, V.; Bracco, L.; Maggi, S.; Grigoletto, F.; Scarlato, G.; Amaducci, L.; Group, I.W. Estrogen-replacement therapy and Alzheimer’s disease in the italian longitudinal study on aging. Neurology 1998, 50, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Alderson, R.F.; Alterman, A.L.; Barde, Y.-A.; Lindsay, R.M. Brain-derived neurotrophic factor increases survival and differentiated functions of rat septal cholinergic neurons in culture. Neuron 1990, 5, 297–306. [Google Scholar] [CrossRef]
- Hagg, T.; Fass-Holmes, B.; Vahlsing, H.L.; Manthorpe, M.; Conner, J.M.; Varon, S. Nerve growth factor (NGF) reverses axotomy-induced decreases in choline acetyltransferase, NGF receptor and size of medial septum cholinergic neurons. Brain Res. 1989, 505, 29–38. [Google Scholar] [CrossRef]
- Gibbs, R.B.; Aggarwal, P. Estrogen and basal forebrain cholinergic neurons: Implications for brain aging and Alzheimer’s disease-related cognitive decline. Horm. Behav. 1998, 34, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Ábrahám, I.M.; Kőszegi, Z.; Tolod-Kemp, E.; Szegő, É.M. Action of estrogen on survival of basal forebrain cholinergic neurons: Promoting amelioration. Psychoneuroendocrinology 2009, 34, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Toran-Allerand, C.D.; Miranda, R.C.; Bentham, W.D.; Sohrabji, F.; Brown, T.J.; Hochberg, R.B.; MacLusky, N.J. Estrogen receptors colocalize with low-affinity nerve growth factor receptors in cholinergic neurons of the basal forebrain. Proc. Natl. Acad. Sci. USA 1992, 89, 4668–4672. [Google Scholar] [CrossRef] [PubMed]
- Miranda, R.C.; Sohrabji, F.; Toran-Allerand, C.D. Presumptive estrogen target neurons express mrnas for both the neurotrophins and neurotrophin receptors: A basis for potential developmental interactions of estrogen with the neurotrophins. Mol. Cell. Neurosci. 1993, 4, 510–525. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, R.B. Effects of ageing and long-term hormone replacement on cholinergic neurones in the medial septum and nucleus basalis magnocellularis of ovariectomized rats. J. Neuroendocrinol. 2003, 15, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Milne, M.R.; Haug, C.A.; Abraham, I.M.; Kwakowsky, A. Estradiol modulation of neurotrophin receptor expression in female mouse basal forebrain cholinergic neurons in vivo. Endocrinology 2015, 156, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Zaborszky, L. The modular organization of brain systems. Basal forebrain: The last frontier. Prog. Brain Res. 2002, 136, 359–372. [Google Scholar] [PubMed]
- Mesulam, M.M.; Mufson, E.J.; Levey, A.I.; Wainer, B.H. Atlas of cholinergic neurons in the forebrain and upper brainstem of the macaque based on monoclonal choline acetyltransferase immunohistochemistry and acetylcholinesterase histochemistry. Neuroscience 1984, 12, 669–686. [Google Scholar] [CrossRef]
- Decker, M.W.; McGaugh, J.L. The role of interactions between the cholinergic system and other neuromodulatory systems in learing and memory. Synapse 1991, 7, 151–168. [Google Scholar] [CrossRef] [PubMed]
- Zaborszky, L.; Csordas, A.; Mosca, K.; Kim, J.; Gielow, M.R.; Vadasz, C.; Nadasdy, Z. Neurons in the basal forebrain project to the cortex in a complex topographic organization that reflects corticocortical connectivity patterns: An experimental study based on retrograde tracing and 3d reconstruction. Cereb. Cortex 2015, 25, 118–137. [Google Scholar] [CrossRef] [PubMed]
- Baxter, M.G.; Chiba, A.A. Cognitive functions of the basal forebrain. Curr. Opin. Neurobiol. 1999, 9, 178–183. [Google Scholar] [CrossRef]
- Page, K.J.; Sofroniew, M.V. The ascending basal forebrain cholinergic system. Prog. Brain Res. 1996, 107, 513–522. [Google Scholar] [PubMed]
- Chiba, A.; Bucci, D.; Holland, P.; Gallagher, M. Basal forebrain cholinergic lesions disrupt increments but not decrements in conditioned stimulus processing. J. Neurosci. 1995, 15, 7315–7322. [Google Scholar] [PubMed]
- Everitt, B.; Robbins, T.; Muir, J. Reversal of visual attentional dysfunction following lesions of the cholinergic basal forebrain by physostigmine and nicotine but not by the 5-HT3 receptor antagonist, ondansetron. Psychopharmacology 1995, 118, 82–92. [Google Scholar] [CrossRef]
- Buzsaki, G.; Bickford, R.; Ponomareff, G.; Thal, L.; Mandel, R.; Gage, F. Nucleus basalis and thalamic control of neocortical activity in the freely moving rat. J. Neurosci. 1988, 8, 4007–4026. [Google Scholar] [PubMed]
- Pirch, J.H.; Corbus, M.J.; Rigdon, G.C.; Lyness, W.H. Generation of cortical event-related slow potentials in the rat involves nucleus basalis cholinergic innervation. Electroencephalogr. Clin. Neurophysiol. 1986, 63, 464–475. [Google Scholar] [CrossRef]
- Bowen, D.M.; Smith, C.B.; White, P.; Davison, A.N. Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain J. Neurol. 1976, 99, 459–496. [Google Scholar] [CrossRef]
- Perry, E.K.; Tomlinson, B.E.; Blessed, G.; Bergmann, K.; Gibson, P.H.; Perry, R.H. Correlation of cholinergic abnormalities with senile plaques and mental test scores in senile dementia. Br. Med. J. 1978, 2, 1457–1459. [Google Scholar] [CrossRef] [PubMed]
- Wenk, G.L. The nucleus basalis magnocellularis cholinergic system: One hundred years of progress. Neurobiol. Learn. Mem. 1997, 67, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Wenk, H.; Bigl, V.; Meyer, U. Cholinergic projections from magnocellular nuclei of the basal forebrain to cortical areas in rats. Brain Res. Rev. 1980, 2, 295–316. [Google Scholar] [CrossRef]
- Bartus, R.T.; Flicker, C.; Dean, R.L.; Pontecorvo, M.; Figueiredo, J.C.; Fisher, S.K. Selective memory loss following nucleus basalis lesions: Long term behavioral recovery despite persistent cholinergic deficiencies. Pharmacol. Biochem. Behav. 1985, 23, 125–135. [Google Scholar] [CrossRef]
- Wenk, G.; Sweeney, J.; Hughey, D.; Carson, J.; Olton, D. Cholinergic function and memory: Extensive inhibition of choline acetyltransferase fails to impair radial maze performance in rats. Pharmacol. Biochem. Behav. 1986, 25, 521–526. [Google Scholar] [CrossRef]
- Olton, D.S.; Wenk, G.L.; Church, R.M.; Meck, W.H. Attention and the frontal cortex as examined by simultaneous temporal processing. Neuropsychologia 1988, 26, 307–318. [Google Scholar] [CrossRef]
- Muir, J.L.; Page, K.J.; Sirinathsinghji, D.J.S.; Robbins, T.W.; Everitt, B.J. Excitotoxic lesions of basal forebrain cholinergic neurons: Effects on learning, memory and attention. Behav. Brain Res. 1993, 57, 123–131. [Google Scholar] [CrossRef]
- Inglis, F.M.; Day, J.C.; Fibiger, H.C. Enhanced acetylcholine release in hippocampus and cortex during the anticipation and consumption of a palatable meal. Neuroscience 1994, 62, 1049–1056. [Google Scholar] [CrossRef]
- Buzsáki, G.; Gage, F. The cholinergic nucleus basalis: A key structure in neocortical arousal. In Central Cholinergic Synaptic Transmission; Frotscher, M., Misgeld, U., Eds.; Birkhäuser: Basel, Switzerland, 1989; Volume 57, pp. 159–171. [Google Scholar]
- Baghdoyan, H.A.; Spotts, J.L.; Snyder, S.G. Simultaneous pontine and basal forebrain microinjections of carbachol suppress rem sleep. J. Neurosci. 1993, 13, 229–242. [Google Scholar] [PubMed]
- Smythies, J. Section I. The cholinergic system. Int. Rev. Neurobiol. 2005, 64, 1–122. [Google Scholar] [PubMed]
- Brazhnik, E.S.; Fox, S.E. Intracellular recordings from medial septal neurons during hippocampal theta rhythm. Exp. Brain Res. 1997, 114, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Vertes, R.P. Hippocampal theta rhythm: A tag for short-term memory. Hippocampus 2005, 15, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Gruber, C.J.; Tschugguel, W.; Schneeberger, C.; Huber, J.C. Production and actions of estrogens. N. Engl. J. Med. 2002, 346, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Kawata, M. Roles of steroid hormones and their receptors in structural organization in the nervous system. Neurosci. Res. 1995, 24, 1–46. [Google Scholar] [CrossRef]
- Boon, W.C.; Chow, J.D.Y.; Simpson, E.R. the multiple roles of estrogens and the enzyme aromatase. In Progress in Brain Research; Luciano, M., Ed.; Elsevier: Amsterdam, Netherlands, 2010; Chapter 12; Volume 181, pp. 209–232. [Google Scholar]
- Zhu, B.T.; Conney, A.H. Functional role of estrogen metabolism in target cells: Review and perspectives. Carcinogenesis 1998, 19, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Callard, G.V.; Petro, Z.; Ryan, K.J. Identification of aromatase in the reptilian brain. Endocrinology 1977, 100, 1214–1218. [Google Scholar] [CrossRef] [PubMed]
- Callard, G.V.; Petro, Z.; Ryan, K.J. Phylogenetic distribution of aromatase and other androgen-converting enzymes in the central nervous system. Endocrinology 1978, 103, 2283–2290. [Google Scholar] [CrossRef] [PubMed]
- Selmanoff, M.K.; Brodkin, L.D.; Weiner, R.I.; Siiteri, P.K. Aromatization and 5α-reduction of androgens in discrete hypothalamic and limbic regions of the male and female rat. Endocrinology 1977, 101, 841–848. [Google Scholar] [CrossRef] [PubMed]
- MacLusky, N.J.; Naftolin, F.; Goldman-Rakic, P.S. Estrogen formation and binding in the cerebral cortex of the developing rhesus monkey. Proc. Natl. Acad. Sci. USA 1986, 83, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Azcoitia, I.; Yague, J.G.; Garcia-Segura, L.M. Estradiol synthesis within the human brain. Neuroscience 2011, 191, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Luchetti, S.; Bossers, K.; Van de Bilt, S.; Agrapart, V.; Morales, R.R.; Frajese, G.V.; Swaab, D.F. Neurosteroid biosynthetic pathways changes in prefrontal cortex in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 1964–1976. [Google Scholar] [CrossRef] [PubMed]
- Ishunina, T.A.; van Beurden, D.; van der Meulen, G.; Unmehopa, U.A.; Hol, E.M.; Huitinga, I.; Swaab, D.F. Diminished aromatase immunoreactivity in the hypothalamus, but not in the basal forebrain nuclei in Alzheimer’s disease. Neurobiol. Aging 2005, 26, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Yague, J.G.; Lavaque, E.; Carretero, J.; Azcoitia, I.; Garcia-Segura, L.M. Aromatase, the enzyme responsible for estrogen biosynthesis, is expressed by human and rat glioblastomas. Neurosci. Lett. 2004, 368, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Ishunina, T.A.; Fischer, D.F.; Swaab, D.F. Estrogen receptor α and its splice variants in the hippocampus in aging and Alzheimer’s disease. Neurobiol. Aging 2007, 28, 1670–1681. [Google Scholar] [CrossRef] [PubMed]
- Yague, J.G.; Azcoitia, I.; DeFelipe, J.; Garcia-Segura, L.M.; Munoz, A. Aromatase expression in the normal and epileptic human hippocampus. Brain Res. 2010, 1315, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Kelicen Ugur, P.; Lule, S.; Cincioglu, M.; Pekiner, C.; Gursoy-Ozdemir, Y. Megestrol acetate inhibits the expression of cytoplasmic aromatase through nuclear C/EBPβ in reperfusion injury-induced ischemic rat hippocampus. Eur. J. Pharmacol. 2011, 654, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, R.J.; Ansar, S.; Duckles, S.P.; Krause, D.N. Androgenic/estrogenic balance in the male rat cerebral circulation: Metabolic enzymes and sex steroid receptors. J. Cereb. Blood Flow Metab. 2007, 27, 1841–1852. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.C. Hormonal regulation of breast development and activity. J. Investig. Dermatol. 1974, 63, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.C. Sex-hormone binding globulin. Clin. Endocrinol. 1974, 3, 69–96. [Google Scholar] [CrossRef]
- Hanukoglu, I. Steroidogenic enzymes: Structure, function, and role in regulation of steroid hormone biosynthesis. J. Steroid Biochem. Mol. Biol. 1992, 43, 779–804. [Google Scholar] [CrossRef]
- White, R.E.; Darkow, D.J.; Lang, J.L. Estrogen relaxes coronary arteries by opening BKCa channels through a cgmp-dependent mechanism. Circ. Res. 1995, 77, 936–942. [Google Scholar] [CrossRef] [PubMed]
- Eriksen, E.F.; Colvard, D.S.; Berg, N.J.; Graham, M.L.; Mann, K.G.; Spelsberg, T.C.; Riggs, B.L. Evidence of estrogen receptors in normal human osteoblast-like cells. Science 1988, 241, 84–86. [Google Scholar] [CrossRef] [PubMed]
- Oursler, M.J.; Cortese, C.; Keeting, P.; Anderson, M.A.; Bonde, S.K.; Riggs, B.L.; Spelsberg, T.C. Modulation of transforming growth factor-β production in normal human osteoblast-like cells by 17 β-estradiol and parathyroid hormone. Endocrinology 1991, 129, 3313–3320. [Google Scholar] [CrossRef] [PubMed]
- Jilka, R.L.; Takahashi, K.; Munshi, M.; Williams, D.C.; Roberson, P.K.; Manolagas, S.C. Loss of estrogen upregulates osteoblastogenesis in the murine bone marrow. Evidence for autonomy from factors released during bone resorption. J. Clin. Investig. 1998, 101, 1942–1950. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, C.; Sanvig Christensen, M.; Transbøl, I. Bone mass in postmenopausal women after withdrawal of oestrogen/gestagen replacement therapy. Lancet 1981, 317, 459–461. [Google Scholar] [CrossRef]
- Green, S.; Walter, P.; Greene, G.; Krust, A.; Goffin, C.; Jensen, E.; Scrace, G.; Waterfield, M.; Chambon, P. Cloning of the human oestrogen receptor cdna. J. Steroid Biochem. 1986, 24, 77–83. [Google Scholar] [CrossRef]
- Smith, E.P.; Boyd, J.; Frank, G.R.; Takahashi, H.; Cohen, R.M.; Specker, B.; Williams, T.C.; Lubahn, D.B.; Korach, K.S. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N. Engl. J. Med. 1994, 331, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Lubahn, D.B.; Moyer, J.S.; Golding, T.S.; Couse, J.F.; Korach, K.S.; Smithies, O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc. Natl. Acad. Sci. USA 1993, 90, 11162–11166. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, G.G.; Enmark, E.; Pelto-Huikko, M.; Nilsson, S.; Gustafsson, J.A. Cloning of a novel receptor expressed in rat prostate and ovary. Proc. Natl. Acad. Sci. USA 1996, 93, 5925–5930. [Google Scholar] [CrossRef] [PubMed]
- Enmark, E.; Pelto-Huikko, M.; Grandien, K.; Lagercrantz, S.; Lagercrantz, J.; Fried, G.; Nordenskjöld, M.; Gustafsson, J.-Å. Human estrogen receptor β-gene structure, chromosomal localization, and expression pattern. J. Clin. Endocrinol. Metab. 1997, 82, 4258–4265. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, J. Estrogen receptor β—A new dimension in estrogen mechanism of action. J. Endocrinol. 1999, 163, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, G.G.; Carlsson, B.; Grandien, K.; Enmark, E.; Haggblad, J.; Nilsson, S.; Gustafsson, J.A. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 1997, 138, 863–870. [Google Scholar] [PubMed]
- Beato, M.; Herrlich, P.; Schütz, G. Steroid hormone receptors: Many actors in search of a plot. Cell 1995, 83, 851–857. [Google Scholar] [CrossRef]
- Drummond, A.E.; Fuller, P.J. The importance of ERβ signalling in the ovary. J. Endocrinol. 2010, 205, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.; Al-Azzawi, F. Immunolocalisation of oestrogen receptor β in human tissues. J. Mol. Endocrinol. 2000, 24, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Shughrue, P.J.; Lane, M.V.; Merchenthaler, I. Comparative distribution of estrogen receptor-α and -β mRNA in the rat central nervous system. J. Comp. Neurol. 1997, 388, 507–525. [Google Scholar] [CrossRef]
- Heldring, N.; Pike, A.; Andersson, S.; Matthews, J.; Cheng, G.; Hartman, J.; Tujague, M.; Ström, A.; Treuter, E.; Warner, M.; et al. Estrogen receptors: How do they signal and what are their targets. Physiol. Rev. 2007, 87, 905–931. [Google Scholar] [CrossRef] [PubMed]
- Mitterling, K.L.; Spencer, J.L.; Dziedzic, N.; Shenoy, S.; McCarthy, K.; Waters, E.M.; McEwen, B.S.; Milner, T.A. Cellular and subcellular localization of estrogen and progestin receptor immunoreactivities in the mouse hippocampus. J. Comp. Neurol. 2010, 518, 2729–2743. [Google Scholar] [CrossRef] [PubMed]
- Waters, E.M.; Yildirim, M.; Janssen, W.G.M.; Lou, W.Y.W.; McEwen, B.S.; Morrison, J.H.; Milner, T.A. Estrogen and aging affect the synaptic distribution of estrogen receptor β-immunoreactivity in the ca1 region of female rat hippocampus. Brain Res. 2011, 1379, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Fuente-Martin, E.; Garcia-Caceres, C.; Morselli, E.; Clegg, D.J.; Chowen, J.A.; Finan, B.; Brinton, R.D.; Tschop, M.H. Estrogen, astrocytes and the neuroendocrine control of metabolism. Rev. Endocr. Metab. Disord. 2013, 14, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Johann, S.; Beyer, C. Neuroprotection by gonadal steroid hormones in acute brain damage requires cooperation with astroglia and microglia. J. Steroid Biochem. Mol. Biol. 2013, 137, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Jesmin, S.; Hattori, Y.; Sakuma, I.; Liu, M.Y.; Mowa, C.N.; Kitabatake, A. Estrogen deprivation and replacement modulate cerebral capillary density with vascular expression of angiogenic molecules in middle-aged female rats. J. Cereb. Blood Flow Metab. 2003, 23, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Alonso de Lecinana, M.; Egido, J.A. Estrogens as neuroprotectants against ischemic stroke. Cerebrovasc. Dis. 2006, 21, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.F.; Toft, D.O. Steroid receptors and their associated proteins. Mol. Endocrinol. 1993, 7, 4–11. [Google Scholar] [PubMed]
- McKenna, N.J.; Lanz, R.B.; O’Malley, B.W. Nuclear receptor coregulators: Cellular and molecular biology. Endocr. Rev. 1999, 20, 321–344. [Google Scholar] [CrossRef] [PubMed]
- Klinge, C.M. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001, 29, 2905–2919. [Google Scholar] [CrossRef] [PubMed]
- Enmark, E.; Gustafsson, J.Å. Oestrogen receptors—An overview. J. Intern. Med. 1999, 246, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Saville, B.; Wormke, M.; Wang, F.; Nguyen, T.; Enmark, E.; Kuiper, G.; Gustafsson, J.-Å.; Safe, S. Ligand-, cell-, and estrogen receptor subtype (α/β)-dependent activation at gc-rich (Sp1) promoter elements. J. Biol. Chem. 2000, 275, 5379–5387. [Google Scholar] [CrossRef] [PubMed]
- Szego, C.M.; Davis, J.S. Adenosine 3′,5′-monophosphate in rat uterus: Acute elevation by estrogen. Proc. Natl. Acad. Sci. USA 1967, 58, 1711–1718. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.S.; Norfleet, A.M.; Pappas, T.C.; Gametchu, B. Rapid actions of estrogens in GH3/B6 pituitary tumor cells via a plasma membrane version of estrogen receptor-α. Steroids 1999, 64, 5–13. [Google Scholar] [CrossRef]
- Mermelstein, P.G.; Micevych, P.E. Nervous system physiology regulated by membrane estrogen receptors. Rev. Neurosci. 2008, 19, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Mermelstein, P.G. Membrane-localised oestrogen receptor alpha and beta influence neuronal activity through activation of metabotropic glutamate receptors. J. Neuroendocrinol. 2009, 21, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Toran-Allerand, C.D.; Guan, X.; MacLusky, N.J.; Horvath, T.L.; Diano, S.; Singh, M.; Connolly, E.S., Jr.; Nethrapalli, I.S.; Tinnikov, A.A. ER-X: A novel, plasma membrane-associated, putative estrogen receptor that is regulated during development and after ischemic brain injury. J. Neurosci. 2002, 22, 8391–8401. [Google Scholar] [PubMed]
- Prossnitz, E.R.; Barton, M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat. Rev. Endocrinol. 2011, 7, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Bosch, M.A.; Tobias, S.C.; Grandy, D.K.; Scanlan, T.S.; Ronnekleiv, O.K.; Kelly, M.J. Rapid signaling of estrogen in hypothalamic neurons involves a novel G-protein-coupled estrogen receptor that activates protein kinase C. J. Neurosci. 2003, 23, 9529–9540. [Google Scholar] [PubMed]
- Carmeci, C.; Thompson, D.A.; Ring, H.Z.; Francke, U.; Weigel, R.J. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics 1997, 45, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Owman, C.; Blay, P.; Nilsson, C.; Lolait, S.J. Cloning of human cdna encoding a novel heptahelix receptor expressed in burkitt’s lymphoma and widely distributed in brain and peripheral tissues. Biochem. Biophys. Res. Commun. 1996, 228, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of hb-egf. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Quinn, J.A.; Frackelton, A.R.; Bland, K.I. Estrogen action via the G protein-coupled receptor, GPR30: Stimulation of adenylyl cyclase and camp-mediated attenuation of the epidermal growth factor receptor-to-mapk signaling axis. Mol. Endocrinol. 2002, 16, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Aronica, S.M.; Kraus, W.L.; Katzenellenbogen, B.S. Estrogen action via the camp signaling pathway: Stimulation of adenylate cyclase and camp-regulated gene transcription. Proc. Natl. Acad. Sci. USA 1994, 91, 8517–8521. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Pang, Y.; Filardo, E.J.; Dong, J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 2005, 146, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Dong, J.; Thomas, P. Estrogen signaling characteristics of atlantic croaker g protein-coupled receptor 30 (GPR30) and evidence it is involved in maintenance of oocyte meiotic arrest. Endocrinology 2008, 149, 3410–3426. [Google Scholar] [CrossRef] [PubMed]
- Miller, V.M.; Duckles, S.P. Vascular actions of estrogens: Functional implications. Pharmacol. Rev. 2008, 60, 210–241. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Dietrich, H.H.; Xiang, C.; Dacey, R.G., Jr. G protein-coupled estrogen receptor agonist improves cerebral microvascular function after hypoxia/reoxygenation injury in male and female rats. Stroke 2013, 44, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; Moss, R.L. 17β-estradiol potentiates kainate-induced currents via activation of the camp cascade. J. Neurosci. 1996, 16, 3620–3629. [Google Scholar] [PubMed]
- Kelly, M.J.; Wagner, E.J. Estrogen modulation of G-protein-coupled receptors. Trends Endocrinol. Metab. 1999, 10, 369–374. [Google Scholar] [CrossRef]
- Manthey, D.; Heck, S.; Engert, S.; Behl, C. Estrogen induces a rapid secretion of amyloid β precursor protein via the mitogen-activated protein kinase pathway. Eur. J. Biochem. 2001, 268, 4285–4291. [Google Scholar] [CrossRef] [PubMed]
- Szegő, É.M.; Barabás, K.; Balog, J.; Szilágyi, N.; Korach, K.S.; Juhász, G.; Ábrahám, I.M. Estrogen induces estrogen receptor α-dependent camp response element-binding protein phosphorylation via mitogen activated protein kinase pathway in basal forebrain cholinergic neurons in vivo. J. Neurosci. 2006, 26, 4104–4110. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S.; Alves, S.E. Estrogen actions in the central nervous system. Endocr. Rev. 1999, 20, 279–307. [Google Scholar] [CrossRef] [PubMed]
- Saenz, C.; Dominguez, R.; de Lacalle, S. Estrogen contributes to structural recovery after a lesion. Neurosci. Lett. 2006, 392, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Woolley, C.; McEwen, B. Estradiol mediates fluctuation in hippocampal synapse density during the estrous cycle in the adult rat. J. Neurosci. 1992, 12, 2549–2554. [Google Scholar] [PubMed]
- Dubal, D.B.; Kashon, M.L.; Pettigrew, L.C.; Ren, J.M.; Finklestein, S.P.; Rau, S.W.; Wise, P.M. Estradiol protects against ischemic injury. J. Cereb. Blood Flow Metab. 1998, 18, 1253–1258. [Google Scholar] [CrossRef] [PubMed]
- Hawk, T.; Zhang, Y.Q.; Rajakumar, G.; Day, A.L.; Simpkins, J.W. Testosterone increases and estradiol decreases middle cerebral artery occlusion lesion size in male rats. Brain Res. 1998, 796, 296–298. [Google Scholar] [CrossRef]
- Dubal, D.B.; Zhu, H.; Yu, J.; Rau, S.W.; Shughrue, P.J.; Merchenthaler, I.; Kindy, M.S.; Wise, P.M. Estrogen receptor α, not β, is a critical link in estradiol-mediated protection against brain injury. Proc. Natl. Acad. Sci. USA 2001, 98, 1952–1957. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Segura, L.M.; Wozniak, A.; Azcoitia, I.; Rodriguez, J.R.; Hutchison, R.E.; Hutchison, J.B. Aromatase expression by astrocytes after brain injury: Implications for local estrogen formation in brain repair. Neuroscience 1999, 89, 567–578. [Google Scholar] [CrossRef]
- Mohamd, E.M.; Ahmed, H.H.; Estefan, S.F.; Farrag, A.E.; Salah, R.S. Windows into estradiol effects in Alzheimer’s disease therapy. Eur. Rev. Med. Pharmacol. Sci. 2011, 15, 1131–1140. [Google Scholar] [PubMed]
- Marin, R.; Guerra, B.; Hernandez-Jimenez, J.G.; Kang, X.L.; Fraser, J.D.; Lopez, F.J.; Alonso, R. Estradiol prevents amyloid-beta peptide-induced cell death in a cholinergic cell line via modulation of a classical estrogen receptor. Neuroscience 2003, 121, 917–926. [Google Scholar] [CrossRef]
- Marin, R.; Guerra, B.; Morales, A.; Diaz, M.; Alonso, R. An oestrogen membrane receptor participates in estradiol actions for the prevention of amyloid-beta peptide1–40-induced toxicity in septal-derived cholinergic sn56 cells. J. Neurochem. 2003, 85, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Kwakowsky, A.; Potapov, K.; Kim, S.; Peppercorn, K.; Tate, W.P.; Abraham, I.M. Treatment of beta amyloid 1–42 (Aβ(1–42))-induced basal forebrain cholinergic damage by a non-classical estrogen signaling activator in vivo. Sci. Rep. 2016, 6, 21101. [Google Scholar] [CrossRef] [PubMed]
- Shumaker, S.A.; Legault, C.; Kuller, L.; Rapp, S.R.; Thal, L.; Lane, D.S.; Fillit, H.; Stefanick, M.L.; Hendrix, S.L.; Lewis, C.E.; et al. Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women. JAMA 2004, 291, 2947–2958. [Google Scholar] [CrossRef] [PubMed]
- Pines, A. Alzheimer’s disease, menopause and the impact of the estrogenic environment. Climacteric 2016, 19, 430–432. [Google Scholar] [CrossRef] [PubMed]
- Pike, C.J.; Carroll, J.C.; Rosario, E.R.; Barron, A.M. Protective actions of sex steroid hormones in Alzheimer’s disease. Front. Neuroendocrinol. 2009, 30, 239–258. [Google Scholar] [CrossRef] [PubMed]
- Goodman, Y.; Bruce, A.J.; Cheng, B.; Mattson, M.P. Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury, and amyloid β-peptide toxicity in hippocampal neurons. J. Neurochem. 1996, 66, 1836–1844. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Gouras, G.K.; Greenfield, J.P.; Vincent, B.; Naslund, J.; Mazzarelli, L.; Fried, G.; Jovanovic, J.N.; Seeger, M.; Relkin, N.R.; et al. Estrogen reduces neuronal generation of Alzheimer beta-amyloid peptides. Nat. Med. 1998, 4, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Lu, M.; Lancaster, T.; Cao, P.; Honda, S.-I.; Staufenbiel, M.; Harada, N.; Zhong, Z.; Shen, Y.; Li, R. Brain estrogen deficiency accelerates aβ plaque formation in an Alzheimer’s disease animal model. Proc. Natl. Acad. Sci. USA 2005, 102, 19198–19203. [Google Scholar] [CrossRef] [PubMed]
- Leissring, M.; Saido, T. Aβ degradation. In Alzheimer’s Disease; Sisodia, S., Tanzi, R., Eds.; Springer: Berlin, Heidelberg, 2007; pp. 157–178. [Google Scholar]
- Lee, J.H.; Jiang, Y.; Han, D.H.; Shin, S.K.; Choi, W.H.; Lee, M.J. Targeting estrogen receptors for the treatment of Alzheimer’s disease. Mol. Neurobiol. 2014, 49, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Luine, V.N. Estradiol increases choline acetyltransferase activity in specific basal forebrain nuclei and projection areas of female rats. Exp. Neurol. 1985, 89, 484–490. [Google Scholar] [CrossRef]
- Gibbs, R.B. Expression of estrogen receptor-like immunoreactivity by different subgroups of basal forebrain cholinergic neurons in gonadectomized male and female rats. Brain Res. 1996, 720, 61–68. [Google Scholar] [CrossRef]
- Mowla, S.J.; Farhadi, H.F.; Pareek, S.; Atwal, J.K.; Morris, S.J.; Seidah, N.G.; Murphy, R.A. Biosynthesis and post-translational processing of the precursor to brain-derived neurotrophic factor. J. Biol. Chem. 2001, 276, 12660–12666. [Google Scholar] [CrossRef] [PubMed]
- Dawbarn, D.; Allen, S.J. Neurotrophins and neurodegeneration. Neuropathol. Appl. Neurobiol. 2003, 29, 211–230. [Google Scholar] [CrossRef] [PubMed]
- Bibel, M.; Hoppe, E.; Barde, Y.-A. Biochemical and functional interactions between the neurotrophin receptors trk and p75NTR. EMBO J. 1999, 18, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Sobreviela, T.; Clary, D.O.; Reichardt, L.F.; Brandabur, M.M.; Kordower, J.H.; Mufson, E.J. Trka-immunoreactive profiles in the central nervous system: Colocalization with neurons containing p75 nerve growth factor receptor, choline acetyltransferase, and serotonin. J. Comp. Neurol. 1994, 350, 587–611. [Google Scholar] [CrossRef] [PubMed]
- Poblete-Naredo, I.; Guillem, A.M.; Juarez, C.; Zepeda, R.C.; Ramirez, L.; Caba, M.; Hernandez-Kelly, L.C.; Aguilera, J.; Lopez-Bayghen, E.; Ortega, A. Brain-derived neurotrophic factor and its receptors in bergmann glia cells. Neurochem. Int. 2011, 59, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Alderson, R.F.; Curtis, R.; Alterman, A.L.; Lindsay, R.M.; DiStefano, P.S. Truncated trkB mediates the endocytosis and release of bdnf and neurotrophin-4/5 by rat astrocytes and schwann cells in vitro. Brain Res. 2000, 871, 210–222. [Google Scholar] [CrossRef]
- Riley, C.P.; Cope, T.C.; Buck, C.R. CNS neurotrophins are biologically active and expressed by multiple cell types. J. Mol. Histol. 2004, 35, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Cragnolini, A.B.; Friedman, W.J. The function of p75NTR in glia. Trends Neurosci. 2008, 31, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Meakin, S.O.; Shooter, E.M. The nerve growth factor family of receptors. Trends Neurosci. 1992, 15, 323–331. [Google Scholar] [CrossRef]
- Kaplan, D.; Hempstead, B.; Martin-Zanca, D.; Chao, M.; Parada, L. The trk proto-oncogene product: A signal transducing receptor for nerve growth factor. Science 1991, 252, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Bothwell, M. Functional interactions of neurotrophins and neurotrophin receptors. Annu. Rev. Neurosci. 1995, 18, 223–253. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.R.; Martin-Zanca, D.; Parada, L.F. Tyrosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature 1991, 350, 158–160. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Martin-Zanca, D.; Barbacid, M.; Parada, L.F. Expression of the tyrosine kinase receptor gene trkB is confined to the murine embryonic and adult nervous system. Development 1990, 109, 845–850. [Google Scholar] [PubMed]
- Cordon-Cardo, C.; Tapley, P.; Jing, S.; Nanduri, V.; O’Rourke, E.; Lamballe, F.; Kovary, K.; Klein, R.; Jones, K.R.; Reichardt, L.F.; et al. The trk tyrosine protein kinase mediates the mitogenic properties of nerve growth factor and neurotrophin-3. Cell 1991, 66, 173–183. [Google Scholar] [CrossRef]
- Berkemeier, L.R.; Winslow, J.W.; Kaplan, D.R.; Nikolics, K.; Goeddel, D.V.; Rosenthal, A. Neurotrophin-5: A novel neurotrophic factor that activates trk and trkB. Neuron 1991, 7, 857–866. [Google Scholar] [CrossRef]
- Chao, M.V. Neurotrophin receptors: A window into neuronal differentiation. Neuron 1992, 9, 583–593. [Google Scholar] [CrossRef]
- Lee, R.; Kermani, P.; Teng, K.K.; Hempstead, B.L. Regulation of cell survival by secreted proneurotrophins. Science 2001, 294, 1945–1948. [Google Scholar] [CrossRef] [PubMed]
- Rabizadeh, S.; Oh, J.; Zhong, L.; Yang, J.; Bitler, C.; Butcher, L.; Bredesen, D. Induction of apoptosis by the low-affinity NGF receptor. Science 1993, 261, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.R.; Miller, F.D. Neurotrophin signal transduction in the nervous system. Curr. Opin. Neurobiol. 2000, 10, 381–391. [Google Scholar] [CrossRef]
- Aloyz, R.S.; Bamji, S.X.; Pozniak, C.D.; Toma, J.G.; Atwal, J.; Kaplan, D.R.; Miller, F.D. P53 is essential for developmental neuron death as regulated by the trkA and p75 neurotrophin receptors. J. Cell Biol. 1998, 143, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Levi-Montalcini, R.; Hamburger, V. Selective growth stimulating effects of mouse sarcoma on the sensory and sympathetic nervous system of the chick embryo. J. Exp. Zool. 1951, 116, 321–361. [Google Scholar] [CrossRef] [PubMed]
- Ehrhard, P.B.; Erb, P.; Graumann, U.; Schmutz, B.; Otten, U. Expression of functional trk tyrosine kinase receptors after T cell activation. J. Immunol. 1994, 152, 2705–2709. [Google Scholar] [PubMed]
- Horigome, K.; Pryor, J.C.; Bullock, E.D.; Johnson, E.M. Mediator release from mast cells by nerve growth factor. Neurotrophin specificity and receptor mediation. J. Biol. Chem. 1993, 268, 14881–14887. [Google Scholar] [PubMed]
- Solomon, A.; Aloe, L.; Pe’er, J.; Frucht-Pery, J.; Bonini, S.; Bonini, S.; Levi-Schaffer, F. Nerve growth factor is preformed in and activates human peripheral blood eosinophils. J. Allergy Clin. Immunol. 1998, 102, 454–460. [Google Scholar] [CrossRef]
- Barde, Y.A.; Edgar, D.; Thoenen, H. Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1982, 1, 549–553. [Google Scholar] [PubMed]
- Leibrock, J.; Lottspeich, F.; Hohn, A.; Hofer, M.; Hengerer, B.; Masiakowski, P.; Thoenen, H.; Barde, Y.A. Molecular cloning and expression of brain-derived neurotrophic factor. Nature 1989, 341, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, C.F. Neurotrophic factors: From structure-function studies to designing effective therapeutics. Trends Biotechnol. 1995, 13, 217–227. [Google Scholar] [CrossRef]
- Borasio, G.D.; Markus, A.; Wittinghofer, A.; Barde, Y.A.; Heumann, R. Involvement of ras p21 in neurotrophin-induced response of sensory, but not sympathetic neurons. J. Cell Biol. 1993, 121, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Vogel, K.S.; Brannan, C.I.; Jenkins, N.A.; Copeland, N.G.; Parada, L.F. Loss of neurofibromin results in neurotrophin-independent survival of embryonic sensory and sympathetic neurons. Cell 1995, 82, 733–742. [Google Scholar] [CrossRef]
- Bartlett, S.E.; Reynolds, A.J.; Weible, M.; Heydon, K.; Hendry, I.A. In sympathetic but not sensory neurones, phosphoinositide-3 kinase is important for NGF-dependent survival and the retrograde transport of 125i-βngf. Brain Res. 1997, 761, 257–262. [Google Scholar] [CrossRef]
- Markus, A.; Holst, A.V.; Rohrer, H.; Heumann, R. NGF-mediated survival depends on p21ras in chick sympathetic neurons from the superior cervical but not from lumbosacral ganglia. Dev. Biol. 1997, 191, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.; Cooper, G. Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science 1995, 267, 2003–2006. [Google Scholar] [CrossRef] [PubMed]
- Vaillant, A.R.; Mazzoni, I.; Tudan, C.; Boudreau, M.; Kaplan, D.R.; Miller, F.D. Depolarization and neurotrophins converge on the phosphatidylinositol 3-kinase-akt pathway to synergistically regulate neuronal survival. J. Cell Biol. 1999, 146, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Blair, L.A.C.; Bence-Hanulec, K.K.; Mehta, S.; Franke, T.; Kaplan, D.; Marshall, J. Akt-dependent potentiation of l channels by insulin-like growth factor-1 is required for neuronal survival. J. Neurosci. 1999, 19, 1940–1951. [Google Scholar] [PubMed]
- Grewal, S.S.; York, R.D.; Stork, P.J.S. Extracellular-signal-regulated kinase signalling in neurons. Curr. Opin. Neurobiol. 1999, 9, 544–553. [Google Scholar] [CrossRef]
- Creedon, D.J.; Johnson, E.M.; Lawrence, J.C. Mitogen-activated protein kinase-independent pathways mediate the effects of nerve growth factor and camp on neuronal survival. J. Biol. Chem. 1996, 271, 20713–20718. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Barker, P.A. Neurotrophin signaling through the p75 neurotrophin receptor. Prog. Neurobiol. 2002, 67, 203–233. [Google Scholar] [CrossRef]
- Carter, B.D.; Kaltschmidt, C.; Kaltschmidt, B.; Offenhauser, N.; Bohm-Matthaei, R.; Baeuerle, P.A.; Barde, Y.A. Selective activation of NF-κB by nerve growth factor through the neurotrophin receptor p75. Science 1996, 272, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Ladiwala, U.; Lachance, C.; Simoneau, S.J.J.; Bhakar, A.; Barker, P.A.; Antel, J.P. P75 neurotrophin receptor expression on adult human oligodendrocytes: Signaling without cell death in response to NGF. J. Neurosci. 1998, 18, 1297–1304. [Google Scholar] [PubMed]
- Kimpinski, K.; Jelinski, S.; Mearow, K. The anti-p75 antibody, MC192, and brain-derived neurotrophic factor inhibit nerve growth factor-dependent neurite growth from adult sensory neurons. Neuroscience 1999, 93, 253–263. [Google Scholar] [CrossRef]
- Yoon, S.O.; Casaccia-Bonnefil, P.; Carter, B.; Chao, M.V. Competitive signaling between trka and p75 nerve growth factor receptors determines cell survival. J. Neurosci. 1998, 18, 3273–3281. [Google Scholar] [PubMed]
- Maggirwar, S.B.; Sarmiere, P.D.; Dewhurst, S.; Freeman, R.S. Nerve growth factor-dependent activation of NF-κB contributes to survival of sympathetic neurons. J. Neurosci. 1998, 18, 10356–10365. [Google Scholar] [PubMed]
- Khursigara, G.; Orlinick, J.R.; Chao, M.V. Association of the p75 neurotrophin receptor with TRAF6. J. Biol. Chem. 1999, 274, 2597–2600. [Google Scholar] [CrossRef] [PubMed]
- Walsh, G.S.; Krol, K.M.; Crutcher, K.A.; Kawaja, M.D. Enhanced neurotrophin-induced axon growth in myelinated portions of the cns in mice lacking the p75 neurotrophin receptor. J. Neurosci. 1999, 19, 4155–4168. [Google Scholar] [PubMed]
- Fahnestock, M.; Michalski, B.; Xu, B.; Coughlin, M.D. The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in Alzheimer’s disease. Mol. Cell. Neurosci. 2001, 18, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Increased prongf levels in subjects with mild cognitive impairment and mild Alzheimer disease. J. Neuropathol. Exp. Neurol. 2004, 63, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Mufson, E.J.; Brashers-Krug, T.; Kordower, J.H. P75 nerve growth factor receptor immunoreactivity in the human brainstem and spinal cord. Brain Res. 1992, 589, 115–123. [Google Scholar] [CrossRef]
- Costantini, C.; Weindruch, R.; Della Valle, G.; Puglielli, L. A TrkA-to-p75NTR molecular switch activates amyloid β-peptide generation during aging. Biochem. J. 2005, 391, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Yaar, M.; Zhai, S.; Pilch, P.F.; Doyle, S.M.; Eisenhauer, P.B.; Fine, R.E.; Gilchrest, B.A. Binding of beta-amyloid to the p75 neurotrophin receptor induces apoptosis. A possible mechanism for Alzheimer’s disease. J. Clin. Investig. 1997, 100, 2333–2340. [Google Scholar] [CrossRef] [PubMed]
- Coulson, E.J. Does the p75 neurotrophin receptor mediate Aβ-induced toxicity in Alzheimer’s disease? J. Neurochem. 2006, 98, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.Y.; Hassenstab, J.; Cruchaga, C.; Goate, A.; Fagan, A.M.; Benzinger, T.L.; Maruff, P.; Snyder, P.J.; Masters, C.L.; Allegri, R.; et al. BDNF Val66Met moderates memory impairment, hippocampal function and tau in preclinical autosomal dominant Alzheimer’s disease. Brain J. Neurol. 2016, 139, 2766–2777. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.Y.; Villemagne, V.L.; Laws, S.M.; Ames, D.; Pietrzak, R.H.; Ellis, K.A.; Harrington, K.D.; Bourgeat, P.; Salvado, O.; Darby, D.; et al. BDNF Val66Met, aβ amyloid, and cognitive decline in preclinical Alzheimer’s disease. Neurobiol. Aging 2013, 34, 2457–2464. [Google Scholar] [CrossRef] [PubMed]
- Korsching, S.; Auburger, G.; Heumann, R.; Scott, J.; Thoenen, H. Levels of nerve growth factor and its mrna in the central nervous system of the rat correlate with cholinergic innervation. EMBO J. 1985, 4, 1389–1393. [Google Scholar] [PubMed]
- Phillips, H.; Hains, J.; Laramee, G.; Rosenthal, A.; Winslow, J. Widespread expression of bdnf but not nt3 by target areas of basal forebrain cholinergic neurons. Science 1990, 250, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, H.; Nihonmatsu, I.; Tsukui, H. Nerve growth factor promotes survival of cultured magnocellular cholinergic neurons from nucleus basalis of meynert in postnatal rats. Neurosci. Lett. 1988, 90, 63–68. [Google Scholar] [CrossRef]
- Hefti, F. Nerve growth factor promotes survival of septal cholinergic neurons after fimbrial transections. J. Neurosci. 1986, 6, 2155–2162. [Google Scholar] [PubMed]
- Kromer, L. Nerve growth factor treatment after brain injury prevents neuronal death. Science 1987, 235, 214–216. [Google Scholar] [CrossRef] [PubMed]
- Koliatsos, V.E.; Price, D.L.; Gouras, G.K.; Cayouette, M.H.; Burton, L.E.; Winslow, J.W. Highly selective effects of nerve growth factor, brain-derived neurotrophic factor, and neurotrophin-3 on intact and injured basal forebrain magnocellular neurons. J. Comp. Neurol. 1994, 343, 247–262. [Google Scholar] [CrossRef] [PubMed]
- Gnahn, H.; Hefti, F.; Heumann, R.; Schwab, M.E.; Thoenen, H. NGF-mediated increase of choline acetyltransferase (ChAT) in the neonatal rat forebrain: Evidence for a physiological role of NGF in the brain? Dev. Brain Res. 1983, 9, 45–52. [Google Scholar] [CrossRef]
- Knusel, B.; Beck, K.; Winslow, J.; Rosenthal, A.; Burton, L.; Widmer, H.; Nikolics, K.; Hefti, F. Brain-derived neurotrophic factor administration protects basal forebrain cholinergic but not nigral dopaminergic neurons from degenerative changes after axotomy in the adult rat brain. J. Neurosci. 1992, 12, 4391–4402. [Google Scholar] [PubMed]
- Sanchez-Ortiz, E.; Yui, D.; Song, D.; Li, Y.; Rubenstein, J.L.; Reichardt, L.F.; Parada, L.F. Trka gene ablation in basal forebrain results in dysfunction of the cholinergic circuitry. J. Neurosci. 2012, 32, 4065–4079. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.J.; Dawbarn, D.; Spillantini, M.G.; Goedert, M.; Wilcock, G.K.; Moss, T.H.; Semenenko, F.M. Distribution of β-nerve growth factor receptors in the human basal forebrain. J. Comp. Neurol. 1989, 289, 626–640. [Google Scholar] [CrossRef] [PubMed]
- Pioro, E.P.; Cuello, A.C. Distribution of nerve growth factor receptor-like immunoreactivity in the adult rat central nervous system. Effect of colchicine and correlation with the cholinergic system—II. Brainstem, cerebellum and spinal cord. Neuroscience 1990, 34, 89–110. [Google Scholar] [CrossRef]
- Yeo, T.T.; Chua-Couzens, J.; Butcher, L.L.; Bredesen, D.E.; Cooper, J.D.; Valletta, J.S.; Mobley, W.C.; Longo, F.M. Absence of p75ntr causes increased basal forebrain cholinergic neuron size, choline acetyltransferase activity, and target innervation. J. Neurosci. 1997, 17, 7594–7605. [Google Scholar] [PubMed]
- Crowley, C.; Spencer, S.D.; Nishimura, M.C.; Chen, K.S.; Pitts-Meek, S.; Armaninl, M.P.; Ling, L.H.; McMahon, S.B.; Shelton, D.L.; Levinson, A.D.; et al. Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell 1994, 76, 1001–1011. [Google Scholar] [CrossRef]
- Volosin, M.; Song, W.; Almeida, R.D.; Kaplan, D.R.; Hempstead, B.L.; Friedman, W.J. Interaction of survival and death signaling in basal forebrain neurons: Roles of neurotrophins and proneurotrophins. J. Neurosci. 2006, 26, 7756–7766. [Google Scholar] [CrossRef] [PubMed]
- Vogels, O.J.; Broere, C.A.; ter Laak, H.J.; Ten Donkelaar, H.J.; Nieuwenhuys, R.; Schulte, B.P. Cell loss and shrinkage in the nucleus basalis meynert complex in Alzheimer’s disease. Neurobiol. Aging 1990, 11, 3–13. [Google Scholar] [CrossRef]
- Lehericy, S.; Hirsch, E.C.; Cervera-Pierot, P.; Hersh, L.B.; Bakchine, S.; Piette, F.; Duyckaerts, C.; Hauw, J.J.; Javoy-Agid, F.; Agid, Y. Heterogeneity and selectivity of the degeneration of cholinergic neurons in the basal forebrain of patients with Alzheimer’s disease. J. Comp. Neurol. 1993, 330, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Cullen, K.M.; Halliday, G.M. Neurofibrillary degeneration and cell loss in the nucleus basalis in comparison to cortical Alzheimer pathology. Neurobiol. Aging 1998, 19, 297–306. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Clark, A.W.; Coyle, J.T.; DeLong, M.R. Alzheimer disease: Evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann. Neurol. 1981, 10, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Grothe, M.; Heinsen, H.; Teipel, S.J. Atrophy of the cholinergic basal forebrain over the adult age range and in early stages of Alzheimer’s disease. Biol. Psychiatry 2012, 71, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Cantero, J.L.; Zaborszky, L.; Atienza, M. Volume loss of the nucleus basalis of meynert is associated with atrophy of innervated regions in mild cognitive impairment. Cereb. Cortex 2016. [Google Scholar] [CrossRef] [PubMed]
- Schliebs, R.; Arendt, T. The cholinergic system in aging and neuronal degeneration. Behav. Brain Res. 2011, 221, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, R.B. Effects of estrogen on basal forebrain cholinergic neurons vary as a function of dose and duration of treatment. Brain Res. 1997, 757, 10–16. [Google Scholar] [CrossRef]
- Gibbs, R.B. Impairment of basal forebrain cholinergic neurons associated with aging and long-term loss of ovarian function. Exp. Neurol. 1998, 151, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Muir, J.L. Acetylcholine, aging, and Alzheimer’s disease. Pharmacol. Biochem. Behav. 1997, 56, 687–696. [Google Scholar] [CrossRef]
- Bartus, R.T. On neurodegenerative diseases, models, and treatment strategies: Lessons learned and lessons forgotten a generation following the cholinergic hypothesis. Exp. Neurol. 2000, 163, 495–529. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, P.; Price, D.; Struble, R.; Clark, A.; Coyle, J.; Delon, M. Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science 1982, 215, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Sohrabji, F.; Miranda, R.C.; Toran-Allerand, C.D. Identification of a putative estrogen response element in the gene encoding brain-derived neurotrophic factor. Proc. Natl. Acad. Sci. USA 1995, 92, 11110–11114. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, R.B.; Wu, D.; Hersh, L.B.; Pfaff, D.W. Effects of estrogen replacement on the relative levels of choline acetyltransferase, trkA, and nerve growth factor messenger RNAs in the basal forebrain and hippocampal formation of adult rats. Exp. Neurol. 1994, 129, 70–80. [Google Scholar] [CrossRef] [PubMed]
- McMillan, P.; Singer, C.; Dorsa, D. The effects of ovariectomy and estrogen replacement on trkA and choline acetyltransferase mrna expression in the basal forebrain of the adult female sprague-dawley rat. J. Neurosci. 1996, 16, 1860–1865. [Google Scholar] [PubMed]
- Singer, C.A.; McMillan, P.J.; Dobie, D.J.; Dorsa, D.M. Effects of estrogen replacement on choline acetyltransferase and trka mRNA expression in the basal forebrain of aged rats. Brain Res. 1998, 789, 343–346. [Google Scholar] [CrossRef]
- Laflamme, N.; Nappi, R.E.; Drolet, G.; Labrie, C.; Rivest, S. Expression and neuropeptidergic characterization of estrogen receptors (ERα and ERβ) throughout the rat brain: Anatomical evidence of distinct roles of each subtype. J. Neurobiol. 1998, 36, 357–378. [Google Scholar] [CrossRef]
- Shughrue, P.J.; Scrimo, P.J.; Merchenthaler, I. Estrogen binding and estrogen receptor characterization (ERα; and ERβ) in the cholinergic neurons of the rat basal forebrain. Neuroscience 2000, 96, 41–49. [Google Scholar] [CrossRef]
- Ishunina, T.A.; Swaab, D.F. Increased expression of estrogen receptor α and β in the nucleus basalis of meynert in Alzheimer’s disease. Neurobiol. Aging 2001, 22, 417–426. [Google Scholar] [CrossRef]
- Mufson, E.J.; Li, J.M.; Sobreviela, T.; Kordower, J.H. Decreased trkA gene expression within basal forebrain neurons in Alzheimer’s disease. Neuroreport 1996, 8, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, S.D.; Che, S.; Wuu, J.; Counts, S.E.; Mufson, E.J. Down regulation of trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer’s disease. J. Neurochem. 2006, 97, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Salehi, A.; Ocampo, M.; Verhaagen, J.; Swaab, D.F. P75 neurotrophin receptor in the nucleus basalis of meynert in relation to age, sex, and Alzheimer’s disease. Exp. Neurol. 2000, 161, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Fisher, B.; Costantino, J.P.; Wickerham, D.L.; Cecchini, R.S.; Cronin, W.M.; Robidoux, A.; Bevers, T.B.; Kavanah, M.T.; Atkins, J.N.; Margolese, R.G.; et al. Tamoxifen for the prevention of breast cancer: Current status of the national surgical adjuvant breast and bowel project P-1 study. J. Natl. Cancer Inst. 2005, 97, 1652–1662. [Google Scholar] [CrossRef] [PubMed]
- Engler-Chiurazzi, E.B.; Singh, M.; Simpkins, J.W. Reprint of: From the 90s to now: A brief historical perspective on more than two decades of estrogen neuroprotection. Brain Res. 2016, 1645, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.K.; Paganini-Hill, A.; Wan, P.C.; Pike, M.C. Effect of hormone replacement therapy on breast cancer risk: Estrogen versus estrogen plus progestin. J. Natl. Cancer Inst. 2000, 92, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Writing Group for the Women’s Health Initiative, I. Risks and benefits of estrogen plus progestin in healthy postmenopausal women. JAMA 2002, 288, 321–333. [Google Scholar] [CrossRef]
- Gurney, E.P.; Nachtigall, M.J.; Nachtigall, L.E.; Naftolin, F. The women’s health initiative trial and related studies: 10 years later: A clinician’s view. J. Steroid Biochem. Mol. Biol. 2014, 142, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.E. Current recommendations: What is the clinician to do? Fertil. Steril. 2014, 101, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Brinton, R.D. Investigative models for determining hormone therapy-induced outcomes in brain: Evidence in support of a healthy cell bias of estrogen action. Ann. N. Y. Acad. Sci. 2005, 1052, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Scharfman, H.E.; MacLusky, N.J. Estrogen-growth factor interactions and their contributions to neurological disorders. Headache 2008, 48, S77–89. [Google Scholar] [CrossRef] [PubMed]
- Kwakowsky, A.; Koszegi, Z.; Cheong, R.Y.; Abraham, I.M. Neuroprotective effects of non-classical estrogen-like signaling activators: From mechanism to potential implications. CNS Neurol. Disord. Drug Targets 2013, 12, 1219–1225. [Google Scholar] [CrossRef] [PubMed]
- Cordey, M.; Gundimeda, U.; Gopalakrishna, R.; Pike, C.J. The synthetic estrogen 4-estren-3α,17β-diol (estren) induces estrogen-like neuroprotection. Neurobiol. Dis. 2005, 19, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Salehi, A.; Verhaagen, J.; Dijkhuizen, P.A.; Swaab, D.F. Co-localization of high-affinity neurotrophin receptors in nucleus basalis of meynert neurons and their differential reduction in Alzheimer’s disease. Neuroscience 1996, 75, 373–387. [Google Scholar] [CrossRef]
- Connor, B.; Young, D.; Lawlor, P.; Gai, W.; Waldvogel, H.; Faull, R.L.; Dragunow, M. Trk receptor alterations in Alzheimer’s disease. Brain Res. Mol. Brain Res. 1996, 42, 1–17. [Google Scholar] [CrossRef]
- Hefti, F.; Weiner, W.J. Nerve growth factor and alzheimer’s disease. Ann. Neurol. 1986, 20, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Seiger, A.; Nordberg, A.; von Holst, H.; Backman, L.; Ebendal, T.; Alafuzoff, I.; Amberla, K.; Hartvig, P.; Herlitz, A.; Lilja, A.; et al. Intracranial infusion of purified nerve growth factor to an Alzheimer patient: The first attempt of a possible future treatment strategy. Behav. Brain Res. 1993, 57, 255–261. [Google Scholar] [CrossRef]
- Eriksdotter-Jonhagen, M.; Linderoth, B.; Lind, G.; Aladellie, L.; Almkvist, O.; Andreasen, N.; Blennow, K.; Bogdanovic, N.; Jelic, V.; Kadir, A.; et al. Encapsulated cell biodelivery of nerve growth factor to the basal forebrain in patients with Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2012, 33, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Wahlberg, L.U.; Lind, G.; Almqvist, P.M.; Kusk, P.; Tornoe, J.; Juliusson, B.; Soderman, M.; Sellden, E.; Seiger, A.; Eriksdotter-Jonhagen, M.; et al. Targeted delivery of nerve growth factor via encapsulated cell biodelivery in Alzheimer disease: A technology platform for restorative neurosurgery. J. Neurosurg. 2012, 117, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.; Westman, E.; Eyjolfsdottir, H.; Almqvist, P.; Lind, G.; Linderoth, B.; Seiger, A.; Blennow, K.; Karami, A.; Darreh-Shori, T.; et al. Brain changes in Alzheimer’s disease patients with implanted encapsulated cells releasing nerve growth factor. J. Alzheimers Dis. 2015, 43, 1059–1072. [Google Scholar] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwakowsky, A.; Milne, M.R.; Waldvogel, H.J.; Faull, R.L. Effect of Estradiol on Neurotrophin Receptors in Basal Forebrain Cholinergic Neurons: Relevance for Alzheimer’s Disease. Int. J. Mol. Sci. 2016, 17, 2122. https://doi.org/10.3390/ijms17122122
Kwakowsky A, Milne MR, Waldvogel HJ, Faull RL. Effect of Estradiol on Neurotrophin Receptors in Basal Forebrain Cholinergic Neurons: Relevance for Alzheimer’s Disease. International Journal of Molecular Sciences. 2016; 17(12):2122. https://doi.org/10.3390/ijms17122122
Chicago/Turabian StyleKwakowsky, Andrea, Michael R. Milne, Henry J. Waldvogel, and Richard L. Faull. 2016. "Effect of Estradiol on Neurotrophin Receptors in Basal Forebrain Cholinergic Neurons: Relevance for Alzheimer’s Disease" International Journal of Molecular Sciences 17, no. 12: 2122. https://doi.org/10.3390/ijms17122122
APA StyleKwakowsky, A., Milne, M. R., Waldvogel, H. J., & Faull, R. L. (2016). Effect of Estradiol on Neurotrophin Receptors in Basal Forebrain Cholinergic Neurons: Relevance for Alzheimer’s Disease. International Journal of Molecular Sciences, 17(12), 2122. https://doi.org/10.3390/ijms17122122