
Macromolecular Interactions Control Structural and Thermal Properties of Regenerated Tri-Component Blended Films

Abstract
:
1. Introduction
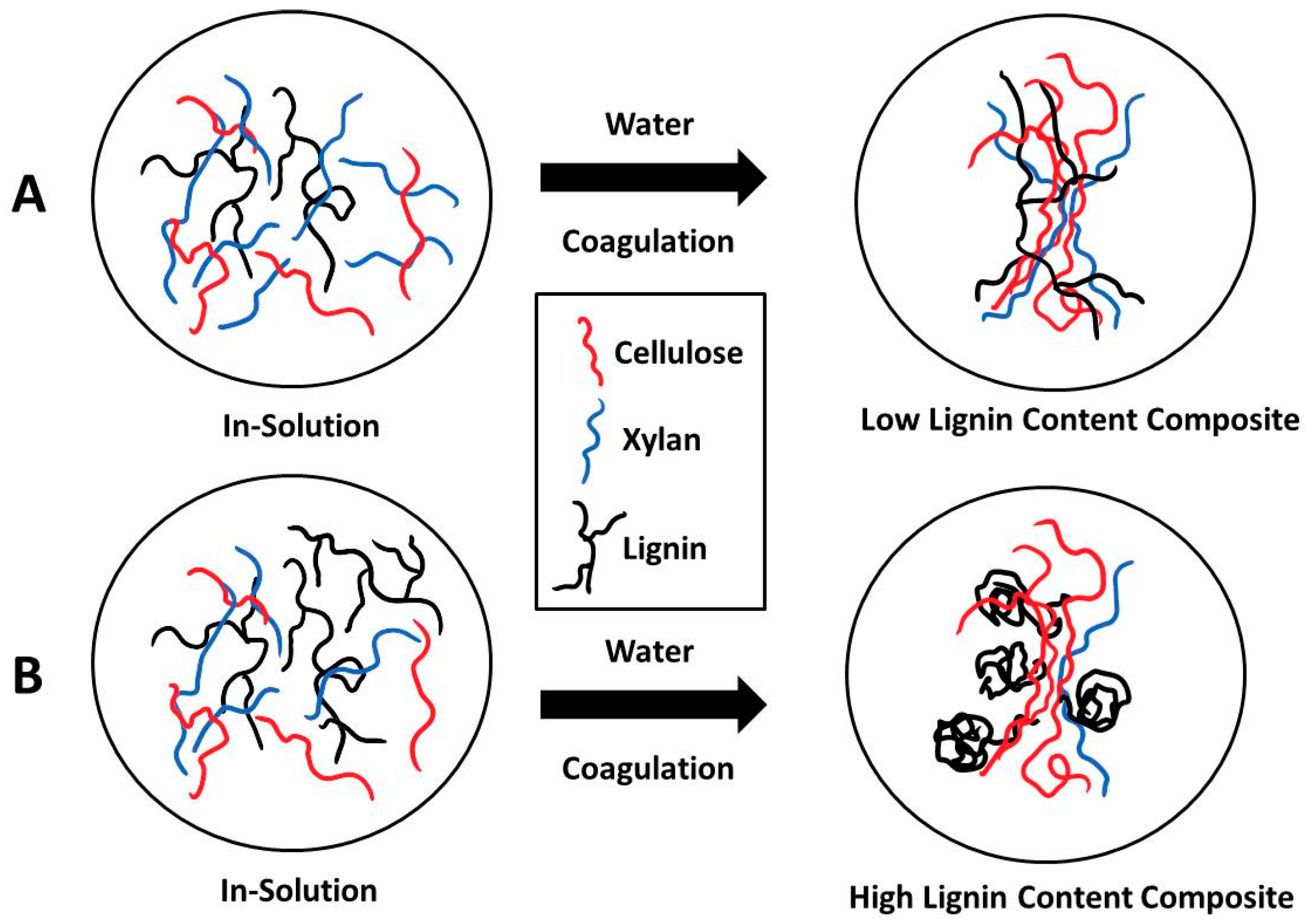
2. Results and Discussion
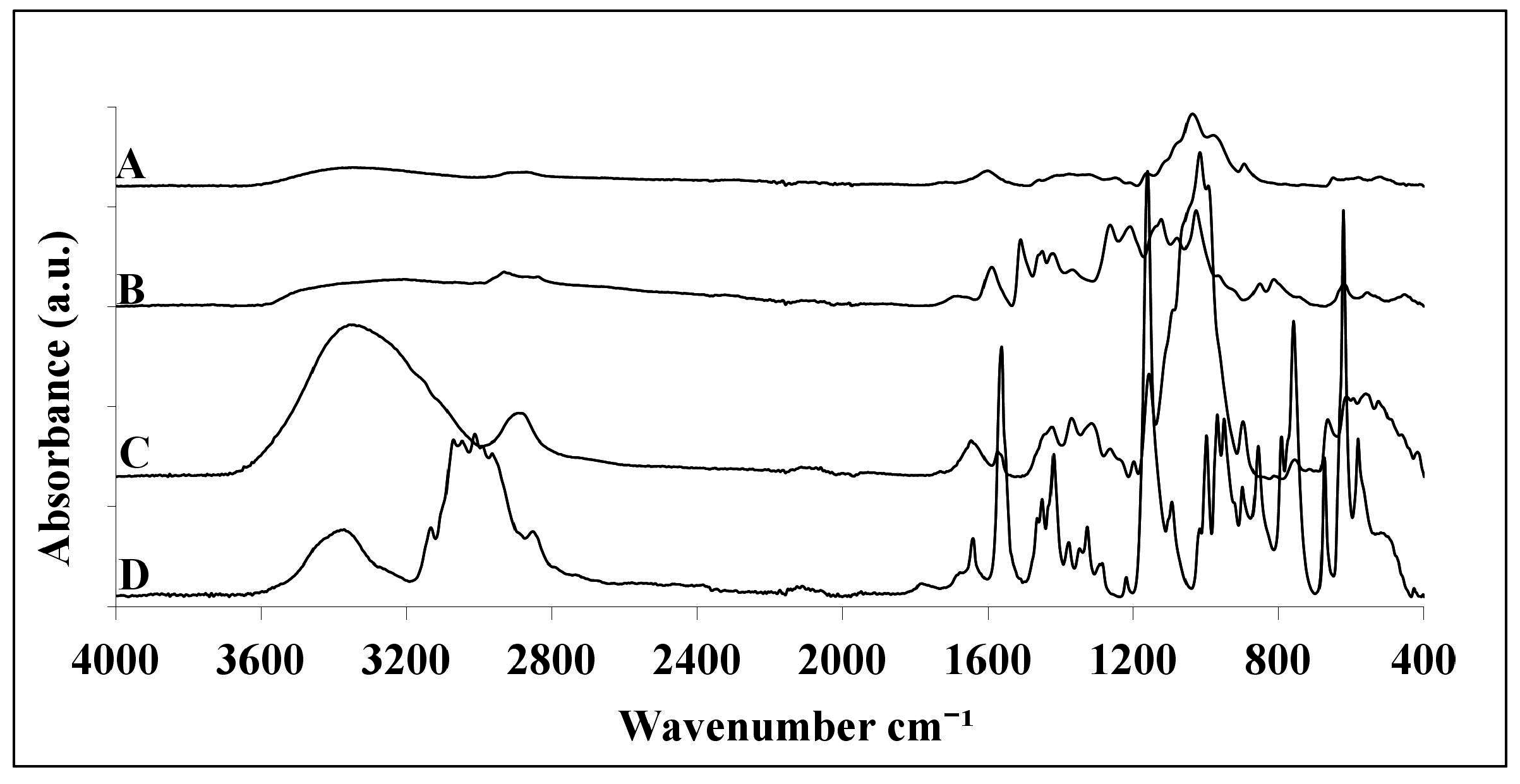
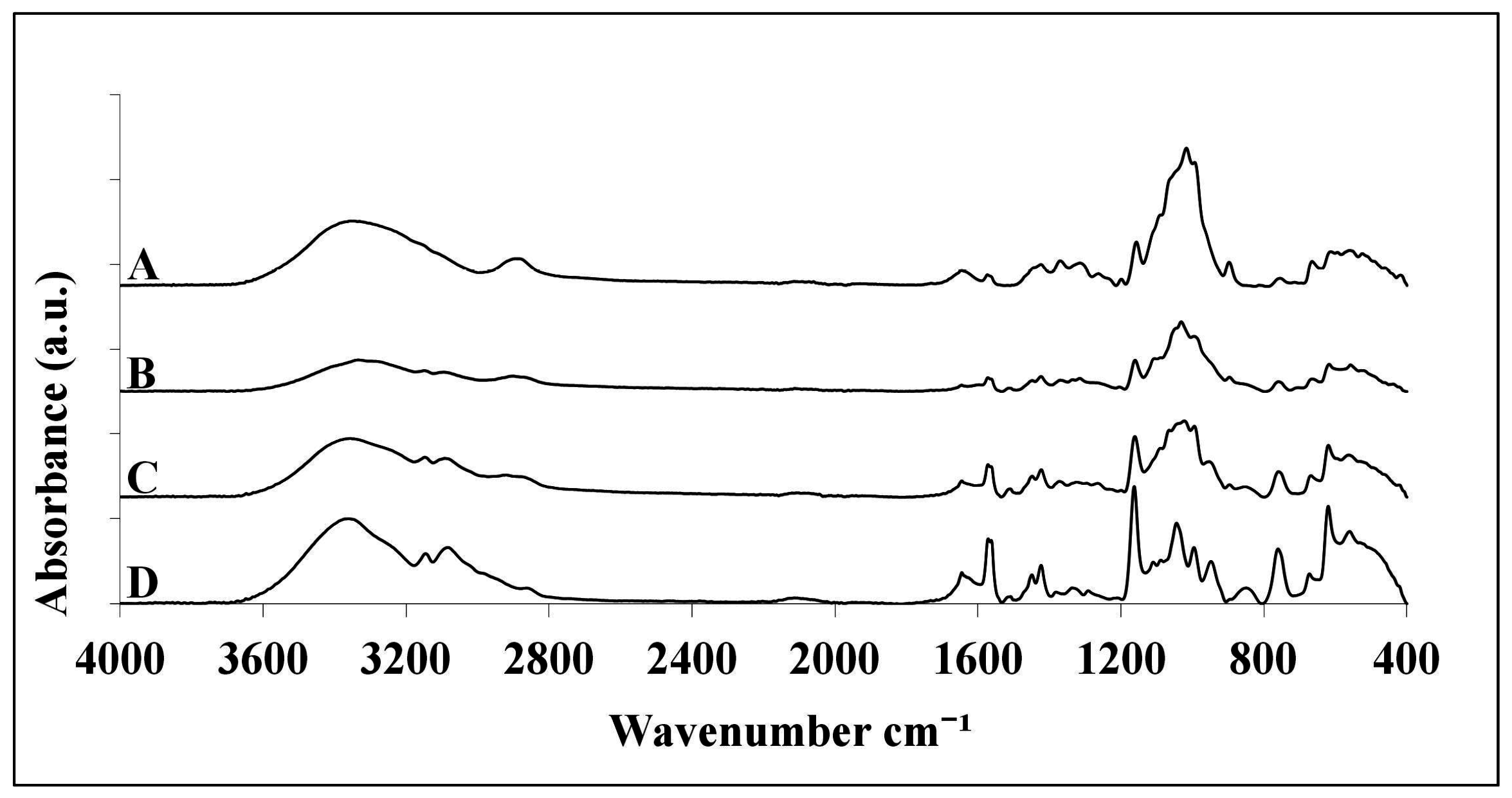
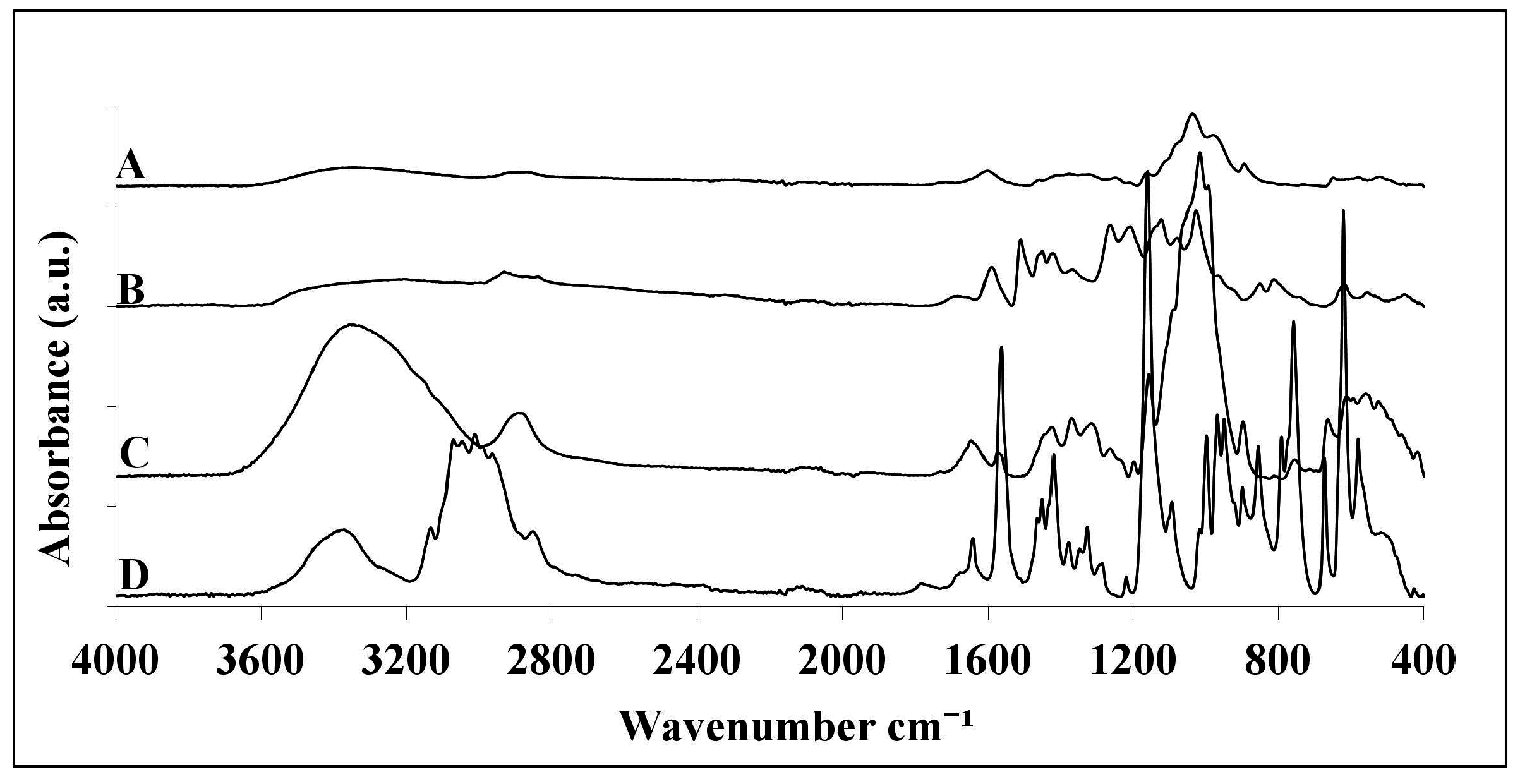
2.1. ATR-FTIR Analysis
2.2. Morphological Analysis
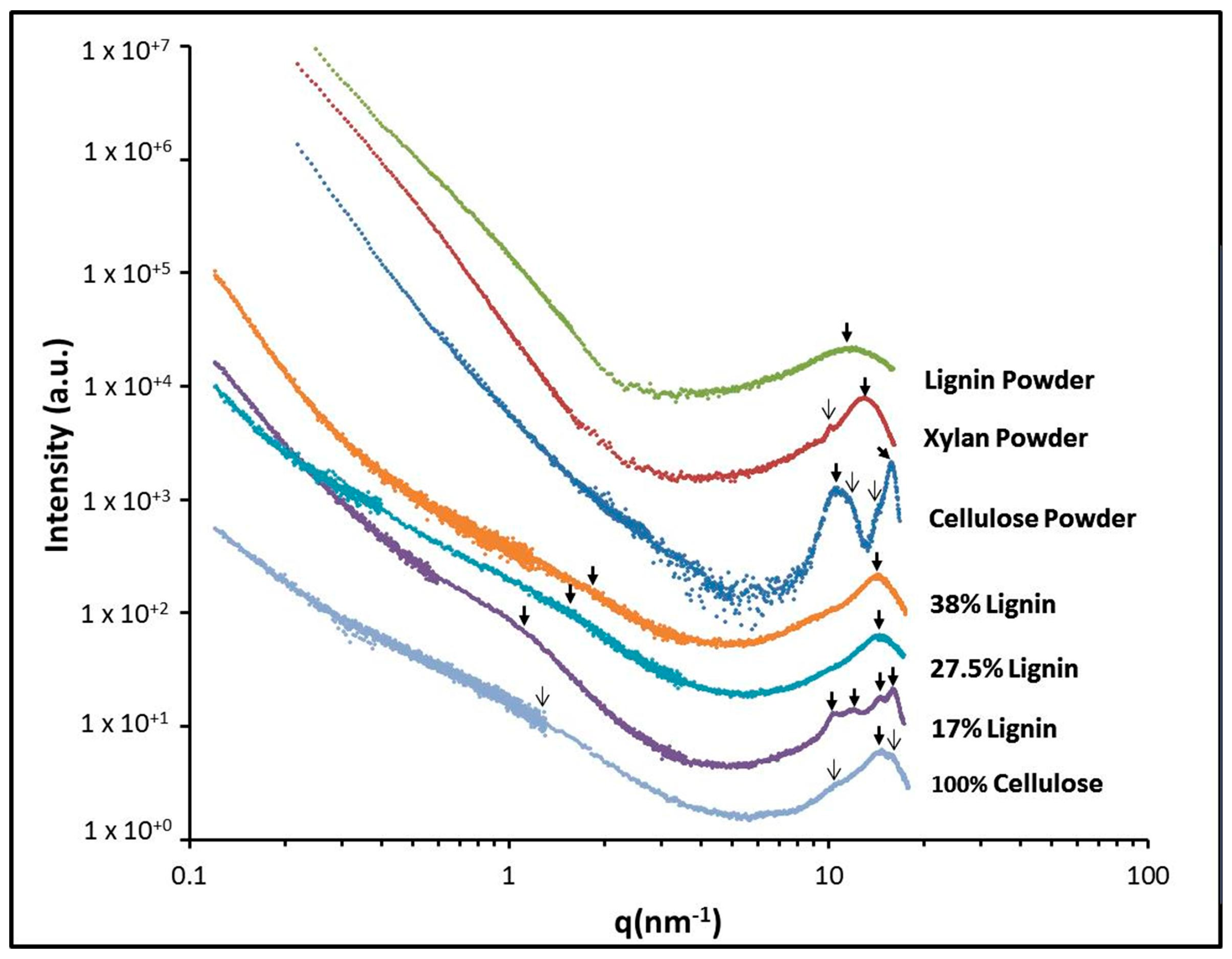
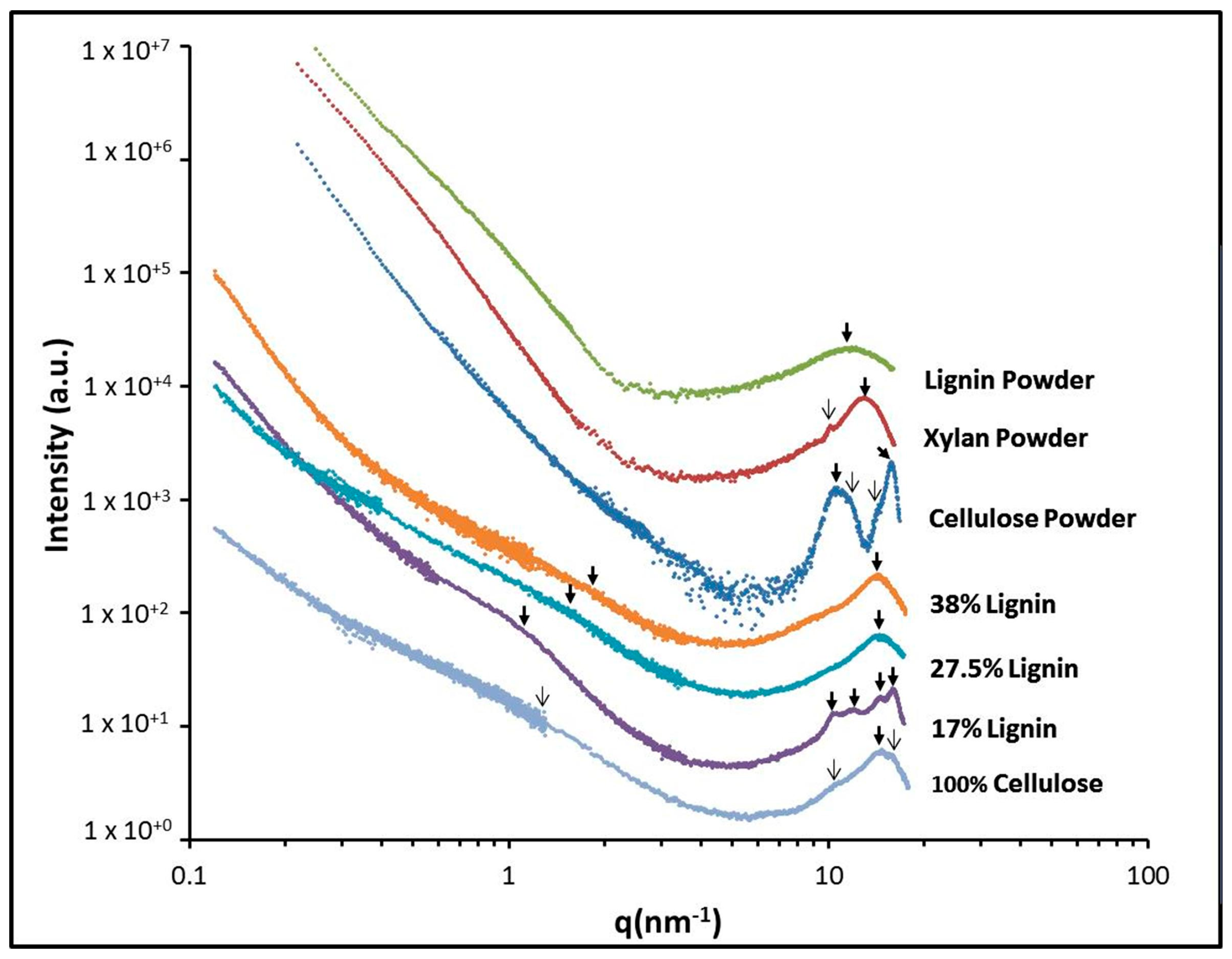
2.3. X-ray Scattering
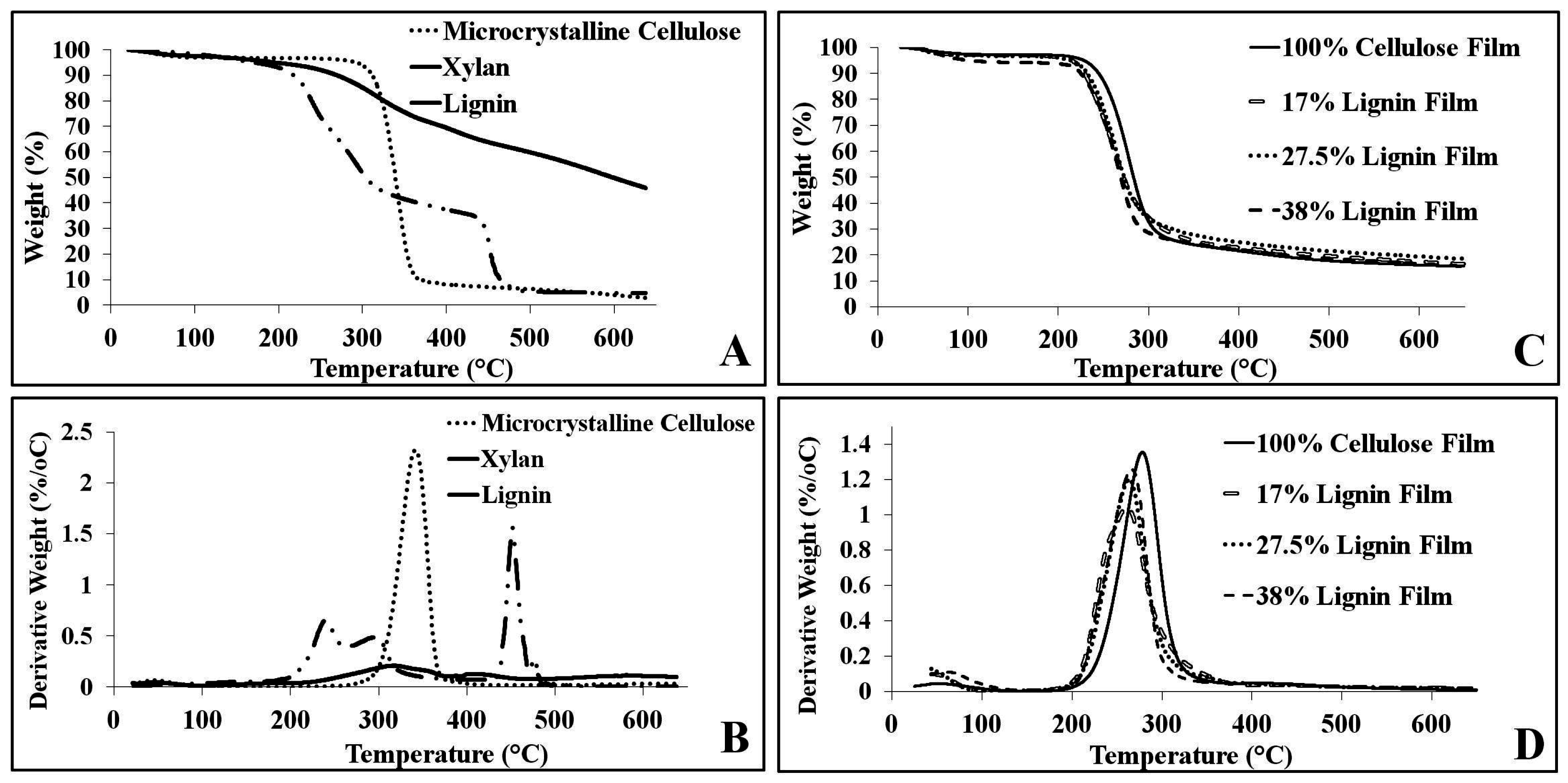
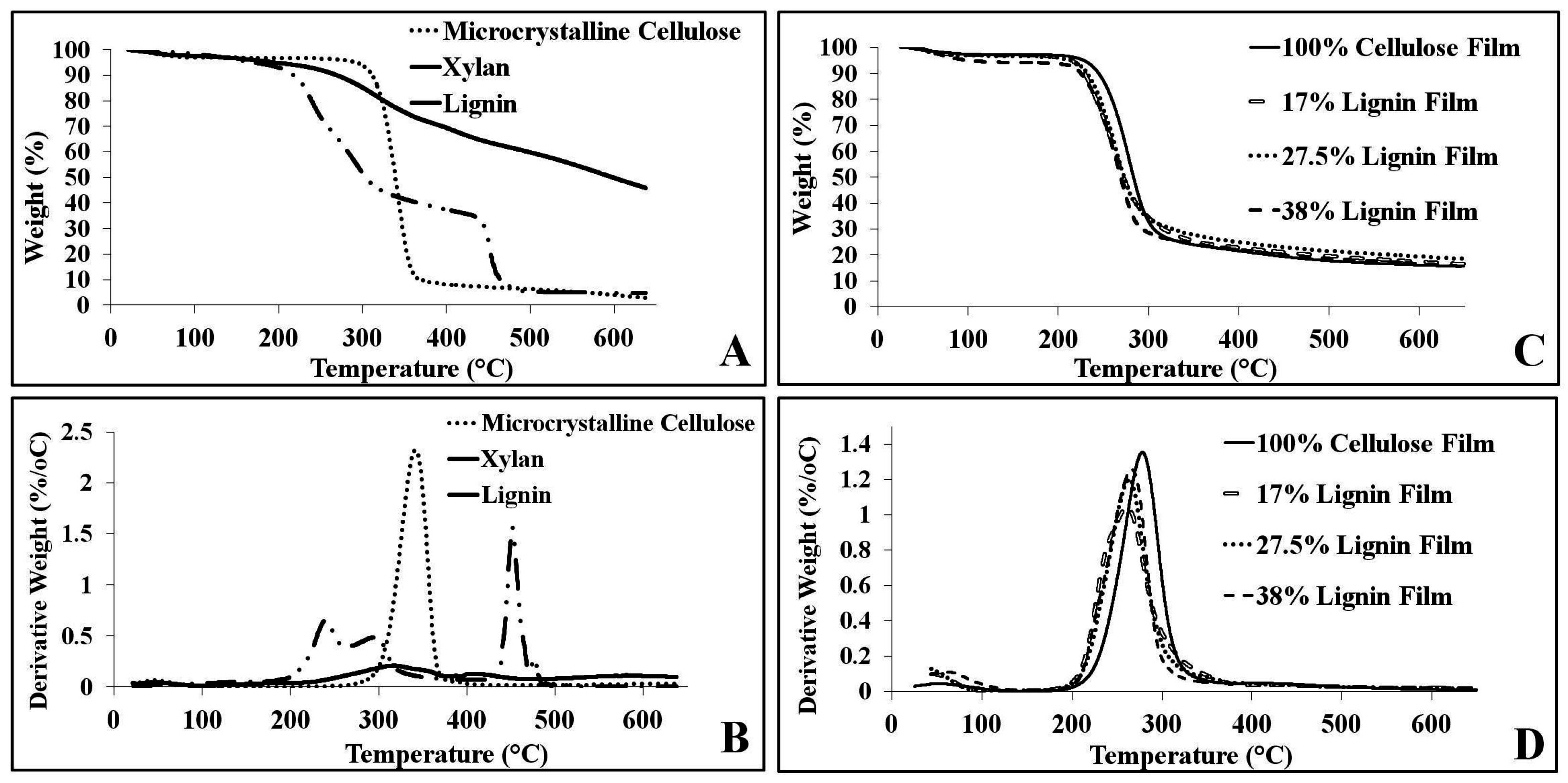
2.4. Thermal Analysis
3. Materials and Methods
3.1. Materials
3.2. Film Regeneration
3.3. Attenuated Total Reflectance Fourier Transform Infrared (ATR-FTIR)
3.4. Scanning Electron Microscope (SEM) and Atomic Force Microscopy (AFM)
3.5. X-ray Scattering
3.6. Thermogravimetric Analysis (TGA)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mohanty, A.K.; Misra, M.; Hinrichsen, G. Biofibres, biodegradable polymers and biocomposites: An overview. Macromol. Mater. Eng. 2000, 276–277, 1–24. [Google Scholar] [CrossRef]
- Ragauskas, A.J.; Williams, C.K.; Davison, B.H.; Britovsek, G.; Cairney, J.; Eckert, C.A.; Frederick, W.J., Jr.; Hallett, J.P.; Leak, D.J.; Liotta, C.L.; et al. The path forward for biofuels and biomaterials. Science 2006, 311, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, H.P.; Tharanathan, R.N. Carbohydrates—The renewable raw materials of high biotechnological value. Crit. Rev. Biotechnol. 2003, 23, 149–173. [Google Scholar] [CrossRef] [PubMed]
- Anwar, Z.; Gulfraz, M.; Irshad, M. Agro-industrial lignocellulosic biomass a key to unlock the future bio-energy: A brief review. J. Radiat. Res. Appl. Sci. 2014, 7, 163–173. [Google Scholar] [CrossRef]
- Kotarska, K.; Świerczyńska, A.; Dziemianowicz, W. Study on the decomposition of lignocellulosic biomass and subjecting it to alcoholic fermentation: Study on the decomposition of lignocellulosic biomass. Renew. Energy 2015, 75, 389–394. [Google Scholar] [CrossRef]
- Solomon, B.D.; Barnes, J.R.; Halvorsen, K.E. Grain and cellulosic ethanol: History, economics, and energy policy. Biomass Bioenergy 2007, 31, 416–425. [Google Scholar] [CrossRef]
- Vancov, T.; Alston, A.-S.; Brown, T.; McIntosh, S. Use of ionic liquids in converting lignocellulosic material to biofuels. Renew. Energy 2012, 45, 1–6. [Google Scholar] [CrossRef]
- Zheng, J.; Rehmann, L. Extrusion pretreatment of lignocellulosic biomass: A review. Int. J. Mol. Sci. 2014, 15, 18967. [Google Scholar] [CrossRef] [PubMed]
- Entcheva, E.; Bien, H.; Yin, L.; Chung, C.Y.; Farrell, M.; Kostov, Y. Functional cardiac cell constructs on cellulose-based scaffolding. Biomaterials 2004, 25, 5753–5762. [Google Scholar] [CrossRef] [PubMed]
- Novotna, K.; Havelka, P.; Sopuch, T.; Kolarova, K.; Vosmanska, V.; Lisa, V.; Svorcik, V.; Bacakova, L. Cellulose-based materials as scaffolds for tissue engineering. Cellulose 2013, 20, 2263–2278. [Google Scholar] [CrossRef]
- Schmer, M.R.; Vogel, K.P.; Mitchell, R.B.; Perrin, R.K. Net energy of cellulosic ethanol from switchgrass. Proc. Natl. Acad. Sci. USA 2008, 105, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Svensson, A.; Nicklasson, E.; Harrah, T.; Panilaitis, B.; Kaplan, D.L.; Brittberg, M.; Gatenholm, P. Bacterial cellulose as a potential scaffold for tissue engineering of cartilage. Biomaterials 2005, 26, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Helbert, W.; Nishiyama, Y.; Okano, T.; Sugiyama, J. Molecular imaging of halocynthia papillosa cellulose. J. Struct. Biol. 1998, 124, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Atalla, R.H.; Vanderhart, D.L. Native cellulose: A composite of two distinct crystalline forms. Science 1984, 223, 283–285. [Google Scholar] [CrossRef] [PubMed]
- Chanliaud, E.; Gidley, M.J. In vitro synthesis and properties of pectin/Acetobacter xylinus cellulose composites. Plant J. 1999, 20, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Marriott, P.E.; Gomez, L.D.; McQueen-Mason, S.J. Unlocking the potential of lignocellulosic biomass through plant science. New Phytol. 2016, 209, 1366–1381. [Google Scholar] [CrossRef] [PubMed]
- Chen, H. Chemical Composition and Structure of Natural Lignocellulose. In Biotechnology of Lignocellulose: Theory and Practice; Springer: Dordrecht, The Netherlands, 2014; pp. 25–71. [Google Scholar]
- Brice, R.E.; Morrison, I.M. The degradation of isolated hemicelluloses and lignin-hemicellulose complexes by cell-free, rumen hemicellulases. Carbohydr. Res. 1982, 101, 93–100. [Google Scholar] [CrossRef]
- Morrison, I.M. Structural investigations on the lignin–carbohydrate complexes of Lolium perenne. Biochem. J. 1974, 139, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Choi, Y.S.; Yoo, C.G.; Kim, T.H.; Brown, R.C.; Shanks, B.H. Cellulose–hemicellulose and cellulose–lignin interactions during fast pyrolysis. ACS Sustain. Chem. Eng. 2015, 3, 293–301. [Google Scholar] [CrossRef]
- Novaes, E.; Kirst, M.; Chiang, V.; Winter-Sederoff, H.; Sederoff, R. Lignin and biomass: A negative correlation for wood formation and lignin content in trees. Plant Physiol. 2010, 154, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Su, J.-F.; Yuan, X.-Y.; Huang, Z.; Wang, X.-Y.; Lu, X.-Z.; Zhang, L.-D.; Wang, S.-B. Physicochemical properties of soy protein isolate/carboxymethyl cellulose blend films crosslinked by Maillard reactions: Color, transparency and heat-sealing ability. Mater. Sci. Eng. C 2012, 32, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Chang, C.; Zhang, L. Properties and applications of biodegradable transparent and photoluminescent cellulose films prepared via a green process. Green Chem. 2009, 11, 177–184. [Google Scholar] [CrossRef]
- Islam, R.; Sparling, R.; Cicek, N.; Levin, D. Optimization of influential nutrients during direct cellulose fermentation into hydrogen by clostridium thermocellum. Int. J. Mol. Sci. 2015, 16, 3116–3132. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Wu, Y.; Chen, Q.; Yu, Z.; Wang, C.; Jin, S.; Ding, Y.; Wu, G. Dissolution of cellulose with ionic liquids and its application: A mini-review. Green Chem. 2006, 8, 325–327. [Google Scholar] [CrossRef]
- Cheng, Y.; Lu, J.; Liu, S.; Zhao, P.; Lu, G.; Chen, J. The preparation, characterization and evaluation of regenerated cellulose/collagen composite hydrogel films. Carbohydr. Polym. 2014, 107, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Pang, J.; Zhang, X.; Wu, Y.; Sun, R. Regenerated cellulose film with enhanced tensile strength prepared with ionic liquid 1-ethyl-3-methylimidazolium acetate (EMIMAc). Cellulose 2013, 20, 1391–1399. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, H.; Li, Z.; Lu, X.; Zhang, X.; Zhang, S.; Zhou, K. Characterization of the regenerated cellulose films in ionic liquids and rheological properties of the solutions. Mater. Chem. Phys. 2011, 128, 220–227. [Google Scholar] [CrossRef]
- Pang, J.-H.; Liu, X.; Wu, M.; Wu, Y.-Y.; Zhang, X.-M.; Sun, R.-C. Fabrication and characterization of regenerated cellulose films using different ionic liquids. J. Spectrosc. 2014, 2014, 214057. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, J.; Zhang, J.; He, J. 1-Allyl-3-methylimidazolium chloride room temperature ionic liquid: A new and powerful nonderivatizing solvent for cellulose. Macromolecules 2005, 38, 8272–8277. [Google Scholar] [CrossRef]
- Mai, N.L.; Koo, Y.-M. Computer-aided design of ionic liquids for high cellulose dissolution. ACS Sustain. Chem. Eng. 2016, 4, 541–547. [Google Scholar] [CrossRef]
- Zhang, J.; Luo, N.; Zhang, X.; Xu, L.; Wu, J.; Yu, J.; He, J.; Zhang, J. All-cellulose nanocomposites reinforced with in situ retained cellulose nanocrystals during selective dissolution of cellulose in an ionic liquid. ACS Sustain. Chem. Eng. 2016, 4, 4417–4423. [Google Scholar] [CrossRef]
- Wang, Q.; Yang, Y.; Chen, X.; Shao, Z. Investigation of rheological properties and conformation of silk fibroin in the solution of AmimCl. Biomacromolecules 2012, 13, 1875–1881. [Google Scholar] [CrossRef] [PubMed]
- Isogai, A.; Atalla, R.H. Dissolution of cellulose in aqueous NaOH solutions. Cellulose 1998, 5, 309–319. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Q.; Yang, Y.; Shao, Z. Effect of various dissolution systems on the molecular weight of regenerated silk fibroin. Biomacromolecules 2013, 14, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Varanasi, P.; Li, C.; Liu, H.; Melnichenko, Y.B.; Simmons, B.A.; Kent, M.S.; Singh, S. Transition of cellulose crystalline structure and surface morphology of biomass as a function of ionic liquid pretreatment and its relation to enzymatic hydrolysis. Biomacromolecules 2011, 12, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Samayam, I.P.; Hanson, B.L.; Langan, P.; Schall, C.A. Ionic-liquid induced changes in cellulose structure associated with enhanced biomass hydrolysis. Biomacromolecules 2011, 12, 3091–3098. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Wang, Q.; Wen, J.; Chen, X.; Shao, Z. Preparation and characterization of transparent silk fibroin/cellulose blend films. Polymer 2013, 54, 5035–5042. [Google Scholar] [CrossRef]
- Chundawat, S.P.S.; Bellesia, G.; Uppugundla, N.; da Costa Sousa, L.; Gao, D.; Cheh, A.M.; Agarwal, U.P.; Bianchetti, C.M.; Phillips, G.N., Jr.; Langan, P.; et al. Restructuring the crystalline cellulose hydrogen bond network enhances its depolymerization rate. J. Am. Chem. Soc. 2011, 133, 11163–11174. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.B.; Spear, S.K.; Holbrey, J.D.; Rogers, R.D. Production of bioactive cellulose films reconstituted from ionic liquids. Biomacromolecules 2004, 5, 1379–1384. [Google Scholar] [CrossRef] [PubMed]
- Paul, U.; Fragouli, D.; Bayer, I.; Athanassiou, A. Functionalized Cellulose Networks for Efficient Oil Removal from Oil–Water Emulsions. Polymers 2016, 8, 52. [Google Scholar] [CrossRef]
- Kathirgamanathan, K.; Grigsby, W.J.; Al-Hakkak, J.; Edmonds, N.R. Two-dimensional FTIR as a tool to study the chemical interactions within cellulose-ionic liquid solutions. Int. J. Polym. Sci. 2015, 2015, 958653. [Google Scholar] [CrossRef]
- Yang, H.; Yan, R.; Chen, H.; Lee, D.H.; Zheng, C. Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel 2007, 86, 1781–1788. [Google Scholar] [CrossRef]
- Yuan, X.; Cheng, G. From cellulose fibrils to single chains: Understanding cellulose dissolution in ionic liquids. Phys. Chem. Chem. Phys. 2015, 17, 31592–31607. [Google Scholar] [CrossRef] [PubMed]
- Vainio, U.; Maximova, N.; Hortling, B.; Laine, J.; Stenius, P.; Simola, L.K.; Gravitis, J.; Serimaa, R. Morphology of dry lignins and size and shape of dissolved kraft lignin particles by X-ray scattering. Langmuir 2004, 20, 9736–9744. [Google Scholar] [CrossRef] [PubMed]
- Cara, P.D.; Pagliaro, M.; Elmekawy, A.; Brown, D.R.; Verschuren, P.; Shiju, N.R.; Rothenberg, G. Hemicellulose hydrolysis catalysed by solid acids. Catal. Sci. Technol. 2013, 3, 2057–2061. [Google Scholar] [CrossRef]
- Ibbett, R.; Gaddipati, S.; Hill, S.; Tucker, G. Structural reorganisation of cellulose fibrils in hydrothermally deconstructed lignocellulosic biomass and relationships with enzyme digestibility. Biotechnol. Biofuels 2013, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ioelovich, M. Nano-structural approach to description of enzymatic hydrolysis of pretreated biomass. J. Sci. Isr. Technol. Adv. 2011. Available online: http://www.polymateltd.com/files/SITA_no.4_Contents.pdf (accessed on 1 July 2016). [Google Scholar]
- Kamel, S. Nanotechnology and its applications in lignocellulosic composites, a mini review. Express Polym. Lett. 2007, 1, 546–575. [Google Scholar] [CrossRef]
- Kennedy, C.J.; Šturcová, A.; Jarvis, M.C.; Wess, T.J. Hydration effects on spacing of primary-wall cellulose microfibrils: A small angle X-ray scattering study. Cellulose 2007, 14, 401–408. [Google Scholar] [CrossRef]
- Gillis, P.; Mark, R.; Tang, R.-C. Elastic stiffness of crystalline cellulose in the folded-chain solid state. J. Mater. Sci. 1969, 4, 1003–1007. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, X.; Ding, X.; Lei, H.; Wang, Z. Preparing spherical lignin from rice husk. Bioprocess Biosyst. Eng. 2013, 36, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- De Silva, R.; Wang, X.; Byrne, N. Tri-component bio-composite materials prepared using an eco-friendly processing route. Cellulose 2013, 20, 2461–2468. [Google Scholar] [CrossRef]
- Pang, J.; Wu, M.; Zhang, Q.; Tan, X.; Xu, F.; Zhang, X.; Sun, R. Comparison of physical properties of regenerated cellulose films fabricated with different cellulose feedstocks in ionic liquid. Carbohydr. Polym. 2015, 121, 71–78. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Relative Concentration of Lignocellulose | ||||
|---|---|---|---|---|
| Experiment Number | 1 | 2 | 3 | 4 |
| Microcrystalline Cellulose | 100% | 45% | 45% | 45% |
| Lignin | 0% | 17% | 27.50% | 38% |
| Xylan | 0% | 38% | 27.50% | 17% |
| Sample | TOnset (°C) | TEnd (°C) | Wt Loss (%) | TΔMax (°C) | Maximum Rate Wt Loss (%/°C) |
|---|---|---|---|---|---|
| Microcrystalline Cellulose | 341.1 | 374.6 | 86.8 | 361.4 | 2.3% |
| Xylan | 221.8 | 458.5 | 89.2 | 451.0 | 1.6% |
| Lignin | 267.7 | 435.2 | 33.2 | 320.0 | 0.21% |
| 100% Cellulose Film | 249.7 | 300.4 | 77.8 | 278.1 | 1.4% |
| 17% Lignin Film | 228.1 | 293.8 | 75.9 | 261.1 | 1.0% |
| 27.5% Lignin Film | 235.5 | 289.1 | 73.7 | 263.5 | 1.2% |
| 38% Lignin Film | 235.7 | 286.9 | 74.2 | 266.5 | 1.3% |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewis, A.; Waters, J.C.; Stanton, J.; Hess, J.; Salas-de la Cruz, D. Macromolecular Interactions Control Structural and Thermal Properties of Regenerated Tri-Component Blended Films. Int. J. Mol. Sci. 2016, 17, 1989. https://doi.org/10.3390/ijms17121989
Lewis A, Waters JC, Stanton J, Hess J, Salas-de la Cruz D. Macromolecular Interactions Control Structural and Thermal Properties of Regenerated Tri-Component Blended Films. International Journal of Molecular Sciences. 2016; 17(12):1989. https://doi.org/10.3390/ijms17121989
Chicago/Turabian StyleLewis, Ashley, Joshua C. Waters, John Stanton, Joseph Hess, and David Salas-de la Cruz. 2016. "Macromolecular Interactions Control Structural and Thermal Properties of Regenerated Tri-Component Blended Films" International Journal of Molecular Sciences 17, no. 12: 1989. https://doi.org/10.3390/ijms17121989
APA StyleLewis, A., Waters, J. C., Stanton, J., Hess, J., & Salas-de la Cruz, D. (2016). Macromolecular Interactions Control Structural and Thermal Properties of Regenerated Tri-Component Blended Films. International Journal of Molecular Sciences, 17(12), 1989. https://doi.org/10.3390/ijms17121989