
Integrins in the Spotlight of Cancer

Abstract
:1. Introduction
Giving Light to Life
2. Hallmarking Cancer
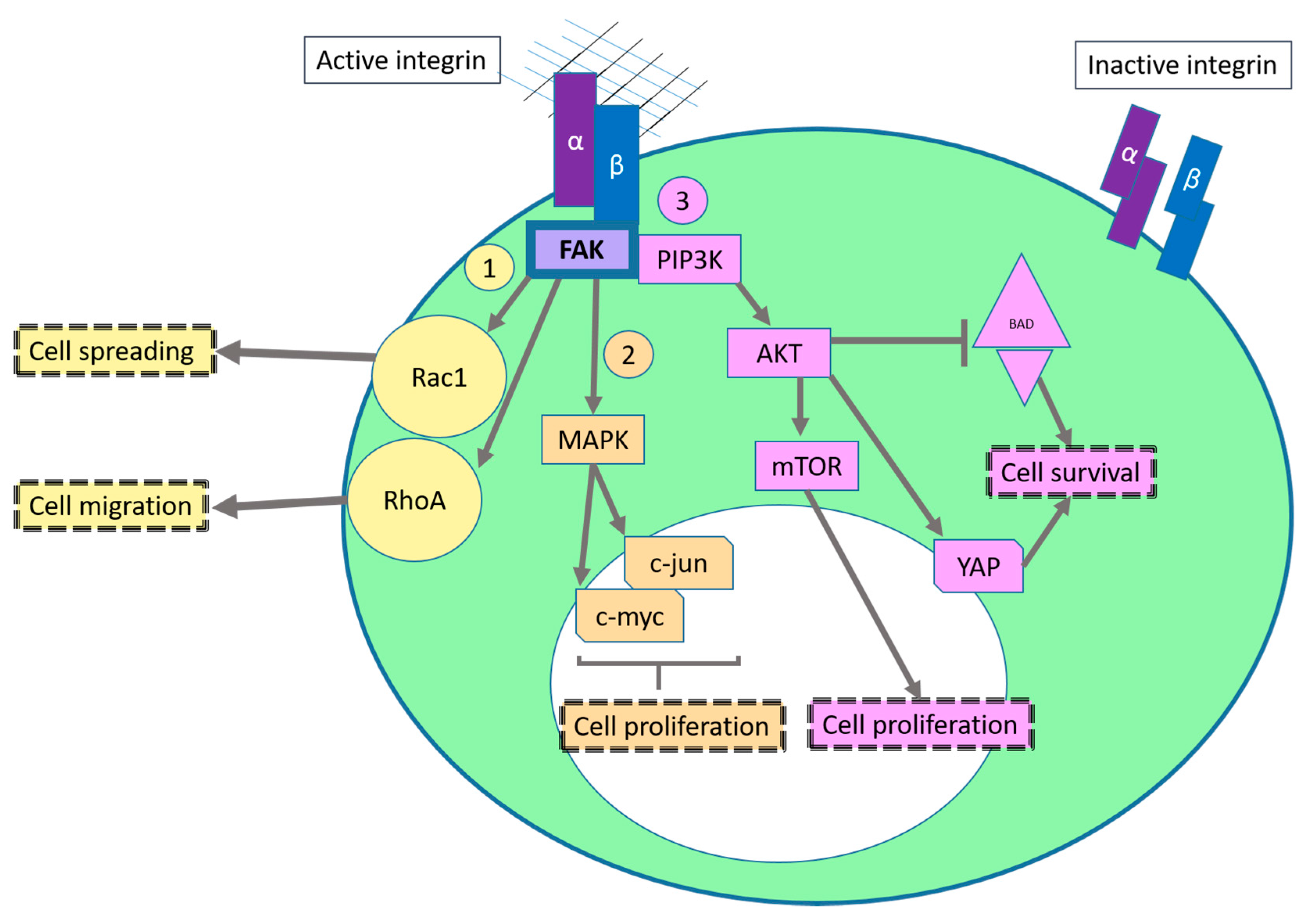
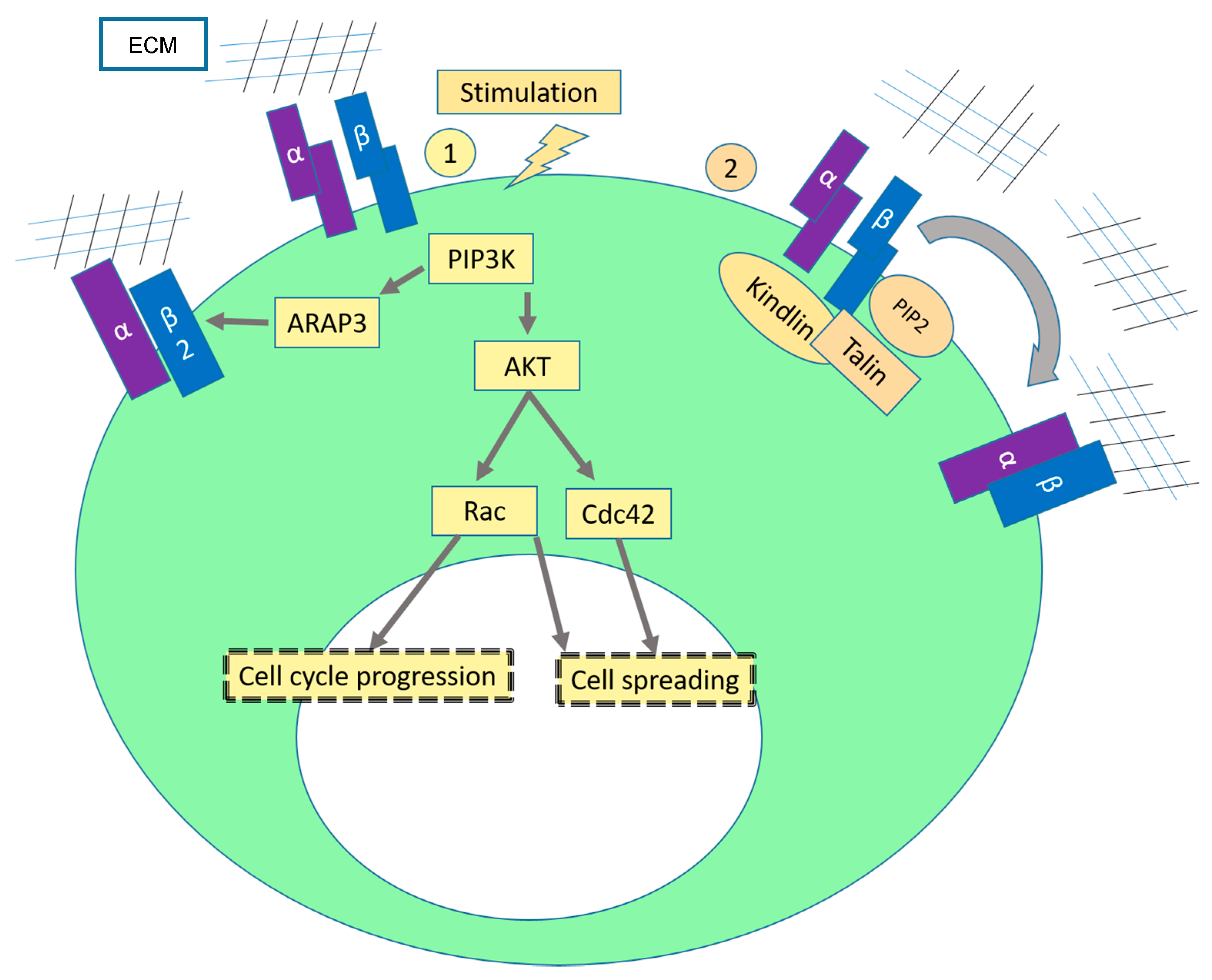
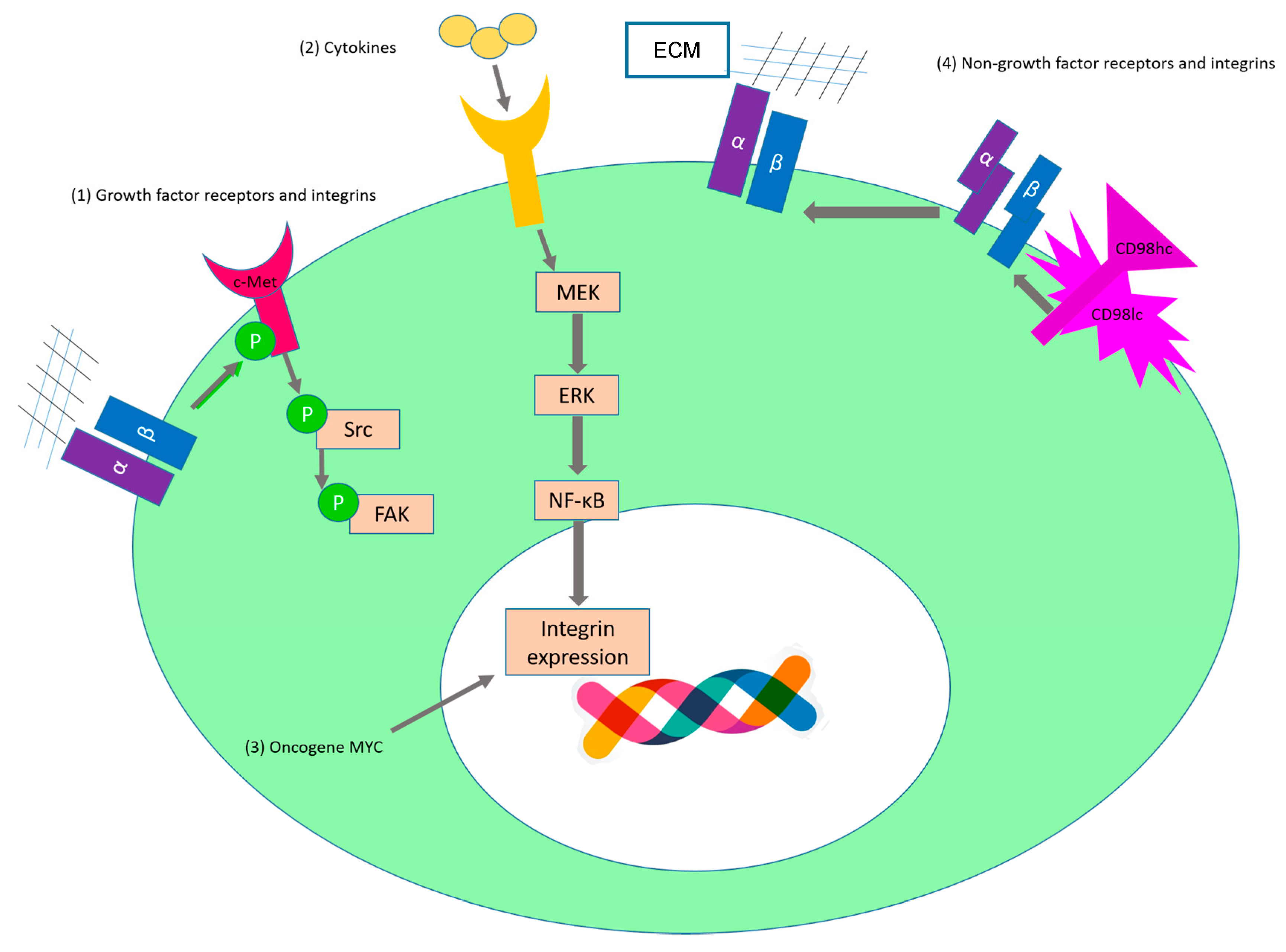
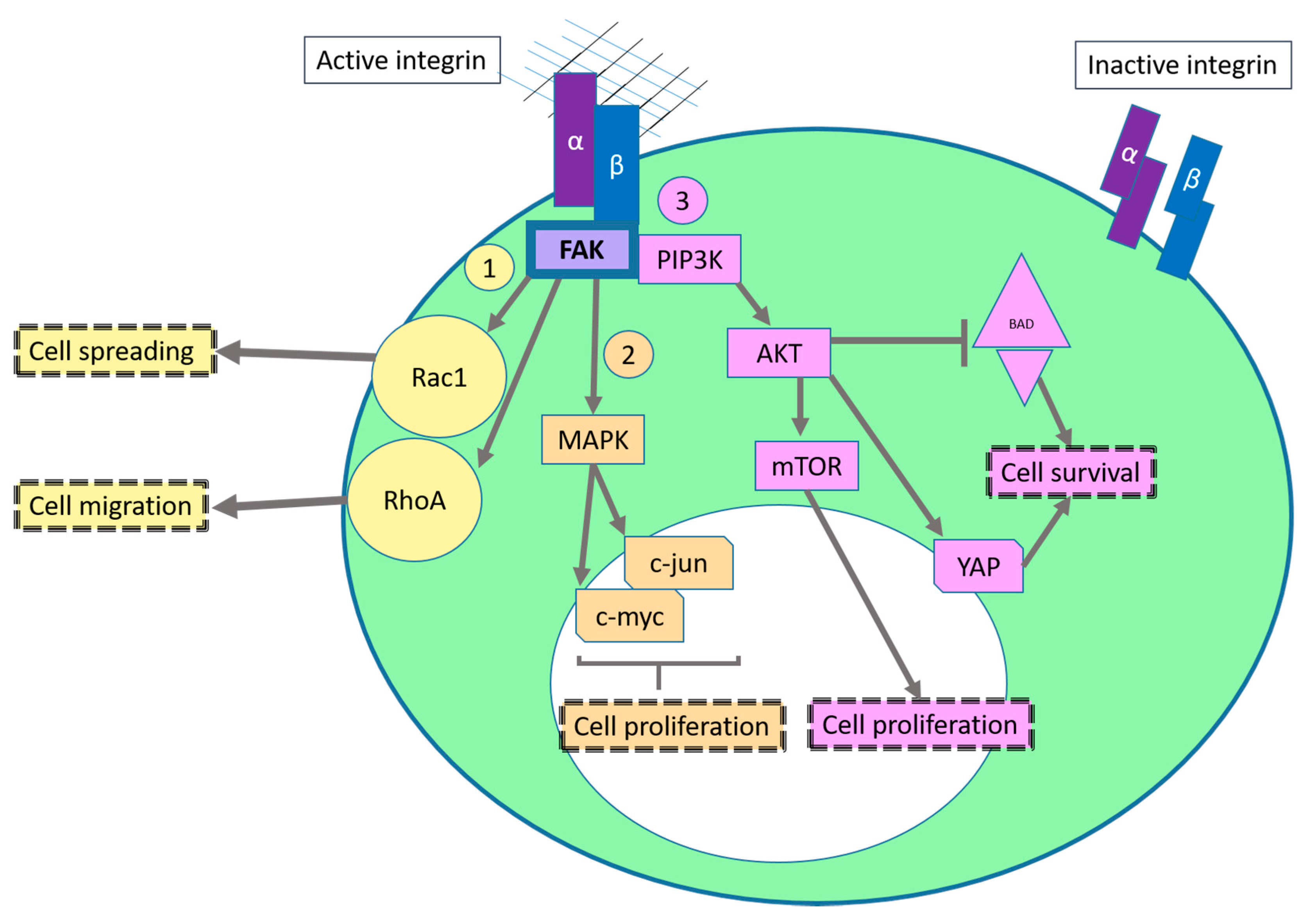
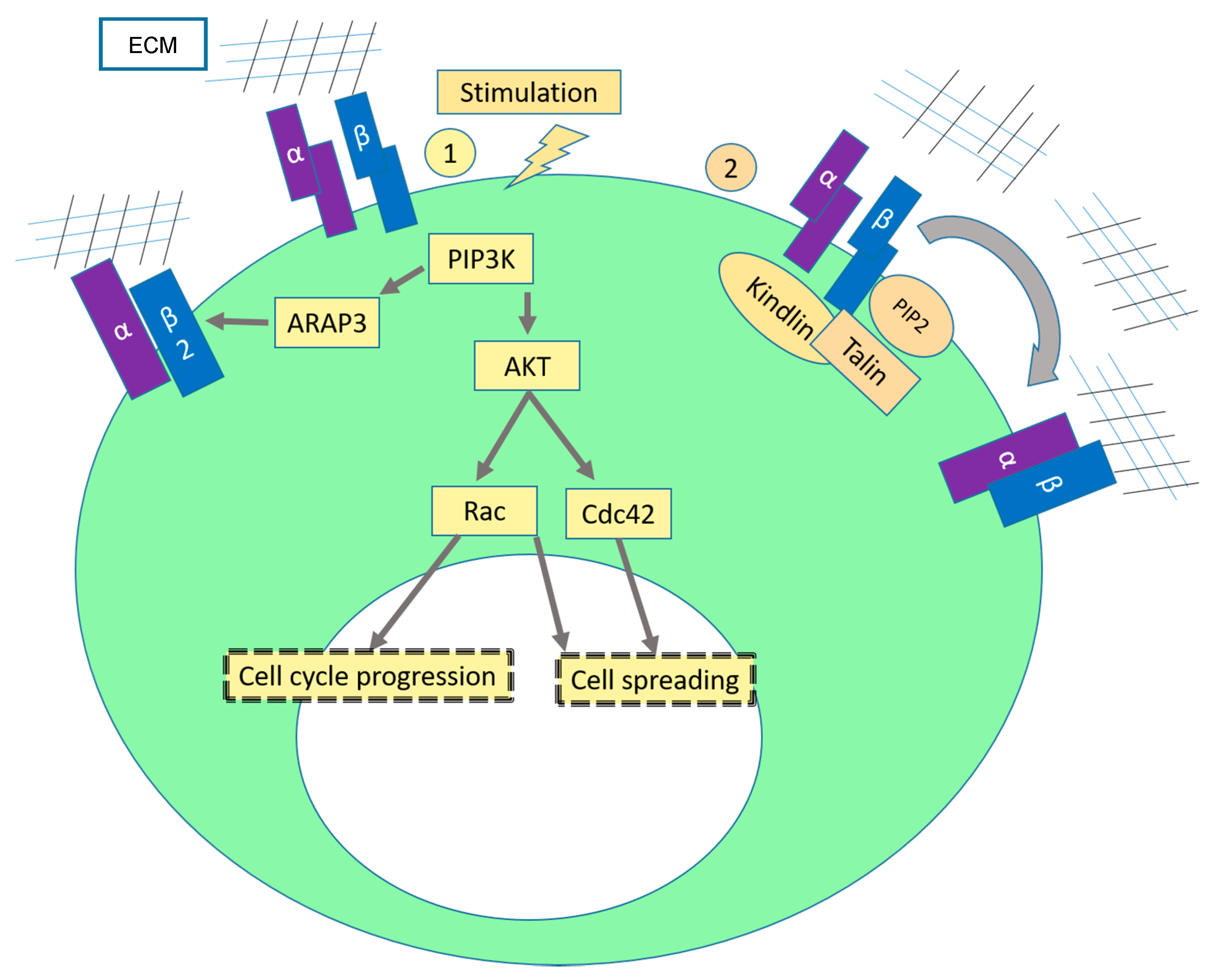
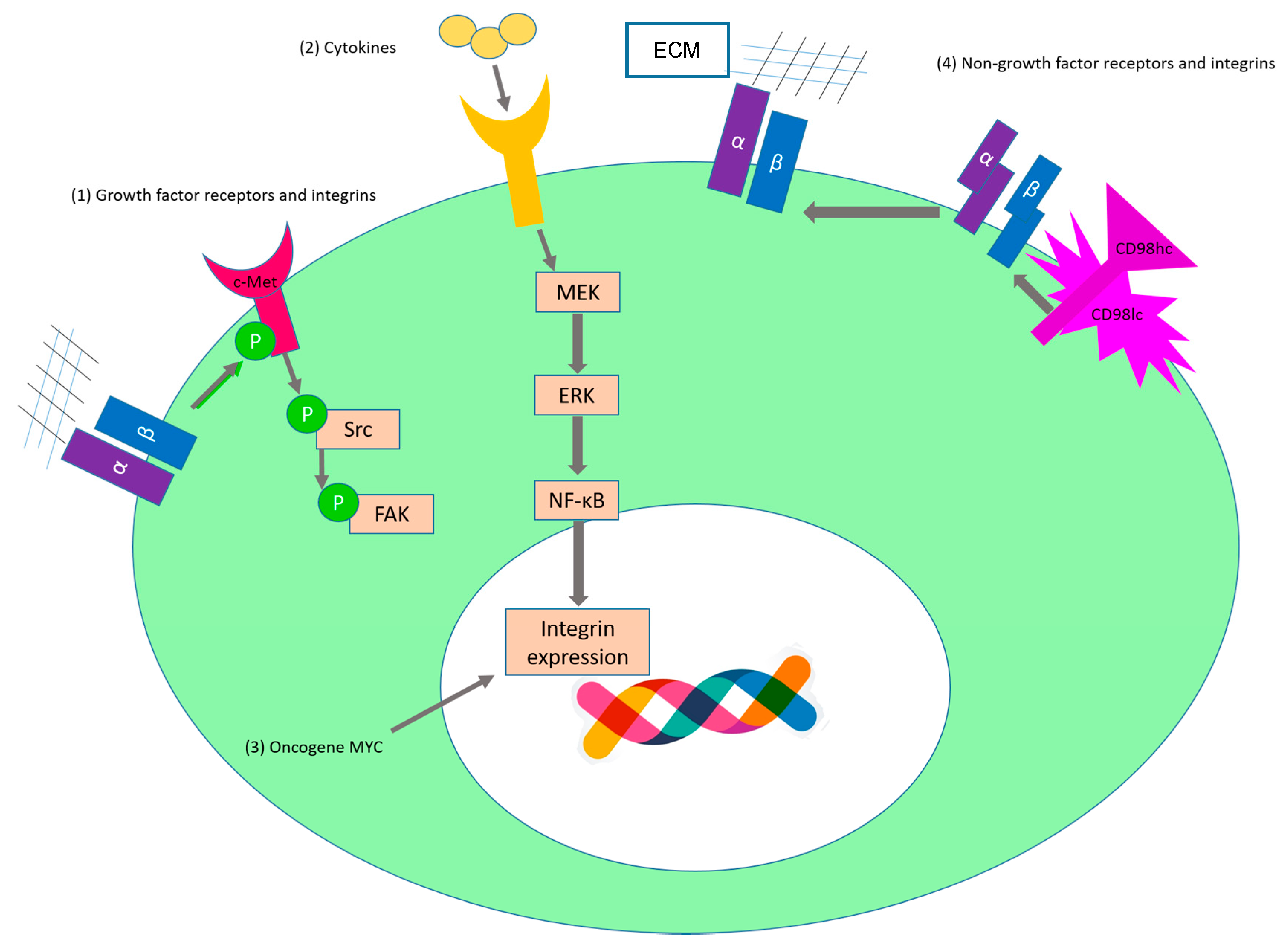
2.1. Sustaining Proliferative Signaling
2.2. Evading Growth Suppressors
2.3. Activating Invasion and Metastasis
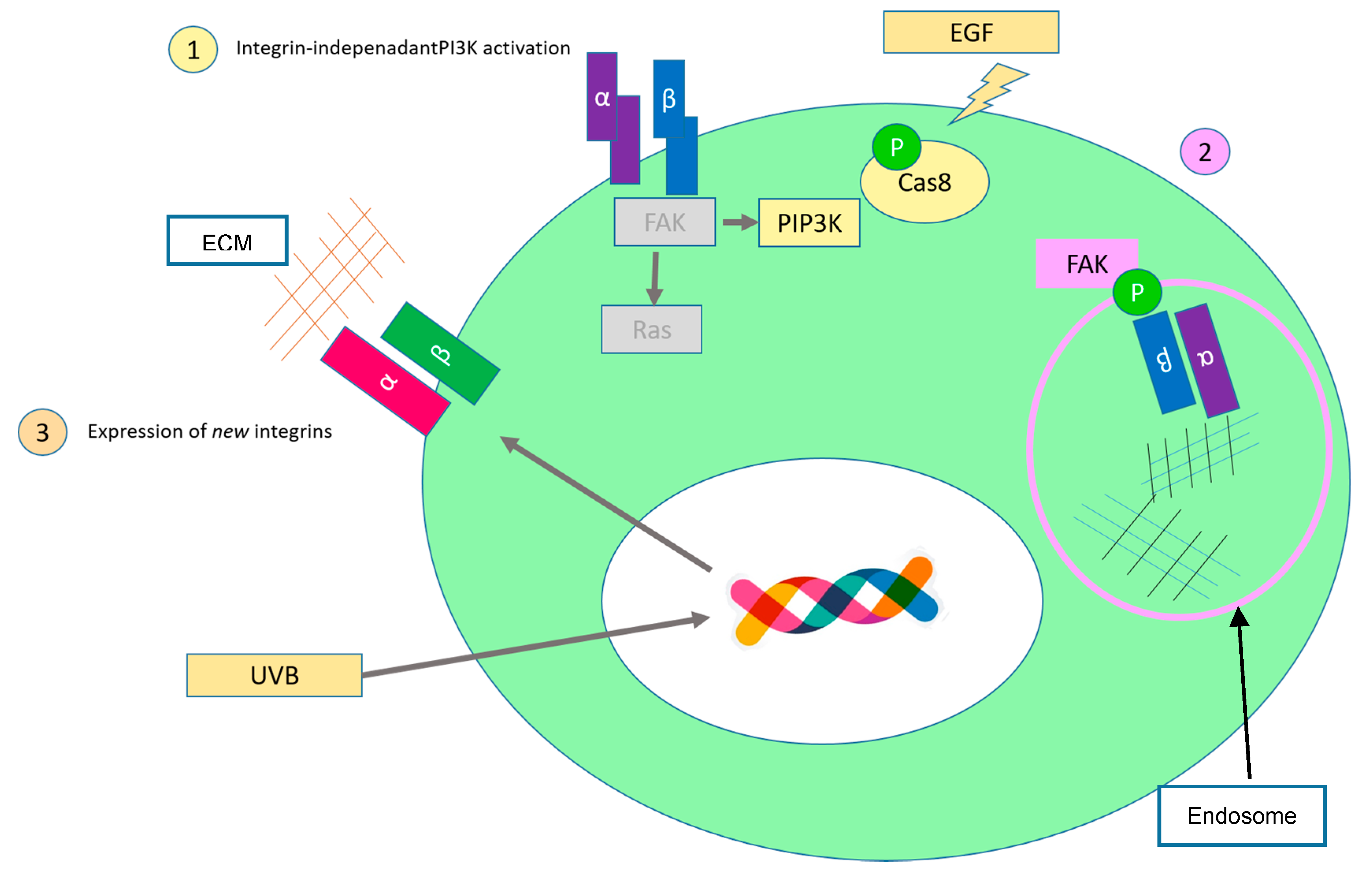
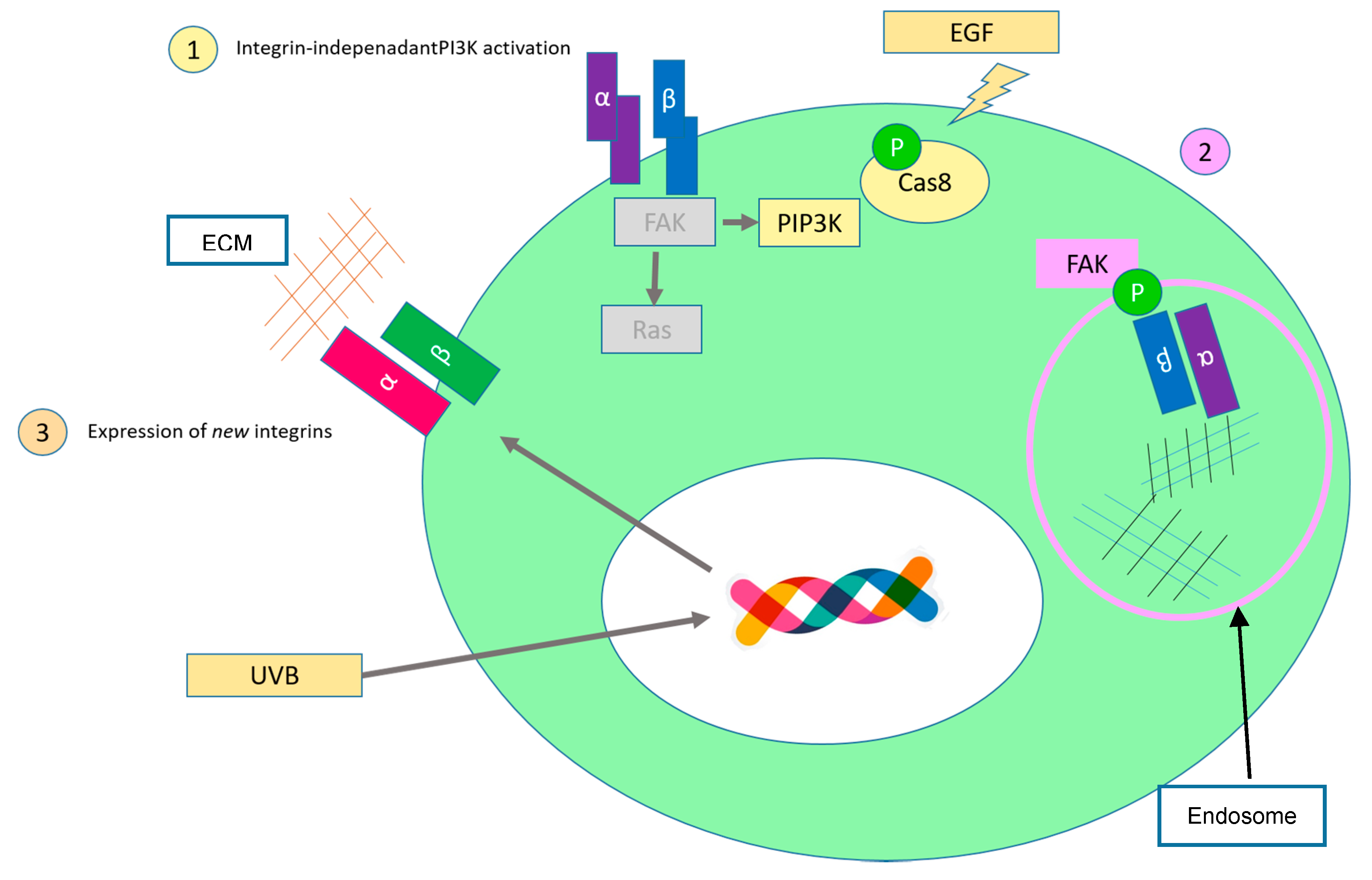
2.4. Limitless Replicative Potential
2.5. Sustained Angiogenesis
2.6. Evading Apoptosis
3. Hotspots for Anticancer Treatment
4. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ECM | Extracellular matrix |
| FAK | Focal adhesion kinase |
| Rac1 | Ras-related C3 botulinum toxin substrate 1 |
| MAPK | MAP kinase pathway |
| EGF | Epidermal growth factor |
| PI3K | Phosphatidylinositol 3-kinase |
| PDK1 | Phosphoinositide-dependent kinase-1 |
| mTOR | Mammalian target of rapamycin |
| VEGF | Vascular endothelial growth factor |
| YAP | Yes-associated protein |
| PIP3 | Phosphatidylinositol-3-phosphate |
| GSK3 | Glycogen synthase kinase 3 |
| PHI-1 | Phosphatase holoenzyme inhibitor 1 |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| CDH17 | Cadherin-17 |
| TNF-α | Tumor necrosis factor-α |
| IGF-IR | Insulin-like growth factor receptor |
| Gab1 | Grb2-associated binder-1 |
| HGF/SF | Hepatocyte growth factor/scatter factor |
| Necl-5 | Nectin-like molecule-5 |
| NF2 | Neurofibromatosis type 2 |
| PAK | p21-activated kinase |
| EMT | Epithelial-mesenchymal transition |
| LAP | Latency-associated peptide |
| LTBP | TGFβ-binding protein |
| BMP-7 | Bone morphogenic protein 7 |
| CAF | Carcinoma associated fibroblasts |
| HREs | Hypoxia response elements |
| CRLR | Calcitonin receptor-like receptor |
| SCF | Stem cell factor |
| ANGPT2 | Angiopoietin 2 |
| uPA | Urokinase plasminogen activator |
| uPAR | Urokinase plasminogen activator receptor |
| MMP | Matrix metalloproteinases |
| SNP | Single-nucleotide polymorphisms |
References
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Ginsberg, M.H. Integrin activation. BMB Rep. 2014, 47, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Sebe-Pedros, A.; Roger, A.J.; Lang, F.B.; King, N.; Ruiz-Trillo, I. Ancient origin of the integrin-mediated adhesion and signaling machinery. Proc. Natl. Acad. Sci. USA 2010, 107, 10142–10147. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.S.; Lu, N.; Denessiouk, K.; Heino, J.; Gullberg, D. Integrins during evolution: Evolutionary trees and model organisms. Biochim. Biophys. Acta 2009, 1788, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Wennerberg, K.; Armulik, A.; Sakai, T.; Karlsson, M.; Fassler, R.; Schaefer, E.M.; Mosher, D.F.; Johansson, S. The cytoplasmic tyrosines of integrin subunit β1 are involved in focal adhesion kinase activation. Mol. Cell. Biol. 2000, 20, 5758–5765. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Guan, J.L. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009, 28, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Horton, E.R.; Humphries, J.D.; Stutchbury, B.; Jacquemet, G.; Ballestrem, C.; Barry, S.T.; Humphries, M.J. Modulation of FAK and Src adhesion signaling occurs independently of adhesion complex composition. J. Cell Biol. 2016, 212, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Chodniewicz, D.; Klemke, R.L. Regulation of integrin-mediated cellular responses through assembly of a CAS/Crk scaffold. Biochim. Biophys. Acta 2004, 1692, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J. Rho GTPases and cell migration. J. Cell Sci. 2001, 114, 2713–2722. [Google Scholar] [PubMed]
- Huveneers, S.; Danen, E.H. Adhesion signaling—Crosstalk between integrins, Src and Rho. J. Cell Sci. 2009, 122, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Moorman, J.P.; Luu, D.; Wickham, J.; Bobak, D.A.; Hahn, C.S. A balance of signaling by rho family small GTPases Rhoa, Rac1 and Cdc42 coordinates cytoskeletal morphology but not cell survival. Oncogene 1999, 18, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Rottner, K.; Hall, A.; Small, J.V. Interplay between Rac and Rho in the control of substrate contact dynamics. Curr. Biol. 1999, 9, 640–648. [Google Scholar] [CrossRef]
- White, D.P.; Caswell, P.T.; Norman, J.C. αvβ3 and α5β1 integrin recycling pathways dictate downstream Rho kinase signaling to regulate persistent cell migration. J. Cell Biol. 2007, 177, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Huttenlocher, A.; Horwitz, A.R. Integrins in cell migration. Cold Spring Harb. Perspect. Biol. 2011, 3, a005074. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. Mapk signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Yee, K.L.; Weaver, V.M.; Hammer, D.A. Integrin-mediated signalling through the MAP-kinase pathway. IET Syst. Biol. 2008, 2, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Bachelot, C.; Rameh, L.; Parsons, T.; Cantley, L.C. Association of phosphatidylinositol 3-Kinase, via the SH2 domains of p85, with focal adhesion kinase in polyoma middle t-transformed fibroblasts. Biochim. Biophys. Acta 1996, 1311, 45–52. [Google Scholar] [CrossRef]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Ouyang, G.; Bao, S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell. Mol. Med. 2005, 9, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. Mtor signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Soung, Y.H.; Korneeva, N.; Kim, T.H.; Chung, J. The role of c-Src in integrin (α6β4) dependent translational control. BMC Cell Biol. 2013, 14, 49. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Moroishi, T.; Guan, K.L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.G.; Moroishi, T.; Guan, K.L. YAP and TAZ: A nexus for hippo signaling and beyond. Trends Cell Biol. 2015, 25, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Pobbati, A.V.; Hong, W. Emerging roles of TEAD transcription factors and its coactivators in cancers. Cancer Biol. Ther. 2013, 14, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Yin, T.; Yang, Q.; Zhang, J.; Wu, Y.I.; Yu, J. Single-molecule tracking of small GTPase Rac1 uncovers spatial regulation of membrane translocation and mechanism for polarized signaling. Proc. Natl. Acad. Sci. USA 2015, 112, E267–E276. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.D.; Burridge, K. The on-off relationship of Rho and Rac during integrin-mediated adhesion and cell migration. Small GTPases 2014, 5, e27958. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhao, B. Integration of mechanical and chemical signals by YAP and TAZ transcription coactivators. Cell Biosci. 2013, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Legate, K.R.; Fassler, R. Mechanisms that regulate adaptor binding to β-integrin cytoplasmic tails. J. Cell Sci. 2009, 122, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Hannigan, G.; Troussard, A.A.; Dedhar, S. Integrin-linked kinase: A cancer therapeutic target unique among its ILK. Nat. Rev. Cancer 2005, 5, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.; Digicaylioglu, M.; Legg, A.T.; Young, C.E.; Young, S.S.; Barr, A.M.; Fletcher, L.; O’Connor, T.P.; Dedhar, S. Role of integrin-linked kinase in nerve growth factor-stimulated neurite outgrowth. J. Neurosci. 2003, 23, 1638–1648. [Google Scholar] [PubMed]
- Li, G.; Li, Y.Y.; Sun, J.E.; Lin, W.H.; Zhou, R.X. ILK-PI3K/Akt pathway participates in cutaneous wound contraction by regulating fibroblast migration and differentiation to myofibroblast. Lab. Investig. 2016, 96, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H. Pull and push: Talin activation for integrin signaling. Cell Res. 2012, 22, 1512–1514. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Kim, C.; Ginsberg, M.H. Molecular mechanism of inside-out integrin regulation. J. Thromb. Haemost. 2011, 9 (Suppl. 1), 20–25. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.; Legate, K.R.; Zent, R.; Fassler, R. The tail of integrins, talin, and kindlins. Science 2009, 324, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Plow, E.F.; Das, M.; Bialkowska, K.; Sossey-Alaoui, K. Of kindlins and cancer. Discoveries 2016, 4, e59. [Google Scholar] [CrossRef] [PubMed]
- Montanez, E.; Ussar, S.; Schifferer, M.; Bosl, M.; Zent, R.; Moser, M.; Fassler, R. Kindlin-2 controls bidirectional signaling of integrins. Genes Dev. 2008, 22, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Gu, Y.; Begum, R.; Chen, H.; Gao, X.; McGrath, J.A.; Parsons, M.; Song, B. Kindlin-1 regulates keratinocyte electrotaxis. J. Investig. Dermatol. 2016, 136, 2229–2239. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Lin, C.; Yan, Z.; Wang, S.; Zhang, Y.; Wang, S.; Wang, J.; Liu, C.; Chen, J. Kindlin-3 is essential for the resting α4β1 integrin-mediated firm cell adhesion under shear flow conditions. J. Biol. Chem. 2016, 291, 10363–10371. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.M.; Isaacs, H.; Hayflick, J.S.; Rogers, K.A.; Sandig, M. The p110Δ isoform of PI3K differentially regulates β1 and β2 integrin-mediated monocyte adhesion and spreading and modulates diapedesis. Microcirculation 2006, 13, 439–456. [Google Scholar] [CrossRef] [PubMed]
- Riaz, A.; Zeller, K.S.; Johansson, S. Receptor-specific mechanisms regulate phosphorylation of AKT at Ser473: Role of RICTOR in β1 integrin-mediated cell survival. PLoS ONE 2012, 7, e32081. [Google Scholar] [CrossRef] [PubMed]
- Zeller, K.S.; Idevall-Hagren, O.; Stefansson, A.; Velling, T.; Jackson, S.P.; Downward, J.; Tengholm, A.; Johansson, S. PI3-Kinase p110α mediates β1 integrin-induced Akt activation and membrane protrusion during cell attachment and initial spreading. Cell Signal. 2010, 22, 1838–1848. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.L. Cell spreading: The power to simplify. Curr. Biol. 2007, 17, R357–R358. [Google Scholar] [CrossRef] [PubMed]
- Raftopoulou, M.; Hall, A. Cell migration: Rho GTPases lead the way. Dev. Biol. 2004, 265, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, L.; Anderson, K.E.; Jakus, Z.; Kovacs, M.; Voigt, S.; Hawkins, P.T.; Stephens, L.; Mocsai, A.; Vermeren, S. Phosphoinositide 3-OH kinase regulates integrin-dependent processes in neutrophils by signaling through its effector ARAP3. J. Immunol. 2013, 190, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, L.; Anderson, K.E.; Nussbaum, C.; Segonds-Pichon, A.; Margarido, T.; Norton, L.; Ludwig, T.; Sperandio, M.; Hawkins, P.T.; Stephens, L.; et al. The GTPase-activating protein ARAP3 regulates chemotaxis and adhesion-dependent processes in neutrophils. Blood 2011, 118, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, J.G. Multiple roles for Arf6: Sorting, structuring, and signaling at the plasma membrane. J. Biol. Chem. 2003, 278, 41573–41576. [Google Scholar] [CrossRef] [PubMed]
- Arthur, W.T.; Burridge, K. RhoA inactivation by p190RhoGAP regulates cell spreading and migration by promoting membrane protrusion and polarity. Mol. Biol. Cell 2001, 12, 2711–2720. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Subbayya Ithychanda, S.; Qin, J.; Plow, E.F. Mechanisms of talin-dependent integrin signaling and crosstalk. Biochim. Biophys. Acta 2014, 1838, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Theodosiou, M.; Widmaier, M.; Bottcher, R.T.; Rognoni, E.; Veelders, M.; Bharadwaj, M.; Lambacher, A.; Austen, K.; Muller, D.J.; Zent, R.; et al. Kindlin-2 cooperates with talin to activate integrins and induces cell spreading by directly binding paxillin. eLife 2016, 5, e10130. [Google Scholar] [CrossRef] [PubMed]
- Shiojima, I.; Walsh, K. Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev. 2006, 20, 3347–3365. [Google Scholar] [CrossRef] [PubMed]
- Shavlakadze, T.; Chai, J.; Maley, K.; Cozens, G.; Grounds, G.; Winn, N.; Rosenthal, N.; Grounds, M.D. A growth stimulus is needed for IGF-1 to induce skeletal muscle hypertrophy in vivo. J. Cell Sci. 2010, 123, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Pohl, T.L.; Seckinger, A.; Spatz, J.P.; Cavalcanti-Adam, E.A. Regulation of integrin and growth factor signaling in biomaterials for osteodifferentiation. Beilstein J. Org. Chem. 2015, 11, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Ivaska, J.; Heino, J. Cooperation between integrins and growth factor receptors in signaling and endocytosis. Annu. Rev. Cell Dev. Biol. 2011, 27, 291–320. [Google Scholar] [CrossRef] [PubMed]
- Alexi, X.; Berditchevski, F.; Odintsova, E. The effect of cell-ECM adhesion on signalling via the ErbB family of growth factor receptors. Biochem. Soc. Trans. 2011, 39, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.M.; Even-Ram, S. Integrin regulation of growth factor receptors. Nat. Cell Biol. 2002, 4, E75–E76. [Google Scholar] [CrossRef] [PubMed]
- Cruet-Hennequart, S.; Maubant, S.; Luis, J.; Gauduchon, P.; Staedel, C.; Dedhar, S. α(v) integrins regulate cell proliferation through integrin-linked kinase (ILK) in ovarian cancer cells. Oncogene 2003, 22, 1688–1702. [Google Scholar] [CrossRef] [PubMed]
- Lossner, D.; Abou-Ajram, C.; Benge, A.; Reuning, U. Integrin αvβ3 mediates upregulation of epidermal growth-factor receptor expression and activity in human ovarian cancer cells. Int. J. Biochem. Cell Biol. 2008, 40, 2746–2761. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Serini, G.; Cecchini, G.; Besati, C.; Ambesi-Impiombato, F.S.; Marchisio, P.C.; de Filippi, R. Growth factor-dependent activation of αvβ3 integrin in normal epithelial cells: Implications for tumor invasion. J. Cell Biol. 1998, 142, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.A.; Brunie, L.; Bacher, A.S.; Kessler, H.; Gottschalk, K.E.; Reuning, U. Cytoplasmic salt bridge formation in integrin αvss3 stabilizes its inactive state affecting integrin-mediated cell biological effects. Cell Signal. 2014, 26, 2493–2503. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Takada, Y.K.; Takada, Y. Insulin-like growth factor (IGF) signaling requires αvβ3-IGF1-IGF type 1 receptor (IGF1R) ternary complex formation in anchorage independence, and the complex formation does not require IGF1R and Src activation. J. Biol. Chem. 2013, 288, 3059–3069. [Google Scholar] [CrossRef] [PubMed]
- Freeman, K.W.; Gangula, R.D.; Welm, B.E.; Ozen, M.; Foster, B.A.; Rosen, J.M.; Ittmann, M.; Greenberg, N.M.; Spencer, D.M. Conditional activation of fibroblast growth factor receptor (FGFR) 1, but not FGFR2, in prostate cancer cells leads to increased osteopontin induction, extracellular signal-regulated kinase activation, and in vivo proliferation. Cancer Res. 2003, 63, 6237–6243. [Google Scholar] [PubMed]
- Mitra, A.K.; Sawada, K.; Tiwari, P.; Mui, K.; Gwin, K.; Lengyel, E. Ligand-independent activation of c-Met by fibronectin and α(5)β(1)-integrin regulates ovarian cancer invasion and metastasis. Oncogene 2011, 30, 1566–1576. [Google Scholar] [CrossRef] [PubMed]
- Morello, V.; Cabodi, S.; Sigismund, S.; Camacho-Leal, M.P.; Repetto, D.; Volante, M.; Papotti, M.; Turco, E.; Defilippi, P. β1 integrin controls EGFR signaling and tumorigenic properties of lung cancer cells. Oncogene 2011, 30, 4087–4096. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, B.L.; Chen, M.; Knifley, T.; Davis, K.A.; Harrison, S.M.; Stewart, R.L.; O’Connor, K.L. Integrin α6β4 promotes autocrine epidermal growth factor receptor (EGFR) signaling to stimulate migration and invasion toward hepatocyte growth factor (HGF). J. Biol. Chem. 2015, 290, 27228–27238. [Google Scholar] [CrossRef] [PubMed]
- Gilcrease, M.Z.; Zhou, X.; Lu, X.; Woodward, W.A.; Hall, B.E.; Morrissey, P.J. α6β4 integrin crosslinking induces EGFR clustering and promotes EGF-mediated Rho activation in breast cancer. J. Exp. Clin. Cancer Res. 2009, 28, 67. [Google Scholar] [CrossRef] [PubMed]
- Ricono, J.M.; Huang, M.; Barnes, L.A.; Lau, S.K.; Weis, S.M.; Schlaepfer, D.D.; Hanks, S.K.; Cheresh, D.A. Specific cross-talk between epidermal growth factor receptor and integrin αvβ5 promotes carcinoma cell invasion and metastasis. Cancer Res. 2009, 69, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.C.; Hogg, N. Integrins take partners: Cross-talk between integrins and other membrane receptors. Trends Cell Biol. 1998, 8, 390–396. [Google Scholar] [CrossRef]
- Petty, H.R.; Worth, R.G.; Todd, R.F., 3rd. Interactions of integrins with their partner proteins in leukocyte membranes. Immunol. Res. 2002, 25, 75–95. [Google Scholar] [CrossRef] [PubMed]
- Cantor, J.M.; Ginsberg, M.H.; Rose, D.M. Integrin-associated proteins as potential therapeutic targets. Immunol. Rev. 2008, 223, 236–251. [Google Scholar] [CrossRef] [PubMed]
- Deves, R.; Boyd, C.A. Surface antigen CD98(4F2): Not a single membrane protein, but a family of proteins with multiple functions. J. Membr. Biol. 2000, 173, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Prager, G.W.; Feral, C.C.; Kim, C.; Han, J.; Ginsberg, M.H. CD98hc (SLC3A2) interaction with the integrin β subunit cytoplasmic domain mediates adhesive signaling. J. Biol. Chem. 2007, 282, 24477–24484. [Google Scholar] [CrossRef] [PubMed]
- Cantor, J.M.; Ginsberg, M.H. CD98 at the crossroads of adaptive immunity and cancer. J. Cell Sci. 2012, 125, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Collis, E.A.; Mackinnon, A.C.; Simpson, K.J.; Haslett, C.; Zent, R.; Ginsberg, M.; Sethi, T. CD98hc (SLC3A2) interaction with β 1 integrins is required for transformation. J. Biol. Chem. 2004, 279, 54731–54741. [Google Scholar] [CrossRef] [PubMed]
- Poettler, M.; Unseld, M.; Braemswig, K.; Haitel, A.; Zielinski, C.C.; Prager, G.W. CD98HC (SLC3A2) drives integrin-dependent renal cancer cell behavior. Mol. Cancer 2013, 12, 169. [Google Scholar] [CrossRef] [PubMed]
- Bianconi, D.H.M.; Gleiss, A.; Unseld, M.; Weigl, R.; Schindl, M.; Scheithauer, W.; Zielienski, C.; Prager, G. Functional role of 4F2hc in pancreatic ductal adenocarcinoma. Pancreas 2016, 45, 1498. [Google Scholar]
- Kaira, K.; Oriuchi, N.; Imai, H.; Shimizu, K.; Yanagitani, N.; Sunaga, N.; Hisada, T.; Kawashima, O.; Kamide, Y.; Ishizuka, T.; et al. CD98 expression is associated with poor prognosis in resected non-small-cell lung cancer with lymph node metastases. Ann. Surg. Oncol. 2009, 16, 3473–3481. [Google Scholar] [CrossRef] [PubMed]
- Kaira, K.; Ohde, Y.; Endo, M.; Nakagawa, K.; Okumura, T.; Takahashi, T.; Murakami, H.; Tsuya, A.; Nakamura, Y.; Naito, T.; et al. Expression of 4F2hc (CD98) in pulmonary neuroendocrine tumors. Oncol. Rep. 2011, 26, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Kaira, K.; Takahashi, T.; Abe, M.; Akamatsu, H.; Nakagawa, K.; Ohde, Y.; Okumura, T.; Murakami, H.; Tsuya, A.; Nakamura, Y.; et al. CD98 expression is associated with the grade of malignancy in thymic epithelial tumors. Oncol. Rep. 2010, 24, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Rietbergen, M.M.; Martens-de Kemp, S.R.; Bloemena, E.; Witte, B.I.; Brink, A.; Baatenburg de Jong, R.J.; Leemans, C.R.; Braakhuis, B.J.; Brakenhoff, R.H. Cancer stem cell enrichment marker CD98: A prognostic factor for survival in patients with human papillomavirus-positive oropharyngeal cancer. Eur. J. Cancer 2014, 50, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Sakata, T.; Ferdous, G.; Tsuruta, T.; Satoh, T.; Baba, S.; Muto, T.; Ueno, A.; Kanai, Y.; Endou, H.; Okayasu, I. L-type amino-acid transporter 1 as a novel biomarker for high-grade malignancy in prostate cancer. Pathol. Int. 2009, 59, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Ichinoe, M.; Mikami, T.; Yoshida, T.; Igawa, I.; Tsuruta, T.; Nakada, N.; Anzai, N.; Suzuki, Y.; Endou, H.; Okayasu, I. High expression of L-type amino-acid transporter 1 (LAT1) in gastric carcinomas: Comparison with non-cancerous lesions. Pathol. Int. 2011, 61, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, M.; Kaira, K.; Ohshima, Y.; Ishioka, N.S.; Shino, M.; Sakakura, K.; Takayasu, Y.; Takahashi, K.; Tominaga, H.; Oriuchi, N.; et al. Prognostic significance of amino-acid transporter expression (LAT1, ASCT2, and xCT) in surgically resected tongue cancer. Br. J. Cancer 2014, 110, 2506–2513. [Google Scholar] [CrossRef] [PubMed]
- Prager, G.W.; Poettler, M.; Schmidinger, M.; Mazal, P.R.; Susani, M.; Zielinski, C.C.; Haitel, A. CD98hc (SLC3A2), a novel marker in renal cell cancer. Eur. J. Clin. Investig. 2009, 39, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Boudjadi, S.; Carrier, J.C.; Groulx, J.F.; Beaulieu, J.F. Integrin α1β1 expression is controlled by c-MYC in colorectal cancer cells. Oncogene 2016, 35, 1671–1678. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, A.; Wary, K.K.; Giancotti, F.G.; Gardner, H.A. Integrin α1β1 mediates a unique collagen-dependent proliferation pathway in vivo. J. Cell Biol. 1998, 142, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Moro, L.; Venturino, M.; Bozzo, C.; Silengo, L.; Altruda, F.; Beguinot, L.; Tarone, G.; Defilippi, P. Integrins induce activation of EGF receptor: Role in MAP kinase induction and adhesion-dependent cell survival. EMBO J. 1998, 17, 6622–6632. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Whiting, C.; Borza, C.; Hu, W.; Mont, S.; Bulus, N.; Zhang, M.Z.; Harris, R.C.; Zent, R.; Pozzi, A. Integrin α1β1 regulates epidermal growth factor receptor activation by controlling peroxisome proliferator-activated receptor gamma-dependent caveolin-1 expression. Mol. Cell. Biol. 2010, 30, 3048–3058. [Google Scholar] [CrossRef] [PubMed]
- Bartolome, R.A.; Barderas, R.; Torres, S.; Fernandez-Acenero, M.J.; Mendes, M.; Garcia-Foncillas, J.; Lopez-Lucendo, M.; Casal, J.I. Cadherin-17 interacts with α2β1 integrin to regulate cell proliferation and adhesion in colorectal cancer cells causing liver metastasis. Oncogene 2014, 33, 1658–1669. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lin, Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol. Sin. 2008, 29, 1275–1288. [Google Scholar] [CrossRef] [PubMed]
- Ezoe, K.; Horikoshi, T. Tumor necrosis factor-α increased the integrin α2β1 expression and cell attachment to type I collagen in human dermal fibroblasts. Biochem. Biophys. Res. Commun. 1993, 192, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Goel, H.L.; Breen, M.; Zhang, J.; Das, I.; Aznavoorian-Cheshire, S.; Greenberg, N.M.; Elgavish, A.; Languino, L.R. β1a integrin expression is required for type 1 insulin-like growth factor receptor mitogenic and transforming activities and localization to focal contacts. Cancer Res. 2005, 65, 6692–6700. [Google Scholar] [CrossRef] [PubMed]
- Goel, H.L.; Underwood, J.M.; Nickerson, J.A.; Hsieh, C.C.; Languino, L.R. β1 integrins mediate cell proliferation in three-dimensional cultures by regulating expression of the sonic hedgehog effector protein, GLI1. J. Cell. Physiol. 2010, 224, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Otte, A.; Mandel, K.; Reinstrom, G.; Hass, R. Abolished adherence alters signaling pathways in phorbol ester-induced human U937 cells. Cell Commun. Signal. 2011, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Sun, X.; Ritzenthaler, J.D.; Roman, J. Fish oil inhibits human lung carcinoma cell growth by suppressing integrin-linked kinase. Mol. Cancer Res. 2009, 7, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Abramovitch, S.; Dahan-Bachar, L.; Sharvit, E.; Weisman, Y.; Ben Tov, A.; Brazowski, E.; Reif, S. Vitamin D inhibits proliferation and profibrotic marker expression in hepatic stellate cells and decreases thioacetamide-induced liver fibrosis in rats. Gut 2011, 60, 1728–1737. [Google Scholar] [CrossRef] [PubMed]
- Larriba, M.J.; Garcia de Herreros, A.; Munoz, A. Vitamin D and the epithelial to mesenchymal transition. Stem Cells Int. 2016, 2016, 6213872. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Mula, R.V.; Li, J.; Weigel, N.L.; Falzon, M. PTHrP contributes to the anti-proliferative and integrin α6β4-regulating effects of 1,25-dihydroxyvitamin D(3). Steroids 2007, 72, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.H.; Yang, R.S.; Hou, S.M.; Tang, C.H. TNF-α increases αvβ3 integrin expression and migration in human chondrosarcoma cells. J. Cell. Physiol. 2011, 226, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Fisher, H.W.; Yeh, J. Contact inhibition in colony formation. Science 1967, 155, 581–582. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, W.; Kakunaga, S.; Itoh, S.; Shingai, T.; Takekuni, K.; Satoh, K.; Inoue, Y.; Hamaguchi, A.; Morimoto, K.; Takeuchi, M.; et al. Tage4/Nectin-like molecule-5 heterophilically trans-interacts with cell adhesion molecule Nectin-3 and enhances cell migration. J. Biol. Chem. 2003, 278, 28167–28172. [Google Scholar] [CrossRef] [PubMed]
- Ogita, H.; Takai, Y. Nectins and nectin-like molecules: Roles in cell adhesion, polarization, movement, and proliferation. IUBMB Life 2006, 58, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Mayor, R.; Carmona-Fontaine, C. Keeping in touch with contact inhibition of locomotion. Trends Cell Biol. 2010, 20, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Astin, J.W.; Batson, J.; Kadir, S.; Charlet, J.; Persad, R.A.; Gillatt, D.; Oxley, J.D.; Nobes, C.D. Competition amongst Eph receptors regulates contact inhibition of locomotion and invasiveness in prostate cancer cells. Nat. Cell Biol. 2010, 12, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- Roycroft, A.; Mayor, R. Molecular basis of contact inhibition of locomotion. Cell. Mol. Life Sci. 2016, 73, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Chiasson-MacKenzie, C.; Morris, Z.S.; Baca, Q.; Morris, B.; Coker, J.K.; Mirchev, R.; Jensen, A.E.; Carey, T.; Stott, S.L.; Golan, D.E.; et al. NF2/merlin mediates contact-dependent inhibition of EGFR mobility and internalization via cortical actomyosin. J. Cell Biol. 2015, 211, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Obremski, V.J.; Hall, A.M.; Fernandez-Valle, C. Merlin, the neurofibromatosis type 2 gene product, and β1 integrin associate in isolated and differentiating Schwann cells. J. Neurobiol. 1998, 37, 487–501. [Google Scholar] [CrossRef]
- Lopez-Lago, M.A.; Okada, T.; Murillo, M.M.; Socci, N.; Giancotti, F.G. Loss of the tumor suppressor gene NF2, encoding merlin, constitutively activates integrin-dependent mTORC1 signaling. Mol. Cell. Biol. 2009, 29, 4235–4249. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Valle, C.; Tang, Y.; Ricard, J.; Rodenas-Ruano, A.; Taylor, A.; Hackler, E.; Biggerstaff, J.; Iacovelli, J. Paxillin binds schwannomin and regulates its density-dependent localization and effect on cell morphology. Nat. Genet. 2002, 31, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, R.; Poernbacher, I.; Buser, N.; Hafen, E.; Stocker, H. The WW domain protein Kibra acts upstream of Hippo in Drosophila. Dev. Cell 2010, 18, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef] [PubMed]
- Gumbiner, B.M.; Kim, N.G. The Hippo-YAP signaling pathway and contact inhibition of growth. J. Cell Sci. 2014, 127, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Goel, H.L.; Gao, H.; Pursell, B.; Shultz, L.D.; Greiner, D.L.; Ingerpuu, S.; Patarroyo, M.; Cao, S.; Lim, E.; et al. A laminin 511 matrix is regulated by TAZ and functions as the ligand for the α6bβ1 integrin to sustain breast cancer stem cells. Genes Dev. 2015, 29, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the light: The growing complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.M.; Truong, T.N.; Schwartz, M.A. Integrins regulate the apoptotic response to DNA damage through modulation of p53. Proc. Natl. Acad. Sci. USA 2002, 99, 3627–3632. [Google Scholar] [CrossRef] [PubMed]
- Bachelder, R.E.; Marchetti, A.; Falcioni, R.; Soddu, S.; Mercurio, A.M. Activation of p53 function in carcinoma cells by the α6β4 integrin. J. Biol. Chem. 1999, 274, 20733–20737. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Janouskova, H.; Maglott, A.; Leger, D.Y.; Bossert, C.; Noulet, F.; Guerin, E.; Guenot, D.; Pinel, S.; Chastagner, P.; Plenat, F.; et al. Integrin α5β1 plays a critical role in resistance to temozolomide by interfering with the p53 pathway in high-grade glioma. Cancer Res. 2012, 72, 3463–3470. [Google Scholar] [CrossRef] [PubMed]
- Iwanicki, M.P.; Chen, H.Y.; Iavarone, C.; Zervantonakis, I.K.; Muranen, T.; Novak, M.; Ince, T.A.; Drapkin, R.; Brugge, J.S. Mutant p53 regulates ovarian cancer transformed phenotypes through autocrine matrix deposition. JCI Insight 2016, 1, e86829. [Google Scholar] [CrossRef] [PubMed]
- Velez-delValle, C.; Marsch-Moreno, M.; Castro-Munozledo, F.; Galvan-Mendoza, I.J.; Kuri-Harcuch, W. Epithelial cell migration requires the interaction between the vimentin and keratin intermediate filaments. Sci. Rep. 2016, 6, 24389. [Google Scholar] [CrossRef] [PubMed]
- Talmadge, J.E.; Fidler, I.J. AACR centennial series: The biology of cancer metastasis: Historical perspective. Cancer Res. 2010, 70, 5649–5669. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7. [Google Scholar] [CrossRef] [PubMed]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin. Transl. Med. 2015, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.; Marshall, J.F. The role of integrins in TGFβ activation in the tumour stroma. Cell Tissue Res. 2016, 365, 657–673. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.G.; Vignjevic, D.M. Modes of cancer cell invasion and the role of the microenvironment. Curr. Opin. Cell Biol. 2015, 36, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Paolillo, M.; Serra, M.; Schinelli, S. Integrins in glioblastoma: Still an attractive target? Pharmacol. Res. 2016, 113, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin α v β 6 binds and activates latent TGF β 1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef]
- Ozawa, A.; Sato, Y.; Imabayashi, T.; Uemura, T.; Takagi, J.; Sekiguchi, K. Molecular basis of the ligand binding specificity of αvβ8 integrin. J. Biol. Chem. 2016, 291, 11551–11565. [Google Scholar] [CrossRef] [PubMed]
- Tatler, A.L.; Goodwin, A.T.; Gbolahan, O.; Saini, G.; Porte, J.; John, A.E.; Clifford, R.L.; Violette, S.M.; Weinreb, P.H.; Parfrey, H.; et al. Amplification of TGFβ induced ITGB6 gene transcription may promote pulmonary fibrosis. PLoS ONE 2016, 11, e0158047. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.S.; Harpel, J.G.; Giancotti, F.G.; Rifkin, D.B. Interactions between growth factors and integrins: Latent forms of transforming growth factor-β are ligands for the integrin αvβ1. Mol. Biol. Cell 1998, 9, 2627–2638. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.Y.; Chiao, C.C.; Kuo, W.Y.; Hsiao, Y.C.; Chen, Y.J.; Wei, Y.Y.; Lai, T.H.; Fong, Y.C.; Tang, C.H. TGF-β1 increases motility and αvβ3 integrin up-regulation via PI3K, Akt and NF-kappab-dependent pathway in human chondrosarcoma cells. Biochem. Pharmacol. 2008, 75, 1292–1301. [Google Scholar] [CrossRef] [PubMed]
- Wendt, M.K.; Smith, J.A.; Schiemann, W.P. Transforming growth factor-β-induced epithelial-mesenchymal transition facilitates epidermal growth factor-dependent breast cancer progression. Oncogene 2010, 29, 6485–6498. [Google Scholar] [CrossRef] [PubMed]
- Wendt, M.K.; Taylor, M.A.; Schiemann, B.J.; Sossey-Alaoui, K.; Schiemann, W.P. Fibroblast growth factor receptor splice variants are stable markers of oncogenic transforming growth factor β1 signaling in metastatic breast cancers. Breast Cancer Res. 2014, 16, R24. [Google Scholar] [CrossRef] [PubMed]
- Galliher, A.J.; Schiemann, W.P. Beta3 integrin and Src facilitate transforming growth factor-β mediated induction of epithelial-mesenchymal transition in mammary epithelial cells. Breast Cancer Res. 2006, 8, R42. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.S.; Tan, L.; Smith, A.; Gray, N.S.; Wendt, M.K. Covalent targeting of fibroblast growth factor receptor inhibits metastatic breast cancer. Mol. Cancer Ther. 2016, 15, 2096–2106. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Kodaira, M.; Ito, A.; Okazaki, M.; Kawaguchi, N.; Hamada, Y.; Takada, Y.; Matsuura, N. Enhanced expression of integrin αvβ3 induced by TGF-β is required for the enhancing effect of fibroblast growth factor 1 (FGF1) in TGF-β-induced epithelial-mesenchymal transition (EMT) in mammary epithelial cells. PLoS ONE 2015, 10, e0137486. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K.; Kusanagi, K.; Inoue, H. Divergence and convergence of TGF-β/BMP signaling. J. Cell. Physiol. 2001, 187, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Maegdefrau, U.; Bosserhoff, A.K. BMP activated Smad signaling strongly promotes migration and invasion of hepatocellular carcinoma cells. Exp. Mol. Pathol. 2012, 92, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, Y.; Hanyu, A.; Kanda, H.; Ishikawa, Y.; Akiyama, F.; Iwase, T.; Ogata, E.; Ehata, S.; Miyazono, K.; Imamura, T. Bone morphogenetic protein signaling enhances invasion and bone metastasis of breast cancer cells through Smad pathway. Oncogene 2008, 27, 6322–6333. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.; Yang, S.T.; Lin, C.Y.; Hsu, C.J.; Tsai, C.H.; Su, J.L.; Tang, C.H. BMP-7 enhances cell migration and αvβ3 integrin expression via a c-Src-dependent pathway in human chondrosarcoma cells. PLoS ONE 2014, 9, e112636. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Lesperance, J.; Seguin, L.; Gozo, M.; Kato, S.; Franovic, A.; Yebra, M.; Shattil, S.J.; Cheresh, D.A. Integrin αvβ3 drives slug activation and stemness in the pregnant and neoplastic mammary gland. Dev. Cell 2014, 30, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Cantor, D.; Slapetova, I.; Kan, A.; McQuade, L.R.; Baker, M.S. Overexpression of αvβ6 integrin alters the colorectal cancer cell proteome in favor of elevated proliferation and a switching in cellular adhesion that increases invasion. J. Proteome Res. 2013, 12, 2477–2490. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Song, S.H.; Kim, H.P.; Han, S.W.; Yi, E.C.; Kim, T.Y. Dynamic cohesin-mediated chromatin architecture controls epithelial-mesenchymal plasticity in cancer. EMBO Rep. 2016, 17, 1343–1359. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. Integrin β1-focal adhesion kinase signaling directs the proliferation of metastatic cancer cells disseminated in the lungs. Proc. Natl. Acad. Sci. USA 2009, 106, 10290–10295. [Google Scholar] [CrossRef] [PubMed]
- Knuchel, S.; Anderle, P.; Werfelli, P.; Diamantis, E.; Ruegg, C. Fibroblast surface-associated FGF-2 promotes contact-dependent colorectal cancer cell migration and invasion through FGFR-SRC signaling and integrin αvβ5-mediated adhesion. Oncotarget 2015, 6, 14300–14317. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Pansino, F.; Clyde, R.; Murthi, P.; Quinn, M.A.; Rice, G.E.; Agrez, M.V.; Mok, S.; Baker, M.S. Overexpression of α(v)β6 integrin in serous epithelial ovarian cancer regulates extracellular matrix degradation via the plasminogen activation cascade. Carcinogenesis 2002, 23, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.J.; Lewis, M.P.; Hart, I.R.; Marshall, J.F.; Speight, P.M. αvβ6 integrin promotes invasion of squamous carcinoma cells through up-regulation of matrix metalloproteinase-9. Int. J. Cancer 2001, 92, 641–650. [Google Scholar] [CrossRef]
- Dutta, A.; Li, J.; Fedele, C.; Sayeed, A.; Singh, A.; Violette, S.M.; Manes, T.D.; Languino, L.R. Alphavβ6 integrin is required for TGFβ1-mediated matrix metalloproteinase2 expression. Biochem. J. 2015, 466, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Li, J.; Lu, H.; Akech, J.; Pratap, J.; Wang, T.; Zerlanko, B.J.; FitzGerald, T.J.; Jiang, Z.; Birbe, R.; et al. Integrin αvβ6 promotes an osteolytic program in cancer cells by upregulating MMP2. Cancer Res. 2014, 74, 1598–1608. [Google Scholar] [CrossRef] [PubMed]
- Del Rosso, M.; Fibbi, G.; Pucci, M.; D’Alessio, S.; del Rosso, A.; Magnelli, L.; Chiarugi, V. Multiple pathways of cell invasion are regulated by multiple families of serine proteases. Clin. Exp. Metastasis 2002, 19, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Stoppelli, M.P. The plasminogen activation system in cell invasion. In Madame Curie Bioscience Database; Landes Bioscience: Austin, TX, USA, 2000–2013. [Google Scholar]
- Del Rosso, M. Upar in angiogenesis regulation. Blood 2011, 117, 3941–3943. [Google Scholar] [CrossRef] [PubMed]
- Del Rosso, M.; Margheri, F.; Serrati, S.; Chilla, A.; Laurenzana, A.; Fibbi, G. The urokinase receptor system, a key regulator at the intersection between inflammation, immunity, and coagulation. Curr. Pharm. Des. 2011, 17, 1924–1943. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Mizukami, I.; Todd, R.F., 3rd; Petty, H.R. Urokinase-type plasminogen activator receptors associate with β1 and β3 integrins of fibrosarcoma cells: Dependence on extracellular matrix components. Cancer Res. 1997, 57, 1682–1689. [Google Scholar] [PubMed]
- Margheri, F.; Luciani, C.; Taddei, M.L.; Giannoni, E.; Laurenzana, A.; Biagioni, A.; Chilla, A.; Chiarugi, P.; Fibbi, G.; del Rosso, M. The receptor for urokinase-plasminogen activator (uPAR) controls plasticity of cancer cell movement in mesenchymal and amoeboid migration style. Oncotarget 2014, 5, 1538–1553. [Google Scholar] [CrossRef] [PubMed]
- Aguirre Ghiso, J.A.; Kovalski, K.; Ossowski, L. Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J. Cell Biol. 1999, 147, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular senescence in cancer and aging. Cell 2007, 130, 223–233. [Google Scholar] [CrossRef] [PubMed]
- McClintock, B. The stability of broken ends of chromosomes in Zea mays. Genetics 1941, 26, 234–282. [Google Scholar] [PubMed]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Collins, K.; Mitchell, J.R. Telomerase in the human organism. Oncogene 2002, 21, 564–579. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Wright, W.E.; Piatyszek, M.A.; Rainey, W.E.; Byrd, W.; Shay, J.W. Telomerase activity in human germline and embryonic tissues and cells. Dev. Genet. 1996, 18, 173–179. [Google Scholar] [CrossRef]
- Riou, L.; Bastos, H.; Lassalle, B.; Coureuil, M.; Testart, J.; Boussin, F.D.; Allemand, I.; Fouchet, P. The telomerase activity of adult mouse testis resides in the spermatogonial α6-integrin-positive side population enriched in germinal stem cells. Endocrinology 2005, 146, 3926–3932. [Google Scholar] [CrossRef] [PubMed]
- Ponnala, S.; Chetty, C.; Veeravalli, K.K.; Dinh, D.H.; Klopfenstein, J.D.; Rao, J.S. MMP-9 silencing regulates hTERT expression via β1 integrin-mediated FAK signaling and induces senescence in glioma xenograft cells. Cell Signal. 2011, 23, 2065–2075. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Crothers, J.; Haqq, C.M.; Blackburn, E.H. Cellular and gene expression responses involved in the rapid growth inhibition of human cancer cells by RNA interference-mediated depletion of telomerase RNA. J. Biol. Chem. 2005, 280, 23709–23717. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M. (Ed.) Angiogenic Switch. In Encyclopedia of Cancer; Springer: Berlin/Heidelberg, Germany, 2012; p. 186.
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Poettler, M.; Unseld, M.; Mihaly-Bison, J.; Uhrin, P.; Koban, F.; Binder, B.R.; Zielinski, C.C.; Prager, G.W. The urokinase receptor (CD87) represents a central mediator of growth factor-induced endothelial cell migration. Thromb. Haemost. 2012, 108, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Demircioglu, F.; Hodivala-Dilke, K. Alphavβ3 integrin and tumour blood vessels-learning from the past to shape the future. Curr. Opin. Cell Biol. 2016, 42, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Avraamides, C.J.; Garmy-Susini, B.; Varner, J.A. Integrins in angiogenesis and lymphangiogenesis. Nat. Rev. Cancer 2008, 8, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Bell, K.; Mousa, S.A.; Varner, J.A. Regulation of angiogenesis in vivo by ligation of integrin α5β1 with the central cell-binding domain of fibronectin. Am. J. Pathol. 2000, 156, 1345–1362. [Google Scholar] [CrossRef]
- Sheppard, D. Endothelial integrins and angiogenesis: Not so simple anymore. J. Clin. Investig. 2002, 110, 913–914. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Ye, G.; Li, J.; Wang, Y. Recent advance in molecular angiogenesis in glioblastoma: The challenge and hope for anti-angiogenic therapy. Brain Tumor Pathol. 2015, 32, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef] [PubMed]
- Wenger, R.H.; Stiehl, D.P.; Camenisch, G. Integration of oxygen signaling at the consensus HRE. Sci. STKE 2005, 2005, re12. [Google Scholar] [CrossRef] [PubMed]
- Cowden Dahl, K.D.; Robertson, S.E.; Weaver, V.M.; Simon, M.C. Hypoxia-inducible factor regulates αvβ3 integrin cell surface expression. Mol. Biol. Cell 2005, 16, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Keely, S.; Glover, L.E.; MacManus, C.F.; Campbell, E.L.; Scully, M.M.; Furuta, G.T.; Colgan, S.P. Selective induction of integrin β1 by hypoxia-inducible factor: Implications for wound healing. FASEB J. 2009, 23, 1338–1346. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.L.; Schwab, L.P.; Krutilina, R.; Parke, D.N.; Sethuraman, A.; Hoogewijs, D.; Schorg, A.; Gotwald, L.; Fan, M.; Wenger, R.H.; et al. ITGA6 is directly regulated by hypoxia-inducible factors and enriches for cancer stem cell activity and invasion in metastatic breast cancer models. Mol. Cancer 2016, 15, 26. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.C.; Chuang, H.C.; Salunke, S.B.; Kulp, S.K.; Chen, C.S. A novel HIF-1α-integrin-linked kinase regulatory loop that facilitates hypoxia-induced HIF-1α expression and epithelial-mesenchymal transition in cancer cells. Oncotarget 2015, 6, 8271–8285. [Google Scholar] [CrossRef] [PubMed]
- Skuli, N.; Monferran, S.; Delmas, C.; Favre, G.; Bonnet, J.; Toulas, C.; Cohen-Jonathan Moyal, E. Alphavβ3/αvβ5 integrins-FAK-RhoB: A novel pathway for hypoxia regulation in glioblastoma. Cancer Res. 2009, 69, 3308–3316. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Shen, S.M.; Zhao, X.Y.; Chen, G.Q. Targeted genes and interacting proteins of hypoxia inducible factor-1. Int. J. Biochem. Mol. Biol. 2012, 3, 165–178. [Google Scholar] [PubMed]
- Han, Z.B.; Ren, H.; Zhao, H.; Chi, Y.; Chen, K.; Zhou, B.; Liu, Y.J.; Zhang, L.; Xu, B.; Liu, B.; et al. Hypoxia-inducible factor (HIF)-1 α directly enhances the transcriptional activity of stem cell factor (SCF) in response to hypoxia and epidermal growth factor (EGF). Carcinogenesis 2008, 29, 1853–1861. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.D.; Hackett, S.F.; Hirota, K.; Oshima, Y.; Cai, Z.; Berg-Dixon, S.; Rowan, A.; Yan, Z.; Campochiaro, P.A.; Semenza, G.L. Cell type-specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ. Res. 2003, 93, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Nikitenko, L.L.; Smith, D.M.; Bicknell, R.; Rees, M.C. Transcriptional regulation of the CRLR gene in human microvascular endothelial cells by hypoxia. FASEB J. 2003, 17, 1499–1501. [Google Scholar] [CrossRef] [PubMed]
- Goel, H.L.; Mercurio, A.M. VEGF targets the tumour cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Vlahakis, N.E.; Young, B.A.; Atakilit, A.; Hawkridge, A.E.; Issaka, R.B.; Boudreau, N.; Sheppard, D. Integrin α9β1 directly binds to vascular endothelial growth factor (VEGF)-a and contributes to VEGF-a-induced angiogenesis. J. Biol. Chem. 2007, 282, 15187–15196. [Google Scholar] [CrossRef] [PubMed]
- Vachon, P.H. Integrin signaling, cell survival, and anoikis: Distinctions, differences, and differentiation. J. Signal Transduct. 2011, 2011, 738137. [Google Scholar] [CrossRef] [PubMed]
- Frisch, S.M.; Ruoslahti, E. Integrins and anoikis. Curr. Opin. Cell Biol. 1997, 9, 701–706. [Google Scholar] [CrossRef]
- Benoit, Y.D.; Groulx, J.F.; Gagne, D.; Beaulieu, J.F. RGD-dependent epithelial cell-matrix interactions in the human intestinal crypt. J. Signal Transduct. 2012, 2012, 248759. [Google Scholar] [CrossRef] [PubMed]
- Benoit, Y.D.; Lussier, C.; Ducharme, P.A.; Sivret, S.; Schnapp, L.M.; Basora, N.; Beaulieu, J.F. Integrin α8β1 regulates adhesion, migration and proliferation of human intestinal crypt cells via a predominant RhoA/ROCK-dependent mechanism. Biol. Cell 2009, 101, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Stupack, D.G.; Puente, X.S.; Boutsaboualoy, S.; Storgard, C.M.; Cheresh, D.A. Apoptosis of adherent cells by recruitment of caspase-8 to unligated integrins. J. Cell Biol. 2001, 155, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Lotti, R.; Marconi, A.; Truzzi, F.; Dallaglio, K.; Gemelli, C.; Borroni, R.G.; Palazzo, E.; Pincelli, C. A previously unreported function of β(1)B integrin isoform in caspase-8-dependent integrin-mediated keratinocyte death. J. Investig. Dermatol. 2010, 130, 2569–2577. [Google Scholar] [CrossRef] [PubMed]
- Buchheit, C.L.; Weigel, K.J.; Schafer, Z.T. Cancer cell survival during detachment from the ECM: Multiple barriers to tumour progression. Nat. Rev. Cancer 2014, 14, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Teitz, T.; Wei, T.; Valentine, M.B.; Vanin, E.F.; Grenet, J.; Valentine, V.A.; Behm, F.G.; Look, A.T.; Lahti, J.M.; Kidd, V.J. Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat. Med. 2000, 6, 529–535. [Google Scholar] [PubMed]
- Helfer, B.; Boswell, B.C.; Finlay, D.; Cipres, A.; Vuori, K.; Bong Kang, T.; Wallach, D.; Dorfleutner, A.; Lahti, J.M.; Flynn, D.C.; et al. Caspase-8 promotes cell motility and calpain activity under nonapoptotic conditions. Cancer Res. 2006, 66, 4273–4278. [Google Scholar] [CrossRef] [PubMed]
- Barbero, S.; Mielgo, A.; Torres, V.; Teitz, T.; Shields, D.J.; Mikolon, D.; Bogyo, M.; Barila, D.; Lahti, J.M.; Schlaepfer, D.; et al. Caspase-8 association with the focal adhesion complex promotes tumor cell migration and metastasis. Cancer Res. 2009, 69, 3755–3763. [Google Scholar] [CrossRef] [PubMed]
- Alanko, J.; Mai, A.; Jacquemet, G.; Schauer, K.; Kaukonen, R.; Saari, M.; Goud, B.; Ivaska, J. Integrin endosomal signalling suppresses anoikis. Nat. Cell Biol. 2015, 17, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Alanko, J.; Ivaska, J. Integrin “endoadhesome” signaling suppresses anoikis. Cell Cycle 2016, 15, 605–606. [Google Scholar] [CrossRef] [PubMed]
- Guadamillas, M.C.; Cerezo, A.; Del Pozo, M.A. Overcoming anoikis—Pathways to anchorage-independent growth in cancer. J. Cell Sci. 2011, 124, 3189–3197. [Google Scholar] [CrossRef] [PubMed]
- Plantefaber, L.C.; Hynes, R.O. Changes in integrin receptors on oncogenically transformed cells. Cell 1989, 56, 281–290. [Google Scholar] [CrossRef]
- Guo, W.; Giancotti, F.G. Integrin signalling during tumour progression. Nat. Rev. Mol. Cell Biol. 2004, 5, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, A.M.; Reisfeld, R.A.; Cheresh, D.A. Integrin α v β 3 rescues melanoma cells from apoptosis in three-dimensional dermal collagen. Proc. Natl. Acad. Sci. USA 1994, 91, 8856–8860. [Google Scholar] [CrossRef] [PubMed]
- Milner, R.; Hung, S.; Erokwu, B.; Dore-Duffy, P.; LaManna, J.C.; del Zoppo, G.J. Increased expression of fibronectin and the α 5 β 1 integrin in angiogenic cerebral blood vessels of mice subject to hypobaric hypoxia. Mol. Cell. Neurosci. 2008, 38, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Jean, C.; Gravelle, P.; Fournie, J.J.; Laurent, G. Influence of stress on extracellular matrix and integrin biology. Oncogene 2011, 30, 2697–2706. [Google Scholar] [CrossRef] [PubMed]
- Bianconi, D.; Schuler, A.; Pausz, C.; Geroldinger, A.; Kaider, A.; Lenz, H.J.; Kornek, G.; Scheithauer, W.; Zielinski, C.C.; Pabinger, I.; et al. Integrin β-3 genetic variants and risk of venous thromboembolism in colorectal cancer patients. Thromb. Res. 2015, 136, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Li, Z.; Jiang, H.; Liu, T. SNPs in microRNA-binding sites in the ITGB1 and ITGB3 3′-UTR increase colorectal cancer risk. Cell Biochem. Biophys. 2014, 70, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Bohanes, P.; Yang, D.; Loupakis, F.; LaBonte, M.J.; Gerger, A.; Ning, Y.; Lenz, C.; Lenz, F.; Wakatsuki, T.; Zhang, W.; et al. Integrin genetic variants and stage-specific tumor recurrence in patients with stage II and III colon cancer. Pharmacogenom. J. 2015, 15, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Kim, D.K.; Oh, I.H.; Song, J.Y.; Kwon, K.H.; Choe, B.K.; Kim, Y.H. A missense polymorphism (rs11895564, Ala380thr) of integrin α 6 is associated with the development and progression of papillary thyroid carcinoma in Korean population. J. Korean Surg. Soc. 2011, 81, 308–315. [Google Scholar] [CrossRef] [PubMed]
- PatentsObserver.com. Chitosan Covalently Linked with Small Molecule Integrin Antagonist for Targeted Delivery. Available online: http://www.patentobserver.com (accessed on 15 September 2016).
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The first anti-angiogenic small molecule drug candidate design, synthesis and clinical evaluation. Anticancer Agents Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.C.; van Zandwijk, N.; Reid, G. Cilengitide inhibits attachment and invasion of malignant pleural mesothelioma cells through antagonism of integrins αvβ3 and αvβ5. PLoS ONE 2014, 9, e90374. [Google Scholar] [CrossRef] [PubMed]
- Alghisi, G.C.; Ponsonnet, L.; Ruegg, C. The integrin antagonist cilengitide activates αvβ3, disrupts VE-cadherin localization at cell junctions and enhances permeability in endothelial cells. PLoS ONE 2009, 4, e4449. [Google Scholar] [CrossRef] [PubMed]
- Chilla, A.; Bianconi, D.; Geetha, N.; Dorda, A.; Poettler, M.; Unseld, M.; Sykoutri, D.; Redlich, K.; Zielinski, C.C.; Prager, G.W. Effects of cilengitide in osteoclast maturation and behavior. Exp. Cell Res. 2015, 337, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Bretschi, M.; Merz, M.; Komljenovic, D.; Berger, M.R.; Semmler, W.; Bauerle, T. Cilengitide inhibits metastatic bone colonization in a nude rat model. Oncol. Rep. 2011, 26, 843–851. [Google Scholar] [PubMed]
- Bauerle, T.; Komljenovic, D.; Merz, M.; Berger, M.R.; Goodman, S.L.; Semmler, W. Cilengitide inhibits progression of experimental breast cancer bone metastases as imaged noninvasively using VCT, MRI and DCE-MRI in a longitudinal in vivo study. Int. J. Cancer 2011, 128, 2453–2462. [Google Scholar] [CrossRef] [PubMed]
- Heiduschka, G.; Lill, C.; Schneider, S.; Seemann, R.; Kornek, G.; Schmid, R.; Kotowski, U.; Thurnher, D. The effect of cilengitide in combination with irradiation and chemotherapy in head and neck squamous cell carcinoma cell lines. Strahlenther. Onkol. 2014, 190, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Christenheit, A.; Heffeter, P.; Selzer, E. A novel small-molecule integrin antagonist inhibits cells adhesion followed by anoikis in endothelial cells—A comparative analysis with cilengitide. Glob. J. Cancer Ther. 2016, 2, 9–18. [Google Scholar]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated mgmt promoter (centric eortc 26071–22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef]
- Reynolds, A.R.; Hart, I.R.; Watson, A.R.; Welti, J.C.; Silva, R.G.; Robinson, S.D.; Da Violante, G.; Gourlaouen, M.; Salih, M.; Jones, M.C.; et al. Stimulation of tumor growth and angiogenesis by low concentrations of RGD-mimetic integrin inhibitors. Nat. Med. 2009, 15, 392–400. [Google Scholar] [CrossRef] [PubMed]
- De Franceschi, N.; Hamidi, H.; Alanko, J.; Sahgal, P.; Ivaska, J. Integrin traffic—The update. J. Cell Sci. 2015, 128, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Hersey, P.; Sosman, J.; O’Day, S.; Richards, J.; Bedikian, A.; Gonzalez, R.; Sharfman, W.; Weber, R.; Logan, T.; Buzoianu, M.; et al. A randomized phase 2 study of etaracizumab, a monoclonal antibody against integrin α(v)β(3), + or—Dacarbazine in patients with stage IV metastatic melanoma. Cancer 2010, 116, 1526–1534. [Google Scholar] [CrossRef] [PubMed]
- O’Day, S.; Pavlick, A.; Loquai, C.; Lawson, D.; Gutzmer, R.; Richards, J.; Schadendorf, D.; Thompson, J.A.; Gonzalez, R.; Trefzer, U.; et al. A randomised, phase II study of intetumumab, an anti-αv-integrin mAb, alone and with dacarbazine in stage IV melanoma. Br. J. Cancer 2011, 105, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Bell-McGuinn, K.M.; Matthews, C.M.; Ho, S.N.; Barve, M.; Gilbert, L.; Penson, R.T.; Lengyel, E.; Palaparthy, R.; Gilder, K.; Vassos, A.; et al. A phase II, single-arm study of the anti-α5β1 integrin antibody volociximab as monotherapy in patients with platinum-resistant advanced epithelial ovarian or primary peritoneal cancer. Gynecol. Oncol. 2011, 121, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Cianfrocca, M.E.; Kimmel, K.A.; Gallo, J.; Cardoso, T.; Brown, M.M.; Hudes, G.; Lewis, N.; Weiner, L.; Lam, G.N.; Brown, S.C.; et al. Phase 1 trial of the antiangiogenic peptide ATN-161 (Ac-PHSCN-NH(2)), a β integrin antagonist, in patients with solid tumours. Br. J. Cancer 2006, 94, 1621–1626. [Google Scholar] [PubMed]
- Mateo, J.; Berlin, J.; de Bono, J.S.; Cohen, R.B.; Keedy, V.; Mugundu, G.; Zhang, L.; Abbattista, A.; Davis, C.; Gallo Stampino, C.; et al. A first-in-human study of the anti-α5β1 integrin monoclonal antibody PF-04605412 administered intravenously to patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 74, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Insider, S. Immunogen (IMGN) to Discontinue IMGN388 Development, Will Refocus Resources. Available online: http://www.streetinsider.com/Corporate+News/ImmunoGen+%28IMGN%29+to+Discontinue+IMGN388+Development%2C+Will+Refocus+Resources/6979519.html (accessed on 30 September 2016).
- Cirkel, G.A.; Kerklaan, B.M.; Vanhoutte, F.; van der Aa, A.; Lorenzon, G.; Namour, F.; Pujuguet, P.; Darquenne, S.; de Vos, F.Y.; Snijders, T.J.; et al. A dose escalating phase I study of GLPG0187, a broad spectrum integrin receptor antagonist, in adult patients with progressive high-grade glioma and other advanced solid malignancies. Investig. New Drugs 2016, 34, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Gao, S.; Wu, M.; Liu, X.; Qiao, J.; Zhou, Q.; Jiang, S.; Niu, Z. Tobacco mosaic virus-based 1D nanorod-drug carrier via the integrin-mediated endocytosis pathway. ACS Appl. Mater. Interfaces 2016, 8, 10800–10807. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.M.; Jiang, H.; Yokoyama, T.; Xu, J.; Bekele, N.B.; Lang, F.F.; Kondo, S.; Gomez-Manzano, C.; Fueyo, J. Δ-24-RGD in combination with RAD001 induces enhanced anti-glioma effect via autophagic cell death. Mol. Ther. 2008, 16, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Clise-Dwyer, K.; Ruisaard, K.E.; Fan, X.; Tian, W.; Gumin, J.; Lamfers, M.L.; Kleijn, A.; Lang, F.F.; Yung, W.K.; et al. Δ-24-RGD oncolytic adenovirus elicits anti-glioma immunity in an immunocompetent mouse model. PLoS ONE 2014, 9, e97407. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Interaction | Interaction Partner | Integrin | Cell Line | Description and References |
|---|---|---|---|---|
| Direct interaction between integrins and growth factor receptors and potentiation of signaling pathways | IGF-1R | α6β4 | MCF-7 | Integrin-IGF1-IGF1 receptor ternary complex formation [63]. |
| FGFR | αvβ3 | K562 | FGFR1-FGF1-integrin αvβ3 ternary complex formation [64]. | |
| c-Met | α5β1 | SKOV3ip1 and HeyA8 | Upon binding to fibronectin, α5β1-integrin interacts directly with the receptor tyrosine kinase c-Met and activates it in a ligand-independent manner [65]. (Figure 3(1)) | |
| Regulation of expression level of growth factor receptors by integrins | EGFR | β1 | A549 | Downregulation of the β1 subunit leads to an increased level of the EGFR at the plasma membrane [66]. |
| EGFR | α6β4 | AsPC1, Suit-2, and Panc-1 | Integrin α6β4 leads to the recruitment of the c-Cbl ubiquitin ligase to the growth factor receptor and this leads to a reduced EGFR degradation [67]. | |
| EGFR | α6β4 | MDA-MB-231 | Crosslinking of integrin induces EGFR clustering and promotes EGF-mediated signaling [68]. | |
| Activation of integrins via growth factor receptor signalling | EGFR | αvβ5 | FG | EGFR ligand binding induces Rap1 activation and this leads to the activation of αvβ5 [69]. |
| Hallmark of Cancer | Integrin’s Contribution |
|---|---|
| Sustaining proliferative signalling |
|
| Evading growth repressors |
|
| Invasion and metastasis |
|
| Limitless replicative potential |
|
| Sustained angiogenesis |
|
| Evasion of apoptosis |
|
| Compound | Target | Stage | Result | Reference |
|---|---|---|---|---|
| Vitaxin (Etaracizumab) | αvβ3 | Phase II | No clinically meaningful improvement in survival. | [225] |
| CTNO 95 (Intetumumab) | αv (αvβ3 and αvβ5) | Phase II | No clinically meaningful improvement in survival. | [226] |
| Volociximab | α5β1 | Phase II | Insufficient clinical activity. | [227] |
| ATN-161 | α5β1 | Phase I | 1/3 of patients manifested stable disease. | [228] |
| Two Phase II trials were discontinued in February 2016. | ||||
| PF-04605412 | α5β1 | Phase I | Trial was prematurely terminated. | [229] |
| IMGN388 | αv integrin-targeting antibody conjugated to a cytotoxic agent | Phase I | Well tolerated but it was discontinued. | [230] |
| GLP0187 | Several αv integrins | Phase I | No signs of monotherapy efficacy. | [231] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bianconi, D.; Unseld, M.; Prager, G.W. Integrins in the Spotlight of Cancer. Int. J. Mol. Sci. 2016, 17, 2037. https://doi.org/10.3390/ijms17122037
Bianconi D, Unseld M, Prager GW. Integrins in the Spotlight of Cancer. International Journal of Molecular Sciences. 2016; 17(12):2037. https://doi.org/10.3390/ijms17122037
Chicago/Turabian StyleBianconi, Daniela, Matthias Unseld, and Gerald W. Prager. 2016. "Integrins in the Spotlight of Cancer" International Journal of Molecular Sciences 17, no. 12: 2037. https://doi.org/10.3390/ijms17122037
APA StyleBianconi, D., Unseld, M., & Prager, G. W. (2016). Integrins in the Spotlight of Cancer. International Journal of Molecular Sciences, 17(12), 2037. https://doi.org/10.3390/ijms17122037