
DNA Damage-Inducible Transcript 4 Is an Innate Surveillant of Hair Follicular Stress in Vitamin D Receptor Knockout Mice and a Regulator of Wound Re-Epithelialization


Abstract
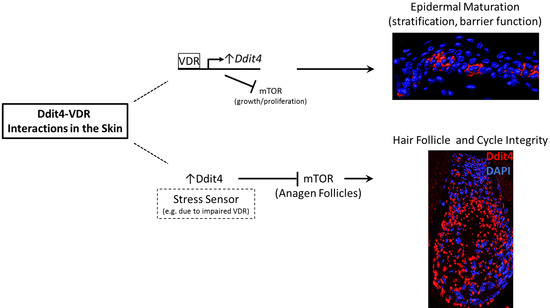
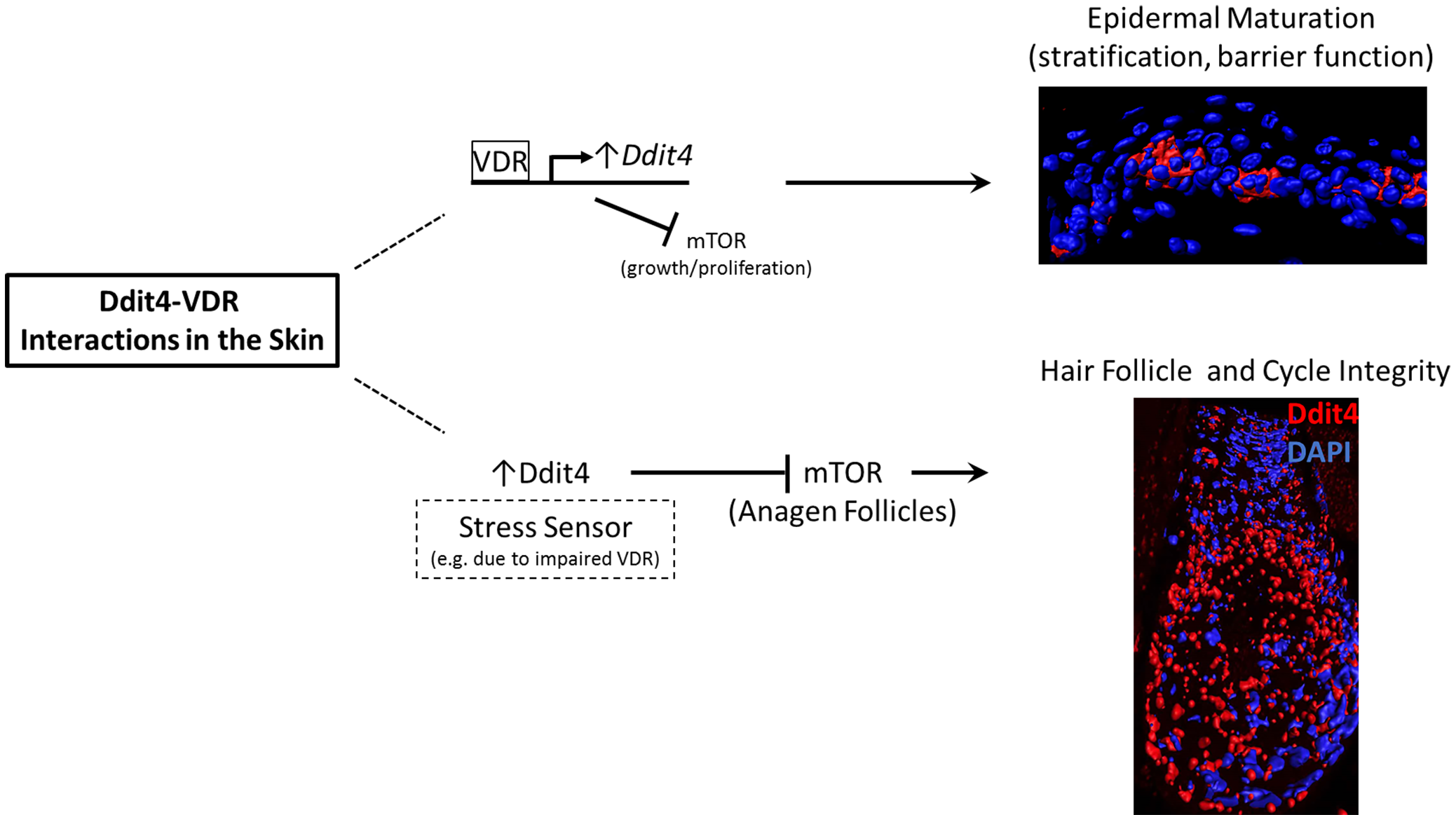
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
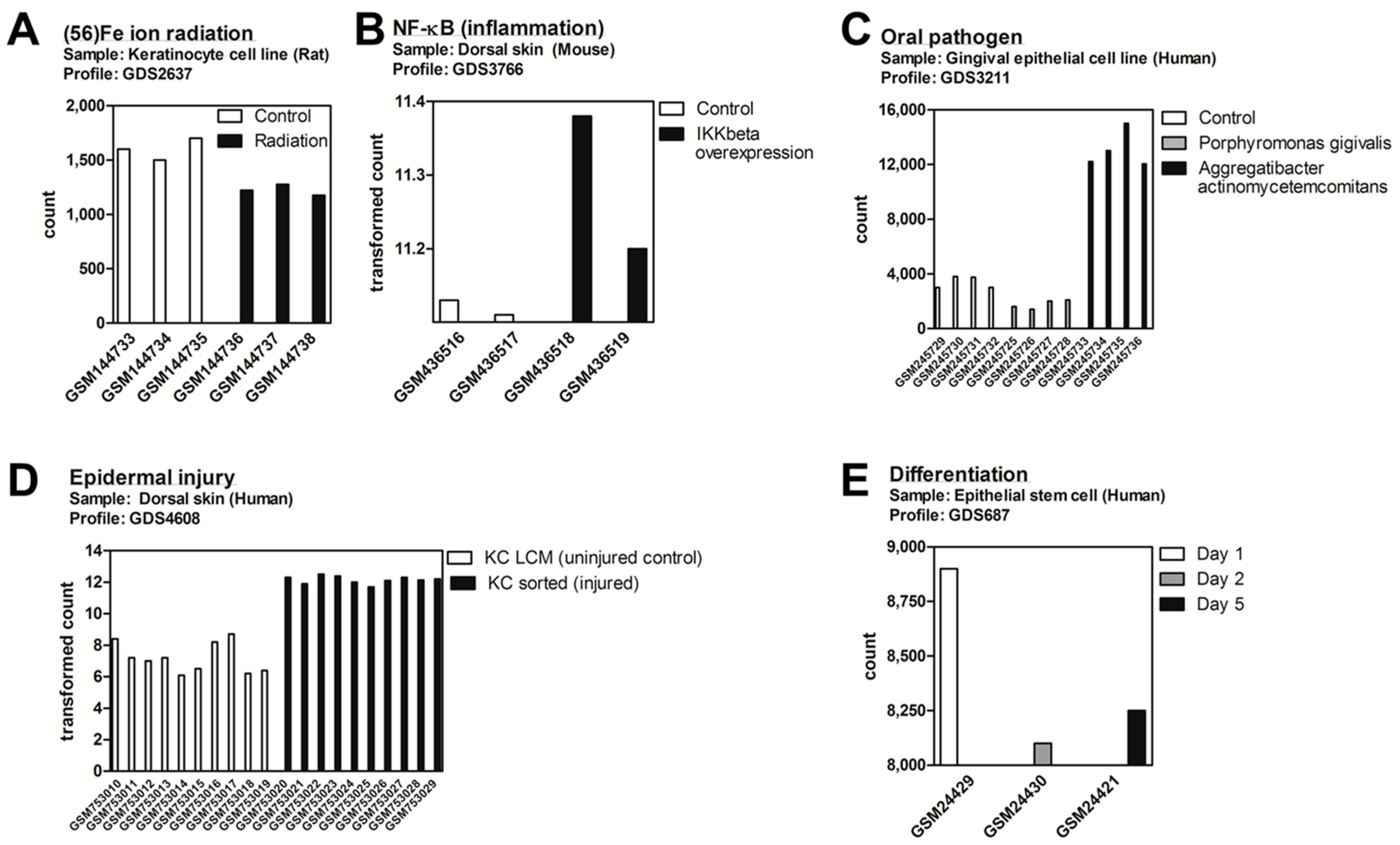
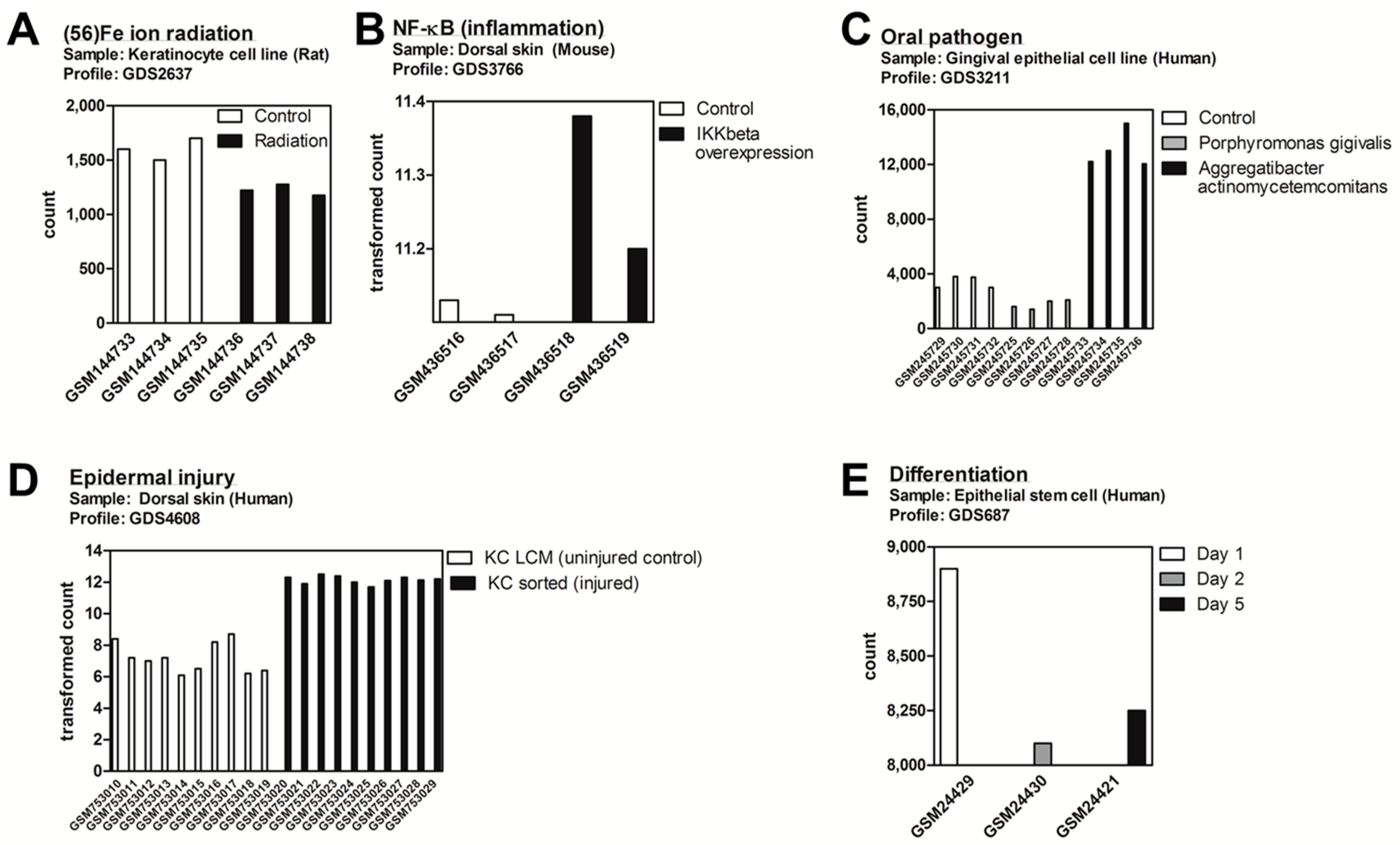
2.1. DDIT4/Ddit4 Is an Acute Phase Effector of Stress Related to Inflammatory and Other Immune Challenges to Physical or Bacterial Complications
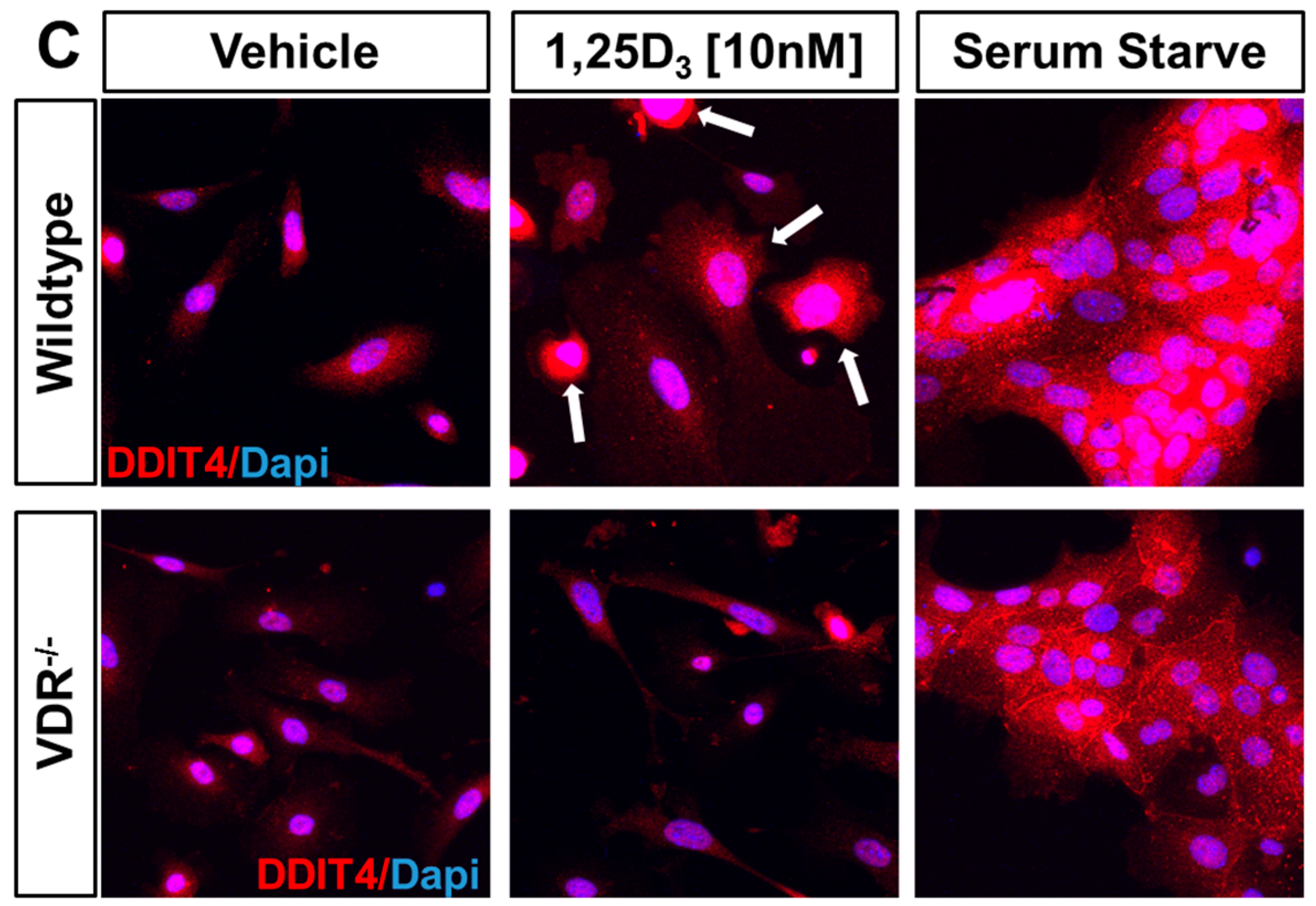
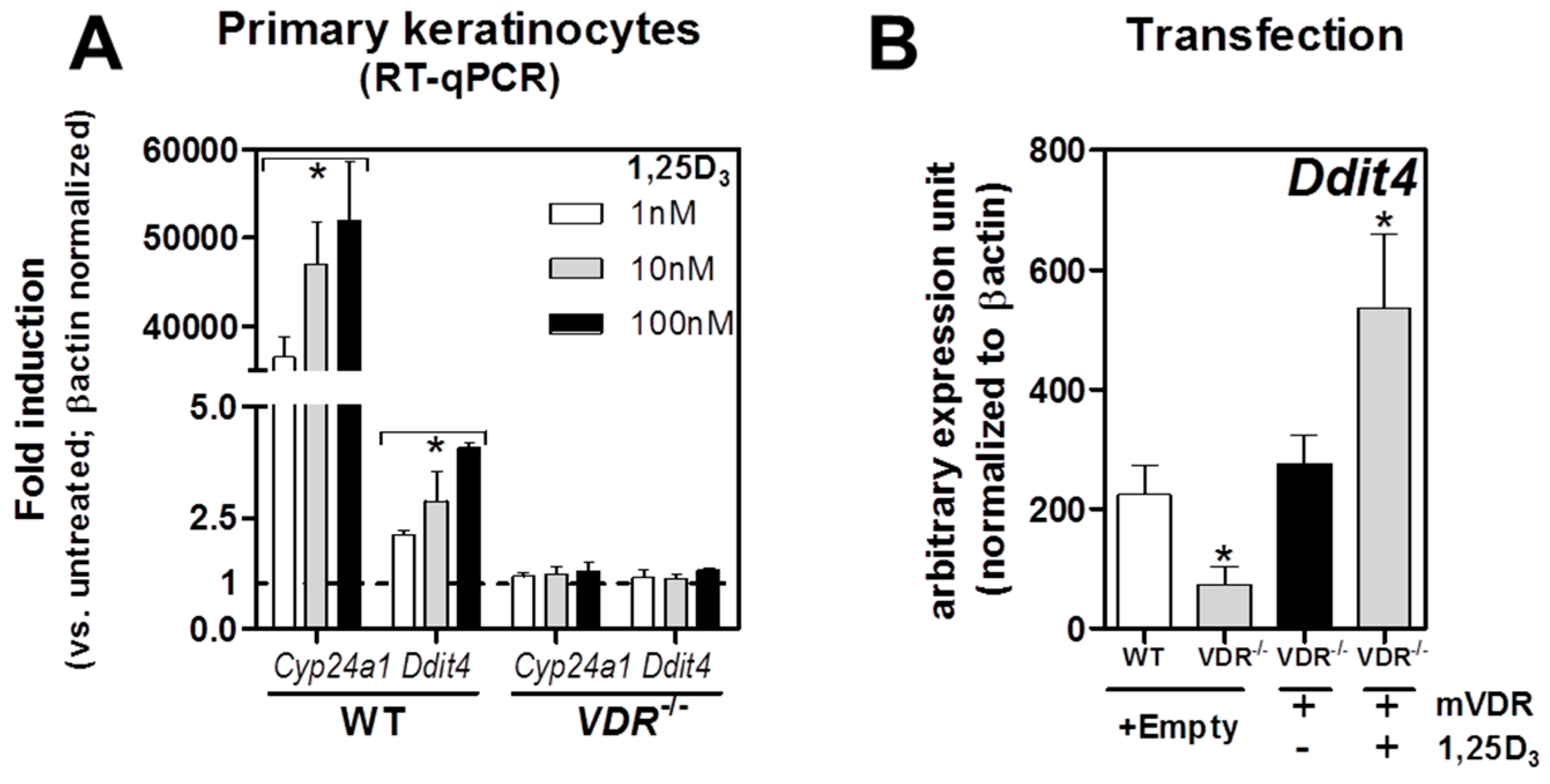
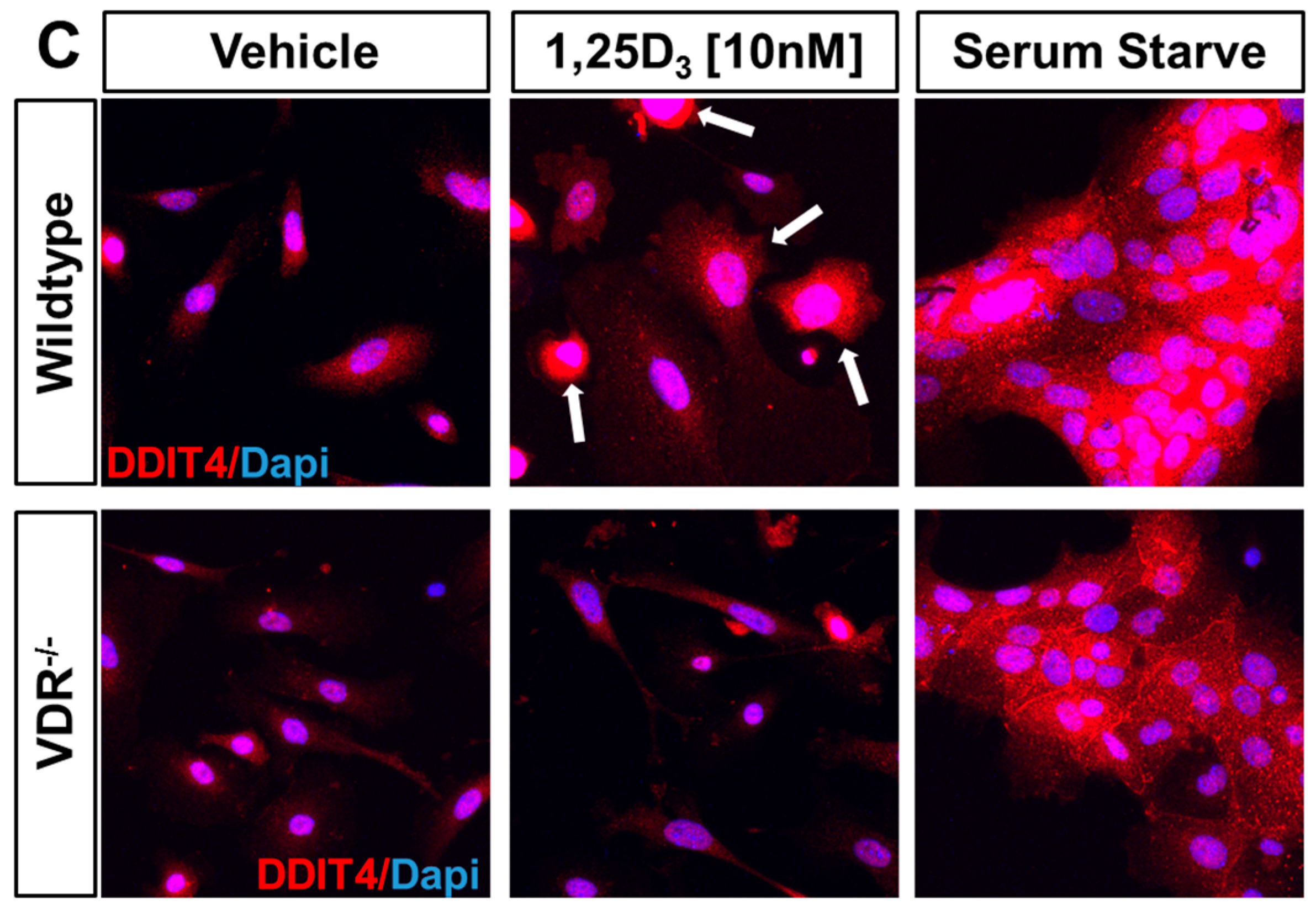
2.2. VDR Positively Regulates Ddit4 mRNA and Protein Expression in Primary Murine Epidermal Keratinocytes
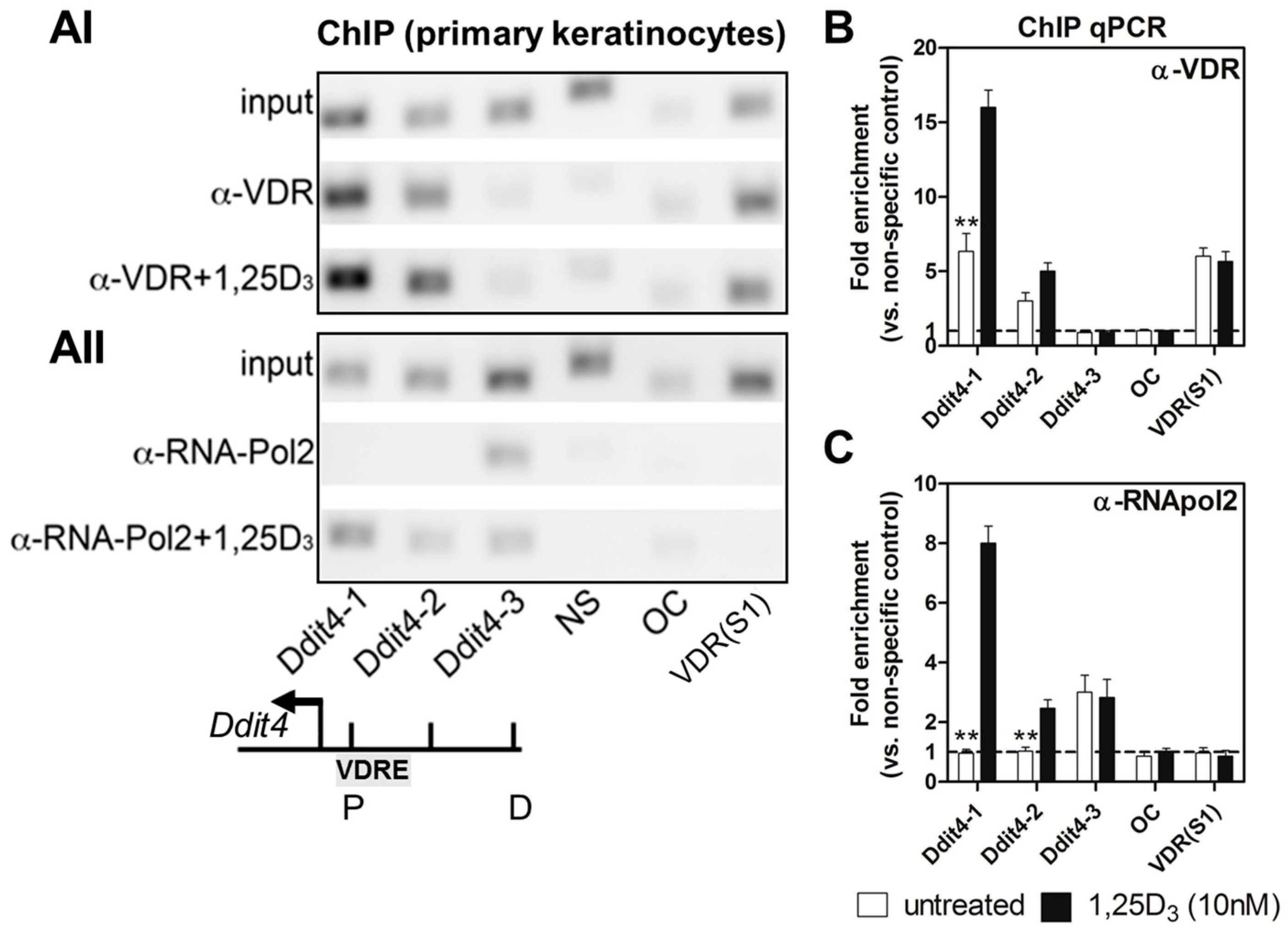
2.3. Ddit4 Is a Direct Transcriptional Effector of the Liganded VDR within Primary Epidermal Keratinocytes
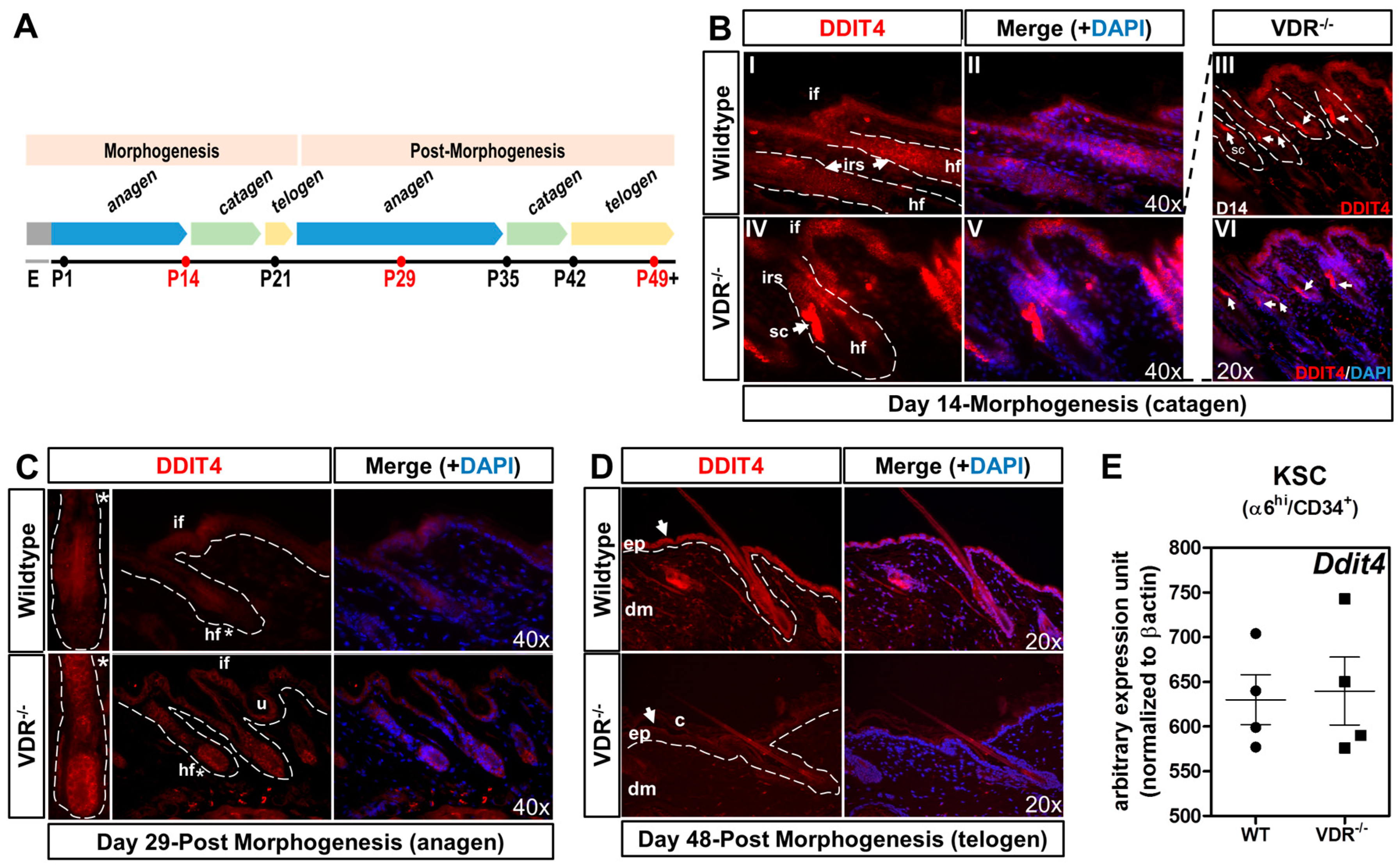
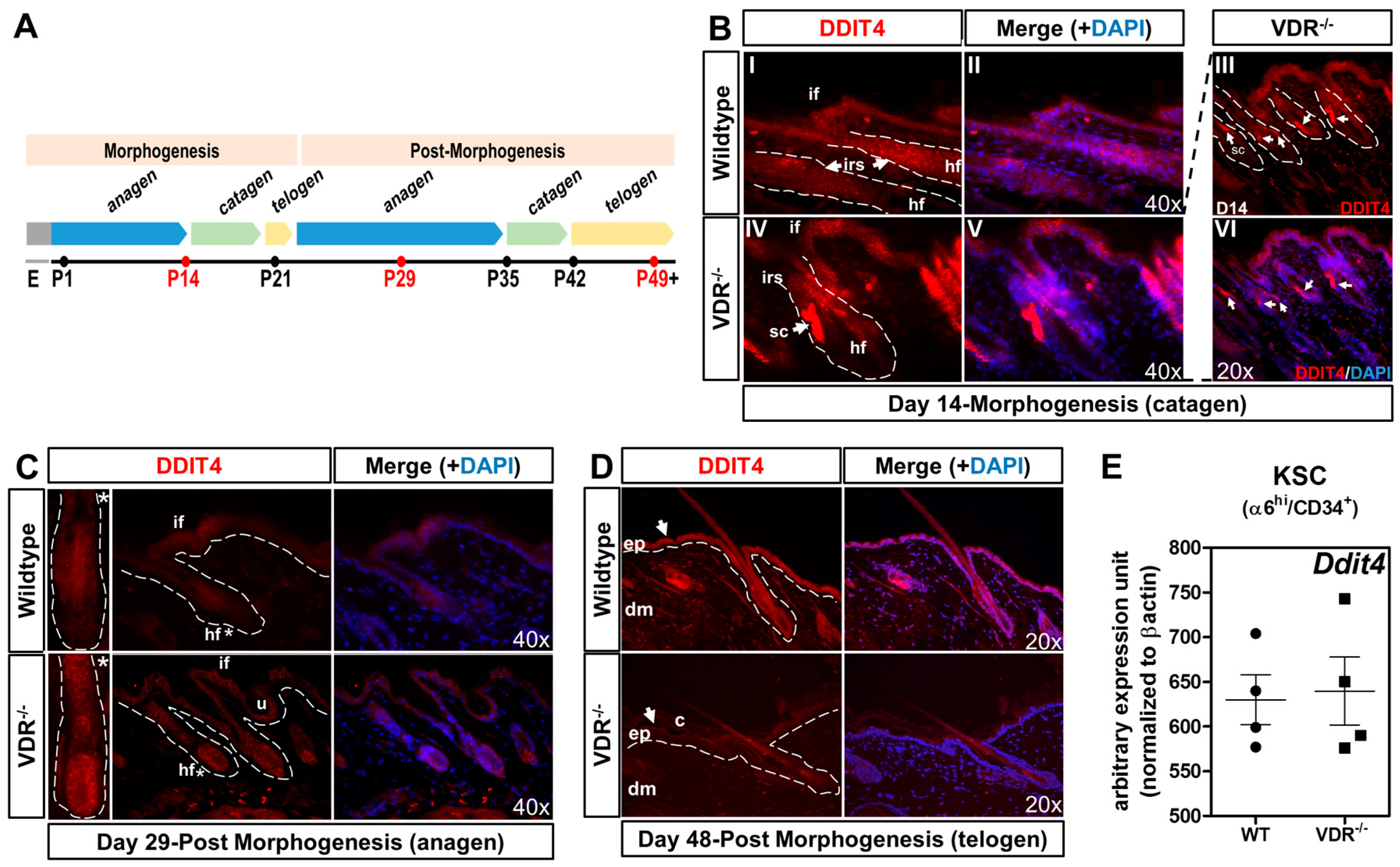
2.4. Formation of Ddit4-Positve Stress Compartments in VDR−/− Morphogenic Follicles and Reduced Ddit4 Epidermal Expression
2.5. The VDR Does Not Regulate Ddit4 within Bulge Keratinocyte Stem Cells
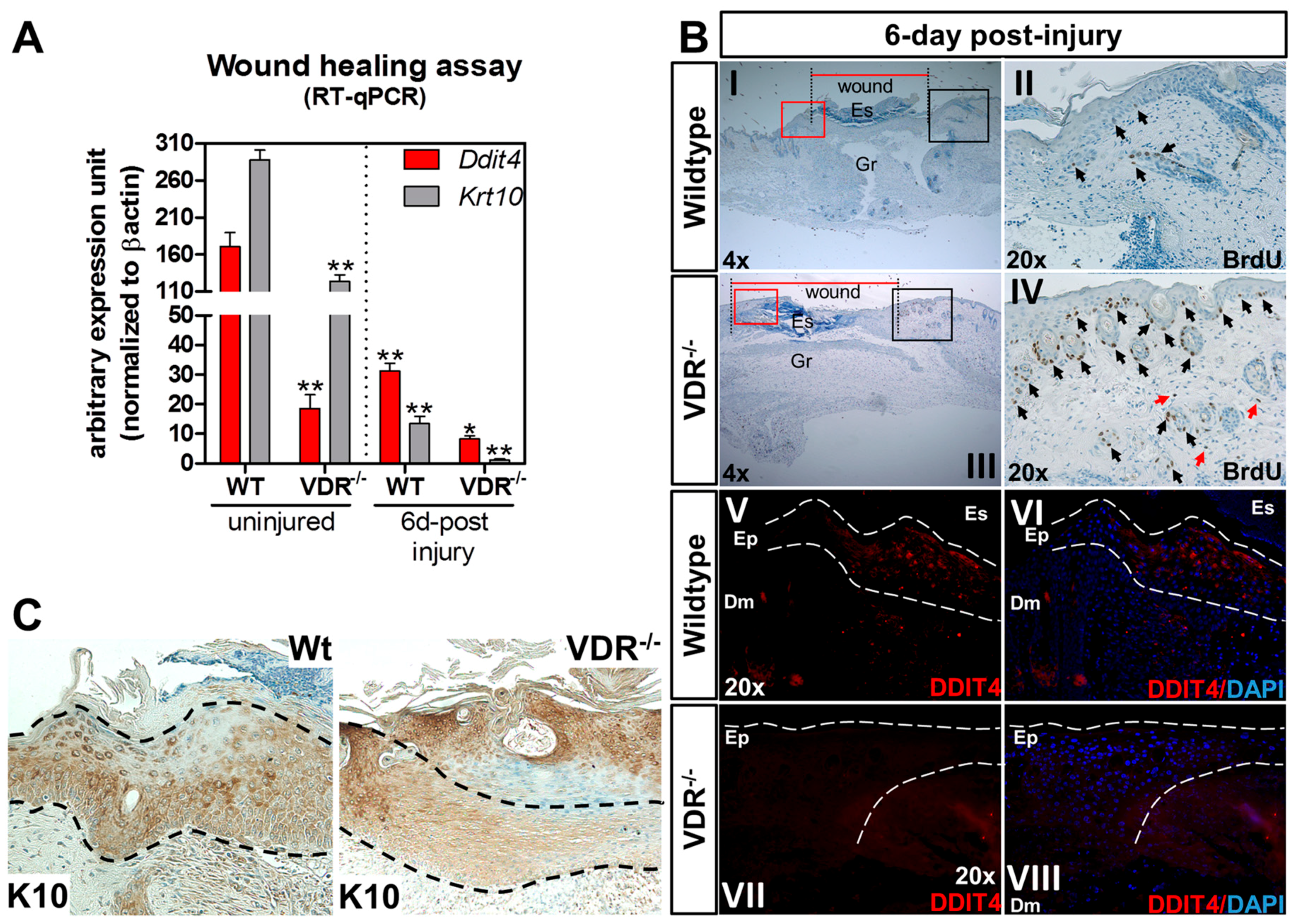
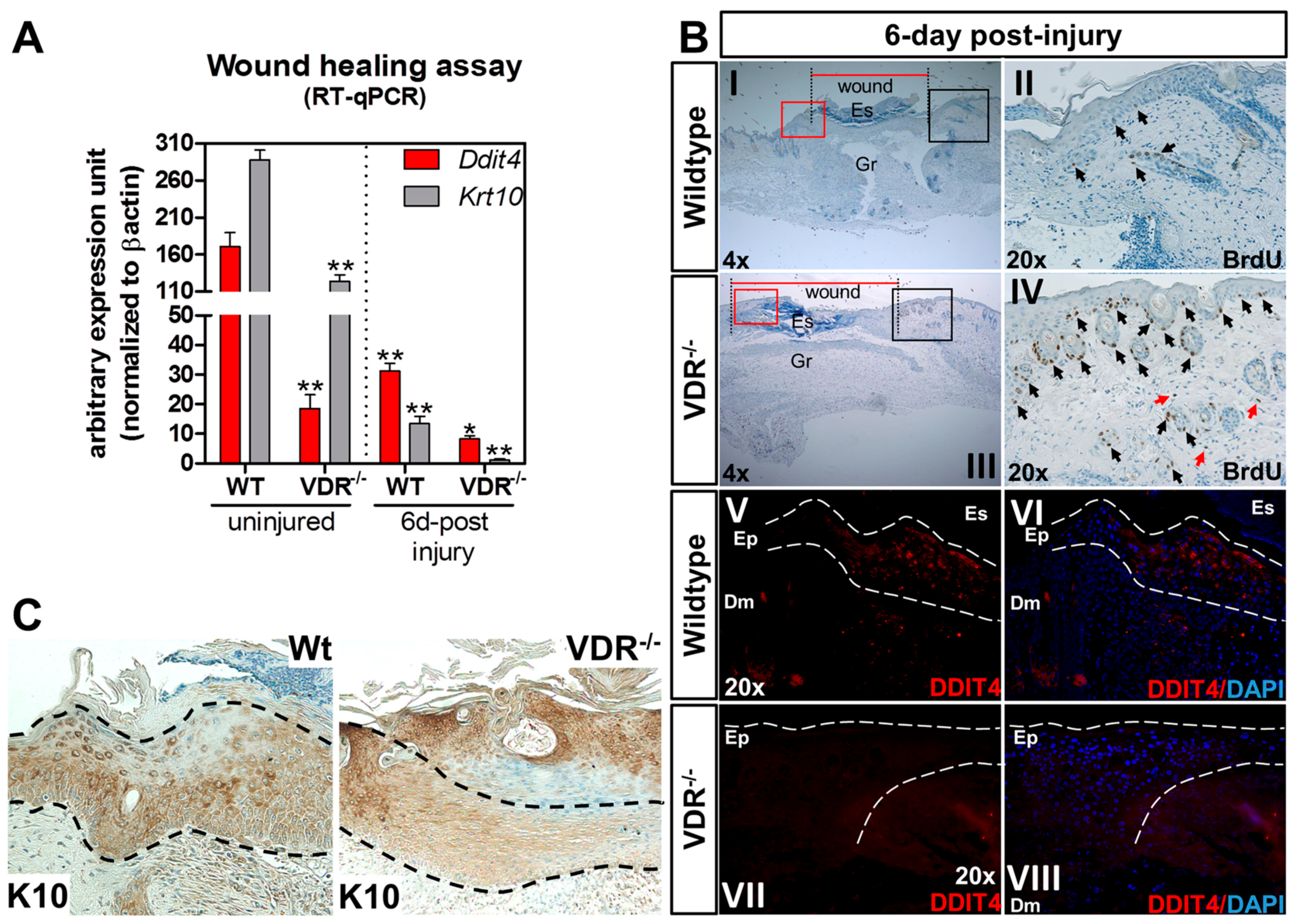
2.6. Impaired Ddit4/Ddit4 and Krt10/Krt10 Expression in the Neo-Epidermis of Wounds from VDR−/− Mice
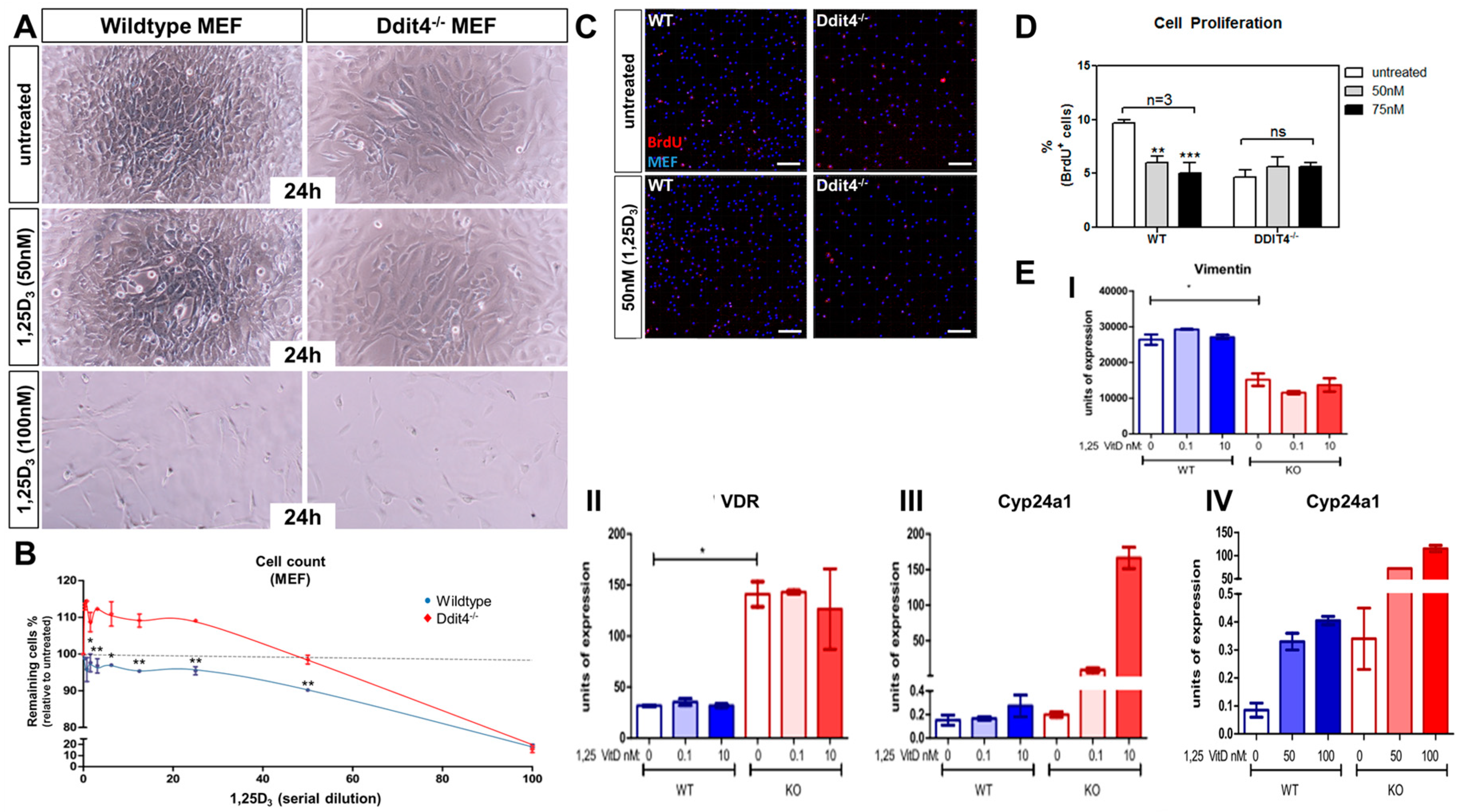
2.7. Ddit4-Deficient Mouse Embryonic Fibroblasts Are Resistant to the Pro-Differentiation Actions of Vitamin D
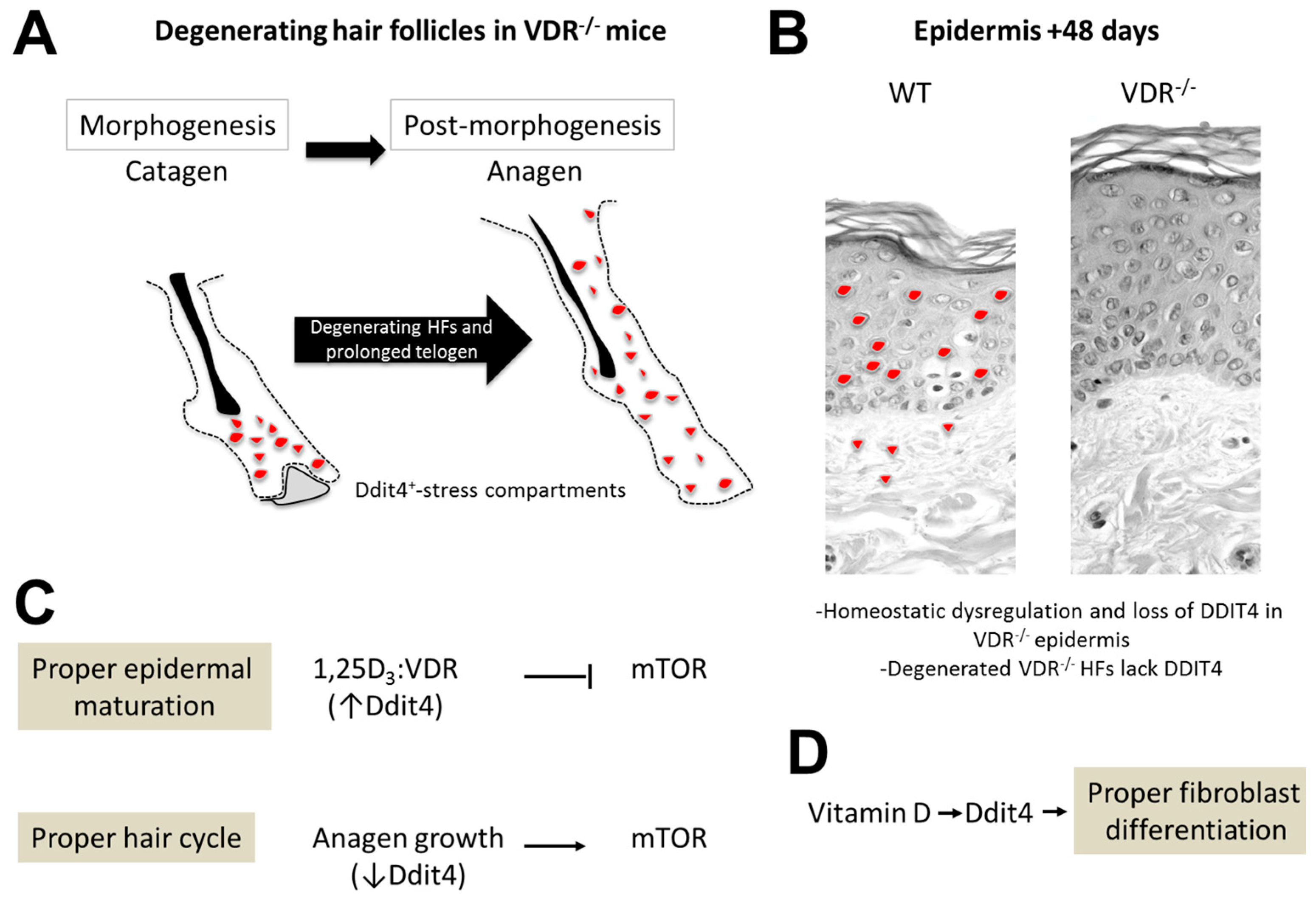
3. Discussion
3.1. Insights into Ddit4-VDR
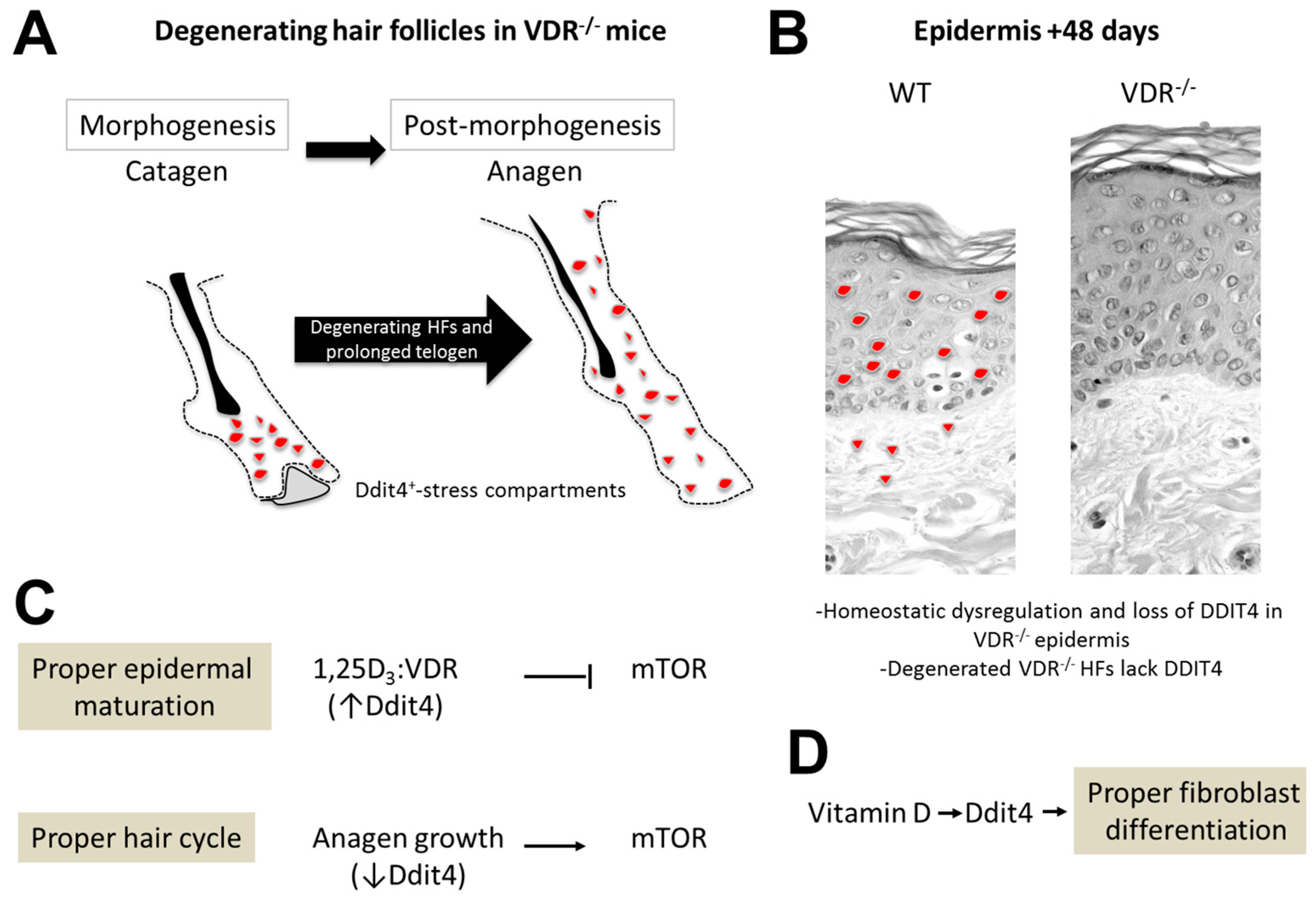
3.2. Hair Follicle Defects in VDR-Deficient Animals
3.3. Epidermal Wounding Defects
4. Materials and Methods
4.1. Animal Maintenance
4.2. Mouse Puncture Assay
4.3. Chromatin Immunoprecipitation (ChIP)
4.4. In Vivo/In Vitro 5-Bromo-2′-deoxyuridine (BrdU) Labeling and Immunocytochemistry
4.5. Fluorescence-Activated Cell Sorting of Hair Follicle Stem Cells
4.6. Immunofluorescent and Immunohistochemical Labeling of Skin
4.7. Ddit4−/− Mouse Embryonic Fibroblasts (MEFs) and Cell Count Measurement
4.8. mRNA Reverse Transcription Quantitative PCR (RT-qPCR) Analysis
4.9. Primary Neonatal Keratinocytes and mVDR Transient Transfection
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations:
| VDR | Vitamin D receptor |
| Ddit4 | DNA damage-inducible transcript 4 |
| VDRE | Vitamin D response element |
| KSC | Keratinocyte stem cell |
| mTOR | Mechanistic/mammalian target of rapamycin |
References
- Hughes, M.R.; Malloy, P.J.; Kieback, D.G.; Kesterson, R.A.; Pike, J.W.; Feldman, D.; O’Malley, B.W. Point mutations in the human vitamin D receptor gene associated with hypocalcemic rickets. Science 1988, 242, 1702–1705. [Google Scholar] [PubMed]
- Luderer, H.F.; Nazarian, R.M.; Zhu, E.D.; Demay, M.B. Ligand-dependent actions of the vitamin D receptor are required for activation of TGF-β signaling during the inflammatory response to cutaneous injury. Endocrinology 2013, 154, 16–24. [Google Scholar] [PubMed]
- Oda, Y.; Tu, C.L.; Menendez, A.; Nguyen, T.; Bikle, D.D. Vitamin D and calcium regulation of epidermal wound healing. J. Steroid Biochem. Mol. Biol. 2015, 164, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Komuves, L.; Yu, Q.C.; Elalieh, H.; Ng, D.C.; Leary, C.; Chang, S.; Crumrine, D.; Yoshizawa, T.; Kato, S.; et al. Lack of the vitamin D receptor is associated with reduced epidermal differentiation and hair follicle growth. J. Investig. Dermatol. 2002, 118, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Sakai, Y.; Demay, M.B. Evaluation of keratinocyte proliferation and differentiation in vitamin D receptor knockout mice. Endocrinology 2000, 141, 2043–2049. [Google Scholar] [CrossRef] [PubMed]
- Demay, M.B.; MacDonald, P.N.; Skorija, K.; Dowd, D.R.; Cianferotti, L.; Cox, M. Role of the vitamin D receptor in hair follicle biology. J. Steroid Biochem. Mol. Biol. 2007, 103, 344–346. [Google Scholar] [CrossRef] [PubMed]
- Sakai, Y.; Kishimoto, J.; Demay, M.B. Metabolic and cellular analysis of alopecia in vitamin D receptor knockout mice. J. Clin. Investig. 2001, 107, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Sakai, Y.; Demay, M.B. Targeting expression of the human vitamin D receptor to the keratinocytes of vitamin D receptor null mice prevents alopecia. Endocrinology 2001, 142, 5386–5389. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Pirro, A.E.; Amling, M.; Delling, G.; Baron, R.; Bronson, R.; Demay, M.B. Targeted ablation of the vitamin D receptor: An animal model of vitamin D-dependent rickets type II with alopecia. Proc. Natl. Acad. Sci. USA 1997, 94, 9831–9835. [Google Scholar] [CrossRef] [PubMed]
- Erben, R.G.; Soegiarto, D.W.; Weber, K.; Zeitz, U.; Lieberherr, M.; Gniadecki, R.; Moller, G.; Adamski, J.; Balling, R. Deletion of deoxyribonucleic acid binding domain of the vitamin D receptor abrogates genomic and nongenomic functions of vitamin D. Mol. Endocrinol. 2002, 16, 1524–1537. [Google Scholar] [CrossRef] [PubMed]
- Skorija, K.; Cox, M.; Sisk, J.M.; Dowd, D.R.; MacDonald, P.N.; Thompson, C.C.; Demay, M.B. Ligand-independent actions of the vitamin D receptor maintain hair follicle homeostasis. Mol. Endocrinol. 2005, 19, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Tumbar, T.; Guasch, G.; Greco, V.; Blanpain, C.; Lowry, W.E.; Rendl, M.; Fuchs, E. Defining the epithelial stem cell niche in skin. Science 2004, 303, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Cianferotti, L.; Cox, M.; Skorija, K.; Demay, M.B. Vitamin D receptor is essential for normal keratinocyte stem cell function. Proc. Natl. Acad. Sci. USA 2007, 104, 9428–9433. [Google Scholar] [CrossRef] [PubMed]
- Palmer, H.G.; Martinez, D.; Carmeliet, G.; Watt, F.M. The vitamin D receptor is required for mouse hair cycle progression but not for maintenance of the epidermal stem cell compartment. J. Investig. Dermatol. 2008, 128, 2113–2117. [Google Scholar] [CrossRef] [PubMed]
- Luderer, H.F.; Gori, F.; Demay, M.B. Lymphoid enhancer-binding factor-1 (LEF1) interacts with the DNA-binding domain of the vitamin D receptor. J. Biol. Chem. 2011, 286, 18444–18451. [Google Scholar] [CrossRef] [PubMed]
- Lisse, T.S.; Saini, V.; Zhao, H.; Luderer, H.F.; Gori, F.; Demay, M.B. The vitamin D receptor is required for activation of cWnt and hedgehog signaling in keratinocytes. Mol. Endocrinol. 2014, 28, 1698–1706. [Google Scholar] [CrossRef] [PubMed]
- Teichert, A.; Elalieh, H.; Bikle, D. Disruption of the hedgehog signaling pathway contributes to the hair follicle cycling deficiency in Vdr knockout mice. J. Cell. Physiol. 2010, 225, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D.D. Vitamin D and the skin: Physiology and pathophysiology. Rev. Endoc. Metab. Disord. 2012, 13, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D.D.; Elalieh, H.; Chang, S.; Xie, Z.; Sundberg, J.P. Development and progression of alopecia in the vitamin D receptor null mouse. J. Cell. Physiol. 2006, 207, 340–353. [Google Scholar] [CrossRef] [PubMed]
- Oda, Y.; Uchida, Y.; Moradian, S.; Crumrine, D.; Elias, P.M.; Bikle, D.D. Vitamin D receptor and coactivators SRC2 and 3 regulate epidermis-specific sphingolipid production and permeability barrier formation. J. Investig. Dermatol. 2009, 129, 1367–1378. [Google Scholar] [CrossRef] [PubMed]
- Zinser, G.M.; Sundberg, J.P.; Welsh, J. Vitamin D3 receptor ablation sensitizes skin to chemically induced tumorigenesis. Carcinogenesis 2002, 23, 2103–2109. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D.D. Vitamin D regulated keratinocyte differentiation. J. Cell. Biochem. 2004, 92, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Schauber, J.; Dorschner, R.A.; Coda, A.B.; Buchau, A.S.; Liu, P.T.; Kiken, D.; Helfrich, Y.R.; Kang, S.; Elalieh, H.Z.; Steinmeyer, A.; et al. Injury enhances TLR2 function and antimicrobial peptide expression through a vitamin D-dependent mechanism. J. Clin. Investig. 2007, 117, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Ellisen, L.W.; Ramsayer, K.D.; Johannessen, C.M.; Yang, A.; Beppu, H.; Minda, K.; Oliner, J.D.; McKeon, F.; Haber, D.A. REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol. Cell 2002, 10, 995–1005. [Google Scholar] [CrossRef]
- Sofer, A.; Lei, K.; Johannessen, C.M.; Ellisen, L.W. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol. Cell. Biol. 2005, 25, 5834–5845. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef] [PubMed]
- McGhee, N.K.; Jefferson, L.S.; Kimball, S.R. Elevated corticosterone associated with food deprivation upregulates expression in rat skeletal muscle of the mTORC1 repressor, REDD1. J. Nutr. 2009, 139, 828–834. [Google Scholar] [CrossRef] [PubMed]
- Baida, G.; Bhalla, P.; Kirsanov, K.; Lesovaya, E.; Yakubovskaya, M.; Yuen, K.; Guo, S.; Lavker, R.M.; Readhead, B.; Dudley, J.T.; et al. REDD1 functions at the crossroads between the therapeutic and adverse effects of topical glucocorticoids. EMBO Mol. Med. 2015, 7, 42–58. [Google Scholar] [CrossRef] [PubMed]
- Lisse, T.S.; Liu, T.; Irmler, M.; Beckers, J.; Chen, H.; Adams, J.S.; Hewison, M. Gene targeting by the vitamin D response element binding protein reveals a role for vitamin D in osteoblast mTOR signaling. FASEB J. 2011, 25, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Lisse, T.S.; Vadivel, K.; Bajaj, S.P.; Chun, R.F.; Hewison, M.; Adams, J.S. The heterodimeric structure of heterogeneous nuclear ribonucleoprotein C1/C2 dictates 1,25-dihydroxyvitamin D-directed transcriptional events in osteoblasts. Bone Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhang, R.; Burns, F.J. Gene expression and cell cycle arrest in a rat keratinocyte line exposed to 56Fe ions. J. Radiat. Res. 2007, 48, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Page, A.; Navarro, M.; Garin, M.; Perez, P.; Casanova, M.L.; Moreno, R.; Jorcano, J.L.; Cascallana, J.L.; Bravo, A.; Ramirez, A. IKKβ leads to an inflammatory skin disease resembling interface dermatitis. J. Investig. Dermatol. 2010, 130, 1598–1610. [Google Scholar] [CrossRef] [PubMed]
- Handfield, M.; Mans, J.J.; Zheng, G.; Lopez, M.C.; Mao, S.; Progulske-Fox, A.; Narasimhan, G.; Baker, H.V.; Lamont, R.J. Distinct transcriptional profiles characterize oral epithelium—Microbiota interactions. Cell. Microbiol. 2005, 7, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Kennedy-Crispin, M.; Billick, E.; Mitsui, H.; Gulati, N.; Fujita, H.; Gilleaudeau, P.; Sullivan-Whalen, M.; Johnson-Huang, L.M.; Suarez-Farinas, M.; Krueger, J.G. Human keratinocytes’ response to injury upregulates CCL20 and other genes linking innate and adaptive immunity. J. Investig. Dermatol. 2012, 132, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Roh, C.; Tao, Q.; Lyle, S. Dermal papilla-induced hair differentiation of adult epithelial stem cells from human skin. Physiol. Genom. 2004, 19, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Milde, P.; Hauser, U.; Simon, T.; Mall, G.; Ernst, V.; Haussler, M.R.; Frosch, P.; Rauterberg, E.W. Expression of 1,25-dihydroxyvitamin D3 receptors in normal and psoriatic skin. J. Investig. Dermatol. 1991, 97, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Lisse, T.S.; Chun, R.F.; Rieger, S.; Adams, J.S.; Hewison, M. Vitamin D activation of functionally distinct regulatory miRNAs in primary human osteoblasts. J. Bone Miner. Res. 2013, 28, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Zella, L.A.; Meyer, M.B.; Nerenz, R.D.; Lee, S.M.; Martowicz, M.L.; Pike, J.W. Multifunctional enhancers regulate mouse and human vitamin D receptor gene transcription. Mol. Endocrinol. 2010, 24, 128–147. [Google Scholar] [CrossRef] [PubMed]
- Reichrath, J.; Schilli, M.; Kerber, A.; Bahmer, F.A.; Czarnetzki, B.M.; Paus, R. Hair follicle expression of 1,25-dihydroxyvitamin D3 receptors during the murine hair cycle. Br. J. Dermatol. 1994, 131, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Squarize, C.H.; Castilho, R.M.; Bugge, T.H.; Gutkind, J.S. Accelerated wound healing by mTOR activation in genetically defined mouse models. PLoS ONE 2010, 5, e10643. [Google Scholar] [CrossRef] [PubMed]
- Castilho, R.M.; Squarize, C.H.; Chodosh, L.A.; Williams, B.O.; Gutkind, J.S. mTOR mediates Wnt-induced epidermal stem cell exhaustion and aging. Cell Stem Cell 2009, 5, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Blanpain, C.; Lowry, W.E.; Geoghegan, A.; Polak, L.; Fuchs, E. Self-renewal, multipotency, and the existence of two cell populations within an epithelial stem cell niche. Cell 2004, 118, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Danielsson, C.; Fehsel, K.; Polly, P.; Carlberg, C. Differential apoptotic response of human melanoma cells to 1α,25-dihydroxyvitamin D3 and its analogues. Cell Death Differ. 1998, 5, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Al-Hendy, A.; Diamond, M.P.; Boyer, T.G.; Halder, S.K. Vitamin D3 inhibits Wnt/β-catenin and mTOR signaling pathways in human uterine fibroid cells. J. Clin. Endocrinol. Metab. 2016, 101, 1542–1551. [Google Scholar] [CrossRef] [PubMed]
- Fingar, D.C.; Salama, S.; Tsou, C.; Harlow, E.; Blenis, J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002, 16, 1472–1487. [Google Scholar] [CrossRef] [PubMed]
- Brafman, A.; Mett, I.; Shafir, M.; Gottlieb, H.; Damari, G.; Gozlan-Kelner, S.; Vishnevskia-Dai, V.; Skaliter, R.; Einat, P.; Faerman, A.; et al. Inhibition of oxygen-induced retinopathy in RTP801-deficient mice. Investig. Ophthalmol. Vis. Sci. 2004, 45, 3796–3805. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Mett, I.; Bhunia, A.K.; Bowman, J.; Perez, M.; Zhang, L.; Gandjeva, A.; Zhen, L.; Chukwueke, U.; Mao, T.; et al. Rtp801, a suppressor of mTOR signaling, is an essential mediator of cigarette smoke-induced pulmonary injury and emphysema. Nat. Med. 2010, 16, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Kamocki, K.; van Demark, M.; Fisher, A.; Rush, N.I.; Presson, R.G., Jr.; Hubbard, W.; Berdyshev, E.V.; Adamsky, S.; Feinstein, E.; Gandjeva, A.; et al. RTP801 is required for ceramide-induced cell-specific death in the murine lung. Am. J. Respir. Cell Mol. Biol. 2013, 48, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Reiling, J.H.; Hafen, E. The hypoxia-induced paralogs Scylla and Charybdis inhibit growth by down-regulating S6K activity upstream of TSC in Drosophila. Genes Dev. 2004, 18, 2879–2892. [Google Scholar] [CrossRef] [PubMed]
- Shoshani, T.; Faerman, A.; Mett, I.; Zelin, E.; Tenne, T.; Gorodin, S.; Moshel, Y.; Elbaz, S.; Budanov, A.; Chajut, A.; et al. Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol. Cell. Biol. 2002, 22, 2283–2293. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Parent, C.A. Review series: TOR kinase complexes and cell migration. J. Cell Biol. 2011, 194, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Lei, M.; Shi, J.; Yu, Y.; Qiu, W.; Lai, X.; Liu, Y.; Yang, T.; Yang, L.; Widelitz, R.B.; et al. Roles of GasderminA3 in catagen-telogen transition during hair cycling. J. Investig. Dermatol. 2015, 135, 2162–2172. [Google Scholar] [CrossRef] [PubMed]
- Ragone, G.; Bresin, A.; Piermarini, F.; Lazzeri, C.; Picchio, M.C.; Remotti, D.; Kang, S.M.; Cooper, M.D.; Croce, C.M.; Narducci, M.G.; et al. The Tcl1 oncogene defines secondary hair germ cells differentiation at catagen-telogen transition and affects stem-cell marker CD34 expression. Oncogene 2009, 28, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Peters, E.M.; Stieglitz, M.G.; Liezman, C.; Overall, R.W.; Nakamura, M.; Hagen, E.; Klapp, B.F.; Arck, P.; Paus, R. p75 Neurotrophin receptor-mediated signaling promotes human hair follicle regression (Catagen). Am. J. Pathol. 2006, 168, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.O.; Kim, J.Y.; Kwack, M.H.; Hong, S.H.; Kim, M.K.; Kim, J.C.; Sung, Y.K. Identification of troglitazone responsive genes: Induction of RTP801 during troglitazone-induced apoptosis in Hep 3B cells. BMB Rep. 2010, 43, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Molitoris, J.K.; McColl, K.S.; Swerdlow, S.; Matsuyama, M.; Lam, M.; Finkel, T.H.; Matsuyama, S.; Distelhorst, C.W. Glucocorticoid elevation of dexamethasone-induced gene 2 (Dig2/RTP801/REDD1) protein mediates autophagy in lymphocytes. J. Biol. Chem. 2011, 286, 30181–30189. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D.D. The vitamin D receptor: A tumor suppressor in skin. Discov. Med. 2011, 11, 7–17. [Google Scholar] [PubMed]
- Bikle, D.D.; Chang, S.; Crumrine, D.; Elalieh, H.; Man, M.Q.; Dardenne, O.; Xie, Z.; Arnaud, R.S.; Feingold, K.; Elias, P.M. Mice lacking 25OHD 1α-hydroxylase demonstrate decreased epidermal differentiation and barrier function. J. Steroid Biochem. Mol. Biol. 2004, 89, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Elizondo, R.A.; Yin, Z.; Lu, X.; Watsky, M.A. Effect of vitamin D receptor knockout on cornea epithelium wound healing and tight junctions. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5245–5251. [Google Scholar] [CrossRef] [PubMed]
- Palmer, H.G.; Anjos-Afonso, F.; Carmeliet, G.; Takeda, H.; Watt, F.M. The vitamin D receptor is a Wnt effector that controls hair follicle differentiation and specifies tumor type in adult epidermis. PLoS ONE 2008, 3, e1483. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, H.; Rieger, S.; Abe, K.; Hewison, M.; Lisse, T.S. DNA Damage-Inducible Transcript 4 Is an Innate Surveillant of Hair Follicular Stress in Vitamin D Receptor Knockout Mice and a Regulator of Wound Re-Epithelialization. Int. J. Mol. Sci. 2016, 17, 1984. https://doi.org/10.3390/ijms17121984
Zhao H, Rieger S, Abe K, Hewison M, Lisse TS. DNA Damage-Inducible Transcript 4 Is an Innate Surveillant of Hair Follicular Stress in Vitamin D Receptor Knockout Mice and a Regulator of Wound Re-Epithelialization. International Journal of Molecular Sciences. 2016; 17(12):1984. https://doi.org/10.3390/ijms17121984
Chicago/Turabian StyleZhao, Hengguang, Sandra Rieger, Koichiro Abe, Martin Hewison, and Thomas S. Lisse. 2016. "DNA Damage-Inducible Transcript 4 Is an Innate Surveillant of Hair Follicular Stress in Vitamin D Receptor Knockout Mice and a Regulator of Wound Re-Epithelialization" International Journal of Molecular Sciences 17, no. 12: 1984. https://doi.org/10.3390/ijms17121984
APA StyleZhao, H., Rieger, S., Abe, K., Hewison, M., & Lisse, T. S. (2016). DNA Damage-Inducible Transcript 4 Is an Innate Surveillant of Hair Follicular Stress in Vitamin D Receptor Knockout Mice and a Regulator of Wound Re-Epithelialization. International Journal of Molecular Sciences, 17(12), 1984. https://doi.org/10.3390/ijms17121984