
Metformin Inhibits TGF-β1-Induced Epithelial-to-Mesenchymal Transition via PKM2 Relative-mTOR/p70s6k Signaling Pathway in Cervical Carcinoma Cells

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
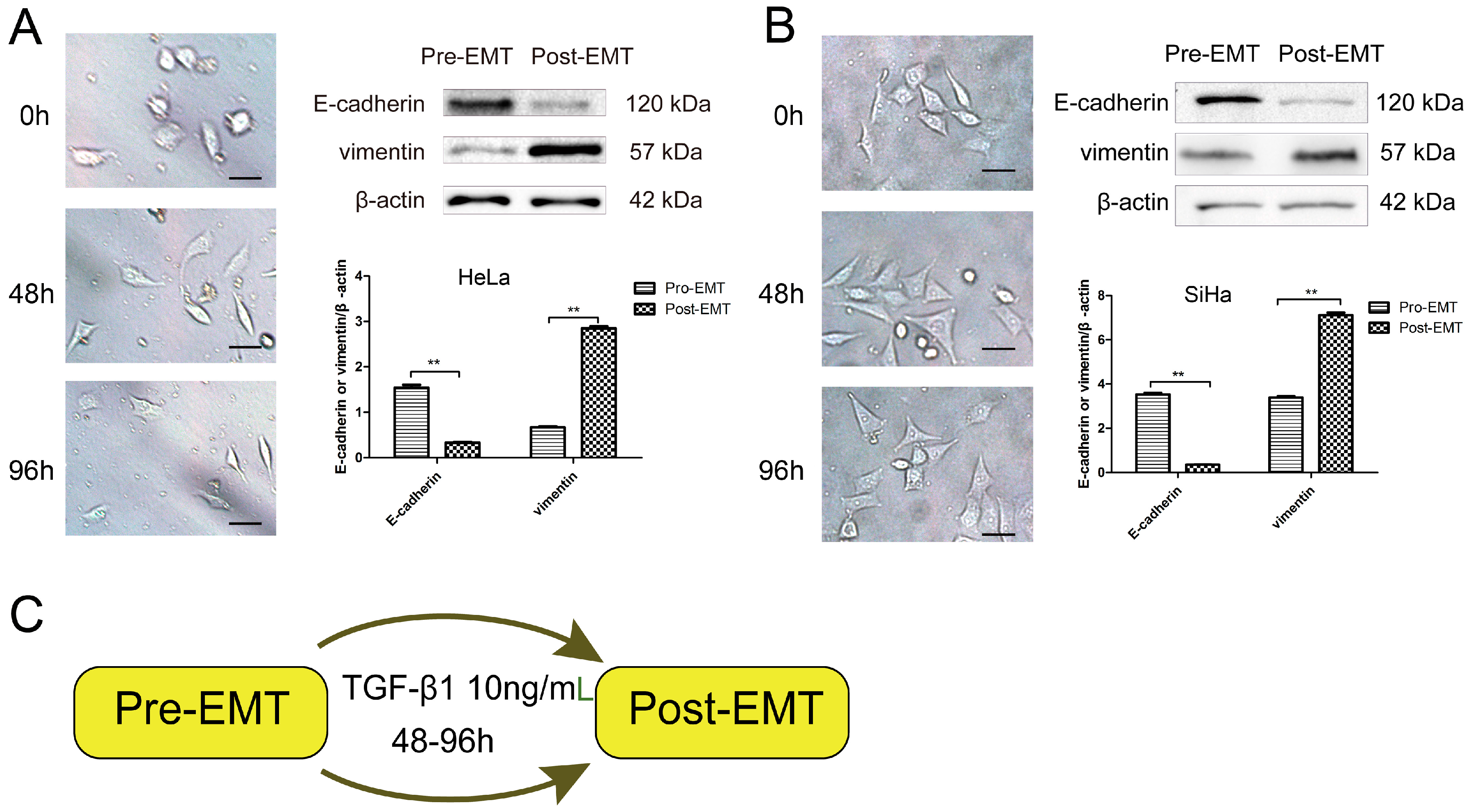
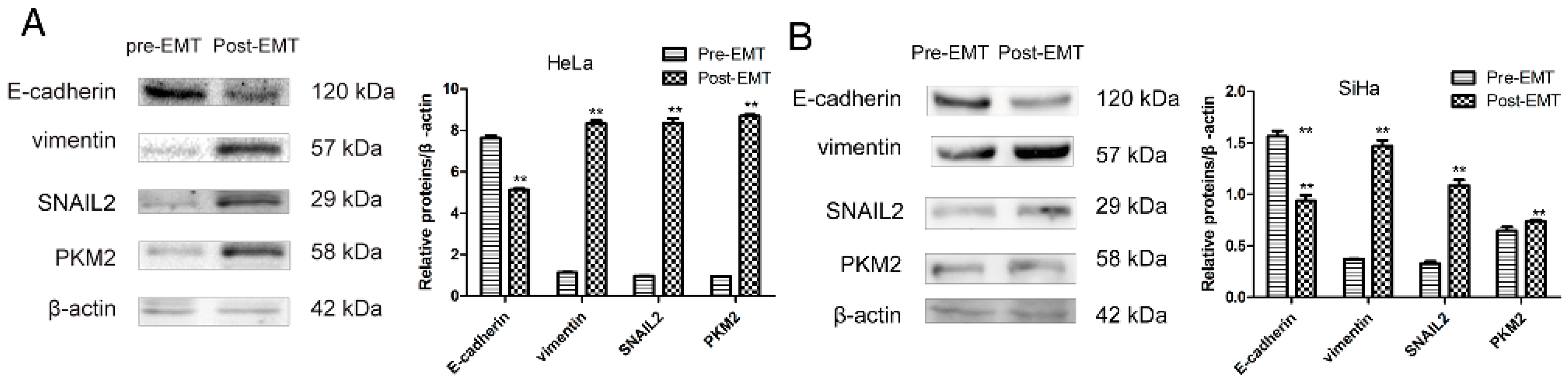
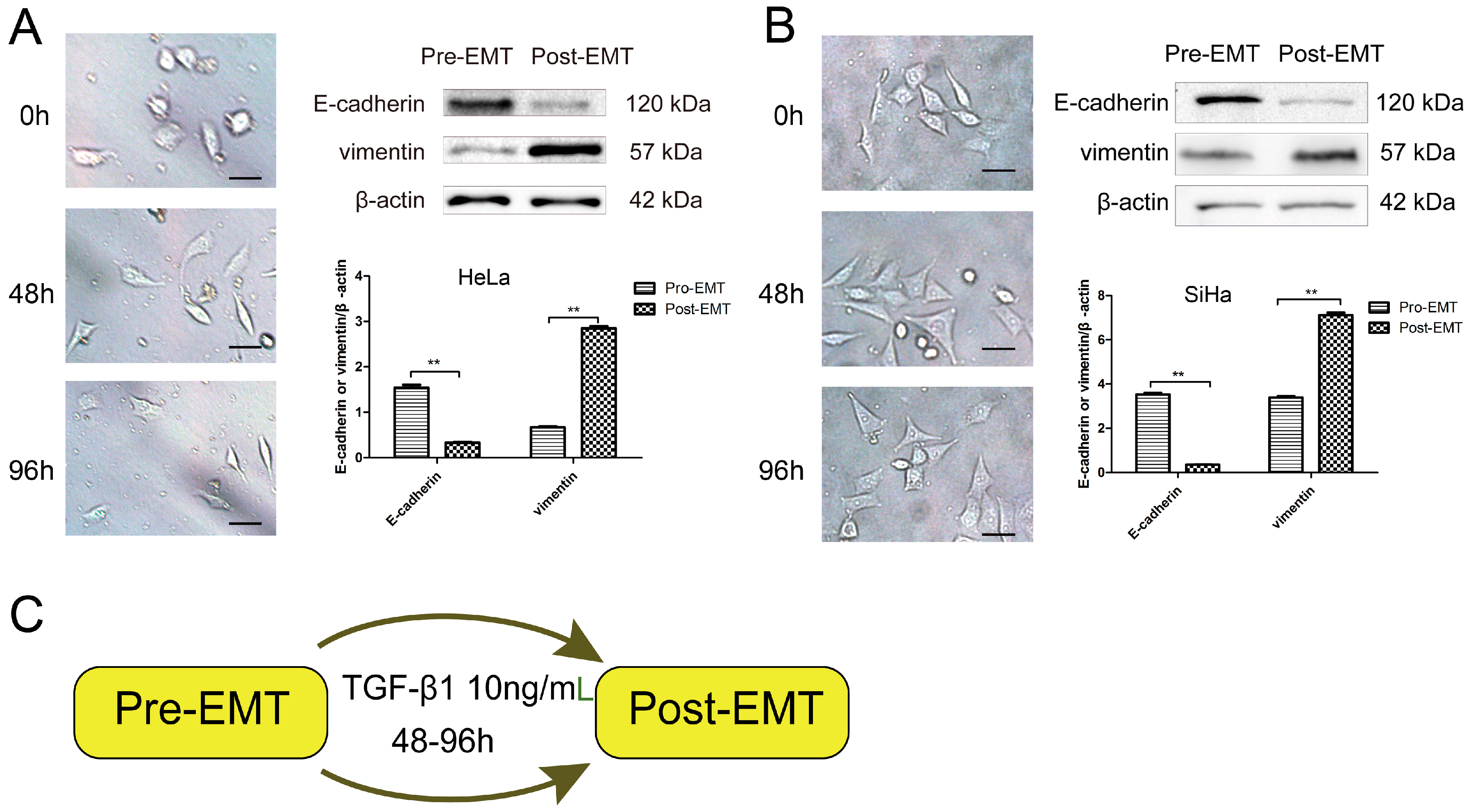
2.1. Transforming Growth Factor Beta 1 (TGF-β1) Induces Epithelial-to-Mesenchymal Transition (EMT) in Cerivical Cancer Cells
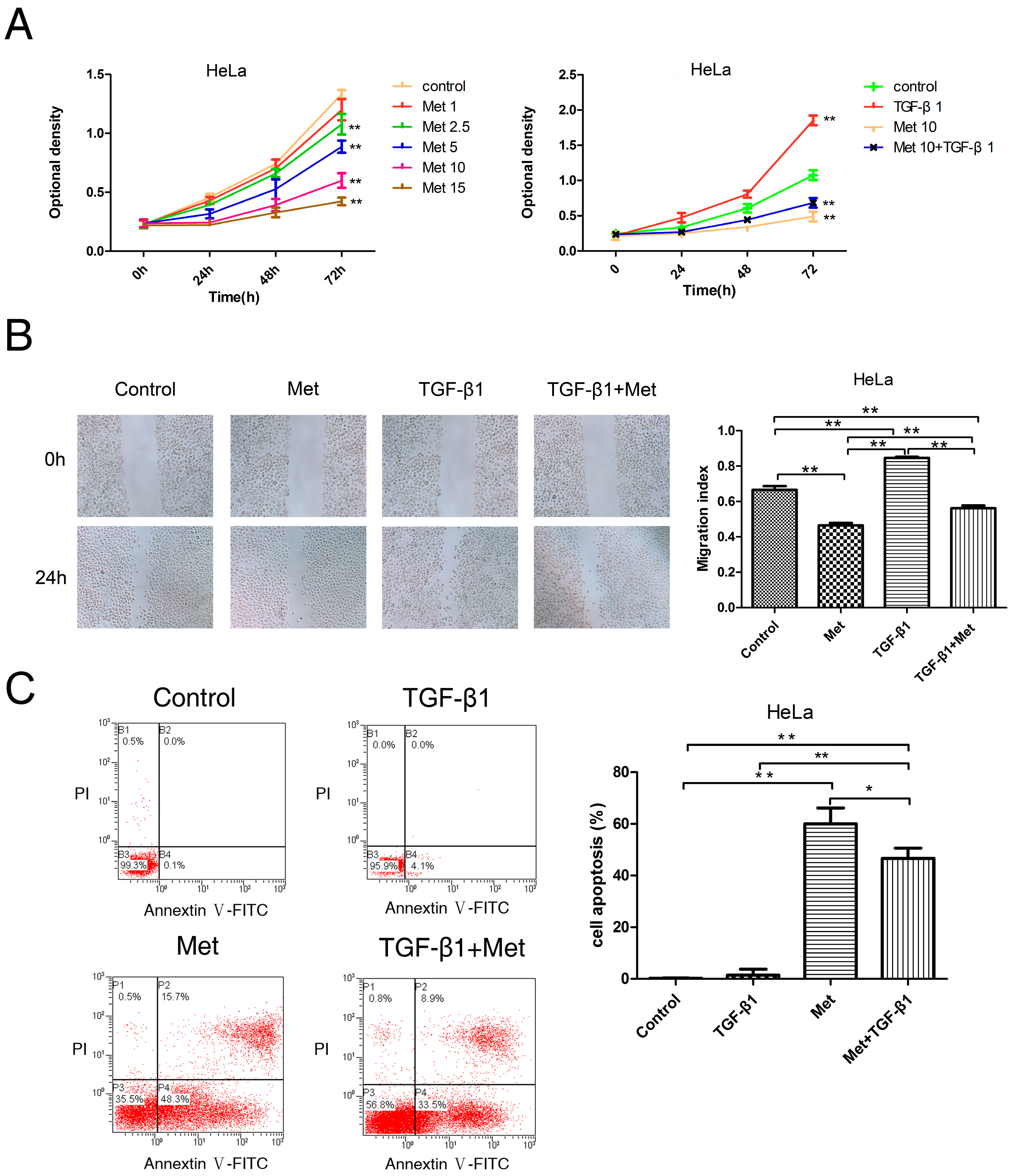
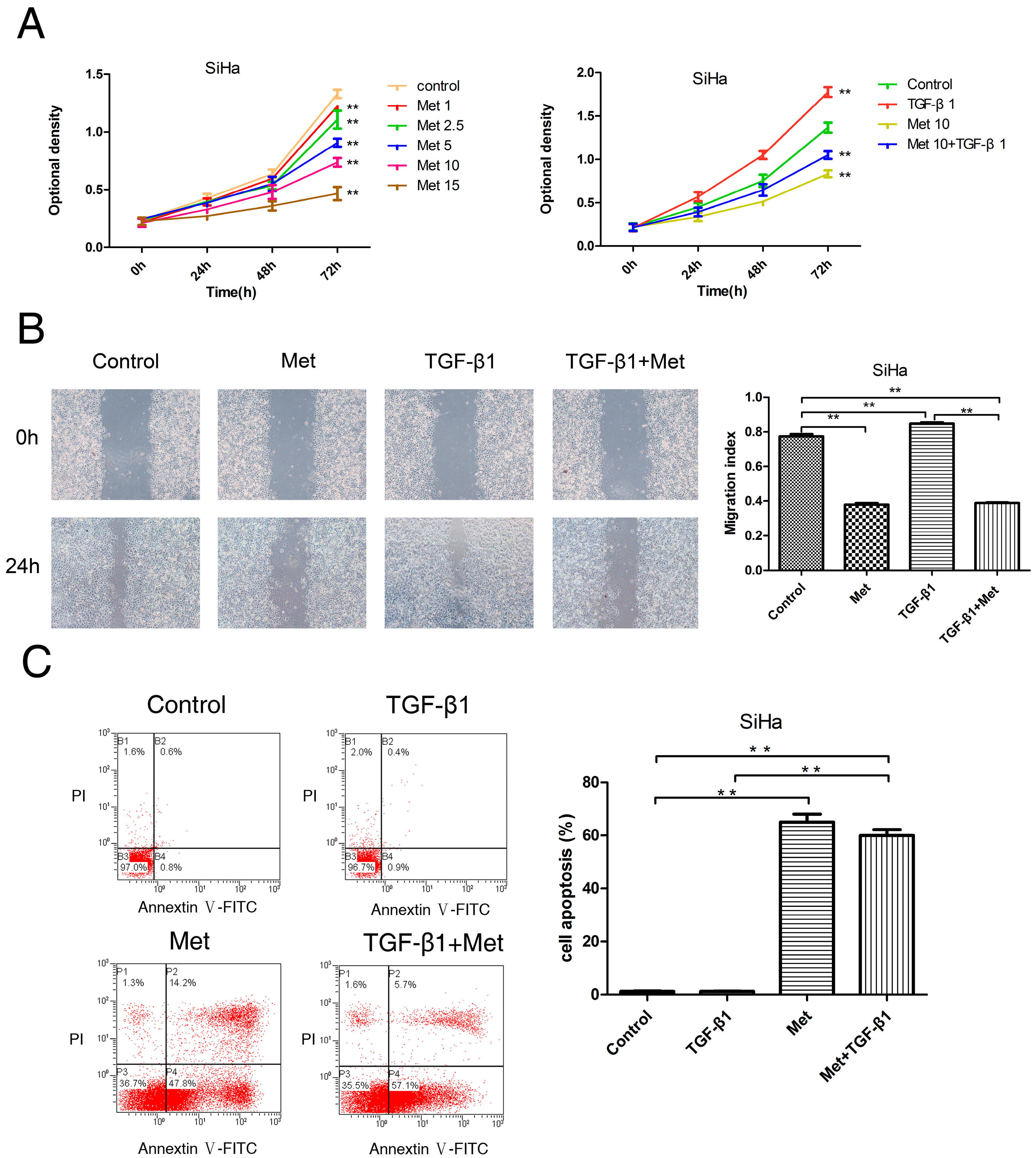
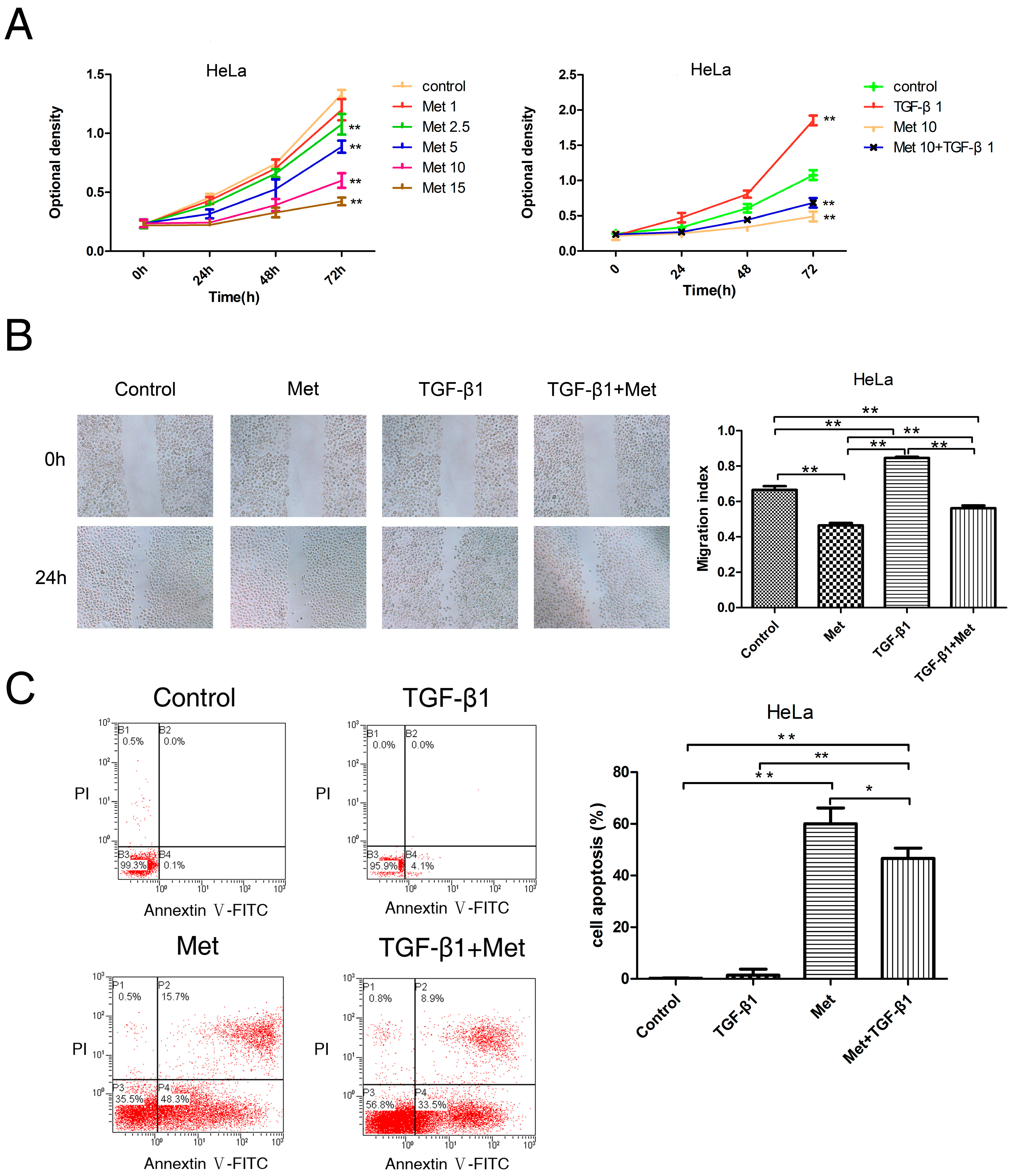
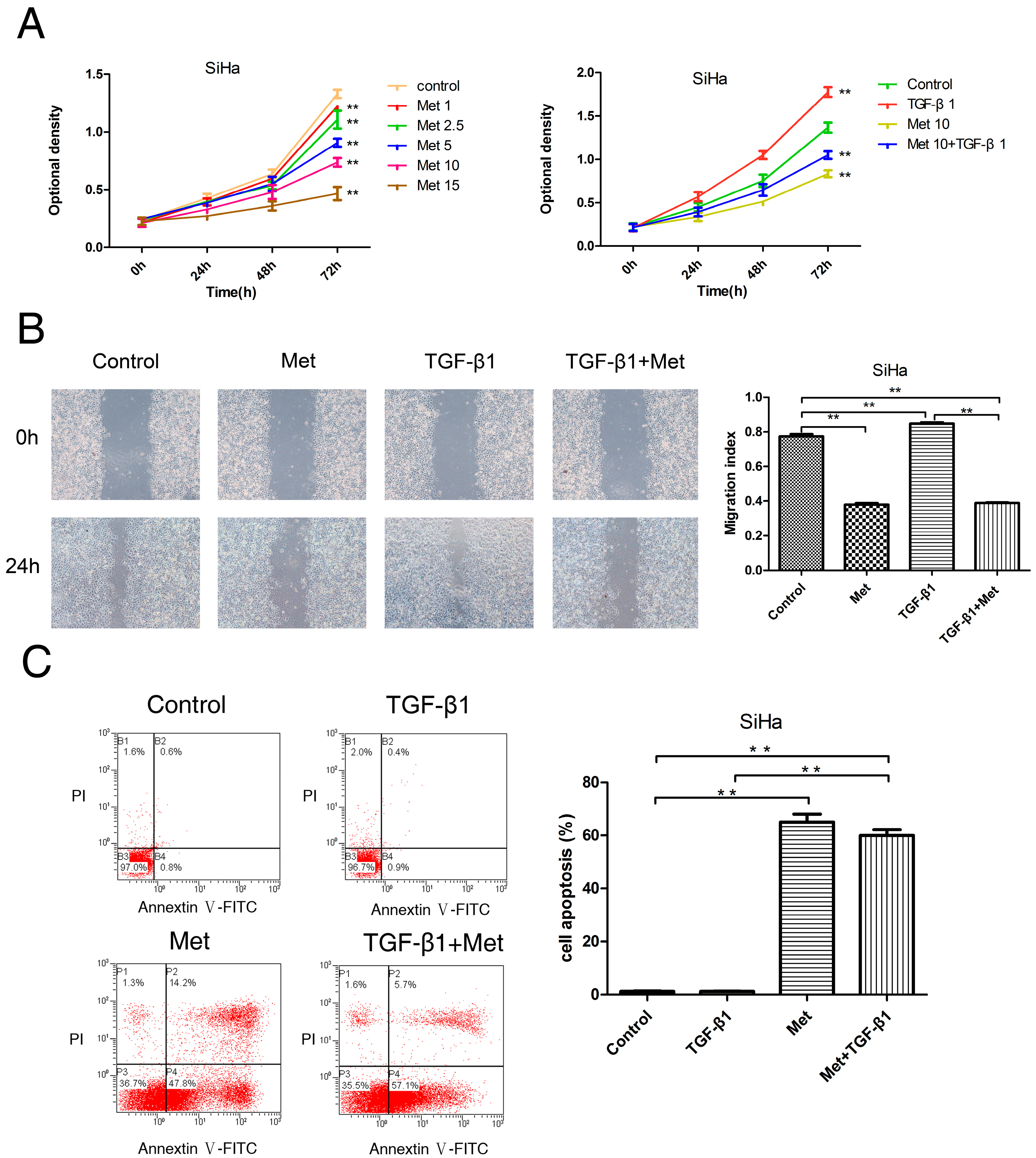
2.2. Metformin Inhibits the TGF-β1-Induced Proliferation, Migration, and Induces Apoptosis
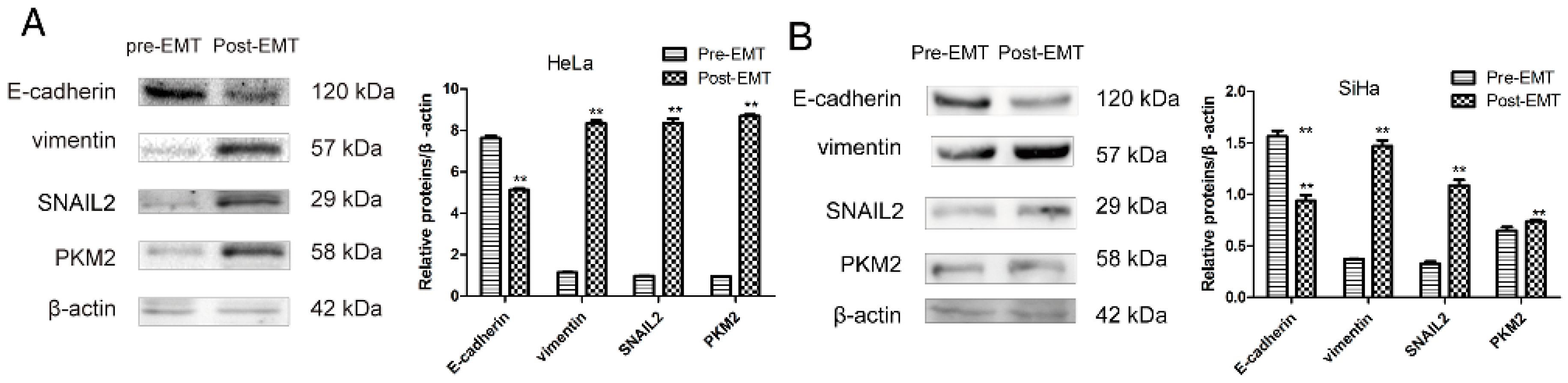
2.3. PKM2 Expression Is Required to Induce EMT
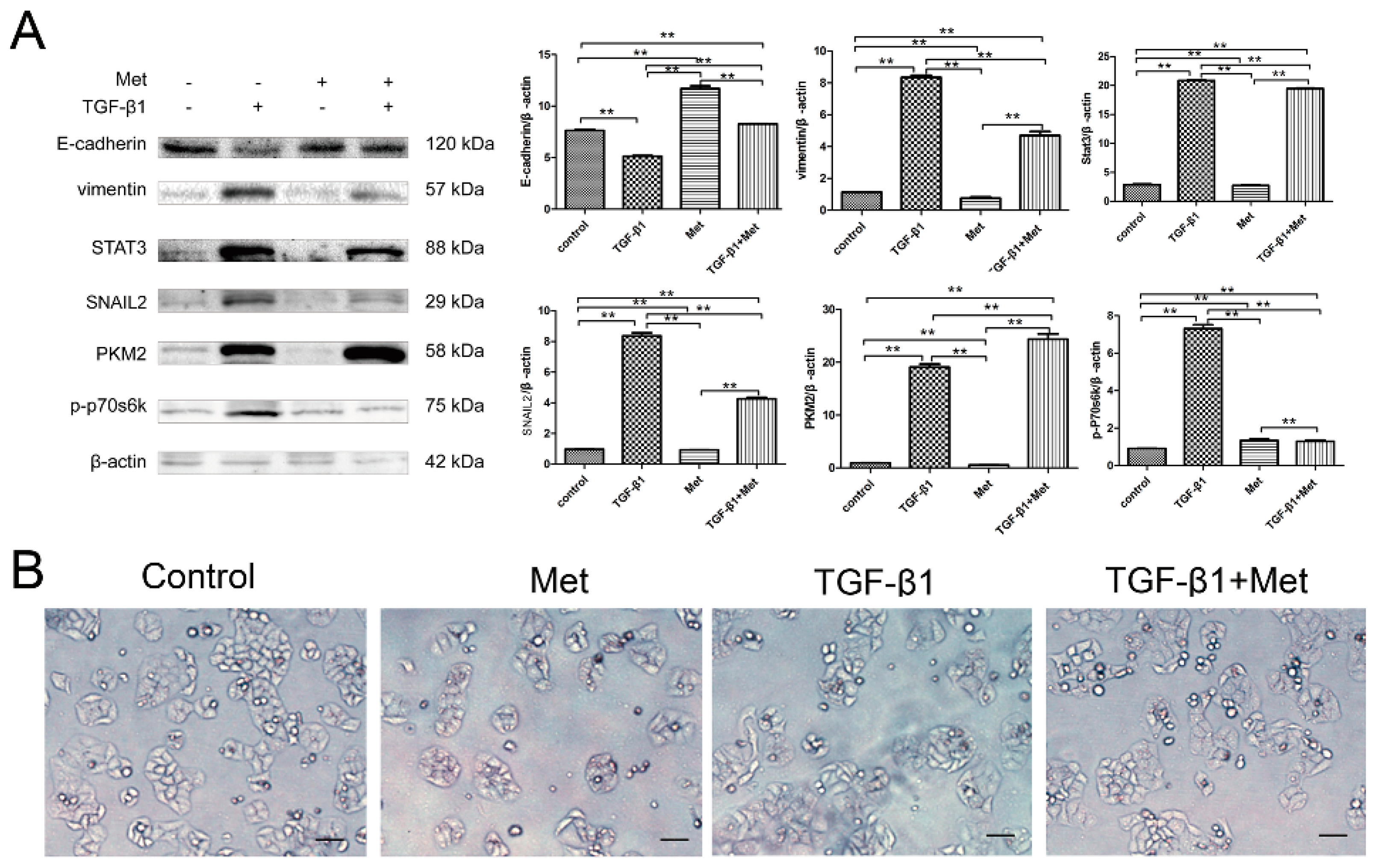
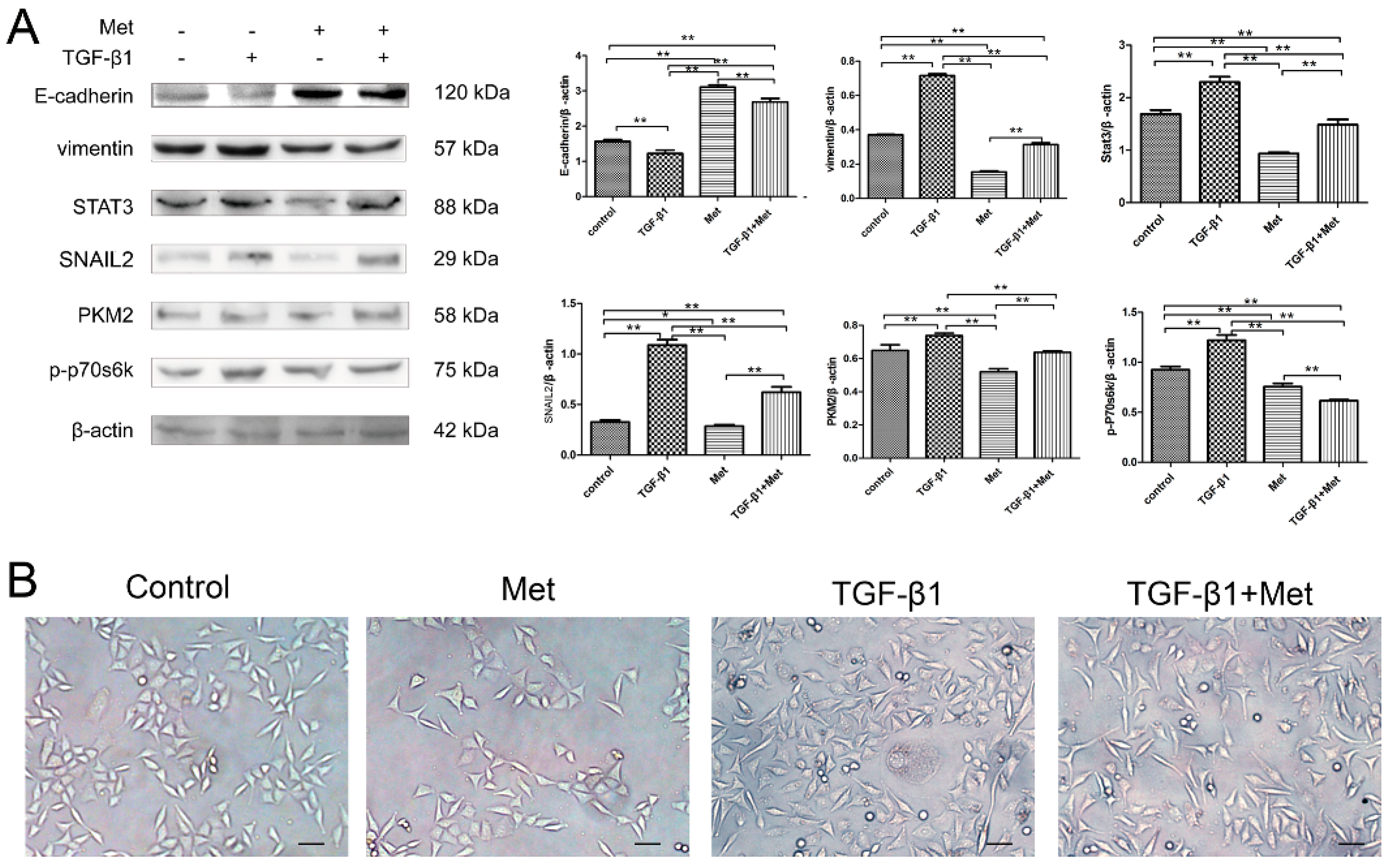
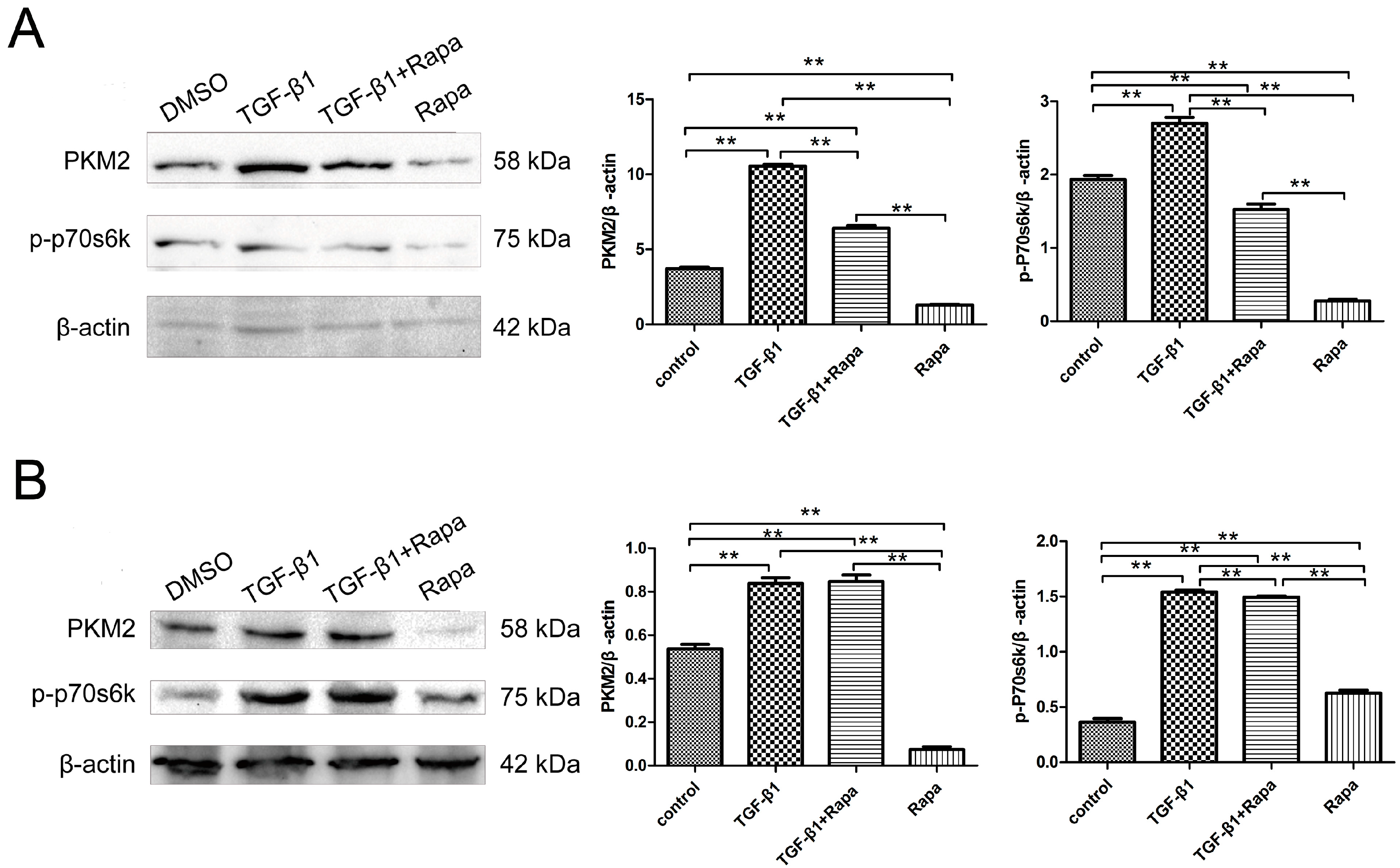
2.4. mTOR/p70s6k Signaling Involved in Regulating PKM2 Expression in the EMT Condition
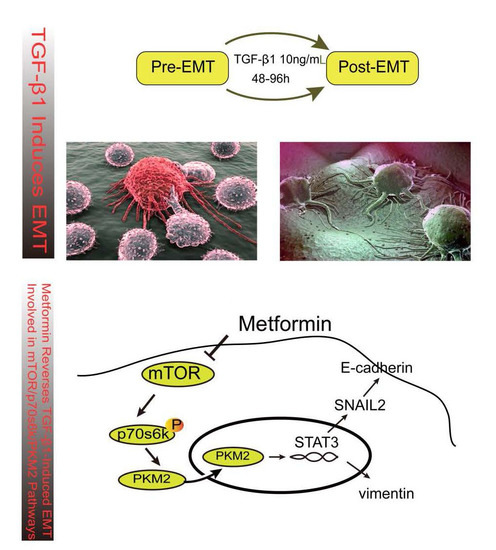
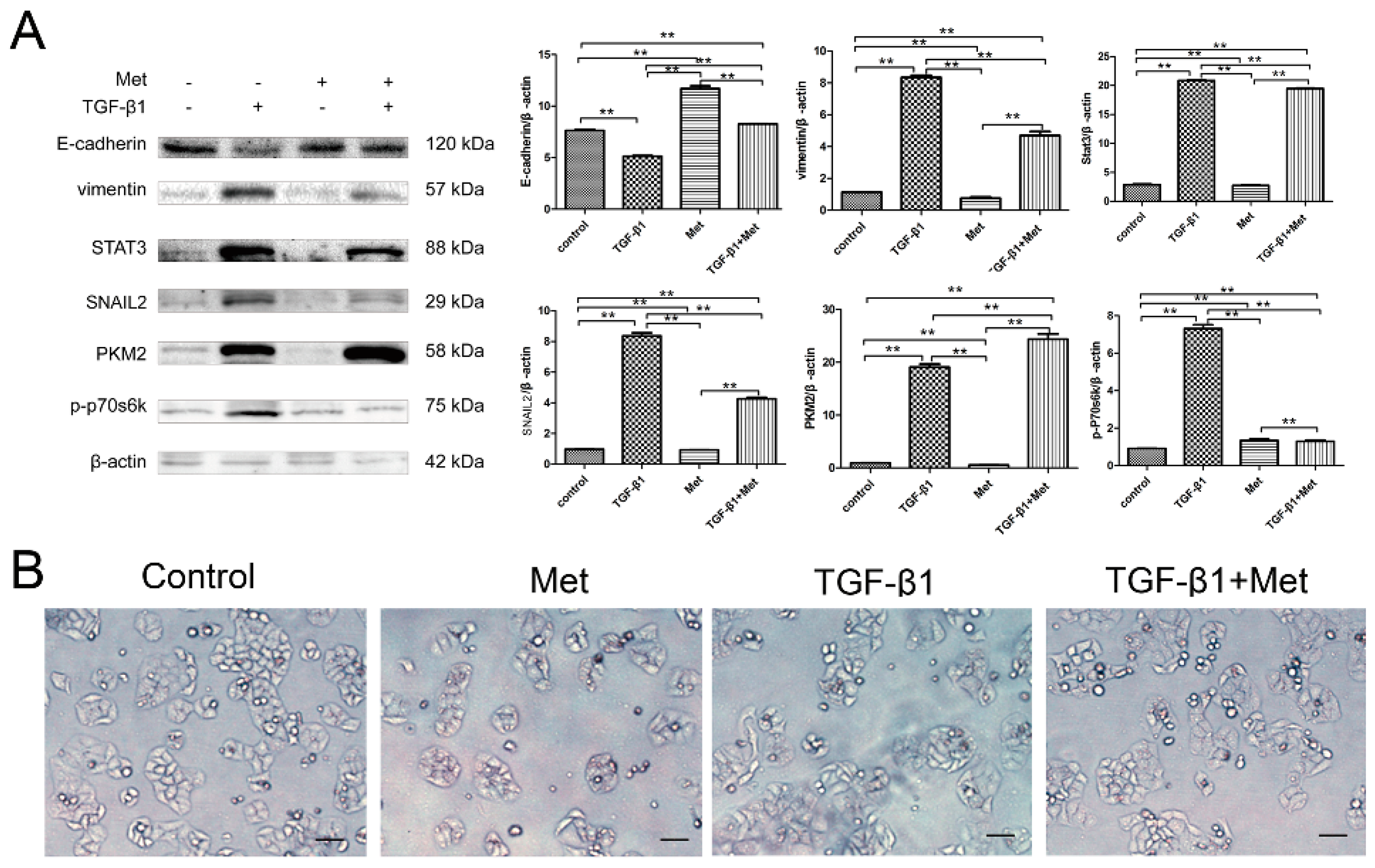
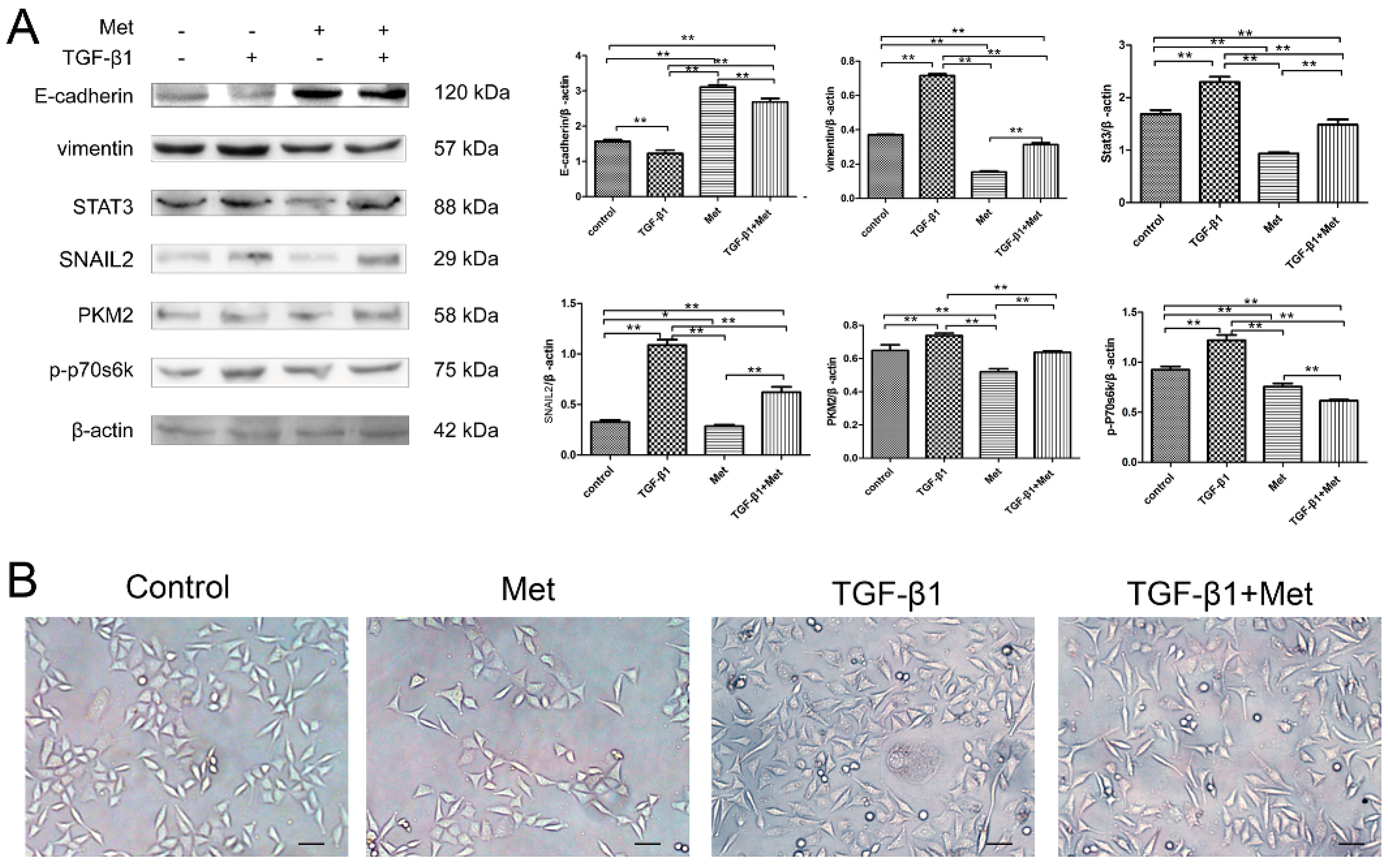
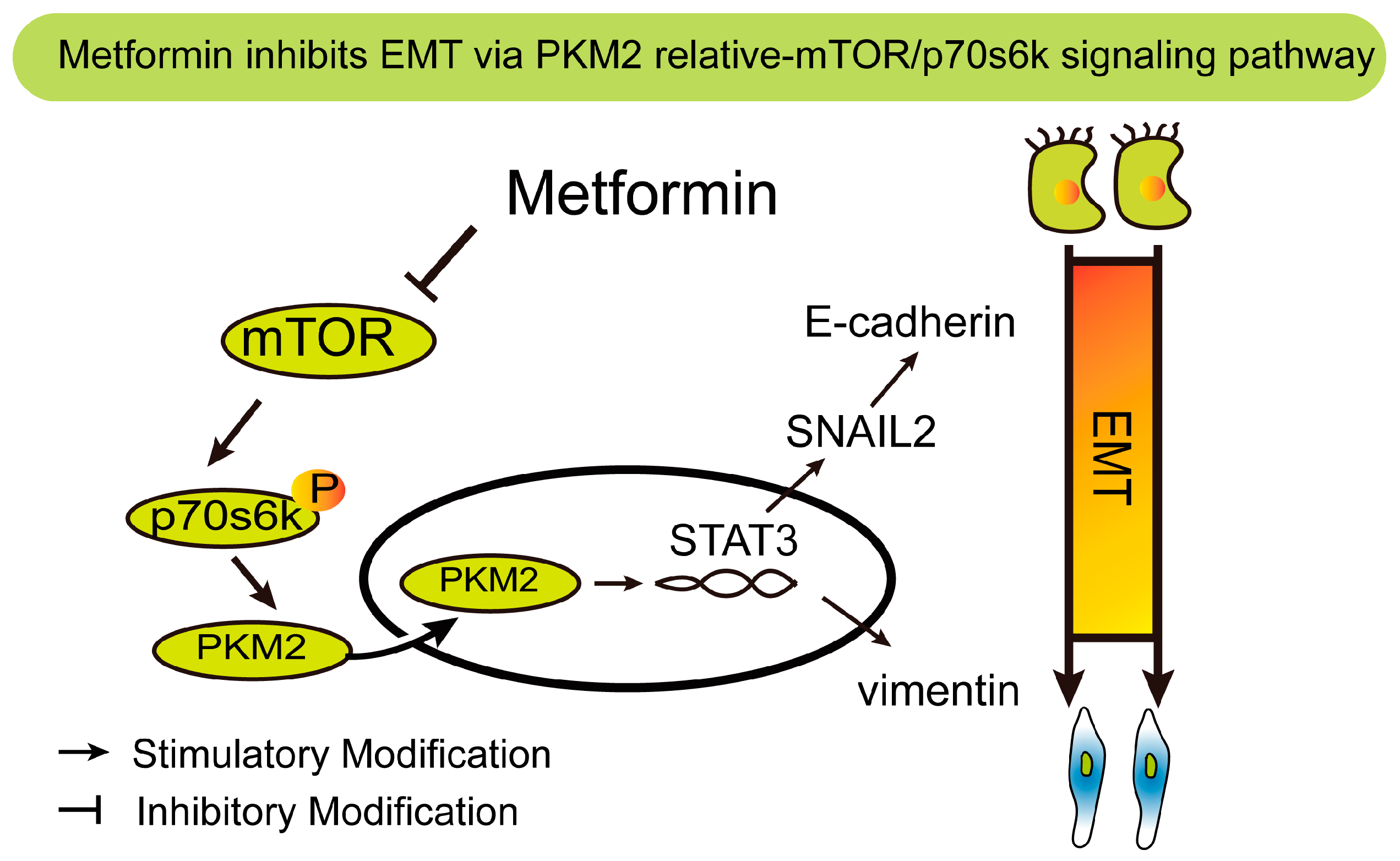
2.5. Metformin Reverses TGF-β1-Induced EMT Involved in mTOR/p70s6k/PKM2 Signaling Pathways in Cervical Carcinoma Cells
3. Discussion
4. Materials and Methods
4.1. Cell Cultures and Treatments
4.2. Cell Viability Assay
4.3. Wound Healing Assay
4.4. Annexin V-FITC Apoptosis Assay
4.5. Western Blot Analysis
4.6. Statistical Analysis
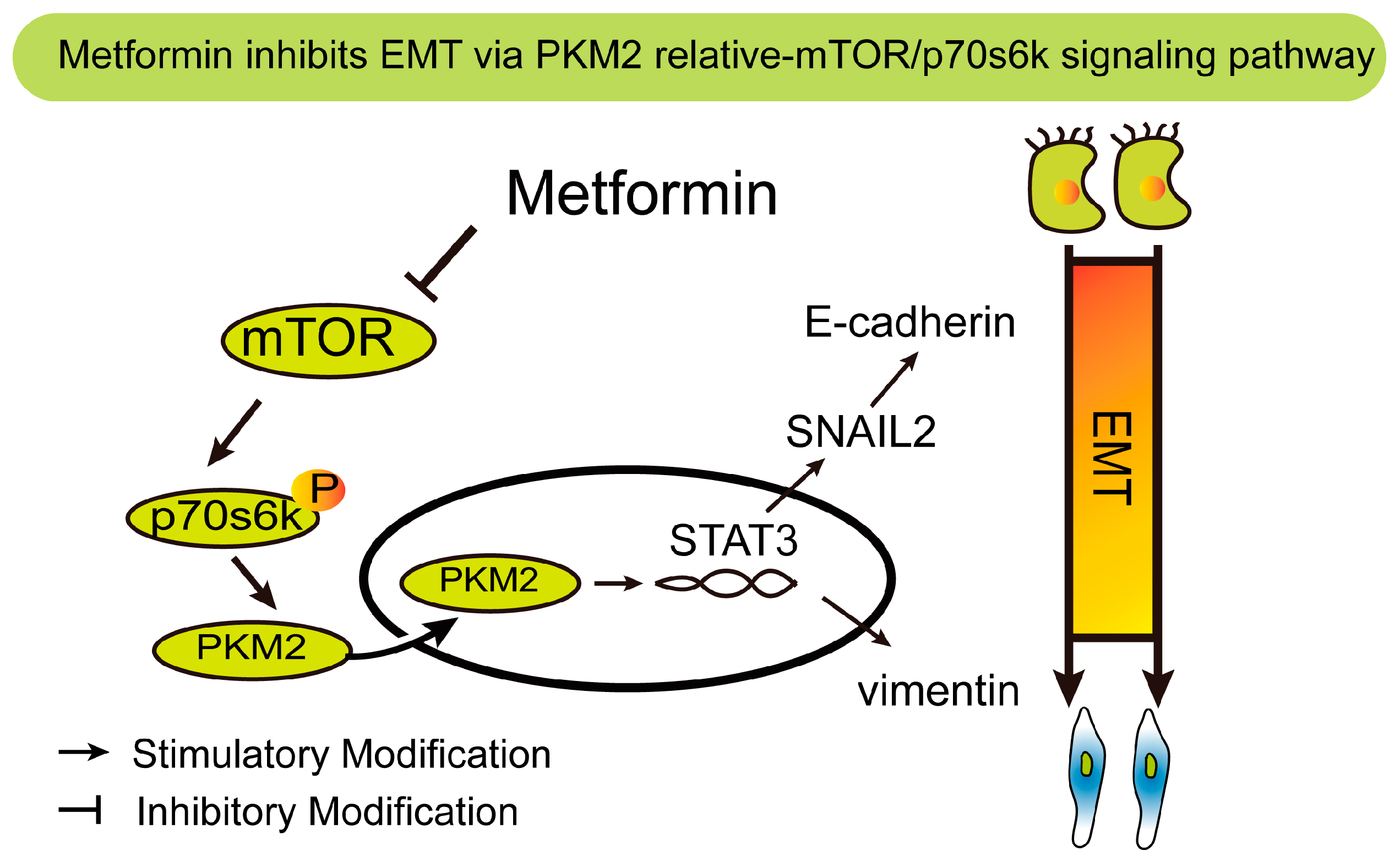
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| TGF-β1 | Recombinant transforming growth factor-β1 |
| mTOR | Mammalian target of rapamycin |
| S6K1 | Ribosomal p70 S6 kinase |
| PKM2 | Pyruvate kinase M2 |
References
- Colombo, N.; Carinelli, S.; Colombo, A.; Marini, C.; Rollo, D.; Sessa, C.; ESMO guidelines working group. Cervical cancer: Esmo clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2012, 23, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Prall, F. Tumour budding in colorectal carcinoma. Histopathology 2007, 50, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.; Buck, E.; Petti, F.; Griffin, G.; Brown, E.; Ramnarine, N.; Iwata, K.K.; Gibson, N.; Haley, J.D. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res. 2005, 65, 9455–9462. [Google Scholar] [CrossRef] [PubMed]
- Kurrey, N.K.; Jalgaonkar, S.P.; Joglekar, A.V.; Ghanate, A.D.; Chaskar, P.D.; Doiphode, R.Y.; Bapat, S.A. Snail and slug mediate radioresistance and chemoresistance by antagonizing P53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells 2009, 27, 2059–2068. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Joo, K.M.; Lee, J.; Yoon, Y.; Nam, D.H. Targeting the epithelial to mesenchymal transition in glioblastoma: The emerging role of Met signaling. Oncol. Targets Ther. 2014, 7, 1933–1944. [Google Scholar]
- Kudo-Saito, C.; Shirako, H.; Takeuchi, T.; Kawakami, Y. Cancer metastasis is accelerated through immunosuppression during snail-induced EMT of cancer cells. Cancer Cell 2009, 15, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Dedhar, S.; Kalluri, R.; Thompson, E.W. The epithelial-mesenchymal transition: New insights in signaling, development, and disease. J. Cell Biol. 2006, 172, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Maeda, M.; Johnson, K.R.; Wheelock, M.J. Cadherin switching: Essential for behavioral but not morphological changes during an epithelium-to-mesenchyme transition. J. Cell Sci. 2005, 118, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Voulgari, A.; Pintzas, A. Epithelial-mesenchymal transition in cancer metastasis: Mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim. Biophys. Acta 2009, 1796, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Quintanilla, M.; Cano, A. Transforming growth factor β-1 induces snail transcription factor in epithelial cell lines: Mechanisms for epithelial mesenchymal transitions. J. Biol. Chem. 2003, 278, 21113–21123. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Venkatasubbarao, K.; Lazor, J.W.; Sperry, J.; Jin, C.; Cao, L.; Freeman, J.W. Inhibition of STAT3 Tyr705 phosphorylation by Smad4 suppresses transforming growth factor β-mediated invasion and metastasis in pancreatic cancer cells. Cancer Res. 2008, 68, 4221–4228. [Google Scholar] [CrossRef] [PubMed]
- Mell, L.K.; Meyer, J.J.; Tretiakova, M.; Khramtsov, A.; Gong, C.; Yamada, S.D.; Montag, A.G.; Mundt, A.J. Prognostic significance of E-cadherin protein expression in pathological stage I–III endometrial cancer. Clin. Cancer Res. 2004, 10, 5546–5553. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Shi, H.; Wang, F.; Hong, D.; Lv, W.; Xie, X.; Cheng, X. Expression of E-, P- and N-Cadherin and its clinical significance in cervical squamous cell carcinoma and precancerous lesions. PLoS ONE 2016, 11, e0155910. [Google Scholar] [CrossRef] [PubMed]
- Hamabe, A.; Konno, M.; Tanuma, N.; Shima, H.; Tsunekuni, K.; Kawamoto, K.; Nishida, N.; Koseki, J.; Mimori, K.; Gotoh, N.; et al. Role of pyruvate kinase M2 in transcriptional regulation leading to epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2014, 111, 15526–15531. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. Origin of cancer cells. Oncologia 1956, 9, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E.; Harlan, D.M.; Archer, M.C.; Bergenstal, R.M.; Gapstur, S.M.; Habel, L.A.; Pollak, M.; Regensteiner, J.G.; Yee, D. Diabetes and cancer: A consensus report. Diabetes Care 2010, 33, 1674–1685. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Guigas, B.; Garcia, N.S.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 2012, 122, 253–270. [Google Scholar] [CrossRef] [PubMed]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Meng, Q.; Chen, X.; Zhao, Y.; Liu, W.; Hu, J.; Xue, F.; Wang, X.; Cai, L. TRIM44 promotes proliferation and metastasis in nonsmall cell lung cancer via mTOR signaling pathway. Oncotarget 2016, 7, 30479–30491. [Google Scholar] [PubMed]
- Li, L.; Han, R.; Xiao, H.; Lin, C.; Wang, Y.; Liu, H.; Li, K.; Chen, H.; Sun, F.; Yang, Z.; et al. Metformin sensitizes EGFR-TKI-resistant human lung cancer cells in vitro and in vivo through Inhibition of IL-6 signaling and EMT reversal. Clin. Cancer Res. 2014, 20, 2714–2726. [Google Scholar] [CrossRef] [PubMed]
- Hosono, K.; Endo, H.; Takahashi, H.; Sugiyama, M.; Uchiyama, T.; Suzuki, K.; Nozaki, Y.; Yoneda, K.; Fujita, K.; Yoneda, M.; et al. Metformin suppresses azoxymethane-induced colorectal aberrant crypt foci by activating AMP-activated protein kinase. Mol. Carcinog. 2010, 49, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Dann, S.G.; Selvaraj, A.; Thomas, G. mTOR complex1-S6K1 Signaling: At the crossroads of obesity, diabetes and cancer. Trends Mol. Med. 2007, 13, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Qi, S.; Zhao, X.; Li, M.; Ding, S.; Lu, J.; Zhang, H. Metformin inhibits 17β-estradiol-induced epithelial-to-mesenchymal transition via βklotho-related ERK1/2 signaling and AMPKα signaling in endometrial adenocarcinoma cells. Oncotarget 2016, 7, 21315–21331. [Google Scholar] [PubMed]
- Wurth, R.; Pattarozzi, A.; Gatti, M.; Bajetto, A.; Corsaro, A.; Parodi, A.; Sirito, R.; Massollo, M.; Marini, C.; Zona, G.; et al. Metformin selectively affects human glioblastoma tumor-initiating cell viability: A role for metformin-induced inhibition of Akt. Cell Cycle 2013, 12, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Janzer, A.; German, N.J.; Gonzalez-Herrera, K.N.; Asara, J.M.; Haigis, M.C.; Struhl, K. Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, 10574–10579. [Google Scholar] [CrossRef] [PubMed]
- Currie, C.J.; Poole, C.D.; Jenkins-Jones, S.; Gale, E.A.; Johnson, J.A.; Morgan, C.L. Mortality after incident cancer in people with and without Type 2 diabetes: Impact of metformin on survival. Diabetes Care 2012, 35, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Cauchy, F.; Mebarki, M.; Albuquerque, M.; Laouirem, S.; Rautou, P.E.; Soubrane, O.; Raymond, E.; Bedossa, P.; Paradis, V. Anti-angiogenic effect of metformin in human liver carcinogenesis related to metabolic syndrome. Gut 2015, 64, 1498–1500. [Google Scholar] [CrossRef] [PubMed]
- Griss, T.; Vincent, E.E.; Egnatchik, R.; Chen, J.; Ma, E.H.; Faubert, B.; Viollet, B.; DeBerardinis, R.J.; Jones, R.G. Metformin antagonizes cancer cell proliferation by suppressing mitochondrial-dependent biosynthesis. PLoS Biol. 2015, 13, e1002309. [Google Scholar] [CrossRef] [PubMed]
- Cabello, P.; Pineda, B.; Tormo, E.; Lluch, A.; Eroles, P. The Antitumor effect of metformin is mediated by miR-26a in breast cancer. Int. J. Mol. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Zhang, J.; Xu, Z.; Feng, Y.; Zhang, M.; Liu, J.; Chen, R.; Shen, J.; Wu, J.; Lu, Z.; et al. Metformin is a novel suppressor for transforming growth factor (TGF)-β1. Sci. Rep. 2016, 6, 28597. [Google Scholar] [CrossRef] [PubMed]
- Kurimoto, R.; Iwasawa, S.; Ebata, T.; Ishiwata, T.; Sekine, I.; Tada, Y.; Tatsumi, K.; Koide, S.; Iwama, A.; Takiguchi, Y. Drug resistance originating from a TGF-β/FGF-2-driven epithelial-to-mesenchymal transition and its reversion in human lung adenocarcinoma cell lines harboring an EGFR mutation. Int. J. Oncol. 2016, 48, 1825–1836. [Google Scholar] [CrossRef] [PubMed]
- You, A.; Cao, M.; Guo, Z.; Zuo, B.; Gao, J.; Zhou, H.; Li, H.; Cui, Y.; Fang, F.; Zhang, W.; et al. Metformin sensitizes sorafenib to inhibit postoperative recurrence and metastasis of hepatocellular carcinoma in orthotopic mouse models. J. Hematol. Oncol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, A.; Palumbo, F.; Rasi, I.; Posca, D.; Pavlidou, T.; Paoluzi, S.; Castagnoli, L.; Cesareni, G. Metformin induces apoptosis and downregulates pyruvate kinase M2 in breast cancer cells only when grown in nutrient-poor conditions. PLoS ONE 2015, 10, e0136250. [Google Scholar] [CrossRef] [PubMed]
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brule, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J.; et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010, 11, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Ben Sahra, I.; Regazzetti, C.; Robert, G.; Laurent, K.; le Marchand-Brustel, Y.; Auberger, P.; Tanti, J.F.; Giorgetti-Peraldi, S.; Bost, F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011, 71, 4366–4372. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Gong, H.; Wang, Y.; Guo, S.; Liu, K. AMPK/mTOR-Mediated inhibition of survivin partly contributes to metformin-induced apoptosis in human gastric cancer cell. Cancer Biol. Ther. 2015, 16, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martinez, J.M.; Moran, J.; Clarke, R.G.; Gray, A.; Cosulich, S.C.; Chresta, C.M.; Alessi, D.R. Ku-0063794 is a specific inhibitor of the mammalian target of Rapamycin (mTOR). Biochem. J. 2009, 421, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Feng, W.; Zhang, S.; Bian, K.; Yang, Y.; Fang, C.; Chen, M.; Yang, J.; Zou, X. Metformin inhibits gastric cancer via the inhibition of HIF1α/PKM2 signaling. Am. J. Cancer Res. 2015, 5, 1423–1434. [Google Scholar] [PubMed]
- Bridges, H.R.; Jones, A.J.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Finlay, D.K.; Rosenzweig, E.; Sinclair, L.V.; Feijoo-Carnero, C.; Hukelmann, J.L.; Rolf, J.; Panteleyev, A.A.; Okkenhaug, K.; Cantrell, D.A. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 2012, 209, 2441–2453. [Google Scholar] [CrossRef] [PubMed]
- Fingar, D.C.; Blenis, J. Target of Rapamycin (TOR): An integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene 2004, 23, 3151–3171. [Google Scholar] [CrossRef] [PubMed]
- Khaleghpour, K.; Pyronnet, S.; Gingras, A.C.; Sonenberg, N. Translational homeostasis: Eukaryotic translation initiation factor 4E control of 4E-binding protein 1 and P70 S6 kinase activities. Mol. Cell. Biol. 1999, 19, 4302–4310. [Google Scholar] [CrossRef] [PubMed]
- Nobukini, T.; Thomas, G. The mTOR/S6k signalling pathway: The role of the TSC1/2 tumour suppressor complex and the proto-oncogene Rheb. Novartis Found. Symp. 2005, 262, 148–154. [Google Scholar]
- Montagne, J.; Stewart, M.J.; Stocker, H.; Hafen, E.; Kozma, S.C.; Thomas, G. Drosophila S6 kinase: A regulator of cell size. Science 1999, 285, 2126–2129. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Chen, X.; Ma, J.; Peng, H.; Wang, F.; Zha, X.; Wang, Y.; Jing, Y.; Yang, H.; Chen, R.; et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc. Natl. Acad. Sci. USA 2011, 108, 4129–4134. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, K.; Hao, M. Metformin Inhibits TGF-β1-Induced Epithelial-to-Mesenchymal Transition via PKM2 Relative-mTOR/p70s6k Signaling Pathway in Cervical Carcinoma Cells. Int. J. Mol. Sci. 2016, 17, 2000. https://doi.org/10.3390/ijms17122000
Cheng K, Hao M. Metformin Inhibits TGF-β1-Induced Epithelial-to-Mesenchymal Transition via PKM2 Relative-mTOR/p70s6k Signaling Pathway in Cervical Carcinoma Cells. International Journal of Molecular Sciences. 2016; 17(12):2000. https://doi.org/10.3390/ijms17122000
Chicago/Turabian StyleCheng, Keyan, and Min Hao. 2016. "Metformin Inhibits TGF-β1-Induced Epithelial-to-Mesenchymal Transition via PKM2 Relative-mTOR/p70s6k Signaling Pathway in Cervical Carcinoma Cells" International Journal of Molecular Sciences 17, no. 12: 2000. https://doi.org/10.3390/ijms17122000
APA StyleCheng, K., & Hao, M. (2016). Metformin Inhibits TGF-β1-Induced Epithelial-to-Mesenchymal Transition via PKM2 Relative-mTOR/p70s6k Signaling Pathway in Cervical Carcinoma Cells. International Journal of Molecular Sciences, 17(12), 2000. https://doi.org/10.3390/ijms17122000